

Background: Dcp1a is a protein involved in mRNA decapping, processing body formation, and signal transduction.
Results: GFP-Dcp1a, through its amino-terminal domain, can activate protein kinase R (PKR).
Conclusion: Dcp1a may play novel roles in translation regulation and innate immunity.
Significance: We present a novel instance of RNA degradation proteins affecting translation regulation.
Keywords: MRNA Decay, P-body, Positive-strand RNA Viruses, Protein Kinase R (PKR), Translation Regulation, Poliovirus
Abstract
We have shown previously that poliovirus infection disrupts cytoplasmic P-bodies in infected mammalian cells. During the infectious cycle, poliovirus causes the directed cleavage of Dcp1a and Pan3, coincident with the dispersion of P-bodies. We now show that expression of Dcp1a prior to infection, surprisingly, restricts poliovirus infection. This inhibition of infection was independent of P-body formation because expression of GFP-Dcp1a mutants that cannot enter P-bodies restricted poliovirus infection similar to wild-type GFP-Dcp1a. Expression of wild-type or mutant GFP-Dcp1a induced phosphorylation of eIF2α through the eIF2α kinase protein kinase R (PKR). Activation of PKR required the amino-terminal EVH1 domain of Dcp1a. This PKR-induced translational inhibition appears to be specific to Dcp1a because the expression of other P-body components, Pan2, Pan3, Ccr4, or Caf1, did not result in the inhibition of poliovirus gene expression or induce eIF2α phosphorylation. The translation blockade induced by Dcp1a expression suggests novel signaling linking RNA degradation/decapping and regulation of translation.
Introduction
Eukaryotic cells form cytoplasmic RNA granules known as processing bodies (P-bodies)2 that contain cellular mRNAs and proteins involved in mRNA decapping, deadenylation, miRNA-mediated mRNA silencing, and mRNA storage (1–3). P-bodies are dynamic foci, with protein and mRNA components readily exchanging between the RNA granules and the cytosol. The size and number of P-bodies roughly correlate with the level of non-translating mRNA within a cell. Conditions that increase the level of non-translating or deadenylated mRNA increase the size and number of P-bodies (4–6). P-bodies are readily identified by their protein constituents, which include the RNA helicase Rck/p54, the cellular 5′-3′ exoribonuclease 1 (Xrn1), components of the decapping complex, mRNA-decapping enzymes 1a and 2 (Dcp1a and Dcp2), and components of the deadenylation complexes, i.e. PABP-dependent poly(A) nucleases 2 and 3, Pan2/Pan3 (1, 7–9). The degradation of cellular mRNA can occur within P-bodies (5, 10) and typically begins with a two-stage shortening of the poly(A) tail mediated by the poly(A) nucleases Ccr4, Caf1, Pan2, and Pan3 (11, 12).
P-body formation is also modulated by several stimuli ranging from stage of the cell cycle to virus infection. Numerous examples of viral modulation of P-body size and number as well as modification or cleavage of P-body constituent proteins have been described (13). Poliovirus infection leads to the loss of P-bodies by 3–4 h post-infection (hpi) and the cleavage or degradation of several key proteins critical for cellular RNA decay (14). Infection with West Nile virus or hepatitis C virus leads to a progressive reduction in the number of P-body foci 24–36 hpi and relocalization of P-body components to viral replication centers (15–17). The genomic RNA of dengue virus interacts with the P-body component Rck/p54 (DDX6) and is presumed to be important for viral replication (18).
Dcp1a is a cofactor involved in the removal of the 5′-methylguanosine cap from eukaryotic mRNA and is a commonly used marker for the analysis of cellular P-bodies (4–6, 19, 20). Following poly(A) tail shortening, the 5′-methylguanosine cap is removed through the action of Dcp1a and Dcp2 (3, 21–24). Subsequent to decapping, Xrn1 degrades mRNA in a 5′-3′ fashion (4, 5, 25–28). In decapping, yeast Dcp2, which contains decapping activity, directly interacts with Dcp1a, which modulates its function. However, the human Dcp1a-Dcp2 interaction is thought to require the cofactors enhancer of decapping 3 and 4 (EDC3 and EDC4) as well as the RNA helicase Rck/p54 (8, 29–31). Dcp1a and other proteins involved in mRNA degradation or translation repression are key factors in messenger ribonucleoprotein granule assembly (20, 32). Conditions that decrease translation rates inside the cell and those that activate mRNA decapping and degradation increase the size and number of P-bodies (4–6, 19).
Previous studies have indicated roles for Dcp1a in P-body formation, maintenance, and regulation (6, 32, 33). Dcp1a in P-bodies constantly and quickly exchanges with Dcp1a in the cytoplasmic pool during fluorescence recovery after photobleaching analysis (34). Phosphorylation at Ser-315 mediates Dcp1a release from P-bodies as well as the regulation of P-body formation during translational stress (32), which suggests that Dcp1a may be involved in modulation of, or signal transduction from, P-bodies. In contrast, Dcp2 has a slower recovery rate during fluorescence recovery after photobleaching experiments, suggesting that it is a core P-body protein (34). The C-terminal domain of Dcp1a is highly conserved in plants and metazoa, folds in an α-helical conformation, and mediates homotrimerization that is required for Dcp1a incorporation into activated decapping complexes and localization to P-bodies (35).
Dcp1a has additional roles in cellular signaling that are not as well studied as its role in decapping. Previous studies have demonstrated the involvement of Dcp1a in cellular signaling pathways through protein-protein interactions (36, 37). All eukaryotic orthologues of Dcp1a contain an N-terminal EVH1 (enabled vasodilator-stimulated protein homology 1) domain (30, 31, 37, 38). EVH1 domains are known to interact with a variety of proline-rich protein ligands (39–41). Dcp1a interacts with Dcp2 and Xrn1 to facilitate decapping reactions through the EVH1 domain (36, 42). A recent report suggests that ERK kinase-mediated phosphorylation of Dcp1a enhances its interaction with Dcp2 (36). The EVH1 domain of Dcp1a also has a role in transforming growth factor β (TGF-β) signaling through a SMAD4 interaction (37, 43). Interestingly, the EVH1 domain of Dcp1a is highly conserved, but the substrate specificity varies across species (42). Recent studies have looked at the status of Dcp1a and P-bodies during stages of the cell cycle and the importance of Dcp1a phosphorylation for its function (32, 33, 36). These studies have highlighted the importance of Dcp1a phosphorylation in several signaling pathways that affect P-body formation and maintenance.
Our previous studies demonstrated that Dcp1a is cleaved during poliovirus infection by poliovirus protease 3Cpro and that P-bodies were disrupted during infection (14). In an effort to understand the importance of Dcp1a cleavage to P-body disruption and further elucidate Dcp1a biology, we utilized the well documented GFP-Dcp1a expression system (32, 33, 36). Previous studies utilizing the GFP-Dcp1a overexpression system have demonstrated that GFP-Dcp1a retains the same functions as endogenous Dcp1a (32, 33, 35, 36) and can enter and function in active decapping complexes and P-bodies (35). Additionally, GFP-Dcp1a expression has been used to determine alterations in the phosphorylation status of Dcp1a throughout the cell cycle and in response to cellular stress (32, 33). These studies demonstrated the phosphorylation of GFP-Dcp1a by JNK or ERK kinases is important for regulating Dcp1a function and localization to P-bodies (32, 36). Expression of GFP-Dcp1a does not significantly alter mRNA degradation or P-body formation, even in the presence of truncations that inhibit GFP-Dcp1a function (32, 33, 35, 36). Taken together, these studies suggest that GFP-Dcp1a expression is a valid tool for studying the role of Dcp1a in cellular biology. In this study, we report the inhibition of cellular translation upon GFP-Dcp1a expression through the activation of double-stranded RNA-dependent protein kinase (PKR) and subsequent phosphorylation of eIF2α. We also demonstrate the importance of the amino-terminal EVH1 domain for translational inhibition. The results presented here demonstrate a new link between the mRNA degradation machinery, the regulation of translation initiation, and cellular stress sensors.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfections
A549, PKR−/−, GCN2−/−, PERK−/−, and PACT−/− mouse embryonic fibroblast (MEF) cells were cultured in DMEM supplemented with 10% FBS and maintained at 37 °C with 5% CO2. PKR−/− MEFs were a gift from Mauro Costa-Mattioli (Baylor College of Medicine, Houston, TX). GCN2−/−, PERK−/−, and corresponding wild-type control MEFs were obtained from the ATCC and were originally developed in the laboratory of David Ron (University of Cambridge, Cambridge, United Kingdom). PACT−/− and corresponding wild-type control MEFs were a gift from Ganes Sen (Cleveland Clinic, Cleveland, Ohio). Cells were seeded at a density of 1.5 × 105 cells/well in a 12-well plate. Eighteen hours later, cells were transfected with the indicated plasmids using Xtremegene-HP transfection reagent (Roche) according to the protocol of the manufacturer. Additionally, A549 cells or MEFs were electroporated with the indicated plasmids utilizing the Neon transfection system (Invitrogen) in accordance with the protocol of the manufacturer for each cell type. Unless indicated otherwise in the figure legends, cells were transfected with 1 μg of DNA/1.5 × 105 cells.
Virus and Infections
Twenty-four hours post-transfection, A549 cells were infected with poliovirus type 1 (Mahoney strain) with a multiplicity of infection of 10 in DMEM containing 2% FBS for the indicated times. Poliovirus was grown in HeLa S3 cells and purified on CsCl gradients as described previously (44). Poliovirus was titrated by plaque assay on HeLa monolayers overlaid with 2× DMEM containing 1% methylcellulose.
Antibodies
The following antibodies were used in immunoblot analyses and immunofluorescence: anti-Dcp1a polyclonal (a gift from A. B. Shyu), anti-Dcp1a monoclonal (Novus), anti-Rck/p54(DDX6) (a gift from C. E. Cameron), anti-α-tubulin (Sigma), anti-V5 tag (Invitrogen), anti-p-eIF2α-S51 (Cell Signaling Technology), anti-PKR (BD Biosciences), anti-P-PKR (Abcam), and anti-GFP (Santa Cruz Biotechnology).
Plasmids and Cloning
Plasmids encoding GFP-Dcp1a (peGFP-Dcp1a and peGFP-Dcp1a-ΔCTD) were provided by M. Kracht (32). The plasmid expressing adenoviral VA1/2 RNA, pAdVAntage, was purchased from Promega. N-terminal truncations and EVH1 expression fragments were created from the plasmid encoding GFP-Dcp1a using the In-Fusion HD cloning system (Clontech). Dcp1a coding sequences were amplified by PCR using the described primers. The PCR products were ligated into the pcDNA4-GFP vector that had been linearized by EcoRV digestion for 2 h at 37 °C. Ligation products were transformed into DH5α Escherichia coli (New England Biolabs), and colonies were selected by growth on ampicillin plates.
GFP-Dcp1a-ΔNTD was generated using the primers TGGAATTCTGCAGATTCCCAGCAAGCTGCTCGGGACAAA (forward) and GCCACTGTGCTGGATTCATAGGTTGTGGTTGTCTTTGTTCTTGGTCAG (reverse). GFP-EVH1 was generated using the primers TGGAATTCTGCAGATATGGAGGCGCTGAGTCGAGCTGGGCAGGAG (forward) and GCCACTGTGCTGGATTCATCGCCGTGTCTCCTCTTCTACCACATC (reverse). EVH1-Cherry was generated using the primers TGGAATTCTGCAGATATGGAGGCGCTGAGTCGAGCTGGGCAGGAG (forward) and GCCACTGTGCTGGATTCGCCGTGTCTCCTCTTCTACCACATC (reverse).
Plasmids for V5-Pan2, V5-Pan3, V5-Ccr4, and V5-Caf1 were provided by A. B. Shyu (11, 45). The PKR expression plasmids pcDNA6-PKR and pcDNA6-PKR-K296W were provided by Charles Samuel (46).
Immunofluorescence
A549 or MEF cells were seeded on glass coverslips and transfected with the appropriate plasmids as described previously. Cells were allowed to express the transgene for 18–24 h, after which the cells were fixed with 4% paraformaldehyde in PEM (80 mm PIPES, 5 mm EGTA, and 2 mm MgCl2). Cells were then permeabilized in 0.5% Triton X-100 and blocked with 5% BSA in Tris-buffered saline-0.1% Tween 20 (TBS-T). The cells were then incubated with the appropriate primary antibodies for two hours at room temperature or overnight at 4 °C. Cover slips were mounted on microscope slides with Vectashield mounting medium with DAPI (Vector Laboratories) to visualize nuclei. The coverslips were analyzed by microscopy on a Nikon TE2000 inverted microscope. Deconvolution microscopy was performed with an Applied Precision DeltaVision image restoration microscope with conservative deconvolution algorithms.
For single cell quantification of eIF2α phosphorylation, images of 50–100 transfected cells/plasmid were assayed for anti-eIF2α P-S51 antibody reactivity using ImageJ to measure the mean cytoplasmic pixel intensity. The results are represented as a percentage of Dcp1a-transfected cells positive for P-S51 staining above a 6% threshold of maximal eIF2α phosphorylation response obtained with arsenite treatment. Experiments were repeated a minimum of three times. Error bars indicate mean ± S.D. p values were determined by Welch's two-sample t test.
Ribopuromycylation Assay (RPA)
Cells were grown, seeded, and transfected as described previously. Ribopuromycylation assays were conducted in accordance with previous studies (47) using the 12D10 antibody raised against puromycin (48) Briefly, on the day following transfection, the medium was replaced with fresh 10% FBS DMEM. 4–6 h later, cells were treated with 50 μg/ml puromycin and 100 μg/ml cycloheximide for 5 min at 37 °C. Cells were then washed with cold PBS and extracted with digitonin containing permeabilization buffer (50 mm Tris-HCl (pH 7.5), 5 mm MgCl2, 25 mm KCl, 100 μg/ml cycloheximide, 1× protease inhibitor mixture (Thermo), 10 units/ml RNase inhibitor (New England Biolabs, Ipswich, MA), and 0.015% digitonin) for 5 min on ice. Cells were then washed again with permeabilization buffer and processed for immunofluorescence as described previously.
Immunoblot Analysis
Following transfection and plasmid expression, the cells were collected and lysed in 1% Nonidet P-40 buffer (10 mm Tris-Cl (pH 7.4), 100 mm NaCl, and 1 mm EDTA) to release cytoplasmic contents. Nuclei were cleared, and the cytoplasmic fraction was added to 2× Laemmli sample buffer prior to 10% SDS-PAGE. Protein was transferred to nitrocellulose membranes and immunoblotted in accordance with standard procedures. Secondary peroxidase-coupled anti-mouse (Pierce) or anti-rabbit (Bio-Rad) antibodies were utilized for the detection of proteins of interest.
RESULTS
GFP-Dcp1a Expression Inhibits Poliovirus Protein Expression
Previous studies have demonstrated that a GFP fusion variant of Dcp1a (GFP-Dcp1a) colocalizes with endogenous P-body markers and is capable of entering active decapping complexes (32, 33, 35). We confirmed that expressed GFP-Dcp1a localized to constitutive P-bodies in A549 cells using the endogenous P-body marker Rck/p54 (Fig. 1A, first panel). Additionally, we detected only rare P-bodies devoid of a GFP-Dcp1a signal, suggesting that GFP-Dcp1a is ubiquitously localized to P-bodies in the transfected cell.
FIGURE 1.

GFP-Dcp1a expression inhibits poliovirus protein expression. A, A549 cells were transfected with 1 μg/1.5 × 105 cells of pcDNA4-GFP-Dcp1a and stained with an antibody against RCK/p54 (first panel). A549 cells were transfected with 1 μg/1.5 × 105 cells of pcDNA4-GFP (second panel), peGFP-Dcp1a (third panel), or peGFP-Dcp1a-ΔCTD (fourth panel) and infected with poliovirus (PV) (multiplicity of infection of 10) for 5 h. Cells were stained with an antibody against poliovirus 3Cpro (red). The highlighted area of the larger merged color image is separated into its constituent colors in the bottom panels. B, quantification of poliovirus titer resulting from infection of A549 cells expressing GFP or GFP-Dcp1a. Error bars indicate mean ± S.D. **, p < 0.0001. PFU, plaque-forming units.
Our previous studies demonstrated that poliovirus infection disrupts endogenous P-bodies by 3–4 hpi and results in the cleavage of endogenous Dcp1a (14). To further explore Dcp1a cleavage during poliovirus infection, GFP-Dcp1a was expressed in A549 cells, and the cells were infected with poliovirus. At 5 hpi, the infected cells were processed for immunofluorescence with antibodies against poliovirus proteins. Interestingly, GFP-Dcp1a containing P-bodies were retained at late time points of infection (Fig. 1A, third panel). Additionally, GFP-Dcp1a was not cleaved during poliovirus infection (data not shown). Furthermore, in cells expressing GFP-Dcp1a, poliovirus gene expression was undetectable by immunofluorescence. In contrast, adjacent non-transfected cells or cells expressing GFP readily expressed poliovirus protein and exhibited cytopathic effects (Fig. 1A, second and third panels). GFP-Dcp1a-ΔCTD contains a deletion of amino acids 515–582 and does not significantly localize to P-bodies. However, GFP-Dcp1a-ΔCTD also blocked poliovirus protein expression (Fig. 1A, fourth panel). In addition to observing a loss of poliovirus protein production in cells expressing GFP-Dcp1a or GFP-Dcp1a-ΔCTD, plaque assays of supernatants from infected cells at 4 and 8 hpi were conducted. We observed a 3-fold loss of poliovirus production in cell populations that were transfected with GFP-Dcp1a compared with cells expressing GFP alone (Fig. 1B). In these experiments, transfection/expression efficiencies averaged 70%, leaving roughly 30% of cells fully permissive for virus growth. Together, these data indicate that expression of GFP-Dcp1a can block virus protein expression and reduce progeny virus production.
GFP-Dcp1a Expression Induces eIF2α Phosphorylation
We next sought to identify a mechanism by which GFP-Dcp1a expression would inhibit poliovirus gene expression. The active translation status of GFP-Dcp1a-transfected cells was examined utilizing an RPA (47, 48), which detects incorporation of puromycin into nascent peptides on ribosomes. Fig. 2A illustrates the effects of expression of control GFP, GFP-Dcp1a, and sodium arsenite treatment, a potent translation inhibitor (49), on nascent peptide generation. Cells expressing GFP alone displayed robust puromycin staining in the RPA, indicating active cellular translation. Sodium arsenite treatment blocked translation and the RPA signal as expected, indicating that the RPA was performing as described previously (47). Cells expressing GFP-Dcp1a did not display significant puromycin staining, similar to cells treated with arsenite, suggesting that translation is repressed in these cells when compared with surrounding non-transfected cells (Fig. 2A). Quantitation of intensity of the RPA stain in single cells revealed that translation activity was reduced in cells expressing Dcp1 over a wide range of levels, including cells with low expression levels (Fig. 2F).
FIGURE 2.

GFP-Dcp1 expression induces eIF2α phosphorylation. A, A549 cells were transfected with 1 μg/1.5 × 105 cells of pcDNA4-GFP (top left panel) and peGFP-Dcp1a (top right panel) or treated with 500 μm sodium arsenite (Ars) (bottom panel) and processed according to the RPA procedure described under “Experimental Procedures.” B, A549 cells were transfected with 1 μg/1.5 × 105 cells OF GFP (left panel) and GFP-Dcp1a (center panel) plasmid DNA or treated with 500 μm sodium arsenite (right panel) and incubated with an antibody against phospho-eIF2α (Ser-51). Corresponding eIF2α phosphorylation is shown in red. The highlighted area of the larger merged color image is separated into its constituent colors in the bottom panels. C, quantification of eIF2α phosphorylation in response to GFP or GFP-Dcp1a expression as described under “Experimental Procedures.” Error bars indicate the mean ± S.D. D, immunoblot analysis of A549 cells transfected with increasing amounts of peGFP-Dcp1a (2 μg, 1 μg, 500 ng, or 250 ng/5 × 105 cells) or treated with 500 μm sodium arsenite measuring the levels of GFP-Dcp1a compared with endogenous Dcp1a with the corresponding induction of P-eIF2α. The ratio of GFP-Dcp1a to endogenous Dcp1a is indicated beneath the immunoblot analysis, as are the ratios of P-eIF2α to untreated cells. E, scattergram showing the levels of eIF2α phosphorylation and Dcp1a expression in individual A549 cells transfected with 2 μg DNA/5 × 105 cells. The range of fluorescence signals was obtained from cell-by-cell analysis with Image J software and plotted as mean cytoplasmic pixel intensity. The controls indicate the cytoplasmic pixel intensities observed in cells with GFP expression and 500 μm sodium arsenite treatment. The red dashed line indicates the mean Dcp1a expression level corresponding to D, calculated to a population 6-fold excess of GFP-Dcp1a over endogenous Dcp1. Blue dashed lines indicate calculated mean expression levels for populations with 2- or 3-fold Dcp1a-GFP expression excess over endogenous Dcp1a. F, scattergram showing the level of translation activity (RPA fluorescence intensity) versus Dcp1a expression level in individual cells and within control cells (untreated, treated with arsenite, or expressing GFP) analyzed as in E.
A restriction of translation induced by GFP-Dcp1a could explain the lack of viral gene products and reduced virus titer during poliovirus infection. Further, this suggested a potential cross-talk role for an RNA decay component with cellular translation regulation. Cellular translation is commonly regulated at the rate-limiting initiation step. Phosphorylation of serine 51 of the α subunit of eukaryotic initiation factor 2 (eIF2α) is a well described method of cellular control of translation initiation (50, 51). eIF2α is phosphorylated by four eIF2α kinases (PERK, PKR, GCN2, and heme-regulated inhibitor (HRI)) (50, 51). The effect of GFP-Dcp1a expression on the induction of eIF2α phosphorylation was examined using immunofluorescence and an antibody specific to eIF2α phosphorylated at Ser-51, as described previously (52). A549 cells expressing a GFP control plasmid did not contain phosphorylated eIF2α as expected (Fig. 2B, left panel). In contrast, cells treated with sodium arsenite, which activates HRI kinase, displayed significantly elevated eIF2α phosphorylation (Fig. 2B, right panel). In the majority of cells expressing GFP-Dcp1a (92.6% of transfected cells), we detected elevated levels of eIF2α phosphorylation (Fig. 2, B (center panel) and C), which suggests that GFP-Dcp1a is inducing the activation of an eIF2α stress kinase. To ensure that the presence of a large protein tag was not substantially enhancing the phosphorylation of eIF2α, we expressed Dcp1a with a small HA tag (HA-Dcp1a) that also induced similar high levels of phospho-eIF2α (Fig. 2C).
To determine which levels of Dcp1a expression were associated with production of phospho-eIF2α, we performed an immunoblot analysis with a Dcp1a-specific antibody from lysates of cells where increasing amounts of peGFP-Dcp1a were transfected into A549 cells. It was found that, in cell populations with a 50% transfection efficiency, approximately three times more GFP-Dcp1a compared with endogenous Dcp1a was required to induce eIF2α phosphorylation to a level detectable by immunoblot analysis in lysates from the whole cell population (Fig. 2D). This suggests an average population activation threshold of near 6-fold in transfected cells. However, quantitation of phospho-eIF2α immunofluorescence levels at a single cell level revealed that widely variable levels of Dcp1a triggered eIF2α phosphorylation, which was often quite significant in cells with low-level expression of the Dcp1a transgene (Fig. 2E).
Analysis of single cells was carried out to determine whether expression of GFP-Dcp1a altered the cytoplasmic distribution or partioning between P-bodies and the remainder of the cytoplasm. A low to moderate/average expression level of Dcp1a associated with eIF2α phosphorylation did not significantly increase the P-body number. Very high expression led to new potential P-body foci in a subset of cells, some of which did not contain p54 (data not shown). A detailed examination of Dcp1a-expressing cells was carried out with deconvolution microscopy to reduce scattered light from the very bright P-body foci. This procedure revealed that GFP-Dcp1a partitioned quite effectively to P-bodies and strongly colocalized with the canonical P-body marker Rck/p54 (Fig. 3C). In some cases the relative concentration of p54 and Dcp1a in individual P-bodies was varied. The degree of partioning of endogenous Dcp1a and GFP-Dcp1a into P-bodies was quite similar (Fig. 3, C and D).
FIGURE 3.

Expression of other P-body proteins does not induce significant eIF2α phosphorylation. A, A549 cells were transfected with plasmids expressing 1 μg/1.5 × 105 cells of V5-Caf1 (top left panel), V5-Ccr4 (top right panel), V5-Pan2 (bottom left panel), and V5-Pan3 (bottom right panel). Cells were incubated with antibodies against V5 (green) and phospho-eIF2α (red). The highlighted area of the larger merged color image is separated into its constituent colors in the bottom panels. B, quantification of eIF2α phosphorylation in response to expression of several P-body contained proteins as described under “Experimental Procedures.” **, p < 0.0001. Error bars indicate mean ± S.E. C, deconvolution microscopic analysis of cytoplasmic distribution of endogenous Dcp1a determined by plotting intensity profiles along the white line (inset shown in grayscale) transecting several P-bodies. D, cytoplasmic distribution of GFP-Dcp1and endogenous Rck/p54 was determined as in C. A.U., arbitrary units.
Expression of Other P-body Proteins Does Not Effectively Induce Phosphorylation of eIF2α
To determine whether expression of other P-body proteins induced eIF2α phosphorylation, V5 epitope-tagged Caf1, Ccr4, Pan2, and Pan3 were expressed in A549 cells. Expression of V5-Caf1 or V5-Pan2 did not significantly induce eIF2α phosphorylation, similar to the results obtained by expressing GFP alone, where up to 10% of transfected cells displayed at least a weak signal (Figs. 2 and 3). Interestingly, expression of V5-Ccr4 or V5-Pan3 induced some low level of eIF2α phosphorylation in slightly more cells. However, expression did not trigger this response in the majority of cells (Fig. 3B). Cells expressing V5-Pan3 that did induce eIF2α phosphorylation also formed large foci containing the transgene, which colocalized with canonical stress granule markers (G3BP-1 and TIA-1, data not shown). Thus, the expression of V5-Pan3 appeared to induce stress granule formation in some cells, which has been shown to trigger eIF2α phosphorylation (52). Taken together, these results suggest that GFP-Dcp1a-induced eIF2α phosphorylation is not a generalized response to protein expression or to an increase in P-body protein concentration but, rather, a specific response to GFP-Dcp1a.
The EVH1 Domain of Dcp1a Is Required to Trigger eIF2α Phosphorylation
To determine which Dcp1a domains were required for induction of eIF2α phosphorylation by GFP-Dcp1a expression, a panel of GFP-tagged truncations of Dcp1a were expressed in A549 cells (Fig. 4A). The C-terminal deletion mutant (GFP-Dcp1a-ΔCTD) (32) lacked the ability to homotrimerize and enter P-bodies but induced a high level of eIF2α phosphorylation (Fig. 4B, first panel) and inhibited poliovirus gene expression, similar to full-length GFP-Dcp1a (Fig. 1A, fourth panel). This suggests that the ability of GFP-Dcp1a to induce eIF2α phosphorylation does not require the trimerization of the Dcp1a constructs, nor does it require localization of Dcp1a to P-bodies.
FIGURE 4.

Mapping the GFP-Dcp1a domain required for the induction of eIF2α phosphorylation. A, layout of GFP-Dcp1 truncations showing full-length Dcp1 (GFP-Dcp1a), C-terminal truncation (GFP-Dcp1a-ΔCTD), amino-terminal truncation (GFP-Dcp1a-ΔNTD), and amino terminus (GFP-EVH1/EVH1-cherry) expression vectors. B, A549 cells were transfected with 1 μg/1.5 × 105 cells of peGFP-Dcp1-ΔCTD (first panel), pcDNA4-GFP-Dcp1-ΔNTD (second panel), pcDNA4-GFP-EVH1 (third panel), or pcDNA4-EVH1-cherry (fourth panel). Cells were incubated with an antibody against phospho-eIF2α. The highlighted area of the larger merged color image is separated into its constituent colors in the bottom panels. C, quantification of eIF2α phosphorylation in response to expression of Dcp1 truncations as described under “Experimental Procedures.” **, p < 0.0001. Error bars indicate mean ± S.E. D, immunoblot illustrating expression of GFP and GFP-Dcp1a along with corresponding phospho-eIF2α.
A truncation of the N-terminal portion of Dcp1a lacking the EVH1 protein interaction domain (GFP-Dcp1a-ΔNTD) was included in P-bodies, although less consistently than the full-length protein. GFP-Dcp1a-ΔNTD poorly induced eIF2α phosphorylation when compared with the full-length protein (18 versus 92.6% of transfected cells) (Fig. 4, B (second panel) and C). This suggested that the N-terminal EVH1 domain is required for induction of eIF2α phosphorylation. To test whether the EVH1 domain was sufficient to induce eIF2α phosphorylation, we expressed the EVH1 domain of Dcp1a with either an N- or C-terminal tag (GFP-EVH1 or EVH1-cherry) in A549 cells and probed for eIF2α phosphorylation. Both fusion proteins containing the EVH1 domain failed to induce eIF2α phosphorylation at a frequency higher than cells expressing GFP (Fig. 4, B (third and fourth panels) and C). Immunoblotting of electroporated A549 lysates illustrates the level of GFP-tagged Dcp1a truncations with the corresponding level of eIF2α phosphorylation, illustrating the reduction in phospho-eIF2α when the EVH1 domain is removed (Fig. 4D). Taken together, these findings suggest that the EVH1 domain of Dcp1a is required, but not sufficient, for the induction of eIF2α phosphorylation in response to GFP-Dcp1a expression.
PKR Is Activated by GFP-Dcp1a Expression
To determine which of the four eIF2α kinases was activated by GFP-Dcp1a, we expressed GFP-Dcp1a in knockout MEF cell lines, each lacking one functional eIF2α kinase. MEFs competent for PKR activity (PKR+/+) phosphorylated eIF2α in response to GFP-Dcp1a expression but not when expressing GFP alone (Fig. 5, A and E). Conversely, MEFs deficient for PKR activity (PKR−/−) did not phosphorylate eIF2α in response to GFP-Dcp1a expression (Fig. 5, B and E). Levels of GFP-Dcp1a-induced eIF2α phosphorylation did not significantly differ between cells deficient for GCN2 or PERK activity and their wild-type counterparts (Fig. 5E and data not shown). These data suggest that PKR is the principle eIF2α kinase activated by GFP-Dcp1a expression.
FIGURE 5.

PKR is activated by GFP-Dcp1a expression. A, WT MEFs electroporated with 1 μg/1.5 × 105 cells of pcDNA4-GFP (left panel) and peGFP-Dcp1a (center panel) or treated with 500 μm sodium arsenite (Ars) for 30 min (right panel). B, PKR-deficient (−/−) MEFs electroporated with 1 μg/1.5 × 105 cells of pcDNA4-GFP (left panel) and peGFP-Dcp1a (right panel). PKR−/− MEFs were coelectroporated with 1 μg/1.5 × 105 cells of pcDNA4-GFP or peGFP-Dcp1a and wild-type PKR (C) or inactive PKR K296W (D). Each set of MEFs was incubated with an antibody against phospho-eIF2α (S51) (red). E, quantification of eIF2α phosphorylation induced in electroporated cells in response to GFP or GFP-Dcp1a expression in several MEF cell lines as described under “Experimental Procedures.” **, p < 0.0001. Error bars indicate mean ± S.E. F, immunoblot of A549 cells transfected with the indicated plasmids and corresponding levels of total and phospho-PKR (Thr-446).
The importance of PKR for eIF2α phosphorylation during GFP-Dcp1a expression was verified by coexpression of either wild-type PKR or catalytically inactive PKR (K296W) with GFP-Dcp1a in PKR−/− MEFs. Coexpression of wild-type PKR rescued GFP-Dcp1a-induced eIF2α phosphorylation in PKR−/− MEFs, similar to the PKR-competent MEFs (Fig. 5, C and E). In contrast, the catalytically inactive PKR (K296W) failed to rescue eIF2α phosphorylation in response to GFP-Dcp1a expression (Fig. 5, D and E). To further demonstrate the activation of PKR by GFP-Dcp1a, we analyzed A549 cell lysates expressing GFP, GFP-Dcp1a, GFP-Dcp1a-ΔNTD, GFP-Dcp1a-ΔCTD, and GFP-EVH1 for total levels of PKR and activated PKR (phospho-T446-PKR) with a specific antibody. Only the constructs shown previously to induce eIF2α phosphorylation, full-length GFP-Dcp1a, and GFP-Dcp1a-ΔCTD induced detectable levels of activated PKR (Fig. 5F).
To further confirm the involvement of PKR in GFP-Dcp1a-induced eIF2α phosphorylation, an expression plasmid encoding the adenoviral VA1/2 RNA (pAdVAantage) was utilized. This viral RNA is a non-coding transcript expressed during adenoviral infection that interacts with and inhibits the function of PKR (53, 54). As shown in Fig. 6A, coexpression of the VA1/2 RNA with either GFP-Dcp1a or GFP-Dcp1a-ΔCTD significantly inhibited the phosphorylation of eIF2α. Fig. 6B demonstrates that, in the presence of VA1/2 RNA, a significant reduction in eIF2α phosphorylation was observed for all of the tested constructs. These results, combined with the results from Fig. 5, demonstrate that catalytically active PKR was required for the phosphorylation of eIF2α induced by GFP-Dcp1a, whereas the activity of other eIF2α kinases was not essential.
FIGURE 6.

Inhibition of cellular PKR reduces Dcp1a-expression-induced eIF2α-phosphorylation. A, A549 cells were transfected with 1 μg/1.5 × 105 cells of pcDNA4-GFP (left panel), peGFP-Dcp1a (center panel), or peGFP-Dcp1a-ΔCTD (right panel). Cells were cotransfected with pAdVAntage (1 μg/1.5 × 105 cells) and pcDNA4-GFP or peGFP-Dcp1a (1 μg/1.5 × 105 cells) and then incubated with an antibody against phospho-eIF2α. B, quantification of eIF2α phosphorylation in response to GFP, GFP-Dcp1, or GFP-Dcp1a-ΔCTD expression as described under “Experimental Procedures.” **, p < 0.0001. Error bars indicate mean ± S.E.
Inhibition of PKR Rescued Poliovirus Gene Expression in Cells Expressing GFP-Dcp1a
To verify that eIF2α phosphorylation and subsequent inhibition of translation initiation was responsible for the observed inhibition of poliovirus gene expression and reduction in viral titer, we attempted to rescue viral replication. A549 cells were electroporated with pcDNA4-GFP or peGFP-Dcp1a with or without coexpression of adenovirus VA1/2 RNA. The cells were infected with poliovirus for 5 h and processed for immunofluorescence. A549 cells that expressed GFP alone or GFP with VA1/2 RNA displayed viral protein production by 5 hpi (Fig. 7A, top and bottom left panels). In contrast, cells expressing GFP-Dcp1a alone displayed significantly reduced poliovirus gene expression (Fig. 7A, top right panel). However, cells expressing both GFP-Dcp1a and adenoviral VA1/2 RNAs produced poliovirus proteins similar to untreated cells and caused partial disruption of P-bodies (Fig. 7A, bottom right panel). Additionally, we conducted plaque assays of supernatants from A549 cells expressing GFP or GFP-Dcp1a with or without adenoviral VA1/2 RNA. Inhibition of PKR activity by VA1/2 RNA expression alleviated the reduction in poliovirus replication (Fig. 7B). These data suggest that the inhibition of PKR rescued poliovirus protein expression and allowed for progression of the viral replication cycle.
FIGURE 7.
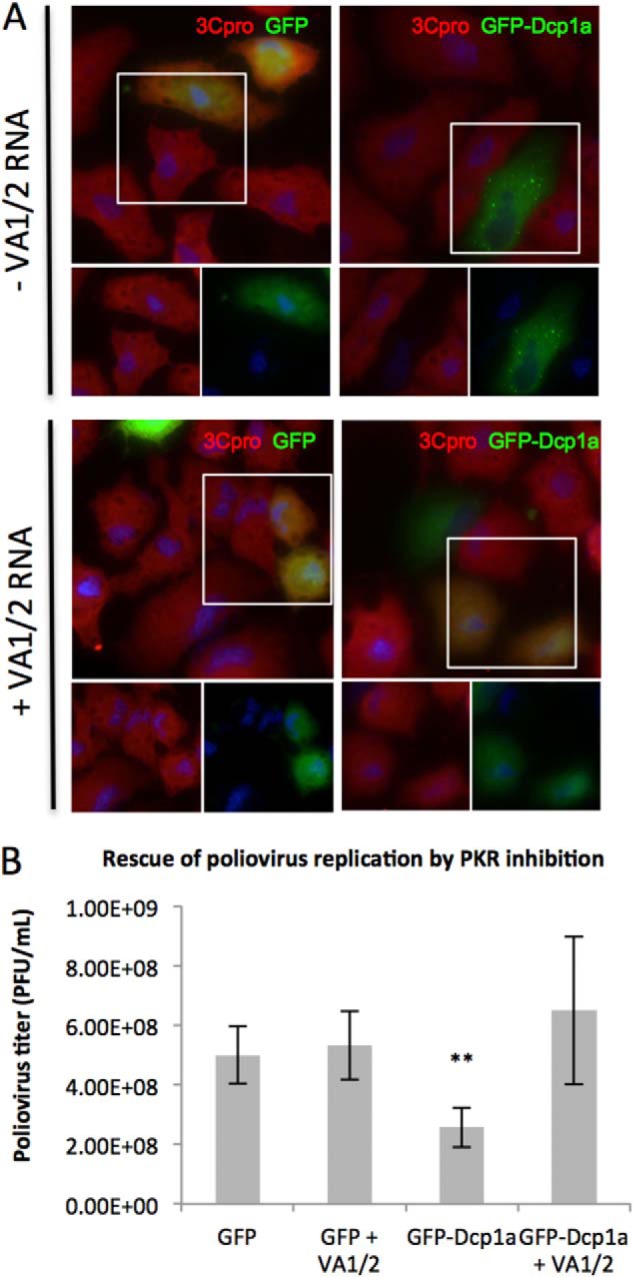
Poliovirus replicates efficiently in GFP-Dcp1a-expressing cells upon PKR inhibition. A, A549 cells were transfected with 1 μg/1. 5 × 105 cells of pcDNA4-GFP alone (top left panel), pcDNA4-GFP and pAdVAntage (bottom left panel), peGFP-Dcp1a alone (top right panel), or peGFP-Dcp1a and pAdVAntage (bottom right panel). Cells were infected with poliovirus (multiplicity of infection of 10) and fixed and processed for immunofluorescence at 5 hpi. Cells were treated with an antibody against poliovirus 3Cpro (red). The highlighted area of the larger merged color image is separated into its constituent colors in the bottom panels. B, quantification of poliovirus titer resulting from infection of A549 cells expressing GFP or GFP-Dcp1a in the absence or presence of PKR inhibitor. **, p < 0.0001. Error bars indicate mean ± S.E.
Activation of PKR by GFP-Dcp1a Requires PACT
PKR is known to exist in an inactive state in unstimulated cells. PKR activation requires the interaction with specific ligands, most notably viral double-stranded RNA. More recently, it has been discovered that a cellular protein, PACT, could stimulate PKR in response to environmental stresses (55, 56). To address the mechanism by which GFP-Dcp1a expression activates PKR and induces eIF2α phosphorylation, we utilized PACT-deficient MEFs (PACT−/−). Wild-type and PACT−/− MEFs were transfected with pcDNA4-GFP or peGFP-Dcp1a and analyzed for eIF2α phosphorylation by immunofluorescence (Fig. 8A). PACT wild-type and PACT−/− cells responded similarly to GFP expression. PACT wild-type MEFs had a significantly elevated number of cells with phospho-eIF2α when expressing GFP-Dcp1a. In contrast, PACT−/− MEFs had substantially reduced levels of phosphorylated eIF2α in response to GFP-Dcp1a expression (Fig. 8B). These data suggest that PACT is involved in GFP-Dcp1a-mediated activation of PKR and subsequent phosphorylation of eIF2α.
FIGURE 8.
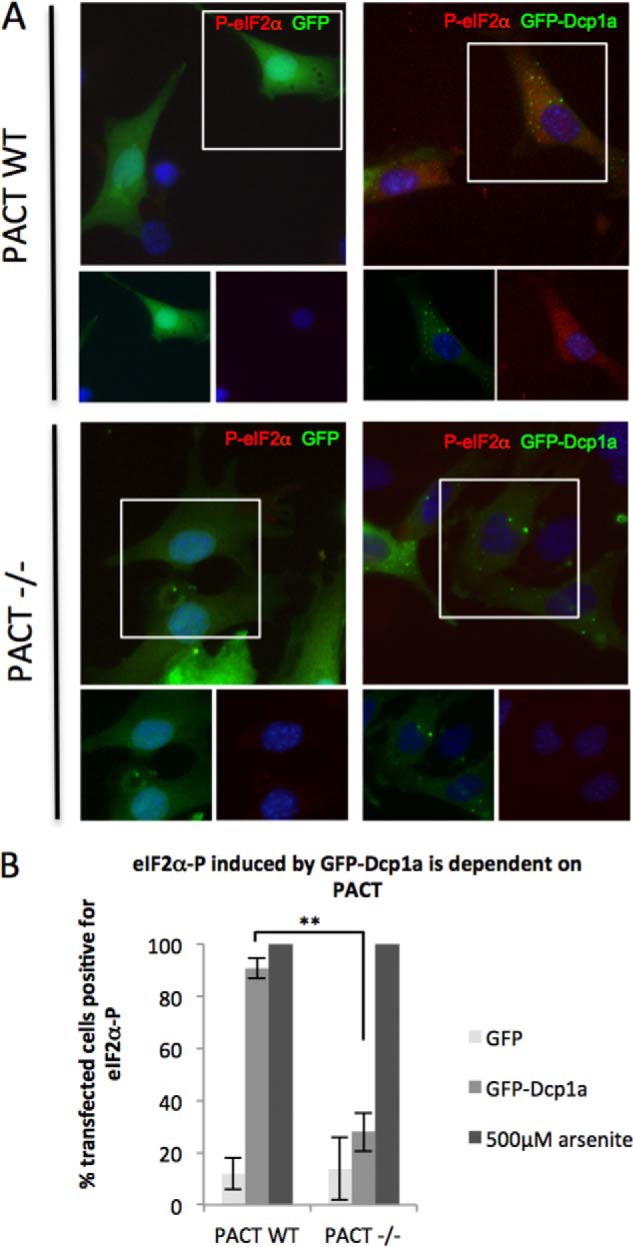
PACT is required for efficient PKR activation in response to GFP-Dcp1a. A, WT and PACT−/− MEFs electroporated with 1 μg/1.5 × 105 cells of pcDNA4-GFP (left panels) or 1 μg/1.5 × 105 cells of peGFP-Dcp1a (right panels) were incubated with an antibody against phospho-eIF2α (S51) (red). B, quantification of eIF2α phosphorylation induced in response to GFP or GFP-Dcp1a expression as described under “Experimental Procedures.” **, p < 0.0001. Error bars indicate mean ± S.D.
DISCUSSION
In a previous study, we showed that poliovirus infection disrupts cellular P-bodies by 3–4 hpi and mediates the cleavage of Dcp1a through the activity of viral 3Cpro (14). Here we demonstrate that poliovirus did not express viral proteins or disrupt P-bodies in cells expressing GFP-Dcp1a (Fig. 1A). The preservation of P-bodies was not due to a modest delay of viral replication kinetics because GFP-Dcp1a-expressing infected cells contained normal amounts of P-bodies at 24 hpi, which is four times longer than the normal poliovirus replication cycle (data not shown). Expression of a C-terminal truncation of Dcp1a (GFP-Dcp1a-ΔCTD), which does not localize to P-bodies, prevented the expression of poliovirus proteins (Fig. 1A). This suggests that GFP-Dcp1a and GFP-Dcp1a-ΔCTD inhibit poliovirus gene expression independent of Dcp1a localization to P-bodies. P-bodies are dynamic and rapidly exchange constituent proteins, such as Dcp1a, with the surrounding cytosol. Concomitant with the inhibition of viral protein expression, we observed a reduction in poliovirus titer produced from infected cells expressing GFP-Dcp1a (Fig. 1B).
RPA analysis revealed reduced translation in cells expressing GFP-Dcp1a compared with non-transfected cells or GFP-expressing cells (Fig. 2A). Global restriction of cellular translation is most often regulated at the rate-limiting initiation step through reducing the availability of the Met-tRNAmeti·eIF2·GTP ternary complex via eIF2α phosphorylation by one of the four eIF2 kinases (50, 51). Phosphorylation of eIF2α also inhibits poliovirus translation at early time points during infection. Poliovirus translation is driven by cap-independent internal ribosome entry site-mediated translation initiation. However, initiation begins on AUG and requires a ternary complex to deliver Met-tRNA to ribosomes (57). However, if viral 3C protease is expressed in sufficient quantity, viral RNA can be translated through an eIF2α-independent pathway that involves production of the truncated form of eIF5B that functionally replaces the ternary complex (58). eIF2α phosphorylation was significantly elevated in GFP-Dcp1a-expressing cells, notably before virus infection was initiated, thus blocking the production of virus protein at the outset of infection and before 3Cpro could be produced at all.
We determined that other P-body proteins, Caf1, Ccr4, Pan2, and Pan3, were not capable of inducing eIF2α phosphorylation comparably to GFP-Dcp1a (Fig. 3). This suggested that the observed increase in phospho-eIF2α was not a result of disruption of P-body protein equilibrium but, rather, a specific response to GFP-Dcp1a expression.
Using eIF2α kinase-deficient MEFs and PKR inhibitors, we determined that functional PKR, but not GCN2 or PERK, was required for GFP-Dcp1a induced eIF2α phosphorylation (Figs. 5 and 6). Finally, we demonstrated that PKR-mediated translational arrest was responsible for the inhibition of poliovirus replication by rescuing poliovirus translation using PKR inhibitors (Fig. 7).
Dcp1a contains several domains, including an N-terminal EVH1 domain, a 14-residue short motif (MI), as well as a C-terminal trimerization domain. The trimerization domain adopts an α-helical conformation and is important for Dcp1a incorporation into active decapping complexes and localization to P-bodies (35). GFP-Dcp1a-ΔCTD inhibited poliovirus gene expression and induced eIF2α phosphorylation similarly to the full-length protein (Figs. 1A and 4), which suggests that localization to P-bodies and inclusion into active decapping complexes is not required to induce eIF2α phosphorylation.
The N-terminal EVH1 domain of Dcp1a is multifunctional because it interacts with Dcp2 and Xrn1 to promote mRNA decapping and also interacts with SMAD4 to mediate TGF-β signaling (30, 31, 36–38, 42, 43). Here we show that the EVH1 domain is involved in a mechanism that triggers PKR activation. Additionally, expression of the EVH1 domain alone did not significantly activate PKR (Fig. 4). This suggests that the EVH1 domain is necessary, but not sufficient, for eIF2α phosphorylation in response to GFP-Dcp1a expression. The EVH1 domain may operate in conjunction with other regions of Dcp1a to mediate protein interactions. One report that indicated EVH1 domain-mediated interactions are affected by phosphorylation of downstream residues in the Dcp1a proline-rich domain (36). As a result, GFP-EVH1 may not be able to interact with PKR or PKR complexes strongly enough to trigger activation without the Dcp1a C-terminal extension. Interestingly, the Dcp1a N terminus has been suggested to be a new class of EVH1 domain, largely because of its longer α-helix (30, 31, 42). As a result of additional conserved residues in the EVH1 domain, Dcp1a is able to bind a variety of proline-rich protein ligands over an extended surface (42). This potentially provides Dcp1a with the ability to interact with numerous protein targets within the mammalian cell, which remain to be elucidated. Modulation of this activity through posttranslational modifications of Dcp1a may provide further understanding of the role of Dcp1a as a coactivator and signal transducer. The data in this study suggest that GFP-Dcp1a may interact with other proteins through the N-terminal domain, either through signaling cascades or directly with PKR.
The cleavage of Dcp1a by poliovirus 3Cpro during viral infection was initially a curious discovery. Poliovirus proteases tend to selectively target proteins involved in cellular gene expression or the antiviral response to make the cellular environment more amenable to viral replication (68). Although Dcp1a has a described role in RNA degradation, no form of poliovirus RNA contains a 5′-methylguanosine cap, and it was not expected to be targeted by Dcp1a for decapping reactions. So why would a poliovirus protease target Dcp1a? This study suggests that Dcp1a may play novel roles in translation regulation and innate immunity because PKR is a central factor in translation regulation and many innate immune pathways. In this regard, it is possible that poliovirus targets Dcp1a for cleavage to control a novel arm of the innate immune response as well as maintaining the cellular translation apparatus in an active configuration. The activation of cellular stress kinase PKR when the levels of Dcp1a are increased indicates that Dcp1a expression must be maintained in stoichiometric balance with other factors. One scenario is that the PKR activation function of Dcp1a is normally repressed by a Dcp1a inhibitor of some sort, which is overcome by increased concentration of Dcp1a. The activation of PKR in response to GFP-Dcp1a expression links Dcp1a to the cellular stress pathways in a way not described previously.
Although PKR autophosphorylates and activates in response to double-stranded RNA, it is also regulated by protein-protein interactions that are independent of double-stranded RNA. Notably, PACT associates with and activates PKR without double-stranded RNA binding (55, 56). In contrast, the human immunodeficiency virus, type 1 transactivation response RNA-binding protein (TRBP) interacts with PKR and inhibits PKR activation (59). TRBP also interacts with PACT and prevents PACT-mediated activation of PKR (60). However, upon cellular stress, PACT is phosphorylated and dissociates from TRBP, allowing PACT-dependent PKR activation (55, 60–64). In this study, we demonstrate a requirement of PACT for the phosphorylation of eIF2α in response to GFP-Dcp1a expression (Fig. 8). This suggests that GFP-Dcp1a expression may mimic a stress signal, leading to the phosphorylation of PACT, disassociation from TRBP, and activation of PKR. Alternatively, Dcp1a may complex with TRBP or PACT, leading to the relief of TRBP-mediated PKR repression. The latter is a possibility because examination of protein lysates immunoprecipitated with a Dcp1a-specific antibody indicated that PACT existed in complexes with the Dcp1a transgene and endogenous Dcp1a (data not shown). Other protein factors activate PKR, although the mechanisms are not as clearly elucidated (65–67). Whether and how GFP-Dcp1a complexes with PACT, TRBP, PKR, or with additional proteins to activate PKR is a subject for future experimentation. The mechanism of PKR activation, the involvement of accessory proteins, and the role of posttranslational modifications of Dcp1a will be interesting areas for future studies.
This work was supported, in whole or in part, by National Institutes of Health Public Health Service Grant AI50237; by NCI, National Institutes of Health Cancer Center Support Grant P30CA1251230; and by National Institutes of Health Grants HD007495, DK56338, and CA125123 (to the Integrated Microscopy Core at Baylor College of Medicine). This work was also supported by the Dan L. Duncan Cancer Center and the John S. Dunn Gulf Coast Consortium for Chemical Genomics.
- P-body
- processing body
- hpi
- hours post-infection
- PKR
- protein kinase R
- MEF
- mouse embryonic fibroblast
- RPA
- ribopuromycylation assay
- PERK
- PKR-like endoplasmic reticulum kinase.
REFERENCES
- 1. Kedersha N., Stoecklin G., Ayodele M., Yacono P., Lykke-Andersen J., Fritzler M. J., Scheuner D., Kaufman R. J., Golan D. E., Anderson P. (2005) Stress granules and processing bodies are dynamically linked sites of mRNP remodeling. J. Cell Biol. 169, 871–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Anderson P., Kedersha N. (2009) RNA granules. Post-transcriptional and epigenetic modulators of gene expression. Nat. Rev. Mol. Cell Biol. 10, 430–436 [DOI] [PubMed] [Google Scholar]
- 3. Sheth U., Parker R. (2003) Decapping and decay of messenger RNA occur in cytoplasmic processing bodies. Science 300, 805–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cougot N., Babajko S., Séraphin B. (2004) Cytoplasmic foci are sites of mRNA decay in human cells. J. Cell Biol. 165, 31–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eulalio A., Behm-Ansmant I., Schweizer D., Izaurralde E. (2007) P-body formation is a consequence, not the cause, of RNA-mediated gene silencing. Mol. Cell. Biol. 27, 3970–3981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Franks T. M., Lykke-Andersen J. (2008) The control of mRNA decapping and P-body formation. Mol. Cell 32, 605–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beelman C. A., Stevens A., Caponigro G., LaGrandeur T. E., Hatfield L., Fortner D. M., Parker R. (1996) An essential component of the decapping enzyme required for normal rates of mRNA turnover. Nature 382, 642–646 [DOI] [PubMed] [Google Scholar]
- 8. Fenger-Grøn M., Fillman C., Norrild B., Lykke-Andersen J. (2005) Multiple processing body factors and the ARE binding protein TTP activate mRNA decapping. Mol. Cell 20, 905–915 [DOI] [PubMed] [Google Scholar]
- 9. Lykke-Andersen J. (2002) Identification of a human decapping complex associated with hUpf proteins in nonsense-mediated decay. Mol. Cell. Biol. 22, 8114–8121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Arribere J. A., Doudna J. A., Gilbert W. V. (2011) Reconsidering movement of eukaryotic mRNAs between polysomes and P bodies. Mol. Cell 44, 745–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zheng D., Ezzeddine N., Chen C. Y., Zhu W., He X., Shyu A. B. (2008) Deadenylation is prerequisite for P-body formation and mRNA decay in mammalian cells. J. Cell Biol. 182, 89–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yamashita A., Chang T. C., Yamashita Y., Zhu W., Zhong Z., Chen C. Y., Shyu A. B. (2005) Concerted action of poly(A) nucleases and decapping enzyme in mammalian mRNA turnover. Nat. Struct. Mol. Biol. 12, 1054–1063 [DOI] [PubMed] [Google Scholar]
- 13. Reineke L. C., Lloyd R. E. (2013) Diversion of stress granules and P-bodies during viral infection. Virology 436, 255–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dougherty J. D., White J. P., Lloyd R. E. (2011) Poliovirus-mediated disruption of cytoplasmic processing bodies. J. Virol. 85, 64–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Emara M. M., Brinton M. A. (2007) Interaction of TIA-1/TIAR with West Nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc. Natl. Acad. Sci. 104, 9041–9046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ariumi Y., Kuroki M., Kushima Y., Osugi K., Hijikata M., Maki M., Ikeda M., Kato N. (2011) Hepatitis C virus hijacks P-body and stress granule components around lipid droplets. J. Virol. 85, 6882–6892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chahar H. S., Chen S., Manjunath N. (2013) P-body components LSM1, GW182, DDX3, DDX6 and XRN1 are recruited to WNV replication sites and positively regulate viral replication. Virology 436, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ward A. M., Bidet K., Yinglin A., Ler S. G., Hogue K., Blackstock W., Gunaratne J., Garcia-Blanco M. A. (2011) Quantitative mass spectrometry of DENV-2 RNA-interacting proteins reveals that the DEAD-box RNA helicase DDX6 binds the DB1 and DB2 3′ UTR structures. RNA Biol. 8, 1173–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Aizer A., Shav-Tal Y. (2008) Intracellular trafficking and dynamics of P bodies. Prion 2, 131–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Erickson S. L., Lykke-Andersen J. (2011) Cytoplasmic mRNP granules at a glance. J. Cell Sci. 124, 293–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bail S., Kiledjian M. (2006) More than 1 + 2 in mRNA decapping. Nat. Struct. Mol. Biol. 13, 7–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Simon E., Camier S., Séraphin B. (2006) New insights into the control of mRNA decapping. Trends Biochem. Sci. 31, 241–243 [DOI] [PubMed] [Google Scholar]
- 23. Long R. M., McNally M. T. (2003) mRNA decay. X (XRN1) marks the spot. Mol. Cell 11, 1126–1128 [DOI] [PubMed] [Google Scholar]
- 24. Jacobson A. (2004) Regulation of mRNA decay. Decapping goes solo. Mol. Cell 15, 1–2 [DOI] [PubMed] [Google Scholar]
- 25. Coller J., Parker R. (2004) Eukaryotic mRNA decapping. Annu. Rev. Biochem. 73, 861–890 [DOI] [PubMed] [Google Scholar]
- 26. Stevens A., Maupin M. K. (1987) A 5′-3′ exoribonuclease of Saccharomyces cerevisiae. Size and novel substrate specificity. Arch. Biochem. Biophys. 252, 339–347 [DOI] [PubMed] [Google Scholar]
- 27. Buchan J. R., Parker R. (2009) Eukaryotic stress granules. The ins and outs of translation. Mol. Cell 36, 932–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stevens A. (2001) 5′-exoribonuclease 1. Xrn1. Methods Enzymol. 342, 251–259 [DOI] [PubMed] [Google Scholar]
- 29. van Dijk E., Cougot N., Meyer S., Babajko S., Wahle E., Séraphin B. (2002) Human Dcp2. A catalytically active mRNA decapping enzyme located in specific cytoplasmic structures. EMBO J. 21, 6915–6924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. She M., Decker C. J., Sundramurthy K., Liu Y., Chen N., Parker R., Song H. (2004) Crystal structure of Dcp1p and its functional implications in mRNA decapping. Nat. Struct. Mol. Biol. 11, 249–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. She M., Decker C. J., Svergun D. I., Round A., Chen N., Muhlrad D., Parker R., Song H. (2008) Structural basis of dcp2 recognition and activation by dcp1. Mol. Cell 29, 337–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rzeczkowski K., Beuerlein K., Müller H., Dittrich-Breiholz O., Schneider H., Kettner-Buhrow D., Holtmann H., Kracht M. (2011) c-Jun N-terminal kinase phosphorylates DCP1a to control formation of P bodies. J. Cell Biol. 194, 581–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Aizer A., Kafri P., Kalo A., Shav-Tal Y. (2013) The P body protein Dcp1a is hyper-phosphorylated during mitosis. PLoS ONE 8, e49783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Aizer A., Brody Y., Ler L. W., Sonenberg N., Singer R. H., Shav-Tal Y. (2008) The dynamics of mammalian P body transport, assembly, and disassembly in vivo. Mol. Biol. Cell 19, 4154–4166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tritschler F., Braun J. E., Motz C., Igreja C., Haas G., Truffault V., Izaurralde E., Weichenrieder O. (2009) DCP1 forms asymmetric trimers to assemble into active mRNA decapping complexes in metazoa. Proc. Natl. Acad. Sci. U.S.A. 106, 21591–21596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chiang P. Y., Shen Y. F., Su Y. L., Kao C. H., Lin N. Y., Hsu P. H., Tsai M. D., Wang S. C., Chang G. D., Lee S. C., Chang C. J. (2013) Phosphorylation of mRNA decapping protein Dcp1a by the ERK signaling pathway during early differentiation of 3T3-L1 preadipocytes. PLoS ONE 8, e61697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Callebaut I. (2002) An EVH1/WH1 domain as a key actor in TGFβ signalling. FEBS Lett. 519, 178–180 [DOI] [PubMed] [Google Scholar]
- 38. Deshmukh M. V., Jones B. N., Quang-Dang D. U., Flinders J., Floor S. N., Kim C., Jemielity J., Kalek M., Darzynkiewicz E., Gross J. D. (2008) mRNA decapping is promoted by an RNA-binding channel in Dcp2. Mol. Cell 29, 324–336 [DOI] [PubMed] [Google Scholar]
- 39. Prehoda K. E., Lee D. J., Lim W. A. (1999) Structure of the enabled/VASP homology 1 domain-peptide complex. A key component in the spatial control of actin assembly. Cell 97, 471–480 [DOI] [PubMed] [Google Scholar]
- 40. Boëda B., Briggs D. C., Higgins T., Garvalov B. K., Fadden A. J., McDonald N. Q., Way M. (2007) Tes, a specific Mena interacting partner, breaks the rules for EVH1 binding. Mol. Cell 28, 1071–1082 [DOI] [PubMed] [Google Scholar]
- 41. Borja M. S., Piotukh K., Freund C., Gross J. D. (2011) Dcp1 links coactivators of mRNA decapping to Dcp2 by proline recognition. RNA 17, 278–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Braun J. E., Truffault V., Boland A., Huntzinger E., Chang C. T., Haas G., Weichenrieder O., Coles M., Izaurralde E. (2012) A direct interaction between DCP1 and XRN1 couples mRNA decapping to 5′ exonucleolytic degradation. Nat. Struct. Mol. Biol. 19, 1324–1331 [DOI] [PubMed] [Google Scholar]
- 43. Ball L. J., Jarchau T., Oschkinat H., Walter U. (2002) EVH1 domains. Structure, function and interactions. FEBS Lett. 513, 45–52 [DOI] [PubMed] [Google Scholar]
- 44. Jones C. L., Ehrenfeld E. (1983) The effect of poliovirus infection on the translation in vitro of VSV messenger ribonucleoprotein particles. Virology 129, 415–430 [DOI] [PubMed] [Google Scholar]
- 45. Chang T. C., Yamashita A., Chen C. Y., Yamashita Y., Zhu W., Durdan S., Kahvejian A., Sonenberg N., Shyu A. B. (2004) UNR, a new partner of poly(A)-binding protein, plays a key role in translationally coupled mRNA turnover mediated by the c-fos major coding-region determinant. Genes Dev. 18, 2010–2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Thomis D. C., Samuel C. E. (1992) Mechanism of interferon action: autoregulation of RNA-dependent P1/eIF-2 α protein kinase (PKR) expression in transfected mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 89, 10837–10841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. David A., Netzer N., Strader M. B., Das S. R., Chen C. Y., Gibbs J., Pierre P., Bennink J. R., Yewdell J. W. (2011) RNA binding targets aminoacyl-tRNA synthetases to translating ribosomes. J. Biol. Chem. 286, 20688–20700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schmidt E. K., Clavarino G., Ceppi M., Pierre P. (2009) SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods 6, 275–277 [DOI] [PubMed] [Google Scholar]
- 49. McEwen E., Kedersha N., Song B., Scheuner D., Gilks N., Han A., Chen J. J., Anderson P., Kaufman R. J. (2005) Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J. Biol. Chem. 280, 16925–16933 [DOI] [PubMed] [Google Scholar]
- 50. Krishnamoorthy T., Pavitt G. D., Zhang F., Dever T. E., Hinnebusch A. G. (2001) Tight binding of the phosphorylated α subunit of initiation factor 2 (eIF2α) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Mol. Cell. Biol. 21, 5018–5030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sudhakar A., Ramachandran A., Ghosh S., Hasnain S. E., Kaufman R. J., Ramaiah K. V. (2000) Phosphorylation of serine 51 in initiation factor 2 α (eIF2 α) promotes complex formation between eIF2 α(P) and eIF2B and causes inhibition in the guanine nucleotide exchange activity of eIF2B. Biochemistry 39, 12929–12938 [DOI] [PubMed] [Google Scholar]
- 52. Reineke L. C., Dougherty J. D., Pierre P., Lloyd R. E. (2012) Large G3BP-induced granules trigger eIF2α phosphorylation. Mol. Biol. Cell 23, 3499–3510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mathews M. B., Shenk T. (1991) Adenovirus virus-associated RNA and translation control. J. Virol. 65, 5657–5662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Thimmappaya B., Weinberger C., Schneider R. J., Shenk T. (1982) Adenovirus VAI RNA is required for efficient translation of viral mRNAs at late times after infection. Cell 31, 543–551 [DOI] [PubMed] [Google Scholar]
- 55. Patel C. V., Handy I., Goldsmith T., Patel R. C. (2000) PACT, a stress-modulated cellular activator of interferon-induced double-stranded RNA-activated protein kinase, PKR. J. Biol. Chem. 275, 37993–37998 [DOI] [PubMed] [Google Scholar]
- 56. Patel R. C., Sen G. C. (1998) Pact, a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 17, 4379–4390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pelletier J., Sonenberg N. (1988) Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature 334, 320–325 [DOI] [PubMed] [Google Scholar]
- 58. White J. P., Reineke L. C., Lloyd R. E. (2011) Poliovirus switches to an eIF2-independent mode of translation during infection. J. Virol. 85, 8884–8893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Benkirane M., Neuveut C., Chun R. F., Smith S. M., Samuel C. E., Gatignol A., Jeang K. T. (1997) Oncogenic potential of TAR RNA binding protein TRBP and its regulatory interaction with RNA-dependent protein kinase PKR. EMBO J. 16, 611–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Daher A., Laraki G., Singh M., Melendez-Peña C. E., Bannwarth S., Peters A. H., Meurs E. F., Braun R. E., Patel R. C., Gatignol A. (2009) TRBP control of PACT-induced phosphorylation of protein kinase R is reversed by stress. Mol. Cell. Biol. 29, 254–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ito T., Yang M., May W. S. (1999) RAX, a cellular activator for double-stranded RNA-dependent protein kinase during stress signaling. J. Biol. Chem. 274, 15427–15432 [DOI] [PubMed] [Google Scholar]
- 62. Bennett R. L., Blalock W. L., Abtahi D. M., Pan Y., Moyer S. A., May W. S. (2006) RAX, the PKR activator, sensitizes cells to inflammatory cytokines, serum withdrawal, chemotherapy, and viral infection. Blood 108, 821–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Singh M., Fowlkes V., Handy I., Patel C. V., Patel R. C. (2009) Essential role of PACT-mediated PKR activation in tunicamycin-induced apoptosis. J. Mol. Biol. 385, 457–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Singh M., Castillo D., Patel C. V., Patel R. C. (2011) Stress-induced phosphorylation of PACT reduces its interaction with TRBP and leads to PKR activation. Biochemistry 50, 4550–4560 [DOI] [PubMed] [Google Scholar]
- 65. Okumura F., Okumura A. J., Uematsu K., Hatakeyama S., Zhang D. E., Kamura T. (2013) Activation of double-stranded RNA-activated protein kinase (PKR) by interferon-stimulated gene 15 (ISG15) modification down-regulates protein translation. J. Biol. Chem. 288, 2839–2847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. George C. X., Thomis D. C., McCormack S. J., Svahn C. M., Samuel C. E. (1996) Characterization of the heparin-mediated activation of PKR, the interferon-inducible RNA-dependent protein kinase. Virology 221, 180–188 [DOI] [PubMed] [Google Scholar]
- 67. Anderson E., Pierre-Louis W. S., Wong C. J., Lary J. W., Cole J. L. (2011) Heparin activates PKR by inducing dimerization. J. Mol. Biol. 413, 973–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lloyd R. (2006) Translational control by viral proteinases. Virus Res. 119, 76–88 [DOI] [PMC free article] [PubMed] [Google Scholar]