

Background: HPK1 is lost in >95% pancreatic cancer through proteasome-mediated degradation.
Results: The ubiquitination and degradation of HPK1 mediated by CUL7/Fbxw8 ubiquitin ligase required HPK1 kinase activity and autophosphorylation.
Conclusion: Targeted HPK1 degradation by CUL7/Fbxw8 ubiquitin ligase constitutes a negative-feedback loop to restrain HPK1 activity.
Significance: Our study revealed a direct link of CUL7/Fbxw8 ubiquitin ligase to the MAPK pathway.
Keywords: E3 Ubiquitin Ligase, MAP Kinases (MAPKs), Pancreatic Cancer, Proteasome, Protein Phosphatase, Serine Threonine Protein Kinase, CUL7 Ubiquitin Ligase, Fbxw8, HPK1, PP4
Abstract
HPK1, a member of mammalian Ste20-like serine/threonine kinases, is lost in >95% pancreatic cancer through proteasome-mediated degradation. However, the mechanism of HPK1 loss has not been defined. The aims of this study are to identify the ubiquitin ligase and to examine the mechanisms that targets HPK1 degradation. We found that the CUL7/Fbxw8 ubiquitin ligase targeted HPK1 for degradation via the 26 S proteasome. The ubiquitination of HPK1 required its kinase activity and autophosphorylation. Wild-type protein phosphatase 4 (PP4), but not the phosphatase-dead PP4 mutant, PP4-RL, inhibits the interaction of Fbxw8 with HPK1 and Fbxw8-mediated ubiquitination of HPK1. In addition, we showed that Thr-355 of HPK1 is a key PP4 dephosphorylation site, through which CUL7/Fbxw8 ubiquitin ligase and PP4 regulates HPK1 stability. Knockdown of Fbxw8 restores endogenous HPK1 protein expression and inhibits cell proliferation of pancreatic cancer cells. Our study demonstrated that targeted degradation of HPK1 by the CUL7/Fbxw8 ubiquitin ligase constitutes a negative-feedback loop to restrain the activity of HPK1 and that CUL7/Fbxw8 ubiquitin ligase promotes pancreatic cancer cell proliferation. CUL7/Fbxw8 ubiquitin ligase-mediated HPK1 degradation revealed a direct link and novel role of CUL7/Fbxw8 ubiquitin ligase in the MAPK pathway, which plays a critical role in cell proliferation and differentiation.
Introduction
Hematopoietic progenitor kinase 1 (HPK1),2 a member of mammalian Ste20-like serine/threonine kinases, functions as a MAP4K to activate MAP3Ks, which signal through the well-established MKK4/MKK7, JNK, or SAPK cascade (1, 2). HPK1 has been shown to form inducible complexes with adaptor proteins, such as Grb2 (3), Gads (4), Nck (5), and Crk (5) and is involved in various signaling systems, including EGF (3, 6), TGF-β (7, 8), erythropoietin (9), protein kinase D (10), prostaglandin E2 (11), T and B cell receptor (4, 5, 12–15) and differentiation of acute myelogenous leukemia cells (16). HPK1 inhibits MEK1/2-mediated Erk activation, which is one of the major Ras effector pathways. HPK1−/− T cells from HPK1 knock-out mice had enhanced TCR-induced activation of Erk2, which can be blocked by a MEK inhibitor, PD98059 (17).
While a number of studies have identified the role of HPK1 in regulating NFκB, Erk, and JNK pathways in hematopoietic cells (2–4,7,9,11–15,18,19), the functions of HPK1 in cancer are unclear. Our previous study showed that HPK1 protein is expressed in normal pancreatic ducts but is lost in >95% of pancreatic cancer, which is one of the most lethal cancers; less than 5% of patients remain alive 5 years after diagnosis. The loss of HPK1 is strongly associated with the progression from low-grade pancreatic intraepithelial neoplasia (PanIN) to invasive pancreatic cancer. More important, restoring HPK1 expression in pancreatic cancer cells causes cell cycle arrest and growth inhibition, which is due in part to the stabilization of p21 and p27 (20). Thus, HPK1 may function as a novel tumor suppressor and loss of HPK1 plays a critical role in the development of pancreatic cancer.
Although phosphorylation and caspase 3-mediated cleavage of HPK1 have been implicated in its regulation (21), the mechanisms of HPK1 regulation remain unclear. HPK1 has been shown to be positively regulated by protein phosphatase 4 (PP4), a serine/threonine phosphatase, which dephosphorylates HPK1 and inhibits HPK1 ubiquitination and degradation (22). Interestingly, our previous study showed that both endogenous HPK1 and exogenously overexpressed HPK1 protein are degraded in pancreatic cancer cells, which can be fully restored by treatment with proteasome inhibitors (20), suggesting that loss of HPK1 in pancreatic cancer is caused by proteasome-mediated protein degradation. However, the ubiquitin ligase and the molecular mechanisms that targets HPK1 for degradation in pancreatic cancer is unknown. Given the novel tumor suppressor function of HPK1 in pancreatic cancer, it is important to identify the ubiquitin ligase and to examine the mechanisms by which HPK1 expression is regulated.
The protein ubiquitination is catalyzed by three enzymes, including a ubiquitin (Ub)-activating enzyme (E1), a Ub-conjugating enzyme (E2), and a Ub ligase (E3) (23). The specificity of this pathway is mainly determined by Ub ligases, which not only interact with the substrates, but also recruit the activated E2 enzymes. The concerted actions between the E3-bound substrate, the Ub ligase, and the Ub-charged E2 produce polyubiquitin chains on targeted proteins, which are then delivered to the 26 S proteasome for degradation (24). Of a few hundreds of Ub ligases, the cullin-RING Ub ligases (CRLs) are grouped as the largest subfamily and are conserved throughout eukaryotes. In these Ub ligases, cullins serve as molecular scaffolds that organize the reactive module to coordinate substrate ubiquitination. Human cells express seven different cullins (CUL1, -2, -3, -4A, -4B, -5, and -7) and each of them can form Ub ligase complexes with other proteins (25). The best characterized CRL is the Skp1-CUL1-F-box protein (SCF) complex (26), which comprises three invariable components: CUL1, Rbx1, and Skp1 as well as one variable component, known as an F-box protein that binds Skp1 through its F-box motif and determines the substrate specificity (23, 24, 26, 27). Skp1 can also form an E3 complex with CUL7, Rbx1, and F-box and WD repeat domain containing 8 (Fbxw8), which is the only known F-box protein in CUL7 E3 complex (28). In this study, we identified CUL7/Fbxw8 complex as the Ub ligase that is responsible for proteasome-mediated HPK1 degradation. Our data revealed novel pathways of HPK1 regulation, a direct link and novel role of CUL7 ubiquitin ligase in the MAPK pathway.
EXPERIMENTAL PROCEDURES
Plasmids
Flag-HPK1, Flag-HGK, Flag-HPK1-M46, Flag-PP4, and HA-PP4-RL were previously described (22, 29). Myc-Fbxw8 and HA-Ub were gifts from Dr. Stephen J. Elledge (Harvard Medical School, Boston, MA). Flag-Fbxw7, His-Ub, His-Ub-K48R, and His-Ub-K63R plasmids were from Dr. Mong-Hong Lee and Dr. Hui-Kuan Lin at our institution. Site-directed mutagenesis of HPK1 was performed using a QuickChange kit (Stratagene) according to the manufacturer's instruction. All mutations were confirmed by DNA sequencing.
Cells and Transfection
Cell lines, HEK293T, Jurkat T cells, Panc-1 and Panc-28 cells were grown in DMEM supplemented with 10% fetal calf serum and 100 units/ml streptomycin/penicillin. Cells were transfected with various amounts of plasmids encoding PP4, PP4-RL, Fbxw8, HPK1, and its mutants, or Ub as indicated in figure legends.
Antibody Array-based Screening for HPK1-interacting Proteins in Pancreatic Cancer Cells
Whole cell lysates were prepared from Panc-1/Flag-HPK1 stable cells treated with 2.0 μm MG132 for 16 h, which stabilized HPK1 protein. The antibody array filter containing 400 different antibodies (HM3000, Hypromatrix, Worcester, MA) was pre-blocked in a buffer containing 5% nonfat milk and then incubated with above-mentioned whole cell lysate. After overnight incubation at 4 °C, the array filter was washed three times with TBST for 15 min and then incubated with the horseradish peroxidase-conjugated anti-Flag antibody in TBST for 2 h at room temperature. The antibody-antigen-HPK1 complex was detected by peroxidase substrate, followed by chemiluminescence.
Immunoprecipitation and Immunoblotting
Immunoprecipitation and immunoblotting were performed as previously described (20). Protein expression was analyzed by 8% or 10% SDS-PAGE, which was electroblotted onto polyvinylidene difluoride membranes (Novex), blocked in 5% skim milk in 1× TBS, and probed with the primary antibodies indicated in the figure legends. Proteins were detected using an enhanced chemiluminescence kit (Amersham Biosciences). Following antibodies were used: anti-HA (Upstate Biotechnology); anti-β-actin and anti-Flag (Sigma-Aldrich); Anti-HPK1 (N-19), anti-PP4, anti-c-Myc (9E10), anti-Skp1, anti-CUL1 and anti-CUL7 (Santa Cruz Biotechnology Inc.), and anti-Fbxw8 (Abgent, Inc.).
Effects of Fbxw8 Overexpression on the Half-life of HPK1 Protein
The half-life of HPK1 protein was measured in 293T cells cotransfected with HPK1 and Fbxw8 constructs compared with those transfected with HPK1 alone. The cells transfected with HPK1 alone or co-transfected with HPK1 and Fbxw8 cDNA constructs were treated with 100 μg of cycloheximide (CHX) for 0, 30 min, 1 h, 2 h, 4 h, and 6 h. The HPK1 protein levels were measured by Western blots.
Immunocomplex Kinase Assays and Ubiquitination Assays
Immunocomplex kinase assays using myelin basic protein (MBP) as a substrate were performed as previously described (20). In vivo ubiquitination assays were performed as described elsewhere (30). Flag-HPK1 and Myc-Fbxw8 were cotransfected with His.Myc-Ub, His.Myc-Ub K48R, or His.Myc-Ub K63R into 293T cells. The cells were treated with 2 μm MG132 for 6 h before harvesting, and lysed in PBS containing 1% nonidet P-40 and 10 mm imidazole. The nickel-nitrilotriacetic acid agarose beads (Ni-NTA, Qiagen Inc.) were then added to the cell extracts and rotated at room temperature for 4 h to pull down the ubiquitinated HPK1 protein. Precipitates were washed three times with PBS containing 1% Nonidet P-40 and 20 mm imidazole and then boiled in SDS-PAGE sample buffer. The protein complexes were separated on 8% SDS-PAGE and probed with anti-HPK1 antibody. The levels of total polyubiquitinated proteins were comparable across the experimental conditions.
Cell Growth Studies
Panc-1 or Panc-28 parental, ShRNA control and Fbxw8 knockdown stable cells were plated at an equal density in triplicates and cultured under normal conditions for 6 days. The cells were harvested daily by trypsinization and washed once with cold PBS. The cell numbers were counted and plotted. The experiments to establish the growth curves were repeated three times.
RESULTS
HPK1 Interacts with Skp1, Rbx1, and CUL7, but not CUL1
To identify HPK1-interacting proteins in pancreatic cancer cells, we performed an antibody-based array screening using the lysates from Panc-1/HPK1 stable clone (C1) treated with MG132, which stabilized HPK1 protein. We found that HPK1 interacted with Skp1 and Rbx1/Rbx2, but not CUL1 (Fig. 1A). To confirm these results, we performed co-immunoprecipitation using the lysates from Panc-1/HPK1 stable clone (C1) treated with MG132. HPK1 antibody could co-precipitate Skp1. No Skp1 was detected in IgG control or Panc-1 parental cells, which had no detectable HPK1 expression. Similar to the results from antibody array, no interaction between HPK1 and CUL1 was detected by immunoprecipitation (Fig. 1B). Since Skp1 can also form an E3 complex with CUL7, we examined the interaction between HPK1 and CUL7 in Panc-1/HPK1 stable cells. CUL7 was specifically co-immunoprecipitated with HPK1 (Fig. 1B). These data suggested that Skp1-CUL7- Rbx1 E3 complex is responsible for proteasome-mediated HPK1 degradation in pancreatic cancer cells.
FIGURE 1.

CUL7/Fbxw8 ubiquitin ligase mediates HPK1 degradation in pancreatic cancer. A and B, analysis of HPK1 protein interactions in Panc-1 cells by antibody array or immunoprecipitation. Panc-1/Flag-HPK1 stable cells (C1) were treated with 2 μm MG132 for 16 h, and then harvested. Cell extracts were used for antibody array analysis (A) or immunoprecipitation with M2 antibody followed by Western blotting using antibodies against HPK1, Skp1, CUL7, CUL1, or Fbxw8 (B). Panc-1 parental cells, which had no detectable HPK1 protein, were used as controls. C, 293T cells were transfected with plasmids expressing Myc-Fbxw8 and Flag-HPK1; 36 h after transfection, the cells were harvested for immunoprecipitation with anti-Myc or anti-Flag antibodies followed by Western blotting as indicated. D, endogenous HPK1 interacts with Fbxw8 in Jurkat T cells. HPK1 from Jurkat T cells was immunoprecipitated with an anti-HPK1 antibody. The immunoprecipitate was then immunoblotted with an anti-Fbxw8 antibody. E, G, and H, overexpression of Fbxw8 results in a dose-dependent decrease of wild-type HPK1 but not the kinase-dead HPK1-M46 and HGK in 293T cells. F, knockdown of Fbxw8 in 293T cells stabilizes HPK1 protein. I, Fbxw8 overexpression decreases the half-life of HPK1 protein in 293T cells.
Fbxw8 Interacts with HPK1 and Promotes HPK1 Degradation
Since Fbxw8 is the only known F-box protein in Skp1-CUL7 E3 complex, we examined the interaction between HPK1 and Fbxw8 and found that endogenous Fbxw8 was specifically co-immunoprecipitated with HPK1 in Panc-1/HPK1 stable cells (Fig. 1B). To further confirm their interactions, Myc-Fbxw8 and Flag-HPK1 were cotransfected into 293T cells and co-immunoprecipitation was performed using either anti-Myc or anti-Flag antibody. HPK1 antibody could co-precipitate Fbxw8 protein and Myc antibody could co-precipitate HPK1 protein (Fig. 1C). In addition, endogenous HPK1 also interacted with Fbxw8 in Jurkat T cells (Fig. 1D). To test whether Fbxw8 promoted HPK1 degradation, both overexpression and knockdown systems of Fbxw8 were used. We found that HPK1 protein levels were inversely correlated with the expression levels of Fbxw8 (Fig. 1, E and F). However, Fbxw8 had little effect on the steady-state level of kinase-dead HPK1-M46 (Fig. 1G), suggesting that Fbxw8-mediated HPK1 degradation requires HPK1 kinase activity. Also we did not observe any effect of Fbxw8 on the expression of HGK, another serine/threonine kinase in STE20/MAP4K family (Fig. 1H), which has been shown to be overexpressed in pancreatic cancer (31). When the half-life of HPK1 protein in 293T cells cotransfected with Fbxw8 was compared with that transfected with HPK1 alone, Fbxw8 accelerated the decay rate of HPK1 protein (Fig. 1I).
Fbxw8 Increases HPK1 Ubiquitination through Lys48
When Fbxw8 was cotransfected with HPK1 and HA-ubiquitin into 293T cells, Fbxw8 markedly increased HPK1 ubiquitination. In contrast, cotransfection of Fbxw7 did not increase HPK1 ubiquitination (Fig. 2A). Fbxw7 is the F-box component of a SCF ubiquitin ligase and acts as a tumor suppressor in many human cancers (32). Since ubiquitination through ubiquitin Lys48 (K48) generally targets proteins for degradation, whereas ubiquitination through K63 plays a critical role in signaling activation and protein trafficking (30), we tested whether HPK1 is ubiquitinated through K48 or K63. Fbxw8-mediated HPK1 ubiquitination was abolished in cells cotransfected with His-Ub K48R. Fbxw8-mediated HPK1 ubiquitination was also reduced in cells cotransfected with His-Ub K63R compared with those cotransfected with His-Ub (Fig. 2B). These data suggest that Fbxw8-mediated HPK1 ubiquitination occurred mainly through K48 but to a lesser extent through K63 of ubiquitin.
FIGURE 2.

Cotransfection of Fbxw8 results in the polyubiquitination of HPK1 protein mainly through K48 of ubiquitin. A, wild-type HPK1 and HA-Ub were cotransfected with either Myc-Fbxw8 or Flag-Fbxw7. Thirty-six hours after transfection, cells were harvested for immunoprecipitation with anti-HPK1 antibody followed by Western blotting as indicated. The inputs for the immunoprecipitation are shown at the bottom. B, Flag-HPK1 and Myc-Fbxw8 were cotransfected with wild-type His.Myc-Ub, His.Myc-Ub K48R, or His.Myc-Ub K63R into 293T cells. Nickel-nitrilotriacetic acid agarose beads were used to pull down the ubiquitinated HPK1 protein.
Fbxw8-mediated HPK1 Ubiquitination Is Dependent upon HPK1 Kinase Activity
Since kinase-dead HPK1-M46 was more stable than wild type HPK1 when cotransfected with Fbxw8, it is possible that HPK1 ubiquitination requires its kinase activity and autophosphorylation. Two serine (Ser-159 and Ser-171) and two threonine residues (Thr-165 and Thr-175) reside in the putative activation loop of HPK1. It has been shown that full activation of HPK1 is dependent on the autophosphorylation of Thr-165 and phosphorylation of Ser-171 (10). We investigated whether phosphorylation of these residues plays a role in Fbxw8-mediated HPK1 degradation by mutating them to alanine. Similar to HPK1-M46, S171A, or T175A mutation abolished HPK1 kinase activity. HPK1-T165A had decreased HPK1 kinase activity, whereas HPK1-S159A had similar kinase activity to wild-type HPK1 (Fig. 3A), suggesting that HPK1 kinase activity requires phosphorylation of Thr-165, Ser-171, and Thr-175, but not Ser-159. When Fbxw8 were cotransfected with HPK1 mutants into 293T cells, Fbxw8 resulted in a dose-dependent decrease in HPK1-S159A expression similar to wild-type HPK1 (Fig. 3B). Fbxw8 degraded HPK1-T165A to a lesser extent than it did to wild-type HPK1 and HPK1-S159A (Fig. 3C). Similar to HPK1-M46, HPK1-S171A and HPK1-T175A were resistant to Fbxw8-mediated degradation (Fig. 3, D and E). Compared with wild-type HPK1, HPK1-M46, HPK1-T165A, and HPK1-S171A had decreased ubiquitination when cotransfected with Fbxw8 (Fig. 3F). Thus Fbxw8-mediated HPK1 ubiquitination are dependent upon HPK1 kinase activity and autophosphorylation.
FIGURE 3.

CUL7/Fbxw8 ubiquitin ligase-mediated degradation and ubiquitination of HPK1 is dependent on HPK1 kinase activity and the autophosphorylation of HPK1. A, kinase activities and autophosphorylation of wild-type HPK1 and HPK1 mutant proteins were measured by immunocomplex kinase assays with the myelin basic protein (MBP) used as a substrate. B–E, overexpression of Fbxw8 results in a dose-dependent decrease in HPK1-S159A and HPK1-T165A protein expression but has no effect on the stability of HPK1 S171A or HPK1 T175A. F, loss of HPK1 kinase activity and autophosphorylation in HPK1-M46 and HPK1-T171A results in a marked decrease in Fbxw8-mediated HPK1 ubiquitination compared with wild-type HPK1. Wild-type or mutant HPK1 and HA-Ub were cotransfected with Myc-Fbxw8. The cells were treated with 2.0 μm MG132 for 6 h before harvesting. Immunoprecipitation with anti-HPK1 antibody followed by Western blot as indicated.
PP4 Inhibits Fbxw8-mediated HPK1 Ubiquitination
To test whether PP4 can inhibit Fbxw8-mediated HPK1 ubiquitination, 293T cells were cotransfected with HA-Ub, Flag-HPK1, Myc-Fbxw8, and Flag-PP4 or Flag-PP4-RL, a phosphatase-dead PP4 mutant and co-immunoprecipitations were performed using anti-HPK1. Cotransfection of PP4 decreased the binding of Fbxw8 to HPK1 and markedly reduced Fbxw8-mediated HPK1 ubiquitination. In contrast, PP4-RL had no significant effect on Fbxw8-mediated HPK1 ubiquitination and their interactions (Fig. 4). Therefore Fbxw8-mediated HPK1 degradation requires serine/threonine phosphorylation of HPK1 and that dephosphorylation of HPK1 by PP4 play a key role in Fbxw8-mediated HPK1 degradation.
FIGURE 4.

PP4, but not the phosphatase-dead mutant PP4-RL, inhibits Fbxw8-mediated HPK1 ubiquitination and the interaction between Fbxw8 and HPK1. HEK293T cells were transfected with Flag-HPK1, HA-ubiquitin, Myc-Fbxw8, Flag-PP4, or HA-PP4-RL as shown, and cell lysates were prepared 42 h after transfection. HPK1 was immunoprecipitated with an anti-HPK1 antibody. The immunoprecipitates were then immunoblotted with an anti-HA antibody. The immunoprecipitated HPK1, Fbxw8, PP4, and PP4-RL were monitored by immunoblotting using an anti-HPK1, anti-Myc, anti-Flag, or anti-HA antibody, respectively. The inputs for the immunoprecipitation were shown at the bottom.
To identify PP4 dephosphorylation sites, we focused on HPK1 proline-rich region (HPK1-PR; amino acids 288–482) since it has been shown that HPK1 interacts with PP4 through the HPK1-PR region (22). In addition, previous study also found that HPK1 autophosphorylation occurs predominantly on threonine residues (10). Using PhosphoSitePlus program, we identified two threonine residues, Thr-349 and Thr-355, in HPK1-PR region (Fig. 5A). We mutated them to alanine and found that both HPK1-T349A and HPK1-T355A retained kinase activity comparable to HPK1 (Fig. 5B). Similar to wild-type HPK1 and HPK1-S159A, HPK1-T349A was stabilized by MG132 (Fig. 5C-5E). In contrast, MG132 had little effect on the stability of HPK1-T355A (Fig. 5F). Thus we focused our study on HPK1-T355A. When HPK1-T355A was cotransfected with Fbxw8 into 293T cells, Fbxw8 failed to destabilize HPK1-T355A (Fig. 5G). To investigate whether Thr-355 was required for PP4-mediated dephosphorylation/stabilization of HPK1, we co-transfected HPK1 or HPK1-T355A with PP4 into HEK293T cells. PP4 stabilized wild-type HPK1 in a dose-dependent manner, but not HPK1-T355A (Fig. 5, H and I). Since there is no phospho-specific antibody for HPK1-Thr355 available, we used the proline-rich region of HPK1, HPK1-PR (aa: 288–482), which contains two putative threonine phosphorylation sites (Thr-349 and Thr-355) to examine the effect of PP4 on HPK1 phosphorylation. We found that inhibition of PP4 using okadaic acid (OA) increased the threonine phosphorylation of HPK1-PR (Fig. 6A), suggesting that Thr-355 of HPK1 is a key PP4 dephosphorylation site, through which Fbxw8 and PP4 regulates HPK1 stability. Consistent with this finding, HPK1-T355A mutant was resistant to Fbxw8-mediated ubiquitination (Fig. 6B).
FIGURE 5.

Threonine 355 of HPK1 is a key residue for PP4 dephosphorylation/stabilization of HPK1 and for CUL7/Fbxw8 ubiquitin ligase-mediated HPK1 degradation. A, schematic drawing showing the possible serine or threonine phosphorylation sites in HPK1 proline-rich region (amino acids 288–482) using PhosphoSitePlus program. B, kinase activities and autophosphorylation of wild-type HPK1, HPK1-T349A and HPK1-T355A were measured by immunocomplex kinase assays with the myelin basic protein (MBP) used as a substrate. C–F, proteasome inhibitor MG132 stabilizes wild-type HPK1, HPK1-S159A, and HPK1-T349A in a dose-dependent manner, but not HPK1-T355A. G, overexpression of Fbxw8 has little effect on the stability of HPK1-T355A. H and I, overexpression of PP4 results in a dose-dependent increase in wild-type HPK1 protein expression, but has little effect on the stability of HPK1-T355A.
FIGURE 6.

PP4 dephosphorylation site is important for CUL7/Fbxw8 ubiquitin ligase-mediated HPK1 ubiquitination. A, okadaic acid (OA) increases threonine phosphorylation of HPK1-PR.HEK293T cells were transfected with wildtype HPK1 and HA-HPK1-PR. Forty-two hours after transfection, the cells were either treated with 50 nm OA or untreated as a control for 4 h before harvest. HA-HPK1-PR were immunoprecipitated with an anti-HA antibody and then immunoblotted with anti-phosphothreonine or anti-HA antibody (top). The input of HPK1 and HA-HPK1-PR were monitored by immunoblotting (bottom). B, Fbxw8 does not increase ubiquitination of HPK1-T355A. HEK293T cells were transfected with HA-ubiquitin and either Flag-HPK1-T355 alone or with Myc-Fbxw8. Forty-two hours after transfection, cell lysates were prepared. HPK1-T355 was immunoprecipitated with an anti-M2 antibody and then immunoblotted with an anti-HA antibody as indicated.
Knockdown of Fbxw8 Restores Endogenous HPK1 Protein Expression and Inhibits Cell Proliferation in Pancreatic Cancer Cells
To examine function of Fbxw8 in HPK1 degradation and pancreatic cancer, we knocked down Fbxw8 using Fbxw8 shRNA, which resulted in dramatic reduction of the expression levels of Fbxw8 mRNA and protein in Panc-1 and Panc-28 cells compared with the control cells by quantitative RT-PCR and Western blots (Fig. 7, A–D). Knockdown of endogenous Fbxw8 effectively restored endogenous HPK1 expression in both Panc-1 and Panc-28 cells (Fig. 7, C and D). Thus we further confirmed the role of Fbxw8 in the proteasome-mediated degradation of HPK1 in pancreatic cancer cells. Fbxw8 knockdown led to increased p27 expression, but had no significant effect on the activation of JNK and Erk, which may be due to the presence of mutant Kras oncogene in these two cell lines (Fig. 7, C and D). Since we previously found that restoring wild-type HPK1 expression in pancreatic cancer cells causes cell-cycle arrest and growth inhibition (20), we examined the effect of Fbxw8 knockdown on cell proliferation. After transduction with Fbxw8 shRNA, the cell proliferation rates were decreased in both Panc-1 and Panc-28 cells compared with cells infected with control shRNA and parental cells, which is due in part to the stabilization of HPK1 (p < 0.05, Fig. 7, E and F).
FIGURE 7.
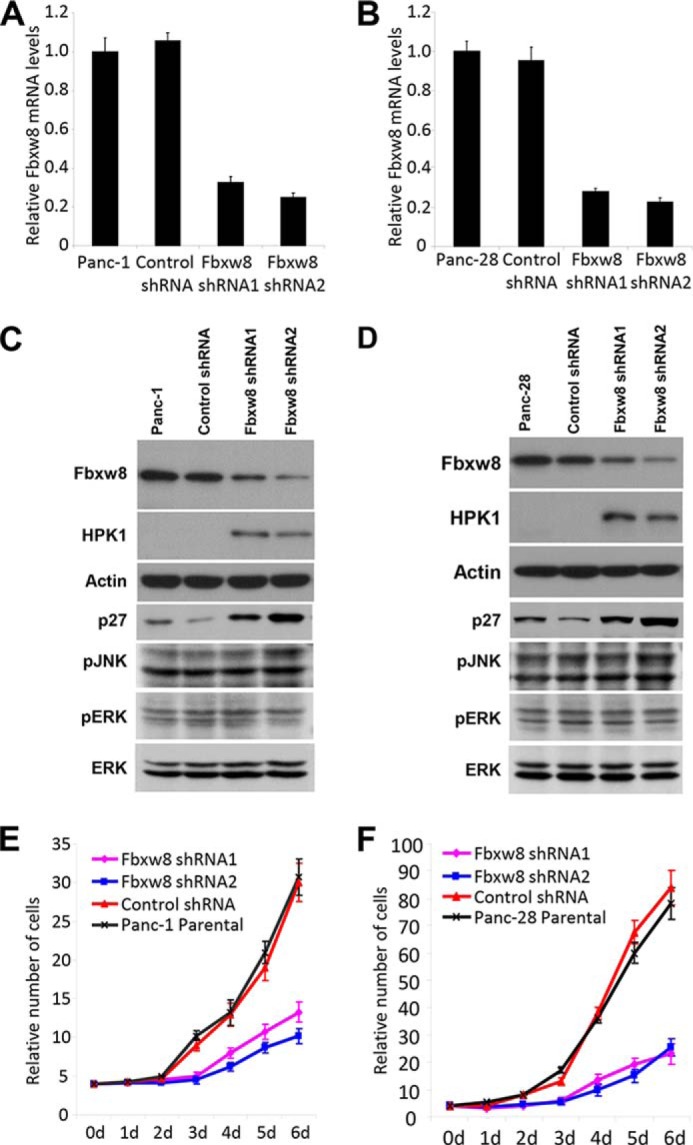
Knockdown of Fbxw8 restores endogenous HPK1 protein expression and inhibits cell proliferation in pancreatic cancer cells. A and B, Fbxw8 knockdown stable cell lines were generated from Panc-1 or Panc-28 cells and screened by QRT-PCR. C and D, knockdown of Fbxw8 restores endogenous HPK1 protein expression in Panc-1 and Panc-28 cells. E and F, Panc-1 or Panc-28 parental, shRNA control, and Fbxw8 knockdown stable cells were plated at an equal density and cultured under normal conditions for the indicated time periods. Cell numbers were counted and plotted. The growth curves have been repeated three times.
DISCUSSION
We previously showed that loss of HPK1 is associated with the progression of PanIN to invasive pancreatic cancer and that loss of HPK1 in pancreatic cancer is due to proteasome-mediated degradation (20). However, the ubiquitin ligase and the molecular mechanism that targets HPK1 for proteasomal degradation had not yet been identified. In this study, we demonstrated that HPK1 is novel proteolytic target of the CUL7/Fbxw8 ubiquitin ligase, which has been show to target cyclin D1 (33), IRS-1 and the Golgi protein Grasp65 for proteasomal degradation (28, 34). Since HPK1 functions as a MAP4K and activates MAP3Ks and their downstream MKK4/MKK7, JNK or SAPK cascade (1, 2), our findings of CUL7/Fbxw8 ubiquitin ligase-mediated HPK1 degradation revealed a direct link and novel role of CUL7/Fbxw8 ubiquitin ligase in MAPK pathway, which plays a critical role in cell proliferation and differentiation.
In this study, we found that HPK1 kinase activity and its autophosphorylation contribute to CUL7/Fbxw8 ubiquitin ligase-mediated HPK1 degradation. Single amino acid substitution of HPK1 Lys-46 to methionine (M46), which abolished HPK1 kinase activity and autophosphorylation, markedly reduced CUL7/Fbxw8 ubiquitin ligase-mediated HPK1 ubiquitination and stabilized HPK1 protein. To further confirm that HPK1 kinase activity and autophosphorylation are required for CUL7/Fbxw8 ubiquitin ligase-mediated HPK1 degradation, we demonstrated that S171A or T175A mutations of HPK1 led to a marked decrease in CUL7/Fbxw8 ubiquitin ligase-mediated ubiquitination of HPK1 and stabilized mutant HPK1 protein compared with wild-type HPK1 when cotransfected with Fbxw8.
PP4 has been shown to stabilize HPK1 through the inhibition of ubiquitination and proteasomal degradation of HPK1 protein (22). However, the mechanism of HPK1 up-regulation by PP4 is unclear. We found that expression of wild-type PP4, but not phosphatase-dead PP4-RL, inhibited the interaction between Fbxw8 and HPK1 and decreased Fbxw8-mediated HPK1 ubiquitination. These data not only suggest that PP4 stabilizes HPK1 by inhibiting CUL7/Fbxw8 ubiquitin ligase-mediated HPK1 degradation, but also suggest that PP4 dephosphorylates the key serine/threonine residues of HPK1 protein, which are required for the binding of Fbxw8 to HPK1 and for CUL7/Fbxw8 ubiquitin ligase-mediated HPK1 degradation. In the light of previous findings that HPK1 interacts with PP4 through HPK1-PR region (22) and that HPK1 autophosphorylation occurs predominantly on threonine residues by phosphoamino acid analysis (10), we identified Thr-355 as a key residue for PP4 dephosphorylation/stabilization of HPK1 protein and for CUL7/Fbxw8 ubiquitin ligase-mediated HPK1 degradation. However, our study did not exclude the possibility that other threonine or serine residues may also been involved in the regulation of HPK1 stability by PP4 and CUL7/Fbxw8 ubiquitin ligase.
On the basis of our findings, we propose that targeted degradation of HPK1 by the CUL7/Fbxw8 ubiquitin ligase constitutes a negative-feedback loop to restrain the activity of HPK1. This ubiquitin ligase recognizes HPK1 in the autophosphorylated forms generated by its own activity and mediates its polyubiquitination and proteasomal destruction. It is possible that HPK1 degradation signal may contain multiple degron motifs that are the targets of other serine/threonine kinases with suboptimal affinity for Fbxw8. The requirement of phosphorylation at multiple sites for the degradation of HPK1 may reflect a biological need to fine-tune MAPK signaling in accordance with the magnitude and duration of the downstream targets. Since lymphocyte receptor signaling can be coupled to SAPK/JNK and NFκB activation by HPK1, the physiological regulatory functions of Fbxw8 in these processes deserve future investigation.
The CUL7/Fbxw8 ubiquitin ligase targets IRS-1 for proteasomal degradation and has been shown to be a key player in growth regulation (28). However the function of the CUL7/Fbxw8 ubiquitin ligase in cancer is unclear. In this study, we found that knockdown Fbxw8 inhibited the proliferation of pancreatic cancer cells, which is in part due to the restoration of HPK1 protein expression. Knockdown Fbxw8 increased the expression of p27, but had no effect on Erk and JNK action in pancreatic cancer cells, which may be due to the presence of active mutant Kras in these cells. Our data suggest that the CUL7/Fbxw8 ubiquitin ligase may have an oncogenic function in pancreatic cancer. Consistent with our findings, Kim et al. showed that CUL7 inhibits Myc-induced apoptosis and promotes Myc-driven cell transformation (35). CUL7/Fbxw8 ubiquitin ligase-mediated degradation of cyclin D1 in the cytoplasm during S phase has been shown to play a critical role in the proliferation of a variety of cancer cells (33). DNA damage-induced upregulation of CUL7 attenuates p53 tumor suppressor functions (36). More recently, Fu et al. showed that the CUL7/Fbxw8 ubiquitin ligase is a key regulator in epithelial-mesenchymal transition in choriocarcinoma cells (37). Using comparative genomic analysis, Paradis et al. demonstrated that CUL7 play a role in liver carcinogenesis (38). The oncogenic function of the CUL7/Fbxw8 ubiquitin ligase in pancreatic cancer needs further investigations. In the light of the recent studies, which demonstrated the major tumor suppressor functions of deubiquitinase USP9X in pancreatic cancer and the recurrent mutations in the components of ubiquitin-dependent pathways in neoplastic pancreatic cysts by whole-exome sequencing (39, 40), our findings of CUL7/Fbxw8 ubiquitin ligase-mediated HPK1 degradation provide new insight into the key functions of proteasomal pathway in the development of pancreatic cancer. Targeting CUL7/Fbxw8 ubiquitin ligase to restore the expression and function of HPK1, in combination with other modalities, may be an effective approach for pancreatic cancer treatment.
Acknowledgments
We thank Dr. Xianzhou Song and Dr. Junli Guo for technical support. We thank Kim-Anh Vu for assistance with the digital images.
This work was supported by the National Institutes of Health though Grant 1R21CA149544-01A1 and by the G. S. Hogan Gastrointestinal Cancer Research Fund at The University of Texas MD Anderson Cancer Center.
- HPK1
- hematopoietic progenitor kinase 1
- CUL
- cullin
- CRL
- the cullin-RING ubiquitin ligase
- PP4
- protein phosphatase 4
- MBP
- myelin basic protein
- OA
- okadaic acid
- SCF
- Skp1-CUL1-F-box protein.
REFERENCES
- 1. Hu M. C.-T., Qiu W. R., Wang X., Meyer C. F., Tan T.-H. (1996) Human HPK1, a novel human hematopoietic progenitor kinase that activates the JNK/SAPK kinase cascade. Genes Dev. 10, 2251–2264 [DOI] [PubMed] [Google Scholar]
- 2. Kiefer F., Tibbles L. A., Anafi M., Janssen A., Zanke B. W., Lassam N., Pawson T., Woodgett J. R., Iscove N. N. (1996) HPK1, a hematopoietic protein kinase activating the SAPK/JNK pathway. EMBO J. 15, 7013–7025 [PMC free article] [PubMed] [Google Scholar]
- 3. Anafi M., Kiefer F., Gish G. D., Mbamalu G., Iscove N. N., Pawson T. (1997) SH2/SH3 adaptor proteins can link tyrosine kinases to a Ste20-related protein kinase, HPK1. J. Biol. Chem. 272, 27804–27811 [DOI] [PubMed] [Google Scholar]
- 4. Liu S. K., Smith C. A., Arnold R., Kiefer F., McGlade C. J. (2000) The adaptor protein Gads (Grb2-related adaptor downstream of Shc) is implicated in coupling hemopoietic progenitor kinase-1 to the activated TCR. J. Immunol. 165, 1417–1426 [DOI] [PubMed] [Google Scholar]
- 5. Ling P., Meyer C. F., Redmond L. P., Shui J.-W., Davis B., Rich R. R., Hu M. C.-T., Wange R. L., Tan T.-H. (2001) Involvement of hematopoietic progenitor kinase 1 (HPK1) in T-cell receptor (TCR) signaling. J. Biol. Chem. 276, 18908–18914 [DOI] [PubMed] [Google Scholar]
- 6. Ling P., Yao Z., Meyer C. F., Wang X. S., Oehrl W., Feller S. M., Tan T.-H. (1999) Interaction of hematopoietic progenitor kinase 1 with adapter proteins Crk and CrkL leads to synergistic activation of c-Jun N-terminal kinase. Mol. Cell. Biol. 19, 1359–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang W., Zhou G., Hu M. C.-T., Yao Z., Tan T.-H. (1997) Activation of the hematopoietic progenitor kinase-1 (HPK1)-dependent, stress-activated c-Jun N-terminal kinase (JNK) pathway by transforming growth factor b (TGF-b)-activated kinase (TAK1), a kinase mediator of TGF b signal transduction. J. Biol. Chem. 272, 22771–22775 [DOI] [PubMed] [Google Scholar]
- 8. Zhou G., Lee S. C., Yao Z., Tan T.-H. (1999) Hematopoietic progenitor kinase 1 is a component of transforming growth factor b-induced c-Jun N-terminal kinase signaling cascade. J. Biol. Chem. 274, 13133–13138 [DOI] [PubMed] [Google Scholar]
- 9. Nagata Y., Kiefer F., Watanabe T., Todokoro K. (1999) Activation of hematopoietic progenitor kinase-1 by erythropoietin. Blood 93, 3347–3354 [PubMed] [Google Scholar]
- 10. Arnold R., Patzak I. M., Neuhaus B., Vancauwenbergh S., Veillette A., Van Lint J., Kiefer F. (2005) Activation of hematopoietic progenitor kinase 1 involves relocation, autophosphorylation, and transphosphorylation by protein kinase D1. Mol. Cell. Biol. 25, 2364–2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sawasdikosol S., Pyarajan S., Alzabin S., Matejovic G., Burakoff S. J. (2007) Prostaglandin E2 activates HPK1 kinase activity via a PKA-dependent pathway. J. Biol. Chem. 282, 34693–34699 [DOI] [PubMed] [Google Scholar]
- 12. Liou J., Kiefer F., Dang A., Hashimoto A., Cobb M. H., Kurosaki T., Weiss A. (2000) HPK1 is activated by lymphocyte antigen receptors and negatively regulates AP-1. Immunity 12, 399–408 [DOI] [PubMed] [Google Scholar]
- 13. Tsuji S., Okamoto M., Yamada K., Okamoto N., Goitsuka R., Arnold R., Kiefer F., Kitamura D. (2001) B cell adaptor containing Src homology 2 domain (BASH) links B cell receptor signaling to the activation of hematopoietic progenitor kinase 1. J. Exp. Med. 194, 529–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yu J., Riou C., Davidson D., Minhas R., Robson J. D., Julius M., Arnold R., Kiefer F., Veillette A. (2001) Synergistic regulation of immunoreceptor signaling by SLP-76-related adaptor Clnk and serine/threonine protein kinase HPK-1. Mol. Cell. Biol. 21, 6102–6112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sauer K., Liou J., Singh S. B., Yablonski D., Weiss A., Perlmutter R. M. (2001) The kinase HPK1 associates physically and functionally with the adaptor proteins BLNK and SLP-76 in lymphocytes. J. Biol. Chem. 276, 45207–45216 [DOI] [PubMed] [Google Scholar]
- 16. Chen-Deutsch X., Studzinski G. P. (2012) Dual role of hematopoietic progenitor kinase 1 (HPK1) as a positive regulator of 1α,25-dihydroxyvitamin D-induced differentiation and cell cycle arrest of AML cells and as a mediator of vitamin D resistance. Cell Cycle 11, 1364–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shui J. W., Boomer J. S., Han J., Xu J., Dement G. A., Zhou G., Tan T. H. (2007) Hematopoietic progenitor kinase 1 negatively regulates T cell receptor signaling and T cell-mediated immune responses. Nat Immunol 8, 84–91 [DOI] [PubMed] [Google Scholar]
- 18. Arnold R., Liou J., Drexler H. C., Weiss A., Kiefer F. (2001) Caspase-mediated cleavage of hematopoietic progenitor kinase 1 (HPK1) converts an activator of NFkB into an inhibitor of NFkB. J. Biol. Chem. 276, 14675–14684 [DOI] [PubMed] [Google Scholar]
- 19. Oehrl W., Kardinal C., Ruf S., Adermann K., Groffen J., Feng G. S., Blenis J., Tan T.-H., Feller S. M. (1998) The germinal center kinase (GCK)-related protein kinases HPK1 and KHS are candidates for highly selective signal transducers of Crk family adapter proteins. Oncogene 17, 1893–1901 [DOI] [PubMed] [Google Scholar]
- 20. Wang H., Song X., Logsdon C., Zhou G., Evans D. B., Abbruzzese J. L., Hamilton S. R., Tan T. H. (2009) Proteasome-mediated degradation and functions of hematopoietic progenitor kinase 1 in pancreatic cancer. Cancer Res. 69, 1063–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen Y.-R., Meyer C. F., Ahmed B., Yao Z., Tan T.-H. (1999) Caspase-mediated cleavage and functional changes of hematopoeitic progenitor kinase 1 (HPK1). Oncogene 18, 7370–7377 [DOI] [PubMed] [Google Scholar]
- 22. Zhou G., Boomer J. S., Tan T. H. (2004) Protein phosphatase 4 is a positive regulator of hematopoietic progenitor kinase 1. J. Biol. Chem. 279, 49551–49561 [DOI] [PubMed] [Google Scholar]
- 23. Hershko A., Ciechanover A. (1998) The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 [DOI] [PubMed] [Google Scholar]
- 24. Deshaies R. J. (1999) SCF and Cullin/Ring H2-based ubiquitin ligases. Annu. Rev. Cell Dev. Biol. 15, 435–467 [DOI] [PubMed] [Google Scholar]
- 25. Petroski M. D., Deshaies R. J. (2005) Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 6, 9–20 [DOI] [PubMed] [Google Scholar]
- 26. Jin J., Cardozo T., Lovering R. C., Elledge S. J., Pagano M., Harper J. W. (2004) Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev. 18, 2573–2580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cardozo T., Pagano M. (2004) The SCF ubiquitin ligase: insights into a molecular machine. Nat. Rev. Mol. Cell Biol. 5, 739–751 [DOI] [PubMed] [Google Scholar]
- 28. Xu X., Sarikas A., Dias-Santagata D. C., Dolios G., Lafontant P. J., Tsai S. C., Zhu W., Nakajima H., Nakajima H. O., Field L. J., Wang R., Pan Z. Q. (2008) The CUL7 E3 ubiquitin ligase targets insulin receptor substrate 1 for ubiquitin-dependent degradation. Mol. Cell 30, 403–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yao Z., Zhou G., Wang X. S., Brown A., Diener K., Gan H., Tan T. H. (1999) A novel human STE20-related protein kinase, HGK, that specifically activates the c-Jun N-terminal kinase signaling pathway. J. Biol. Chem. 274, 2118–2125 [DOI] [PubMed] [Google Scholar]
- 30. Yang W. L., Wang J., Chan C. H., Lee S. W., Campos A. D., Lamothe B., Hur L., Grabiner B. C., Lin X., Darnay B. G., Lin H. K. (2009) The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science 325, 1134–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liang J. J., Wang H., Rashid A., Tan T. H., Hwang R. F., Hamilton S. R., Abbruzzese J. L., Evans D. B., Wang H. (2008) Expression of MAP4K4 is associated with worse prognosis in patients with stage II pancreatic ductal adenocarcinoma. Clin. Cancer Res. 14, 7043–7049 [DOI] [PubMed] [Google Scholar]
- 32. Mansour M. R., Sanda T., Lawton L. N., Li X., Kreslavsky T., Novina C. D., Brand M., Gutierrez A., Kelliher M. A., Jamieson C. H., von Boehmer H., Young R. A., Look A. T. (2013) The TAL1 complex targets the FBXW7 tumor suppressor by activating miR-223 in human T cell acute lymphoblastic leukemia. J. Exp. Med. 210, 1545–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Okabe H., Lee S. H., Phuchareon J., Albertson D. G., McCormick F., Tetsu O. (2006) A critical role for FBXW8 and MAPK in cyclin D1 degradation and cancer cell proliferation. PloS one 1, e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Litterman N., Ikeuchi Y., Gallardo G., O'Connell B. C., Sowa M. E., Gygi S. P., Harper J. W., Bonni A. (2011) An OBSL1-Cul7Fbxw8 ubiquitin ligase signaling mechanism regulates Golgi morphology and dendrite patterning. PLoS Biol. 9, e1001060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kim S. S., Shago M., Kaustov L., Boutros P. C., Clendening J. W., Sheng Y., Trentin G. A., Barsyte-Lovejoy D., Mao D. Y., Kay R., Jurisica I., Arrowsmith C. H., Penn L. Z. (2007) CUL7 is a novel antiapoptotic oncogene. Cancer Res. 67, 9616–9622 [DOI] [PubMed] [Google Scholar]
- 36. Jung P., Verdoodt B., Bailey A., Yates J. R., 3rd, Menssen A., Hermeking H. (2007) Induction of cullin 7 by DNA damage attenuates p53 function. Proc. Natl. Acad. Sci. U. S. A. 104, 11388–11393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang Z., Inuzuka H., Fukushima H., Wan L., Gao D., Shaik S., Sarkar F. H., Wei W. (2011) Emerging roles of the FBW7 tumour suppressor in stem cell differentiation. EMBO Rep. 13, 36–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Paradis V., Albuquerque M., Mebarki M., Hernandez L., Zalinski S., Quentin S., Belghiti J., Soulier J., Bedossa P. (2013) Cullin7: a new gene involved in liver carcinogenesis related to metabolic syndrome. Gut 62, 911–919 [DOI] [PubMed] [Google Scholar]
- 39. Pérez-Mancera P. A., Rust A. G., van der Weyden L., Kristiansen G., Li A., Sarver A. L., Silverstein K. A., Grützmann R., Aust D., Rümmele P., Knösel T., Herd C., Stemple D. L., Kettleborough R., Brosnan J. A., Morgan R., Knight S., Yu J., Stegeman S., Collier L. S., ten Hoeve J. J., de Ridder J., Klein A. P., Goggins M., Hruban R. H., Chang D. K., Biankin A. V., Grimmond S. M., Wessels L. F., Wood S. A., Iacobuzio-Donahue C. A., Pilarsky C., Largaespada D. A., Adams D. J., Tuveson D. A. (2012) The deubiquitinase USP9X suppresses pancreatic ductal adenocarcinoma. Nature 486, 266–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wu J., Jiao Y., Dal Molin M., Maitra A., de Wilde R. F., Wood L. D., Eshleman J. R., Goggins M. G., Wolfgang C. L., Canto M. I., Schulick R. D., Edil B. H., Choti M. A., Adsay V., Klimstra D. S., Offerhaus G. J., Klein A. P., Kopelovich L., Carter H., Karchin R., Allen P. J., Schmidt C. M., Naito Y., Diaz L. A., Jr., Kinzler K. W., Papadopoulos N., Hruban R. H., Vogelstein B. (2011) Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc. Natl. Acad. Sci. U. S. A. 108, 21188–21193 [DOI] [PMC free article] [PubMed] [Google Scholar]