

Background: Clusterin is a highly conserved glycoprotein with broad tissue distribution but enigmatic biological functions.
Results: Clusterin signals via the lipoprotein receptors ApoER2 and VLDLR and stimulates cell proliferation in subventricular zone explants enabling neuronal outgrowth.
Conclusion: Clusterin-mediated signaling is essential for neuronal chain formation in vitro.
Significance: Discovery of a novel function of clusterin in neurogenesis.
Keywords: Apolipoproteins, Cell Proliferation, Cell Signaling, Lipoprotein Receptor, Neurogenesis, apoE Receptor 2, Clusterin, Reelin, Very Low Density Lipoprotein Receptor
Abstract
Clusterin, also known as apolipoprotein J, is a multifunctional glycoprotein with the capacity to interact with a wide range of molecules. Although clusterin has been implicated in a broad spectrum of physiological and pathological processes, such as Alzheimer disease or cancer, its precise functions remain elusive. Here we report, that clusterin binds to apolipoprotein E receptor 2 (ApoER2) and very low density lipoprotein receptor (VLDLR) and is internalized by cells expressing either one of these receptors. Binding of clusterin to these receptors triggers a Reelin-like signal in cells expressing disabled-1 (Dab1). It induces phosphorylation of Dab1, which leads to activation of PI3K/Akt and n-cofilin. Cell proliferation and neuroblast chain formation in subventricular zone (SVZ) explants are compromised when clusterin, which is present in the subventricular zone, is blocked in vitro. These data suggest that in the subventricular zone where Reelin is not present but ApoER2, VLDLR, and Dab1, clusterin might be involved in maintaining neurogenesis in vivo.
Introduction
Correct positioning of neurons in laminated structures of the central nervous system (CNS)2 such as the cortex, the cerebellum, the hippocampus, and the olfactory bulb (OB) depends on Reelin, an extracellular matrix protein (1), on ApoER2 and VLDLR, both cell surface receptors present on migrating neuroblasts (2), and on the intracellular adaptor protein Dab1 (3). These proteins cooperate in a linear signal cascade that results in the phosphorylation of Dab1 leading to the ultimate cell responses required for the correct positioning of newly generated neurons (4). Receptor-mediated clustering of Dab1 (5) activates Src-family tyrosine kinases that phosphorylate Dab1 (6, 7). Phosphorylated Dab1 activates phosphatidylinositide 3-kinase (PI3K) and subsequently PKB/Akt, which in turn inhibits the activity of GSK3β (8). Other downstream events are still poorly understood (9), but involve cullin-5 to regulate the degradation of Dab1 (10) and activation of cofilin which links the Reelin-signaling cascade to the dynamics of actin filaments (11). In addition, Reelin signals via the small GTPase Rap1 to effect the localization of N-cadherin which polarizes migrating neuroblasts toward the radial morphology zone of the cerebral cortex (12). The function of this complex signaling network of Reelin via two receptors in the lamination of the cortex has been recently summarized in the “detach and go” (13) and the “polarity model” model (14). Besides Reelin, thrombospondin-1, which is present in certain areas of the brain, is another ligand for ApoER2 and VLDLR and signals via these receptors (15).
ApoER2 and VLDLR are both members of the LDL receptor family and share modules and protein motifs present in the LDL receptor (16). ApoER2 is a multifaceted receptor. Because of extensive differential splicing a variety of tissue- and species-specific transcripts varying in the number of ligand-binding repeats and the presence of unique modules in the extracellular as well as in the intracellular domain exist (17, 18). VLDLR differs from the LDL receptor by the presence of an eighth ligand binding repeat and in sharp contrast to the LDL receptor, which binds apo B100 and apo E, VLDLR binds many different unrelated ligands (19). Depending on the physiological context, VLDLR functions either in receptor-mediated endocytosis of macromolecules or as signaling receptor as described above. The best understood system where VLDLR acts as transport receptor is the chicken oocyte (20). Functional mutations in the vldlr gene compromise oocyte growth and lead to female sterility in chicken (21). Expressed in the growing oocyte, VLDLR binds and internalizes yolk components produced by the liver such as vitellogenin and VLDL (22), α2-macroglobulin (23), vitamin-binding proteins (24), and clusterin (25) into the growing oocytes.
Clusterin is a heterodimeric glycoprotein of 75–80 kDa which is highly conserved throughout species exhibiting 70–80% identity at the amino acid level between mammals and 45% identity with chicken clusterin (25). In mammals clusterin is proteolytically processed into a α- and a β-chain after cleavage of the signal peptide. These two chains become linked in an antiparallel fashion by 5 disulfide bonds resulting in a heterodimeric protein that contains three amphipathic helices and two domains characterized by a coiled-coil α-helical structure (for review on structural features of clusterin, see Refs. 26). In addition to the secreted soluble form of clusterin, an alternative transcript starting from an ATG present in exon 3 produces an insoluble variant of clusterin (27), which appears to be targeted to the nucleus (28).
Despite its discovery almost 30 years ago the biological functions of clusterin are still enigmatic. Clusterin has been implicated in many physiological and pathological processes such as cancer development, sperm maturation, apoptosis, cell proliferation, complement regulation, lipid transport, and many more (for reviews see Refs. 29–31). In the CNS it is presumed to play an anti-apoptotic role and to promote cell survival and neuroplasticity after ischemic damage (for review see Ref. 32). With respect to neurodegenerative disorders clusterin may play a role in Alzheimer disease promoting sequestering and clearance of amyloid-β (33).
Here we demonstrate that the secreted form of clusterin is expressed in the same brain structures as ApoER2 and VLDLR and signals via these receptors to induce a Reelin-like signaling cascade. Clusterin signaling via ApoER2 or VLDLR has a cell proliferative effect on migrating neuronal precursors of SVZ explants in vitro and therefore may play a role in neurogenesis in the SVZ in mice.
EXPERIMENTAL PROCEDURES
Animals
Wild-type (WT) mice on a C57BL6/J background were housed under standard conditions.
Reagents and Antibodies
Native clusterin purified from human plasma (BioVendor) was used. A mouse monoclonal anti-clusterin antibody (clone 41D; IgG1) was a kind gift from Mark Wilson, University of Wollongong, Australia. The mouse monoclonal anti-triMethyl-Histone H4 antibody (triMe-Lys20; clone 6F8-D9; IgG1) was a kind gift from Stefan Schüchner, Max F. Perutz Laboratories, Vienna. The mouse monoclonal anti-Dab1 antibody (D4) was a kind gift from André Goffinet, University of Louvain, Belgium. A polyclonal anti-Dab1 antibody (Ab54) was raised in rabbits against a glutathione S-transferase fusion protein containing the first 180 amino acids of the short splice variant of murine Dab1 (5). The following antibodies were purchased from the indicated sources: rabbit anti-clusterin (H-330), mouse anti-phosphotyrosine (PY99), goat anti-EEA1 (N-19), and rabbit anti-phospho-cofilin 1 (p-Cofilin 1 (mSer3)-R) Santa Cruz Biotechnology, Inc.; rabbit anti-Akt (9272) and rabbit anti-phospho-Akt (Ser-473; 9271), Cell Signaling Technology; mouse anti-GAPDH (GAPDH-71.1), Sigma-Aldrich.
Preparation of myc-RAP
Myc-RAP was prepared as described in Ref. 34. Briefly, BL21 competent Escherichia coli (Invitrogen) were transformed with a pET-15b (Novagen) vector carrying the Rap-myc/His sequence. Bacteria were grown until they reached mid-log phase and 0.5 mm isopropyl-1-thio-β-galactopyranoside was added to induce expression for 2 h at 30 °C. The cells were harvested, resuspended in TBS-C (TBS pH 7.4, 2 mm CaCl2) with protease inhibitor mix (Complete, Roche Applied Science) and lysed by sonication. After centrifugation, the supernatant was incubated with 2 ml of Ni-NTA Sepharose (Qiagen) rotating at 4 °C overnight. After washing twice with TBS-C, the protein was eluted with 250 mm imidazole in TBS-C.
Solid Phase Binding Assay
The solid-phase binding assay was essentially performed as described in (15). A 96-well plate (MaxiSorp, Nunc) was coated with 100 μl of TBS-C containing 10 μg/ml ApoER2 1–5-MBP/His or VLDLR 1–8-MBP/His overnight at 4 °C. All further incubation steps were carried out at room temperature for 1 h. Ligands and antibodies were incubated in blocking solution (2% BSA in TBS-C, 0.05% Tween). After blocking and binding of clusterin, mouse anti-clusterin antibody (41D) followed by a corresponding HRP-conjugated secondary antibody was used for detection of bound clusterin. For the color reaction, 0.1 mg/ml 3,3′,5,5′-tetramethylbenzidine (TMB) in 0.1 m sodium acetate, pH 6.0 containing 10 mm H2O2 was used. The reaction was stopped after 2 min by addition of 0.3 m H2SO4. The resulting yellow product was photometrically quantified at 450 nm with a multi-label plate reader (Wallac Victor2, Perkin Elmer). For the competition assay, 96-well plates were coated with 10 μg/ml ApoER2 1–5-MBP/His or VLDLR 1–8-MBP/His as described above. Plates were incubated with 25 nm clusterin or BSA in the presence of increasing amounts of either RCM or myc-RAP. Bound clusterin was detected by addition of mouse anti-clusterin antibody (41D) followed by a corresponding HRP-conjugated secondary antibody.
Cell Lines and Preparation of Conditioned Media
NIH 3T3, NIH 3T3 expressing murine Dab1 (Dab1 3T3) and 293 HEK cells were cultivated in Dulbecco's modified Eagle's medium (DMEM; PAA) supplemented with 10% fetal calf serum (Invitrogen), and penicillin/streptomycin (Invitrogen) at 37 °C and 7.5% CO2. Stable NIH 3T3-based cell lines expressing murine ApoER2 harboring LA repeats 1–3, 7, and 8 and containing the proline-rich cytoplasmic insert (ApoER2 3T3), murine VLDLR lacking the O-linked sugar domain (VLDLR 3T3), or either receptor and murine Dab1 (ApoER2/Dab1 3T3 and VLDLR/Dab1 3T3) (35) were kept under puromycin selection (0.75 μg/ml). Reelin-expressing 293 HEK cells were cultivated and used for production of Reelin-conditioned medium (RCM) as described before (17). Briefly, 293 HEK cells stably carrying the full-length mouse Reelin expression construct pCrl (a kind gift of Tom Curran, Perelman School of Medicine at the University of Pennsylvania, Philadelphia) were cultivated in DMEM supplemented with 10% fetal calf serum (Invitrogen), penicillin/streptomycin (Invitrogen) and 0.2 mg/ml G418 at 37 °C and 7.5% CO2. When the cells reached 70% confluency the culture medium was replaced by serum-free medium (OptiMEM). After two more days the conditioned medium was collected, sterile filtered and stored at −80 °C until use. Mock-conditioned medium (MCM) was prepared from untransfected 293 HEK cells using the same procedure. Primary rat neuronal cultures were prepared from embryonic day 16.5 rat brains and were kept in neuronal base medium (PAA) containing B27 supplement (Invitrogen) and penicillin/streptomycin at 37 °C and 5% CO2 for 72 h before use as described (5).
Clusterin Binding and Uptake Assays
For the binding assay clusterin was fluorescently labeled with the DyLightTM 488 microscale antibody labeling kit according to supplier's instructions (Thermo Scientific). ApoER2 3T3, VLDLR 3T3, and 3T3 cells were grown in 8-well culture slides (BD Falcon) for 24h. Cells were precooled on ice for 30 min, washed with ice-cold PBS and incubated with DyLightTM 488 labeled clusterin in OptiMEM on ice for 1h to allow clusterin binding to the cells. After extensive washing with PBS, cells were fixed with 2% formaldehyde solution. Fixed cells were washed again with PBS and nuclei were counterstained with 5 μg/ml DAPI in PBS for 1 h. Cells were washed again with PBS and embedded in Dako fluorescent mounting medium (Dako). For the uptake assay clusterin was fluorescently labeled with the DyLightTM 594 microscale antibody labeling kit according to supplier's instructions (Thermo Scientific). ApoER2 3T3, VLDLR 3T3, and 3T3 cells were precooled on ice for 30 min, washed with ice-cold PBS and incubated with DyLight 594TM-labeled clusterin in OptiMEM on ice for 1 h. Cells were shifted to 37 °C for 20 min, washed with PBS and fixed with 2% formaldehyde solution. After another PBS wash the cells were permeabilized with 0.2% Triton X-100 in PBS for 5 min and blocked in 1% BSA in PBS. Goat anti-EEA1 antibody (N-19; 1:500 in blocking solution) was used to detect early endosome antigen 1. Cells were washed with PBS and incubated with Alexa Fluor® 488 donkey anti-goat IgG (Invitrogen). Nuclei were counterstained with 5 μg/ml DAPI in PBS, and cells were embedded in fluorescent mounting medium (Dako).
Preparation of Cell Extracts, SDS-PAGE, and Western Blotting
Cells were washed twice with ice-cold PBS and lysed in RIPA buffer (Cell Signaling Technology) supplemented with protease inhibitor mix and phosphatase inhibitors (50 mm NaF and 2 mm Na3VO4). Cell lysates were kept on ice for 15 min before and after sonication for 5 × 1s at 70% power (Bandelin Sonopuls HD 70). Cell debris was removed by centrifugation for 15 min at 21460 × g. Proteins were separated by reducing SDS-PAGE and transferred onto nitrocellulose membranes by wet blotting. Membranes were blocked in PBS containing 0.1% Tween-20 and 5% bovine serum albumin (PAA) and incubated with primary and HRP-conjugated secondary antibodies. For detection, enhanced chemiluminescence solution (Pierce) was used.
Dab1 Phosphorylation and Degradation Assay
Primary rat neurons, ApoER2/Dab1 3T3, VLDLR/Dab1 3T3, or Dab1 3T3 cells were starved for 5h in OptiMEM (Invitrogen) and incubated for 20 min with MCM, RCM, OptiMEM, or OptiMEM plus 2.5 nm clusterin. Cell extracts were prepared in RIPA buffer containing protease inhibitor mix and phosphatase inhibitors (50 mm NaF and 2 mm Na3VO4) and used for immunoprecipitation. Extracts were incubated with rabbit anti-Dab1 antiserum (Ab54) overnight at 4 °C, 40 μl of a protein A-Sepharose 4B conjugate (Invitrogen) was added, and samples were further incubated for 1h at 4 °C. Beads were collected by centrifugation at 500 × g for 1 min and washed three times using Hunt buffer (20 mm Tris pH 8.0, 150 mm NaCl, 0.5% Nonidet P-40). Samples were analyzed by Western blotting. A mouse anti-Dab1 antibody (D4) was used to detect total Dab1 levels. A mouse anti-phosphotyrosine antibody (PY99) was used to detect corresponding tyrosine-phosphorylated Dab1. To analyze degradation of Dab1, primary rat neurons were washed with PBS and incubated with MCM, RCM, OptiMEM or OptiMEM plus clusterin (1.25 or 6.25 nm) for 5 h. Cell extracts were prepared as described above and analyzed by Western blotting. As a loading control mouse anti-GAPDH antibody (GAPDH-71.1) was used.
Akt Phosphorylation Assay
Primary rat neurons were starved for 5 h in OptiMEM and incubated for 15 min with MCM, RCM, OptiMEM, or OptiMEM plus 2.5 nm clusterin. Cell extracts were prepared as described above and analyzed by Western blotting. A rabbit anti-Akt antibody (9272) was used to detect total Akt levels. A rabbit anti-phospho-Akt antibody (Ser-473; 9271) was used to detect Akt phosphorylated at serine 473.
Cofilin Phosphorylation Assay
Embryonic day 16.5 rat brains were dissociated and directly stimulated for 15min with MCM, RCM, OptiMEM, or OptiMEM plus 2.5 nm clusterin. Cell extracts were prepared as described above and analyzed by Western blotting. A rabbit anti-phospho-cofilin 1 (p-Cofilin 1 (mSer3)-R) was used to detect cofilin phosphorylated at serine 3.
Histology and Immunohistochemistry
Postnatal day 17 (P17) WT mice were anesthetized with 10 mg/kg xylazine and 75 mg/kg ketamine in 0.9% NaCl and immediately perfused with 4% paraformaldehyde in PBS. Brains were dehydrated and embedded in paraffin according to standard protocols. Serial sagittal sections (5 μm) were obtained. After dehydration the paraffin sections were boiled with citrate buffer (10 mm sodium citrate, 0.05% Tween-20, pH 6.0) for 20 min for antigen retrieval. Endogenous peroxidase activity was blocked by incubation with 3% H2O2 for 10 min in the dark. Slides were blocked with 3% heat inactivated goat serum (PAA) in PBS and incubated with rabbit anti-clusterin antibody (H-330) in blocking solution to detect endogenous clusterin. As a negative control blocking solution without antibody was used. Sections were incubated with a goat-anti rabbit biotinylated antibody (Dako), and the primary antibody was visualized using the VECTASTAIN Elite ABC Kit and the Peroxidase Substrate Kit DAB (both from Vector Laboratories).
SVZ Explants
Brains from 4–5 days old WT mice were placed in ice-cold neuronal base medium (PAA). 500-μm coronal brain slices were obtained using a young mouse brain slicer matrix (Zivic Instruments). The SVZ was dissected from the lateral wall of the anterior horn of the lateral ventricle and cut into pieces of around 300 μm in diameter. The SVZ explants were mixed with Matrigel (BD Biosciences) and allowed to polymerize in a μ-Slide 8-well chamber (Ibidi) at 37 °C for 30 min. After polymerization, 300 μl of serum-free neuronal base medium supplemented with B-27 (Invitrogen), GlutaMAX (Invitrogen) and penicillin/streptomycin (Invitrogen) containing either clusterin (2.5 nm) mouse anti-clusterin antibody (41D; 5 μg) or mouse monoclonal anti-triMethyl-Histone H4 antibody (triMe-Lys20; 5 μg) was added. Cultures were maintained in a humidified 5% CO2, 37 °C Incubator. After 24 and 72 h, the explants were monitored using a JuLi Smart Fluorescence Cell Imager (PAA).
EdU Incorporation Assay for Imaging
SVZ explants of 6 days old WT mice were prepared and cultivated as described above. For the analysis of cell proliferation of explants cultivated for 48 h 50 μm 5-ethynyl-2′-deoxyuridine (EdU) was added for 20 h. EdU-labeled cells were stained using Click-iT® EdU Alexa Fluor 488 Imaging Kit (Invitrogen) according to supplier's instructions. Briefly, explants were washed in PBS and fixed with 4% paraformaldehyde in PBS for 30 min. After two times washing with 3% BSA/PBS cells were permeabilized in 0.5% Triton X-100 in PBS for 20 min. Cells were washed again, and the Click-iT reaction mixture was added for 30 min in the dark. The explants were washed again and embedded in fluorescence mounting medium (ibidi).
Microscopy
Confocal images were acquired using the LSM 510 Meta system (Zeiss) and ZEN software. DIC and phase contrast images were acquired using an Axiovert 135 system and AxioVision software (Zeiss).
EdU Incorporation Assay for Flow Cytometry
SVZ explants of 6 days old WT mice were prepared and cultivated as described above. One group of explants was treated with anti-clusterin mouse monoclonal antibody (41D; 5 μg) for 48 h. The other group was left untreated for 48 h. For the analysis of cell proliferation explants were incubated with 50 μm 5-ethynyl-2′-deoxyuridine (EdU) for 19 h before harvest. Thereafter Matrigel was dispase digested for 45 min (15 units, 37 °C) to obtain single cells. EdU-labeled cells were stained using Click-iT® EdU Alexa Fluor 594 Imaging Kit (Invitrogen) according to supplier's instructions. Cells were analyzed by flow cytometry (FACSAria, BD Biosciences).
Caspase-3 Intracellular Activity Assay (PhiPhiLux® G1D2)
SVZ explants of 4-day-old WT mice were prepared and cultivated Matrigel as described above. One group of explants was treated with anti-clusterin mouse monoclonal antibody (41D; 5 μg) for 72 h. The other group was left untreated for 72 h. Thereafter Matrigel was dispase digested for 45 min (15 units, 37 °C) to obtain single cells. The percentage of apoptotic cells in each group of cells was determined by application of a caspase-3 intracellular activity assay kit (PhiPhiLux® G1D2, Calbiochem). Briefly, cells were pelleted and resuspended in 10 μm peptide substrate in RPMI 1640 medium containing 25 mm HEPES. The suspension was mixed and incubated in a 5% CO2 incubator at 37 °C for 60 min. 1 ml of flow cytometry dilution buffer was added, and the cells were analyzed via flow cytometry (FACSCalibur, BD Biosciences).
RESULTS
Evidence that clusterin might be a general ligand for VLDLR and ApoER2 was corroborated by demonstrating that chicken clusterin binds to VLDLR expressed in chicken oocytes (25) and that clusterin mediates binding and uptake of leptin by both receptors in mice (36). Since chicken clusterin is not cleaved into a disulfide-linked heterodimer, like the mammalian protein, we evaluated the binding of native human clusterin to VLDLR and ApoER2 by quantitative ELISA. As demonstrated in Fig. 1, A and D, clusterin binds with high affinity to both receptors. The Kd values (9 nm for ApoER2 and 16 nm for VLDLR) are in the same range as those determined for thrombospondin-1 and Reelin (15). Binding to both receptors is inhibited by an excess of Reelin (Fig. 1, B and E) and receptor associated protein (RAP) (Fig. 1, C and F), an intracellular chaperon, which prevents premature interaction of these receptors with their cognate ligands in the secretory pathway (37). Therefore clusterin binds to the same binding site as the other known ligands.
FIGURE 1.

Clusterin binds to ApoER2 and VLDLR. A–C, 96-well plates were coated with recombinant ligand-binding domains of ApoER2 (ApoER2 1–5-MBP/His) and D–F, VLDLR (VLDLR 1–8-MBP/His) and incubated with the indicated amounts of clusterin (A, D) or with 25 nm clusterin in the presence of increasing amounts of Reelin-conditioned medium (RCM) (B, E) or Myc-tagged RAP (myc-RAP) (C, F). Bound clusterin was detected with a mouse monoclonal anti-clusterin antibody and an appropriate HRP-conjugated secondary antibody. Absorbance at 450 nm was measured. Kd, dissociation constant. Experiments were repeated three times with similar results. Error bars represent S.E. derived from duplicate (A, D) and triplicate determinations (B, C, E, F).
To confirm these binding data at the cellular level we incubated mouse 3T3 fibroblasts stably expressing ApoER2 (ApoER2 3T3) or VLDLR (VLDLR 3T3) (35) at 4 °C with green fluorescent-labeled clusterin (DyLight 488). As demonstrated in Fig. 2, cells expressing ApoER2 (A) or VLDLR (B) bind significant amounts of labeled clusterin nicely decorating the entire cell membrane. Because of the experimental conditions (4 °C), the bound ligand remained on the cell surface. This effect is receptor specific, since control experiments with cells not expressing either of the receptors did not result in clusterin binding to the cell membrane (Fig. 2C). To test whether clusterin is internalized by ApoER2 and VLDLR we used red fluorescent-labeled clusterin (DyLight 594) and after binding at 4 °C the cells were shifted to 37 °C for 20 min. The cells were fixed and the endosomal compartment was stained using an antibody against the endosomal marker EEA1 (green signal). Under these conditions, clusterin co-localizes with EEA1 demonstrating that ApoER2 (Fig. 2, D–F) and VLDLR (Fig. 2, G–I) internalize clusterin via the endosomal compartment. Again, in control cells not expressing the receptors no clusterin signal was detected within the cells (Fig. 2, J–L). Taken together, these results demonstrate that clusterin binds to both receptors in a similar way as Reelin does and becomes internalized via both receptors.
FIGURE 2.

ApoER2 and VLDLR internalize clusterin via the early endosomal compartment. A, 3T3 cells expressing ApoER2 (ApoER2 3T3), (B) VLDLR (VLDLR 3T3), or (C) no receptor (3T3) were incubated with DyLightTM 488 labeled clusterin (green) at 4 °C to allow binding of clusterin to the receptors. ApoER2 3T3 (D–F), VLDLR 3T3 (G–I), or 3T3 (J–L) were incubated with DyLightTM 594-labeled clusterin (red) at 4 °C. After 1 h, the clusterin-containing medium was replaced with normal medium, and the cells were shifted to 37 °C to allow uptake of the ligand. Immunostaining with a goat anti-EEA1 antibody to visualize the early endosomal compartment is shown in green (E, H, K). Clusterin (red) colocalizes with the early endosome (green) in ApoER2 3T3 (F) and VLDLR 3T3 (I) but not in 3T3 cells (L). Experiments were repeated three times (A–C) or four times (D–L) with similar results. More than 30 cells were analyzed per condition. Scale bars represent 10 μm.
A prerequisite to generate a signal via ApoER2 and VLDLR is a ligand that, like Reelin, is able to cluster the receptors (5). Reelin achieves this task by forming homodimers, which are able to bind at least two receptors (38). Secreted clusterin itself is a heterodimer formed by a α- and a β-chain generated by proteolytic cleavage of a common precursor. At physiological pH, clusterin exists as a mixture of dimers and tetramers (39) and thus might be able to trigger the Reelin signaling cascade via clustering of both receptors. To test whether clusterin is indeed able to signal via the Reelin pathway we used the fibroblast cell system again. These cells express Dab1 in combination with either VLDLR (VLDLR/Dab1 3T3) or ApoER2 (ApoER2/Dab1 3T3) (35). We treated both cell lines with clusterin and Reelin-conditioned medium (RCM) as a control. As shown in Fig. 3A cells expressing ApoER2 and Dab1 respond to clusterin with a robust phosphorylation of Dab1 similar to that induced by Reelin. The reaction is dose-dependent reaching a maximum at around 12.5 nm. At higher concentrations of clusterin (25 nm) the signal is significantly reduced suggesting that clusterin-receptor complexes become dissolved at higher concentrations of the ligand (data not shown). Interestingly, the clusterin-effect is less pronounced in cells expressing VLDLR and Dab1 (Fig. 3B). In this case phosphorylation levels are only slightly increased which was not statistically significant. In control experiments using a fibroblast cell line expressing Dab1 only (Dab1 3T3) Dab1 phosphorylation levels were not increased by the ligands (data not shown). To confirm these results obtained with the fibroblast cell model we performed the corresponding experiment with primary rat neurons (Fig. 3C). Again, clusterin significantly induces Dab1 phosphorylation like Reelin does it. A negative feedback mechanism in the Reelin signaling pathway is the degradation of the critical intracellular component of the pathway, Dab1. Dab1 degradation depends on Dab1 phosphorylation and the E3 ubiquitin ligase cullin 5 (10). As shown in Fig. 3D, addition of RCM to primary rat neurons leads to a significant loss of total Dab1 after 5 h. Addition of clusterin (6.25 nm) also leads to a dramatic reduction in total Dab1 levels.
FIGURE 3.

Clusterin induces Dab1 phosphorylation and degradation. A, 3T3 cells expressing ApoER2 and Dab1 (ApoER2/Dab1 3T3), (B) VLDLR and Dab1 (VLDLR/Dab1 3T3), and (C) primary rat E16.5 WT neurons were incubated with mock-conditioned medium (MCM), Reelin-conditioned medium (RCM), OptiMEM or clusterin (A and B: 2.5 nm and 12.5 nm clusterin; C: 2.5 nm clusterin) Cells were processed for immunoprecipitation of Dab1. Anti-Dab1 antibody was used to detect total Dab1 levels, and anti-phosphotyrosine (pTyr) antibody was used to detect tyrosine-phosphorylated Dab1 (pDab1). Western blots from three independent experiments per cell line were quantified by densitometry using ImageJ 1.48 and normalized with total Dab1 levels (n = 3; plots show mean ± S.E.; n.s. not significant; *, p < 0.05 versus control; ***, p < 0.001 versus control; unpaired Student's t test was performed in GraphPad Prism 6). D, primary rat E16.5 WT neurons were incubated with MCM (lane 1), RCM (lane 2), OptiMEM (lane 3), 1.25 nm clusterin (lane 4), or 6.25 nm clusterin (lane 5) for 5 h. Total cell extracts were prepared, and Western blotting was performed using anti-Dab1 and anti-GAPDH antibodies. Experiments were repeated three times with similar results.
Phosphorylated Dab1 acts as a signaling platform binding a variety of proteins involved in further down-stream signaling events (40). A central knot in this Reelin signaling network is the activation of PI3K (8), which leads to the phosphorylation of PKB/Akt (41) and activation of cofilin via LIMK1 (11). As demonstrated in Fig. 4, clusterin activates this axis in the same way as Reelin does resulting in a robust activation of PKB/Akt (A) and cofilin (B) in primary neurons.
FIGURE 4.
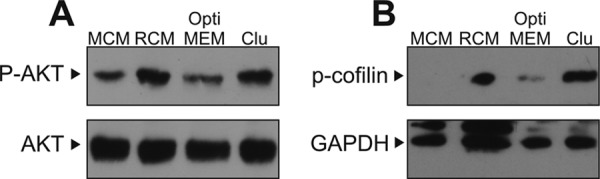
Clusterin activates PI3K/Akt and cofilin. A, primary rat E16.5 WT neurons were incubated with mock-conditioned medium (MCM, lane 1), Reelin-conditioned medium (RCM, lane 2), OptiMEM (lane 3), or 2.5 nm clusterin (lane 4) for 15 min. Total cell extracts were prepared, and Western blotting was performed using anti-Akt and anti-phospho-Akt(Ser473) antibodies. B, E16.5 rat brains were dissociated and directly stimulated with MCM (lane 1), RCM (lane 2), OptiMEM (lane 3), or 2.5 nm clusterin (lane 4) for 15 min. A rabbit anti-phospho-cofilin 1 (p-Cofilin 1 (mSer3)-R) antibody was used to detect cofilin phosphorylated at serine 3. A mouse anti-GAPDH antibody (GAPDH-71.1) was used to detect GAPDH. Experiments were repeated three times with similar results.
Genome-wide in situ hybridization (ISH) studies demonstrated significant expression of clusterin within regions where ApoER2 and VLDLR are also expressed (42) (Fig. 5, columns 1–3). In particular, clusterin is expressed throughout the SVZ, rostral migratory stream (RMS), OB, and cortex (Fig. 5, column 3). To evaluate the presence of clusterin in these regions at the protein level we performed immunohistochemical analyses (IHC) in wild-type (WT) mice (Fig. 5, column 4). Clusterin-positive neurons do not stand out from the background with a high contrast, since clusterin is secreted and is present in the extracellular matrix of these structures as well. Control staining without primary antibody demonstrates that the signal is specific (data not shown). Cells present in the SVZ (Fig. 5D) and RMS (Fig. 5H) strongly express clusterin, which is abundantly present throughout these structures. The same expression patterns were revealed by ISH (Fig. 5, C and G). In the OB (Fig. 5L), presence of clusterin closely follows the expression pattern revealed by ISH (Fig. 5K) highlighting the laminated structures of the OB. From the outside to the inside of the OB most neurons present in the glomerular layer (I, juxtaglomerular neurons), external plexiform layer (II), mitral cell layer (III), and internal plexiform layer (IV) are characterized by strong clusterin expression. The same is true for neurons present in the cortex. Fig. 5O (ISH) and Fig. 5P (IHC) display the outermost part of the cortex including the plexiform layer (I), outer granular layer (II), and pyramidal cell layer (III).
FIGURE 5.

Clusterin is expressed in the same brain regions as ApoER2 and VLDLR. A–C, in situ hybridization studies from the Allen Brain Atlas comparing the expression of ApoER2 (column 1), VLDLR (column 2), and clusterin (column 3) in the subventricular zone (SVZ), (E–G) rostral migratory stream (RMS), (I–K) the outermost layers of the olfactory bulb (OB) and (M–O) cerebral cortex. To confirm the expression of clusterin on protein level sagittal sections (5 μm) of the forebrain from WT mice were immunostained with an antibody against clusterin (column 4). D, clusterin is expressed in cells within the SVZ and the proximal RMS. H, neuroblasts in the distal RMS express clusterin. L, in the OB clusterin is expressed in juxtaglomerular neurons of the glomerular layer (I), and neurons within the external plexiform layer (II), mitral cell layer (III), and internal plexiform layer (IV). P, in the cortex clusterin is expressed in neurons within the plexiform layer (I), outer granular layer (II), and pyramidal cell layer (III). Scale bars represent 100 μm.
Having demonstrated that (i) clusterin activates the central axis of the Reelin-signaling cascade (ApoER2/VLDLR/Dab1/PI3K) and (ii) clusterin is present in brain regions where neurons express ApoER2 and VLDLR we tested whether clusterin signaling might have a physiological relevance. To this end we turned our focus to the SVZ which is experimentally accessible since SVZ explants can be grown and manipulated in vitro. When placed into a three-dimensional extracellular matrix substrate (Matrigel) these explants produce neuroblasts, which form chains together with glia cells and migrate away from the explant (43). Here, we cultivated SVZ explants for 72 h and these explants extended robust chains as expected (Fig. 6, A and B). To test whether neurons forming the chains were newly generated during cultivation of the explants in vitro or derived from an already existing cell population we cultivated the explants for 48 h including a 19 h EdU pulse and analyzed the explants by confocal microscopy (Fig. 6, C and D). All cells in chains which are just emerging from the explants are EdU-positive (Fig. 6C), whereas regions which have already developed long intermingling chains are composed of EdU-positive as well as negative cells (Fig. 6D) indicating that chains of migrating cells are formed by proliferating neuronal precursors just as in the in vivo situation. Addition of clusterin (Fig. 6, E and F) did not result in significant alteration of chain length or individual cells per field. As demonstrated above, clusterin is present in the SVZ and thus also in the explants. Additional clusterin apparently did not alter chain formation. Thus, we cultivated the explants in the presence of an antibody against clusterin, and as demonstrated in Fig. 6, G and H, the explants did not grow out under these conditions. When blocking clusterin in the explants, neither neuroblast chains were formed nor did single neuroblasts migrate out of the explants. Removal of the antibody after 24 h of incubation and addition of soluble clusterin led to partial recovery of the explants (Fig. 6, I and J). Control experiments using an unrelated antibody of the same class did not alter production of neuroblasts and formation of chains (Fig. 6, K and L).
FIGURE 6.

Clusterin is required for chain formation of proliferating migratory neuronal precursors from SVZ explants of WT mice. A, SVZ explants were prepared from P4–5 WT mice and treated with mock medium for 24 h or (B) 72 h. C and D, SVZ explants were prepared from P6 WT mice and cultivated for 48 h. After 20 h of EdU incorporation proliferating nuclei were detected with an Alexa Fluor 488 (green)-conjugated azide (Click-iT EdU imaging kit, Invitrogen). Representative images of the latest arising chains (C) or fully developed chains of migrating neuronal precursors (D) show a large number of proliferating cells. E and F, SVZ explants from P4–5 WT mice were cultivated in the presence of 2.5 nm clusterin or (G, H) a mouse monoclonal anti-clusterin antibody (41D; 5 μg) for 24 h (E, G) or 72 h (F, H). I, SVZ explants from P4–5 WT mice were cultivated in the presence of a mouse anti-clusterin antibody (41D; 5 μg) for 24 h followed by (J) incubation with medium containing 2.5 nm clusterin for 48 h. K and L, as a negative control for clusterin blocking, SVZ explants from P5 WT mice were cultivated in the presence of a mouse monoclonal anti-triMethyl-Histone H4 antibody (triMe-Lys20; 5 μg) for 24 h (K) or 72 h (L). M, 15 explants were analyzed per condition by measuring the chain length at 3 random positions after 72 h in culture (n = 45 for each condition; plot shows mean ± S.E.; n.s. not significant; ****, p < 0.0001; one-way ANOVA and Tukey's Post Hoc Test was performed in GraphPad Prism 6). Representative explants are shown. Individual explants were derived from at least four different WT mice per preparation. Experiments were repeated five times with similar results. Scale bars represent 250 μm (A, B, E–L) or 20 μm (C, D).
The fact that SVZ explants do not produce neuroblast chains in Matrigel when clusterin is blocked raised the question whether proliferation of neuronal precursors is inhibited or apoptosis of these cells prevail under these circumstances. To answer this question we measured cell proliferation and apoptosis directly in the SVZ explants by FACS analysis as described in detail in “Experimental Procedures.” Briefly, explants were cultivated together in the presence or absence of the anti-clusterin antibody and in the presence of EdU. Cultivation of the explants was stopped by the addition of dispase, which dissolved the Matrigel and the extracellular matrix of the explants. The resulting cell suspension was washed and the percentage of EdU-positive cells was determined by FACS analysis. As demonstrated in Fig. 7A, 14.1% of all cells were EdU positive when the explants were grown under standard conditions. In the presence of the anti-clusterin antibody, however, a situation where the explants did not produce neuroblast chains, only 3.2% of the cells were EdU positive (Fig. 7B). Thus, blocking clusterin led to a more than 4-fold reduction of cell proliferation within the explants. In contrast to this, apoptosis, which was also measured by FACS analysis based on caspase-3 activity of the cells present in the explants, clearly demonstrated no significant difference in the presence or absence of the anti-clusterin antibody (Fig. 7, C and D).
FIGURE 7.

Blocking clusterin in SVZ explants of WT mice decreases proliferation, but does not affect apoptosis. A, SVZ explants of 6-day-old WT mice were kept in mock medium (− antibody) or (B) cultivated in the presence of a mouse anti-clusterin antibody (41D; 5 μg) for 48 h (+ antibody). 19 h before harvesting the cells 50 μm EdU was added. Cells were processed according to the Invitrogen Click-IT protocol. The DNA content was stained with propidium iodide (PI). Cells were analyzed by flow cytometry (FACSAria, BD Biosciences). The dual parameter plots show DNA content labeling (DNA content (PI) PE-A) with the labeling of proliferating cells that have incorporated EdU (EdU APC-A). A morphologic gate was set, and 10.000 events were collected. EdU-positive cells were gated and representative dot plot graphs and percentages of EdU-positive cells (red) in each gate are shown. C, SVZ explants of 4-day-old WT mice were kept in mock medium (−antibody, blue) or cultivated in the presence of a mouse anti-clusterin antibody (41D; 5 μg) for 72 h (+ antibody, red). The percentage of apoptotic cells in each group of cells was determined by application of a caspase-3 intracellular activity assay kit (PhiPhiLux® G1D2, Calbiochem). Cells were analyzed by flow cytometry (FACSCalibur, BD Biosciences). The dual parameter plot shows the intensity of PhiPhiLux fluorescence with its corresponding cell count. A morphologic gate was set, and 10,000 events were collected. The caspase-3-positive subpopulation is generally one to two orders of magnitude brighter than the caspase-negative fraction. Cells with a fluorescence signal stronger than 101 were gated, and percentages of PhiPhiLux-positive cells are shown. D, cumulative distribution plot for the Kolmogorov-Smirnov test (K-S) showing a significant overlap of the PhiPhiLux fluorescence distributions of cells kept in mock medium (−antibody, blue) and cells cultivated in the presence of an mouse anti-clusterin antibody (+ antibody, red) (**, p < 0.01).
DISCUSSION
Here we describe clusterin as a novel signaling-ligand for ApoER2 and VLDLR. Native human clusterin binds to ApoER2 and VLDLR with high affinity, and the Kd-values are well within the range of other known ligands such as Reelin and thrombospondin-1. The fact that RAP inhibits clusterin binding suggests that the quality of this binding is similar to that of all other known cognate ligands for these receptors since RAP interferes with all these interactions in a similar way (37). Uptake studies using the fibroblast cell model show that clusterin not only binds to ApoER2 and VLDLR but is internalized as well. Thus, clusterin behaves like Reelin in respect to its interaction with these receptors (44) since endocytosis removes the signaling ligand from the extracellular matrix which results in a temporal reduction of the signal if no further signaling proteins are provided.
A prerequisite of ligands of ApoER2 and VLDLR to elicit a cell signal via phosphorylation of Dab1 is their ability to cluster the receptors inducing the formation of higher order complexes of Dab1 that are a substrate for Src-family kinases (5). Reelin achieves this task by forming homodimers, which are able to bind at least two receptors (38). Thrombospondin-1, another functional ligand for ApoER2 and VLDLR, forms homotrimers and is also able to signal along the same pathway (15). Soluble clusterin is a heterodimer and at physiological conditions forms dimers and tetramers (39) and thus is expected to trigger Dab1 phosphorylation via clustering of both receptors. Here we show that this is indeed the case. Clusterin induces Dab1 phosphorylation in primary neurons and 3T3 fibroblasts expressing ApoER2 and Dab1 in the same way as Reelin does. In fibroblasts expressing VLDLR and Dab1 this effect is less pronounced. The reason for this difference is not yet clear but might be caused by differential sorting of the receptors to rafts and non-raft domains in the fibroblast model (44).
Reelin induces a complex network of events, the most prominent of which is the activation of PI3K. As a consequence, activated Akt regulates phosphorylation of tau, MAP1B mediated microtubule remodeling, as well as cell proliferation and survival. Another branch activated by PI3K engages LIMK1 and modulates cofilin, which acts on actin. Phosphorylation of cofilin at serine 3 stabilizes the cytoskeleton by preventing depolymerization of F-actin. Clusterin-mediated inactivation of cofilin by phosphorylation might therefore affect actin dynamics, cell migration, and morphology.
According to expression data, clusterin is present throughout the CNS. In particular, clusterin is expressed in significant amounts in the SVZ and the RMS. Previous experiments demonstrated that SVZ explants derived from apoer2−/−/vldlr−/− mice do not produce neuroblast chains in vitro, while explants derived from reeler mice form neuronal chains just like in the WT situation (45). This finding together with the fact, that Reelin is not expressed in the SVZ clearly demonstrates that Reelin in contrast to ApoER2 and VLDLR is not necessary for chain formation. Thus, the presence of clusterin might account for the receptor-mediated chain formation in SVZ explants. To this end, we could show that blocking clusterin prevents formation of neuroblast chains in SVZ explants in vitro. Clusterin could account for this effect via two downstream events: First, the activation of the PI3K/Akt pathway might result in proliferation of neuronal precursors, a potential necessity for chain formation. Second, clusterin-mediated inactivation of cofilin might stabilize the actin cytoskeleton therefore altering neuronal migration and chain formation. This does not seem to be the case, since addition of clusterin neither influences chain formation in SVZ explants, nor does it dissolve already existent chains as Reelin does it (46). A prerequisite for the first assumption is to prove that neuronal precursors proliferate in SVZ explants in vitro. Indeed, we could demonstrate that migrating neuronal precursors proliferate in these explants. Detailed analyses on cell proliferation and apoptosis in WT SVZ explants and explants where clusterin was blocked revealed that clusterin promotes proliferation, but does not affect apoptosis of cells within the explants. This is a novel function of clusterin which was hitherto known to have a cell protective or anti-apoptotic function (47). In particular, soluble clusterin was described to protect cells from heat shock and TNF-α by interfering with the apoptotic pathway (48, 49). Detailed studies on the anti-apoptotic effect of clusterin on TNF-α and actinomycin-induced cell death revealed that this effect is mediated via the PI3K/Akt pathway, possibly activated via LRP2 (50). LRP2 belongs to the LDL receptor family just as ApoER2 and VLDLR and it remains to be established whether these different effects of clusterin have more in common than the activation of PI3K/Akt.
The divergent roles of clusterin and Reelin in the brain are supported by the reeler phenotype which results from a mutation in the reelin gene (51) and leads to a migration/position defect of neurons but does not compromise neurogenesis. Obviously, lack of functional Reelin cannot be compensated for by clusterin. In contrast to the drastic phenotype of reeler mice, clusterin−/− mice do not show obvious abnormalities in the development of the brain although clusterin is already expressed in early embryonic development in the vast majority of CNS neurons (52). This phenomenon is often observed when other factors compensate for the lack of single proteins thus concealing the exact function(s) of a pleiotropic molecule especially in the brain (32). Even though the lack of clusterin does not seem to provoke a drastic phenotype in the brain it causes a mild phenotype under defined circumstances. In clusterin−/− mice ischemic damage resulting from the occlusion of the middle cerebral artery is more severe than in WT mice (53, 54). Thus, clusterin has neuroprotective properties in vivo after permanent focal cerebral ischemia, a mouse model of human stroke. Hence, taking a closer look at neurogenesis upon injury in this mouse model might reveal new functions of clusterin in vivo. In addition, the fact that ApoER2 and VLDLR exhibit an extreme wide spectrum of potential ligands (20) further complicates the interpretation of existing data. Most likely other hitherto undefined ligands are present in the SVZ which might compensate for each other in vivo.
From the results of the current study, we propose a new role of clusterin as signaling molecule triggering a Reelin-like signal in target cells expressing ApoER2 or VLDLR and Dab1. Clusterin binds to the lipoprotein receptors ApoER2 and VLDLR. The resulting ligand-receptor complex is not only taken up via endocytosis but initiates phosphorylation of Dab1. This leads to activation of the PI3K/Akt pathway ultimately resulting in a cell proliferative effect. This effect is essential for neuronal chain formation in SVZ explants in vitro and may play a role in neurogenesis in the SVZ in vivo.
Acknowledgments
We thank Harald Rumpler, Thomas Sauer, and Philipp Tondl for technical assistance. We wish to acknowledge fruitful discussions with Raimund Bauer.
This work was supported by the Austrian Science Fund (FWF) Grant P24767-B21 (to J. N.).
- CNS
- central nervous system
- ApoER2
- apolipoprotein E receptor 2
- Dab1
- disabled-1
- EdU
- 5-ethynyl-2′-deoxyuridine
- EEA1
- early endosome antigen
- IHC
- immunohistochemistry
- ISH
- in situ hybridization
- Kd
- dissociation constant
- LDL
- low density lipoprotein
- MCM
- mock-conditioned medium
- OB
- olfactory bulb
- PI3K
- phosphatidylinositide 3-kinase
- RAP
- receptor-associated protein
- RCM
- Reelin-conditioned medium
- RMS
- rostral migratory stream
- SVZ
- subventricular zone
- VLDLR
- very low density lipoprotein receptor.
REFERENCES
- 1. Curran T., D'Arcangelo G. (1998) Role of reelin in the control of brain development. Brain Res. Brain Res. Rev. 26, 285–294 [DOI] [PubMed] [Google Scholar]
- 2. Trommsdorff M., Gotthardt M., Hiesberger T., Shelton J., Stockinger W., Nimpf J., Hammer R. E., Richardson J. A., Herz J. (1999) Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell 97, 689–701 [DOI] [PubMed] [Google Scholar]
- 3. Rice D. S., Sheldon M., D'Arcangelo G., Nakajima K., Goldowitz D., Curran T. (1998) Disabled-1 acts downstream of Reelin in a signaling pathway that controls laminar organization in the mammalian brain. Development 125, 3719–3729 [DOI] [PubMed] [Google Scholar]
- 4. Howell B. W., Herrick T. M., Cooper J. A. (1999) Reelin-induced tryosine phosphorylation of disabled 1 during neuronal positioning. Genes Dev. 13, 643–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Strasser V., Fasching D., Hauser C., Mayer H., Bock H. H., Hiesberger T., Herz J., Weeber E. J., Sweatt J. D., Pramatarova A., Howell B., Schneider W. J., Nimpf J. (2004) Receptor clustering is involved in Reelin signaling. Mol. Cell Biol. 24, 1378–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arnaud L., Ballif B. A., Förster E., Cooper J. A. (2003) Fyn Tyrosine Kinase Is a Critical Regulator of Disabled-1 during Brain Development. Curr. Biol. 13, 9–17 [DOI] [PubMed] [Google Scholar]
- 7. Bock H. H., Herz J. (2003) Reelin activates SRC family tyrosine kinases in neurons. Curr. Biol. 13, 18–26 [DOI] [PubMed] [Google Scholar]
- 8. Bock H. H., Jossin Y., Liu P., Förster E., May P., Goffinet A. M., Herz J. (2003) PI3-Kinase interacts with the adaptor protein Dab1 in response to Reelin signaling and is required for normal cortical lamination. J. Biol. Chem. 278, 38772–38779 [DOI] [PubMed] [Google Scholar]
- 9. Förster E., Bock H. H., Herz J., Chai X., Frotscher M., Zhao S. (2010) Emerging topics in Reelin function. Eur. J. Neurosci. 31, 1511–1518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Feng L., Allen N. S., Simo S., Cooper J. A. (2007) Cullin 5 regulates Dab1 protein levels and neuron positioning during cortical development. Genes Dev. 21, 2717–2730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chai X., Förster E., Zhao S., Bock H. H., Frotscher M. (2009) Reelin stabilizes the actin cytoskeleton of neuronal processes by inducing n-cofilin phosphorylation at serine3. J. Neurosci. 29, 288–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jossin Y., Cooper J. A. (2011) Reelin, Rap1 and N-cadherin orient the migration of multipolar neurons in the developing neocortex. Nat. Neurosci. 14, 697–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cooper J. A. (2008) A mechanism for inside-out lamination in the neocortex. Trends Neurosci. 31, 113–119 [DOI] [PubMed] [Google Scholar]
- 14. Jossin Y. (2011) Polarization of migrating cortical neurons by Rap1 and N-cadherin: Revisiting the model for the Reelin signaling pathway. Small GTPases 2, 322–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Blake S. M., Strasser V., Andrade N., Duit S., Hofbauer R., Schneider W. J., Nimpf J. (2008) Thrombospondin-1 binds to ApoER2 and VLDL receptor and functions in postnatal neuronal migration. EMBO J. 27, 3069–3080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schneider W. J. (1989) The low density lipoprotein receptor. Biochim. Biophys. Acta 988, 303–317 [DOI] [PubMed] [Google Scholar]
- 17. Brandes C., Kahr L., Stockinger W., Hiesberger T., Schneider W. J., Nimpf J. (2001) Alternative splicing in the ligand binding domain of mouse ApoE receptor-2 produces receptor variants binding reelin but not α-2-macroglobulin. J. Biol. Chem. 276, 22160–22169 [DOI] [PubMed] [Google Scholar]
- 18. Kim D.-H., Magoori K., Inoue T. R., Mao C. C., Kim H.-J., Suzuki H., Fujita T., Endo Y., Saeki S., Yamamoto T. T. (1997) Exon/intron organization, chromosome localization, alternative splicing, and transcription units of the human apolipoprotein E receptor 2. J. Biol. Chem. 272, 8498–8504 [DOI] [PubMed] [Google Scholar]
- 19. Nimpf J., Schneider W. J. (1998) The VLDL receptor: an LDL receptor relative with eight ligand binding repeats, LR8. Atherosclerosis 141, 191–202 [DOI] [PubMed] [Google Scholar]
- 20. Nimpf J., Schneider W. J. (2000) From cholesterol transport to signal transduction: low density lipoprotein receptor, very low density lipoprotein receptor, and apolipoprotein E receptor-2. Biochim. Biophys. Acta 1529, 287–298 [DOI] [PubMed] [Google Scholar]
- 21. Bujo H., Yamamoto T., Hayashi K., Hermann M., Nimpf J., Schneider W. J. (1995) Mutant oocytic low density lipoprotein receptor gene family member causes atherosclerosis and female sterility. Proc. Natl. Acad. Sci. U.S.A. 92, 9905–9909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bujo H., Hermann M., Kaderli M. O., Jacobsen L., Sugawara S., Nimpf J., Yamamoto T., Schneider W. J. (1994) Chicken oocyte growth is mediated by an eight ligand binding repeat member of the LDL receptor family. EMBO J. 13, 5165–5175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jacobsen L., Hermann M., Vieira P. M., Schneider W. J., Nimpf J. (1995) The chicken oocyte receptor for lipoprotein deposition recognizes α2-macroglobulin. J. Biol. Chem. 270, 6468–6475 [DOI] [PubMed] [Google Scholar]
- 24. MacLachlan I., Nimpf J., Schneider W. J. (1994) Avian riboflavin binding protein binds to lipoprotein receptors in association with vitellogenin. J. Biol. Chem. 269, 24127–24132 [PubMed] [Google Scholar]
- 25. Mahon M. G., Lindstedt K. A., Hermann M., Nimpf J., Schneider W. J. (1999) Multiple involvement of clusterin in chicken ovarian follicle development. Binding to two oocyte-specific members of the low density lipoprotein receptor gene family. J. Biol. Chem. 274, 4036–4044 [DOI] [PubMed] [Google Scholar]
- 26. Jones S. E., Jomary C. (2002) Clusterin. Int. J. Biochem. Cell Biol. 34, 427–431 [DOI] [PubMed] [Google Scholar]
- 27. Yang C. R., Leskov K., Hosley-Eberlein K., Criswell T., Pink J. J., Kinsella T. J., Boothman D. A. (2000) Nuclear clusterin/XIP8, an x-ray-induced Ku70-binding protein that signals cell death. Proc. Natl. Acad. Sci. U.S.A. 97, 5907–5912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leskov K. S., Klokov D. Y., Li J., Kinsella T. J., Boothman D. A. (2003) Synthesis and functional analyses of nuclear clusterin, a cell death protein. J. Biol. Chem. 278, 11590–11600 [DOI] [PubMed] [Google Scholar]
- 29. Trougakos I. P., Djeu J. Y., Gonos E. S., Boothman D. A. (2009) Advances and challenges in basic and translational research on clusterin. Cancer Res. 69, 403–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Trougakos I. P., Gonos E. S. (2006) Regulation of clusterin/apolipoprotein J, a functional homologue to the small heat shock proteins, by oxidative stress in ageing and age-related diseases. Free Radic. Res. 40, 1324–1334 [DOI] [PubMed] [Google Scholar]
- 31. Wilson M. R., Easterbrook-Smith S. B. (2000) Clusterin is a secreted mammalian chaperone. Trends Biochem. Sci. 25, 95–98 [DOI] [PubMed] [Google Scholar]
- 32. Charnay Y., Imhof A., Vallet P. G., Kovari E., Bouras C., Giannakopoulos P. (2012) Clusterin in neurological disorders: molecular perspectives and clinical relevance. Brain Res. Bull. 88, 434–443 [DOI] [PubMed] [Google Scholar]
- 33. Narayan P., Orte A., Clarke R. W., Bolognesi B., Hook S., Ganzinger K. A., Meehan S., Wilson M. R., Dobson C. M., Klenerman D. (2012) The extracellular chaperone clusterin sequesters oligomeric forms of the amyloid-β(1–40) peptide. Nat. Struct. Mol. Biol. 19, 79–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Koch S., Strasser V., Hauser C., Fasching D., Brandes C., Bajari T. M., Schneider W. J., Nimpf J. (2002) A secreted soluble form of ApoE receptor 2 acts as a dominant-negative receptor and inhibits Reelin signaling. EMBO J. 21, 5996–6004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mayer H., Duit S., Hauser C., Schneider W. J., Nimpf J. (2006) Reconstitution of the Reelin signaling pathway in fibroblasts demonstrates that Dab1 phosphorylation is independent of receptor localization in lipid rafts. Mol. Cell Biol. 26, 19–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bajari T. M., Strasser V., Nimpf J., Schneider W. J. (2003) A model for modulation of leptin activity by association with clusterin. FASEB J. 17, 1505–1507 [DOI] [PubMed] [Google Scholar]
- 37. Willnow T. E. (1998) Receptor-associated protein (RAP): a specialized chaperone for endocytic receptors. Biol. Chem. 379, 1025–1031 [PubMed] [Google Scholar]
- 38. Yasui N., Kitago Y., Beppu A., Kohno T., Morishita S., Gomi H., Nagae M., Hattori M., Takagi J. (2011) Functional importance of covalent homodimer of reelin protein linked via its central region. J. Biol. Chem. 286, 35247–35256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Poon S., Rybchyn M. S., Easterbrook-Smith S. B., Carver J. A., Pankhurst G. J., Wilson M. R. (2002) Mildly acidic pH activates the extracellular molecular chaperone clusterin. J. Biol. Chem. 277, 39532–39540 [DOI] [PubMed] [Google Scholar]
- 40. Gao Z., Godbout R. (2013) Reelin-Disabled-1 signaling in neuronal migration: splicing takes the stage. Cell Mol. Life Sci. 70, 2319–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Beffert U., Morfini G., Bock H. H., Reyna H., Brady S. T., Herz J. (2002) Reelin-mediated signaling locally regulates PKB/Akt and GSK-3b. J. Biol. Chem. 277, 49958–49964 [DOI] [PubMed] [Google Scholar]
- 42. Lein E. S., Hawrylycz M. J., Ao N., Ayres M., Bensinger A., Bernard A., Boe A. F., Boguski M. S., Brockway K. S., Byrnes E. J., Chen L., Chen L., Chen T. M., Chin M. C., Chong J., Crook B. E., Czaplinska A., Dang C. N., Datta S., Dee N. R., Desaki A. L., Desta T., Diep E., Dolbeare T. A., Donelan M. J., Dong H. W., Dougherty J. G., Duncan B. J., Ebbert A. J., Eichele G., Estin L. K., Faber C., Facer B. A., Fields R., Fischer S. R., Fliss T. P., Frensley C., Gates S. N., Glattfelder K. J., Halverson K. R., Hart M. R., Hohmann J. G., Howell M. P., Jeung D. P., Johnson R. A., Karr P. T., Kawal R., Kidney J. M., Knapik R. H., Kuan C. L., Lake J. H., Laramee A. R., Larsen K. D., Lau C., Lemon T. A., Liang A. J., Liu Y., Luong L. T., Michaels J., Morgan J. J., Morgan R. J., Mortrud M. T., Mosqueda N. F., Ng L. L., Ng R., Orta G. J., Overly C. C., Pak T. H., Parry S. E., Pathak S. D., Pearson O. C., Puchalski R. B., Riley Z. L., Rockett H. R., Rowland S. A., Royall J. J., Ruiz M. J., Sarno N. R., Schaffnit K., Shapovalova N. V., Sivisay T., Slaughterbeck C. R., Smith S. C., Smith K. A., Smith B. I., Sodt A. J., Stewart N. N., Stumpf K. R., Sunkin S. M., Sutram M., Tam A., Teemer C. D., Thaller C., Thompson C. L., Varnam L. R., Visel A., Whitlock R. M., Wohnoutka P. E., Wolkey C. K., Wong V. Y., Wood M., Yaylaoglu M. B., Young R. C., Youngstrom B. L., Yuan X. F., Zhang B., Zwingman T. A., Jones A. R. (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176 [DOI] [PubMed] [Google Scholar]
- 43. Wichterle H., Garcia-Verdugo J. M., Alvarez-Buylla A. (1997) Direct evidence for homotypic, glia-independent neuronal migration. Neuron 18, 779–791 [DOI] [PubMed] [Google Scholar]
- 44. Duit S., Mayer H., Blake S. M., Schneider W. J., Nimpf J. (2010) Differential functions of ApoER2 and very low density lipoprotein receptor in Reelin signaling depend on differential sorting of the receptors. J. Biol. Chem. 285, 4896–4908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Andrade N., Komnenovic V., Blake S. M., Jossin Y., Howell B., Goffinet A., Schneider W. J., Nimpf J. (2007) ApoER2/VLDL receptor and Dab1 in the rostral migratory stream function in postnatal neuronal migration independently of Reelin. Proc. Natl. Acad. Sci. U.S.A. 104, 8508–8513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hack I., Bancila M., Loulier K., Carroll P., Cremer H. (2002) Reelin is a detachment signal in tangential chain-migration during postnatal neurogenesis. Nat. Neurosci. 5, 939–945 [DOI] [PubMed] [Google Scholar]
- 47. Klock G., Baiersdörfer M., Koch-Brandt C. (2009) Chapter 7: Cell protective functions of secretory Clusterin (sCLU). Adv. Cancer Res. 104, 115–138 [DOI] [PubMed] [Google Scholar]
- 48. Miyake H., Nelson C., Rennie P. S., Gleave M. E. (2000) Testosterone-repressed prostate message-2 is an antiapoptotic gene involved in progression to androgen independence in prostate cancer. Cancer Res. 60, 170–176 [PubMed] [Google Scholar]
- 49. Sensibar J. A., Sutkowski D. M., Raffo A., Buttyan R., Griswold M. D., Sylvester S. R., Kozlowski J. M., Lee C. (1995) Prevention of cell death induced by tumor necrosis factor alpha in LNCaP cells by overexpression of sulfated glycoprotein-2 (clusterin). Cancer Res. 55, 2431–2437 [PubMed] [Google Scholar]
- 50. Ammar H., Closset J. L. (2008) Clusterin activates survival through the phosphatidylinositol 3-kinase/Akt pathway. J. Biol. Chem. 283, 12851–12861 [DOI] [PubMed] [Google Scholar]
- 51. D'Arcangelo G., Miao G. G., Chen S. C., Soares H. D., Morgan J. I., Curran T. (1995) A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature 374, 719–723 [DOI] [PubMed] [Google Scholar]
- 52. Charnay Y., Imhof A., Vallet P. G., Hakkoum D., Lathuiliere A., Poku N., Aronow B., Kovari E., Bouras C., Giannakopoulos P. (2008) Clusterin expression during fetal and postnatal CNS development in mouse. Neuroscience 155, 714–724 [DOI] [PubMed] [Google Scholar]
- 53. Imhof A., Charnay Y., Vallet P. G., Aronow B., Kovari E., French L. E., Bouras C., Giannakopoulos P. (2006) Sustained astrocytic clusterin expression improves remodeling after brain ischemia. Neurobiol. Dis. 22, 274–283 [DOI] [PubMed] [Google Scholar]
- 54. Wehrli P., Charnay Y., Vallet P., Zhu G., Harmony J., Aronow B., Tschopp J., Bouras C., Viard-Leveugle I., French L. E., Giannakopoulos P. (2001) Inhibition of post-ischemic brain injury by clusterin overexpression. Nat. Med. 7, 977–979 [DOI] [PubMed] [Google Scholar]