

Background: Hypoxia is proposed as a mediator of anthrax lethal factor (LT)-induced pathology.
Results: LT inhibits hypoxia-inducible factor 1α (HIF-1α) translation and causes increased cellular toxicity in response to hypoxic stress.
Conclusion: LT reduces HIF-1α translation, dysregulating host responses to hypoxia.
Significance: Inhibition of HIF-1α translation is a novel mechanism underlying LT-induced pathology.
Keywords: Bacillus, Bacterial Pathogenesis, Hypoxia, Hypoxia-inducible Factor, Infectious Diseases, MAP kinases (MAPKs), mTOR, Protein Stability, Toxins, Translation
Abstract
Hypoxia is considered to be a contributor to the pathology associated with administration of anthrax lethal toxin (LT). However, we report here that serum lactate levels in LT-treated mice are reduced, a finding inconsistent with the anaerobic metabolism expected to occur during hypoxia. Reduced lactate levels are also observed in the culture supernatants of LT-treated cells. LT inhibits the accumulation of hypoxia-inducible factor (HIF)-1α, a subunit of HIF-1, the master regulator directing cellular responses to hypoxia. The toxin has no effect on the transcription or protein turnover of HIF-1α, but instead it acts to inhibit HIF-1α translation. LT treatment diminishes phosphorylation of eIF4B, eIF4E, and rpS6, critical components of the intracellular machinery required for HIF-1α translation. Moreover, blockade of MKK1/2-ERK1/2, but not p38 or JNK signaling, lowers HIF-1α protein levels in both normoxic and hypoxic conditions, consistent with a role for MKK1 and MKK2 as the major targets of LT responsible for the inhibition of HIF-1α translation. The physiological importance of the LT-induced translation blockade is demonstrated by the finding that LT treatment decreases the survival of hepatocyte cell lines grown in hypoxic conditions, an effect that is overcome by preinduction of HIF-1α. Taken together, these data support a role for LT in dysregulating HIF-1α and thereby disrupting homeostatic responses to hypoxia, an environmental characteristic of certain tissues at baseline and/or during disseminated infection with Bacillus anthracis.
Introduction
Anthrax is a life-threatening disease caused by infection with Bacillus anthracis (1, 2). B. anthracis expresses lethal factor (LF)2 and the receptor-binding protective antigen (PA) on its pXO1 virulence plasmid (3). The combination of LF and PA is termed lethal toxin (LT). LF is a zinc-dependent metalloprotease with specific activity against mitogen-activated protein kinase kinases (MKKs) (4, 5) and NACHT leucine-rich repeat protein 1 (NALP1) (6, 7), which are involved in the regulation of metabolism, immunological responses, cellular proliferation, and other critical cellular functions (8, 9). Administration of LT in vivo leads to a shock-like clinical syndrome that parallels the clinical features of late-stage anthrax infection (10, 11). Although the proximal targets of LT are well described, identification of the downstream mediators of toxicity remains an area of active investigation. Current models support the conclusion that the etiology of LT-induced cardiovascular collapse is multifactorial; LT has direct toxic effects on cardiac and smooth muscle (12), which may be exacerbated by disrupted endothelial cell function (12) and/or abrogation of cell-cell adhesion and barrier function (11, 13). These factors combine to induce cardiovascular collapse and death.
Other pathological features of LT-treated animals appear to support this scenario. For example, LT administration to mice results in a pattern of pathology that is characterized by cytotoxicity in tissues that are relatively hypoxic, including the metaphyseal bone marrow and centrilobar regions of the liver (14). These findings have been interpreted to suggest that LT induces tissue hypoxia via cardiovascular compromise, which then leads to cell death in susceptible tissues that are characterized by relatively low baseline oxygen concentrations (14). Although hypoxia has been considered as a contributor to LT-induced pathology, it is possible that LT could affect homeostatic responses to hypoxia. In this regard, LT has been shown to block hypoxia-driven retinal neovascularization in a neonatal mouse model (15).
The master transcription factor that regulates these protective responses is hypoxia-inducible factor (HIF)-1. HIF-1 is composed of two subunits, the O2-labile α subunit (HIF-1α) and the stable β subunit (HIF-1β) (16, 17). The heterodimeric HIF-1 complex binds hypoxia-responsive elements (HREs) containing a conserved RCGTG core sequence. HIF-1 directly controls expression of genes crucial for adaptation to hypoxia, encoding proteins regulating angiogenesis, glucose metabolism, cell growth and survival, tumor metastasis, and immune responses (18, 19).
HIF-1 activity is controlled primarily through post-translational modification and stabilization of the HIF-1α subunit (16, 20, 21). Under normoxic conditions, HIF-1α undergoes post-translational hydroxylation of two proline residues (Pro-402 and Pro-564) within its oxygen-dependent degradation domain, mediated by prolyl hydroxylases (22). Hydroxylated HIF-1α is subsequently bound by the von Hippel-Lindau protein, which recruits the elongin-C/elongin-B/cullin-2/E3-ubiquitin-ligase complex, thus targeting HIF-1α for degradation by the 26 S proteasome (23). During hypoxia, the oxygen-dependent prolyl hydroxylases are inactive. Therefore, HIF-1α escapes ubiquitination and proteasomal degradation and can be transported to the nucleus where it forms a heterodimeric complex with HIF-1β and recruits other co-activators to induce expression of its target genes. HIF-1α has also been reported to be degraded by calpain and the 20 S proteasome (24, 25). In addition to the regulation of protein stability, HIF-1α is regulated at the levels of transcription and translation (26). HIF-1α gene transcription has been shown to be modulated by hypoxia and nonhypoxic stimuli such as LPS, angiotensin II, thrombin, inflammatory cytokines, and reactive oxygen species (26–28). In normoxic conditions, HIF-1α mRNA is translated in a cap-dependent manner that requires activation of the mTOR signaling pathway (29–31). Under hypoxic conditions, mTOR is deactivated (32, 33), and HIF-1α mRNA is translated via a cap-independent mechanism involving an internal ribosome entry site (IRES) located in its 5′-untranslated region (5UTR) (34, 35). Whether anthrax LT modulates HIF-1α protein levels during transcription, translation, and/or following translation was previously unknown.
Here, we provide data that LT directly blocks the translation of the master regulator of cellular responses to hypoxia, HIF-1α. This activity renders tissues more vulnerable to hypoxia-induced cell death. The effects of anthrax LT both to promote hypoxia and to inhibit homeostatic responses to hypoxia implicate a synergistic effect underlying host pathology.
EXPERIMENTAL PROCEDURES
Mice
All animal experiments were performed in accordance with animal protocol WO2011-16, which was approved by the United States Food and Drug Administration Center for Biologics Evaluation and Research (CBER) Institutional Animal Care and Use Committee, in accordance with the United States Public Health Service Policy on Humane Care and Use of Laboratory Animals (Assurance A4295-01). Eight-to-10-week-old C57BL/6J mice purchased from The Jackson Laboratory were allowed at least 1 week to acclimatize to the animal facility prior to the initiation of experiments. For in vivo intoxication experiments, mice were injected intravenously with 200 μg of PA and 80 μg of LF. Mice were sacrificed either 24 or 48 h following LT administration. Blood samples were collected for serum lactate measurements.
Reagents
Lyophilized recombinant PA and LF were purchased from the List Biological Laboratories, Inc. (Campbell, CA), and reconstituted in sterile water to make stock solutions with final concentrations of 1 mg/ml. MG132 (inhibitor of 26 S proteasome), U0126 (inhibitor of MKK1/2), SB203580 (inhibitor of p38), and SP600125 (inhibitor of JNK) were purchased from EMD Millipore (Billerica, MA). ALLN (inhibitor of calpain) was purchased from Enzo Life Sciences (Farmingdale, NY). Lactacystin (inhibitor of 20 S proteasome), rapamycin (inhibitor of mTOR), PP242 (inhibitor of mTOR), and cycloheximide (CHX, inhibitor of mRNA translation) were purchased from Sigma. Antibodies against MKK1, MKK2, MKK3, MKK4, MKK6, and MKK7 were purchased from Santa Cruz Biotechnology (Dallas, TX). Antibodies against p-ERK1/2, ERK1/2, p-p38, p38, p-MNK1, MNK1, p-RSK (Ser-380, Ther-359/Ser-363, and Thr-573), RSK1/2/3, p-eIF4E, eIF4E, p-eIF4B, eIF4B, p-p70S6K, p70S6K, p-rpS6, and rpS6 were purchased from Cell Signaling (Danvers, MA). In addition, the following antibodies were used: anti-human HIF-1α (BD Biosciences), anti-mouse HIF-1α (Novus Biologicals; Littleton, CO), and anti-β-actin (Sigma).
Lactate Assay
The levels of lactate in serum or cell culture supernatants were assessed using the lactate colorimetric/fluorometric assay kit purchased from BioVision (Milpitas, CA) following the manufacturer's instructions. In brief, 46 μl of lactate assay buffer, 2 μl of lactate probe, and 2 μl of lactate enzyme mixture were added into each well of a 96-well plate containing 50 μl of testing samples or lactate standard. Absorbance at 570 nm was measured after incubation of the reaction at room temperature for 20 min. The concentration of lactate in the test samples was calculated based on comparison with a standard curve.
Glucose Uptake Assay
The levels of glucose in cell culture supernatants were assessed using the glucose colorimetric/fluorometric assay kit purchased from BioVision following the manufacturer's instructions. In brief, 46 μl of glucose assay buffer, 2 μl of glucose probe, and 2 μl of glucose enzyme mixture were added into each well of a 96-well plate containing 50 μl of testing samples or glucose standard. Absorbance at 570 nm was measured after incubation of the reaction at room temperature for 20 min. The concentration of glucose in the test samples was calculated based on comparison with a standard curve. Glucose uptake was calculated by subtracting the glucose concentrations in test samples of cell culture supernatants from the glucose concentration in fresh medium.
Gene Expression Profiling
RNA was isolated from HepG2 cells cultured under normoxic or hypoxic conditions with or without LT using TRIzol according to the manufacturer's instructions (Invitrogen) and sent to the Phalanx Biotech Group (San Diego) for expression microarray analysis. Triplicate hybridizations for each sample were performed using the Human Whole-Genome OneArray. Microarray data were normalized using the median scaling approach. Data analysis was focused on HIF-1 direct targets. Heat maps were created using Excel software.
Western Blotting
Cells were lysed with NuPAGE LDS sample buffer (Invitrogen). Cell extracts were then separated on 4–12% NuPAGE BisTris gels (Invitrogen) and transferred to nitrocellulose membranes (Bio-Rad). The membranes were probed with antibodies of interest using standard Western blotting techniques as described previously (36). Western blotting films were scanned, and the intensity of protein bands was quantified using FluorChemTM Q software (Protein Simple Inc., San Jose, CA).
Quantitative PCR
Total RNA was extracted from cells with TRIzol (Invitrogen) following the manufacturer's instructions. RNA was then reverse-transcribed to cDNA using the Omniscript RT kit manufactured by Qiagen (Valencia, CA). The levels of HIF-1α mRNA were measured by quantitative PCR performed using standard techniques with a TaqMan probe (Hs00153153_m1) purchased from Applied Biosystems (Foster City, CA) and normalized to the levels of β-actin (Hs99999903_m1).
Polysome Analysis
Polysome analysis was performed as described previously (37). In brief, HepG2 cells were pretreated with or without LT for 3 h and then cultured under hypoxic conditions for 4 h. The treated cells were then incubated with 0.1 mg/ml CHX for 5 min, detached by scraping into 1 ml of polysome extraction buffer (PEB, 0.3 m NaCl, 15 mm MgCl2, 15 mm Tris-HCl, pH 7.6, 1% Triton X-100, 1 mg/ml heparin, and 0.1 mg/ml CHX), and lysed on ice for 10 min. Nuclei were pelleted (10,000 × g for 10 min), and the resulting supernatant was fractionated through a 10–50% linear sucrose gradient to fractionate cytoplasmic components according to their molecular weights. The eluted fractions were prepared using a fraction collector (Brandel, Gaithersburg, MD), and their quality was monitored at 254 nm by using a UV-6 detector (ISCO, Lincoln, NE). RNA in each fraction was isolated using TRIzol LS (Invitrogen) and reverse-transcribed to cDNA. The levels of HIF-1α and β-actin mRNA in each of the fractions were measured by quantitative PCR, and their abundance was represented as a percent of the total mRNA in the gradient.
Cytotoxicity Assay
HepG2 and Hepa1c1c7 cells were cultured in 96-well plates with or without LT either 3 h prior to or at the initiation of a 24-h incubation under hypoxic conditions. The culture supernatants were assessed for lactate dehydrogenase release using the lactate dehydrogenase-cytotoxicity assay kit II (BioVision; Milpitas, CA). In brief, 2 μl of WST substrate and 100 μl of lactate dehydrogenase assay buffer were added to 10-μl test samples in 96-well plates. Absorbance at 450 nm was measured after a 30-min incubation of the reaction. Medium and supernatants collected from cells extracted with the kit-provided lysis solution were used as minimum (Mini) and maximum (Maxi) lactate dehydrogenase (LDH) release controls, respectively. Cytotoxicity was calculated using Equation 1.
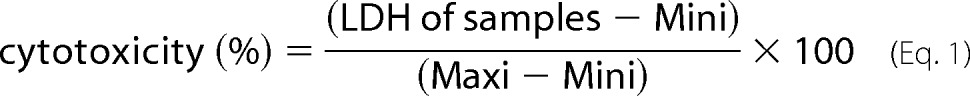 |
RESULTS
Lethal Toxin Treatment Reduces Lactate Production
Anthrax LT has been proposed to induce tissue damage by causing hypoxia (14). To investigate how LT affects the host's ability to respond to hypoxic stress, we first determined lactate levels in the serum of LT-injected mice, which would be expected to increase due to heightened anaerobic metabolism during hypoxia. As shown in Fig. 1A, the levels of lactate in the serum of mice treated with LT for 24 h were paradoxically lower than in control mice. Serum lactate levels were even further reduced following LT treatment for 48 h (Fig. 1A). We next investigated whether LT treatment would similarly reduce lactate production in cultured cells. To address this, we treated HepG2 cells with LT under normoxic or hypoxic conditions for 24 h. As shown in Fig. 1B, HepG2 cells release lactate in normoxic conditions, indicating the propensity of this transformed line to conduct glycolysis in normoxic conditions. Supernatants from LT-treated HepG2 cells had significantly lower concentrations of lactate compared with untreated cells under both normoxic and hypoxic conditions. Moreover, LT treatment reduced glucose uptake by HepG2 cells in both normoxic and hypoxic conditions (Fig. 1C). Taken together, these findings support the conclusion that LT decreases anaerobic metabolism both in vitro and in vivo.
FIGURE 1.
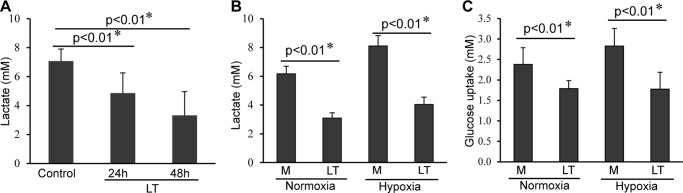
Anthrax LT treatment causes a reduction in lactate accumulation. A, levels of serum lactate in untreated mice (control, n = 13) or mice treated with LT for 24 h (n = 9) and 48 h (n = 7). B, levels of lactate in the supernatants collected from HepG2 cells (5 × 103) cultured under normoxic or hypoxic conditions for 24 h in the presence (LT) or absence (M) of LT. C, uptake of glucose by HepG2 cells (5 × 103) pretreated with (LT) or without (M) LT for 3 h, and then subsequently cultured in normoxic or hypoxic conditions for 24 h. Error bars shown in A–C denote standard deviation generated from three independent experiments. *, statistical analysis was performed using the two-tailed Student's t test.
Lethal Toxin Causes a Reduction in HIF-1α Protein Levels
To gain further insight into the effects of LT on cellular responses to hypoxia, we determined the gene expression profile of HepG2 cells cultured in normoxic or hypoxic conditions, in the presence or absence of LT. We focused our gene profiling analysis on the direct targets of HIF-1, the master regulatory transcription factor of host responses to hypoxia. As shown in Fig. 2A, expression of 17 direct HIF-1 target genes (19) was down-regulated in LT-treated HepG2 cells in both normoxic and hypoxic conditions. These genes encode proteins regulating glucose metabolism, cell growth and survival, and tumor metastasis. For example, Slc2a1 and Slc2a3 encode the glucose transporters (GLUT) 1 and GLUT3, respectively. These findings were also paradoxical when considering the increased glucose requirements during anaerobic metabolism.
FIGURE 2.

Anthrax LT treatment blocks the induction of HIF-1α and its target genes. A, differential expression of HIF-1α target genes in HepG2 cells pretreated for 3 h with (LT) or without (M) LT, and then cultured in normoxic or hypoxic conditions for 20 h. Gene expression is indicated by a color code that ranges from light green (4 or more fold lower than reference levels in normoxic cells cultured in medium alone) to bright red (4 or more fold greater than the reference levels). B, Western blotting analysis of HIF-1α expression in various cell lines treated with (LT) or without (M) LT in normoxic or hypoxic conditions as indicated. Shown is one representative of three independent experiments showing the same trend. C, HepG2 cells were cultured for 3 h in medium alone (M) or with medium containing a combination of wild-type PA and mutated lethal factor lacking protein cleavage activity (LTm), and they were then cultured in normoxic or hypoxic conditions for an additional 4 h. Shown are Western blotting results from one of two independent experiments exhibiting the same trend. The intensity of protein bands in B and C was quantified using Fluorchem Q software; relative amounts are shown below each lane.
HIF-1 activity is primarily controlled through regulation of its α subunit, HIF-1α (16). We next investigated whether LT affects HIF-1α protein levels. As shown in Fig. 2B, the levels of HIF-1α protein were markedly reduced in LT-treated HepG2 cells, in both normoxic and hypoxic conditions. LT treatment also caused a reduction of hypoxia-induced HIF-1α protein in murine hepatocyte Hepa1c1c7 and in human colon adenocarcinoma HT-29 cells, indicating that this activity was not species- or cell line-specific (Fig. 2B). The inhibitory effect of LT on HIF-1α was dependent on the toxin's protease function, as a mutant of LT lacking proteolytic activity failed to reduce HIF-1α protein in HepG2 cells (Fig. 2C).
Lethal Toxin Does Not Affect HIF-1α Gene Transcription or Protein Stability
Cellular levels of HIF-1α protein are regulated via transcription, translation, and protein degradation, which were each considered as possible targets for the activity of LT. We first determined whether LT inhibits HIF-1α gene transcription in HepG2 cells treated with or without LT under conditions of normoxia or hypoxia. As shown in Fig. 3A, LT treatment had negligible effects on HIF-1α gene transcription, as determined by quantitative RT-PCR.
FIGURE 3.

Anthrax LT treatment does not affect HIF-1α gene transcription and protein stability. A, quantitative PCR analysis of HIF-1α mRNA expression in HepG2 cells cultured in medium alone (M) or supplemented with LT for 1–3 h in normoxic conditions (three left columns) or pretreated with LT or medium alone (M) for 3 h, and then cultured in hypoxic conditions for an additional 4 h (two right columns). B and C, Western blotting analysis of HIF-1α protein levels in HepG2 cells pretreated in normoxic conditions with DMSO, MG132, ALLN, and/or lactacystin (Lact) for 30 min and then incubated with or without LT for 7 h as indicated. D, turnover of HIF-1α protein in HepG2 and Hepa1c1c7 cells cultured in the presence or absence of LT in hypoxic conditions. Cells were cultured in hypoxic conditions for 4 h with or without LT during the last 2 h. Cells were subsequently treated with CHX for 0, 0.5, and 1–4 h as shown. HIF-1α protein levels were determined by Western blotting. E, quantification of HIF-1α protein levels. β-Actin was used as a loading control. The intensity of protein bands in B and C was quantified using Fluorchem Q software; relative amounts are shown below each lane. Data shown are representative of three (A–C) or two (D and E) independent experiments.
Next, we assessed post-translational modes of regulation. In normoxic conditions, HIF-1α undergoes oxygen-dependent hydroxylation and ubiquitination, which promotes its degradation by the 26 S proteasome (23). In addition, HIF-1α has been reported to be degraded by calpain and the 20 S proteasome (24, 25). To investigate whether LT reduces HIF-1α through promoting its degradation, we added MG132 (a 26 S proteasome inhibitor), lactacystin (a 20 S proteasome inhibitor), or ALLN (a calpain inhibitor) individually or in combination to the cell cultures 30 min prior to LT treatment to block protein degradation. Western blotting showed that inhibition of protein degradation failed to correct fully the LT-induced HIF-1α reduction (Fig. 3, B and C), suggesting that LT does not reduce HIF-1α levels by promoting its degradation. Chemical inhibition of calpain or the 20 S proteasome had no restorative effect on HIF-1α levels in LT-treated cells, and blockade of the 26 S proteasome only partially enhanced HIF-1α protein levels in the presence of LT. Moreover, a protein turnover study showed similar HIF-1α turnover rates in LT-treated compared with untreated HepG2 and Hepa1c1c7 cells (Fig. 3, D and E), further substantiating that LT treatment does not enhance HIF-1α protein degradation. Collectively, these data show that the LT-induced reduction of HIF-1α protein levels cannot be attributed to the toxin's effect on either HIF-1α gene transcription or protein stability.
Lethal Toxin Inhibits HIF-1α Protein Translation
In addition to the levels of gene transcription and protein stability, HIF-1α is also regulated at the level of translation (26). To investigate whether LT treatment causes a reduction in HIF-1α protein levels by inhibiting its translation, we first addressed the kinetics of protein accumulation in time course experiments in which protein degradation was blocked with a 26 S proteasome inhibitor, MG132. Measurement of HIF-1α protein in HepG2 treated with MG132 for 0 and 1–3 h by Western blotting showed that LT treatment completely blocks MG132-induced HIF-1α protein accumulation (Fig. 4A). We next investigated whether the blockade of HIF-1α accumulation was due to defective de novo protein synthesis. To this end, we treated HepG2 cells with CHX for 4 h to block HIF-1α translation (with or without wild-type LT or protease-deficient mutant LT for the last 2 h), washed away CHX, and then cultured the cells in fresh medium to resume protein synthesis. As shown in Fig. 4B, MG132-induced accumulation of de novo synthesized HIF-1α was markedly reduced in LT-treated HepG2 cells. To confirm the effect of LT on HIF-1α mRNA translation, we next examined the relative distribution of HIF-1α mRNA in individual fractions collected from polysome sucrose gradients. In the sucrose gradient experiments (from top to bottom), fractions 1–4 include mRNAs that are not associated with components of the translational machinery or co-sediment with individual ribosomal subunits (monosomes), so they are not considered to be undergoing translation. Fractions 5–7 include mRNAs that bind to single ribosomes or formed polysomes of low molecular weight, and they are considered to be translated at low-to-moderate levels. Fractions 8–10 include the mRNAs that are associated with polysomes of high molecular weight, and they are thus considered to be actively translated. As shown in Fig. 4C, LT treatment led to a shift of HIF-1α mRNA distribution from fraction 9 to fraction 4, further supporting the conclusion that HIF-1α mRNA translation in hypoxic conditions is inhibited by LT treatment. Of note, LT treatment also changed the polysome distribution of β-actin mRNA (Fig. 4C), suggesting that LT may have broader effects on protein synthesis.
FIGURE 4.

Anthrax LT treatment inhibits HIF-1α protein synthesis. A, Western blotting analysis of HIF-1α protein levels in HepG2 cells cultured in the presence or absence of LT and/or MG132 for the indicated periods of time. B, Western blotting analysis of HIF-1α protein levels in HepG2 cells, which were treated with CHX for 4 h in the presence or absence of wild-type LT or a protease-deficient LT mutant (LTm) for the last 2 h. Cells were subsequently washed twice with PBS and then incubated with MG132 for 0–3 h in the absence of LT or LT mutant as indicated. The intensity of protein bands in A and B was quantified using Fluorchem Q software; relative amounts are shown below each lane. C, polysomal distribution of HIF-1α (left panel) and β-actin (right panel) mRNA in HepG2 cells cultured in the presence or absence of LT for 3 h and then cultured in hypoxic conditions for 4 h. The treated cells were extracted with polysome extraction buffer, and the supernatants were fractioned through a 10–50% sucrose gradient. HIF-1α and β-actin mRNA levels in each gradient fraction were determined by quantitative PCR and plotted as a percentage of the total HIF-1α and β-actin mRNA levels in each sample. The peak in fraction 4 corresponds to monosomes, whereas the peak in fractions 8–10 corresponds to polysomes with active translation.
Lethal Toxin Inactivates Signaling Pathways Critical for Protein Synthesis
Having shown that LT-induced HIF-1α reduction requires the toxin's proteolytic activity, which leads to cleavage of MKKs and inactivation of MAPK signal pathways (Fig. 2C), we next confirmed the proximal targets of LT activity in HepG2 cells. Consistent with our previous study (36), LT treatment caused cleavage of MKK1, MKK2, MKK3, MKK4, MKK6, but not MKK7 (Fig. 5A). Moreover, disruption of MKKs in HepG2 cells cultured under both normoxic and hypoxic conditions led to inactivation of several of their downstream signaling pathways, including ERK1/2-MNK, ERK1/2-RSK, and p38-MNK (Fig. 5, B and C). It should be noted that phosphorylation of RSK at multiple sites is required for normal kinase function (38), and its phosphorylation at Thr-573 depends on both ERK1/2 and ERK5 activity (39). It is likely that the more modest effect of LT in reducing phosphorylation at the Thr-573 site is due to persistent ERK5 signaling, as its upstream signaling component, MKK5, is resistant to the proteolytic effect of LT (36).
FIGURE 5.

Anthrax LT treatment disrupts signal pathways critical for protein synthesis. HepG2 cells were pretreated with (LT) or without (M) LT for 3 h and then cultured under normoxic or hypoxic conditions for an additional 4 h. Cell lysates were subjected to Western blotting analysis for assessing protein levels of various MKKs (A), total and phosphorylated ERK1/2 and p38 (B), total and phosphorylated MNK and RSK (C), total and phosphorylated eIF4E and eIF4B (D), total and phosphorylated p70S6K and rpS6 (E), and total and phosphorylated eIF2A (F). The intensity of protein bands in C–F was quantified using Fluorchem Q software; relative amounts are shown below each lane. Data shown are representative of three independent experiments.
Translation of HIF-1α mRNA under normoxic and hypoxic conditions is regulated differently. HIF-1α mRNA is translated in a cap-dependent manner in normoxic conditions, whereas it is translated in a cap-independent manner through an internal ribosome entry site (IRES) in its 5UTR under hypoxic conditions (34). The blockade of signaling to RSK and MNK caused by LT has implications for a potential effect on cap-dependent translation. In normoxic conditions, eukaryotic mRNA translation is initiated by assembling the eIF4F translation initiation complex on the 7-methylguanosine 5′ cap of mRNAs (40). The eIF4F complex includes eIF4E, eIF4B, eIF4A, and eIF4G, and its formation is promoted by activation of the MAPK and mTOR signaling pathways. Activation of MAPK induces phosphorylation of MNK and RSK, which subsequently phosphorylates eIF4E and eIF4B, thereby promoting formation of eIF4F. Phosphorylation of 4E-BP1 by mTOR leads to release of eIF4E, which also promotes eIF4F formation. In addition, activation of p70S6K by mTOR induces phosphorylation of the ribosomal protein S6 (rpS6) that promotes translation as well. Previous studies have shown that activation of the mTOR-p70S6K signal pathway is critical for HIF-1α translation (29–31). In line with the inactivation of the MAPK signal transduction pathways, phosphorylation of eIF4E and eIF4B in HepG2 was modestly reduced by LT treatment (Fig. 5D). Moreover, as shown in Fig. 5E, LT treatment slightly inhibited p70S6K phosphorylation and downstream rpS6 phosphorylation in normoxic conditions. In contrast, hypoxia nearly completely blocked phosphorylation of p70S6K with or without LT. Interestingly, LT blocked phosphorylation of rpS6 in hypoxic but not normoxic conditions, suggesting that rpS6 phosphorylation is regulated independently of p70S6K in hypoxic conditions. It should also be noted that hyperphosphorylation of eIF2A induced by stress is well known to inhibit mRNA translation. In this regard, suppression of HIF-1α by tirapazamine has been attributed to eIF2α phosphorylation rather than inactivation of the mTORC1/4E-BP1 pathway (41). However, LT treatment does not affect eIF2A phosphorylation in HepG2 cells (Fig. 5F), ruling it out as a causative factor of LT-induced inhibition of HIF-1α mRNA translation. Taken together, these results indicate that anthrax LT has specific inhibitory effects on pathways critical for HIF-1α translation.
Regulation of HIF-1α Translation by mTOR and MAPK Pathways under Normoxic and Hypoxic Conditions
The signal transduction pathways regulating the translation of HIF-1α mRNA during normoxia and hypoxia differ markedly. To dissect the upstream pathways involved in mediating HIF-1α mRNA translation in normoxic and/or hypoxic conditions, we treated HepG2 cells with pharmacological inhibitors of various signal transduction pathways implicated in HIF-1α mRNA translation. As shown in Fig. 6A, the MKK1/2 inhibitor U0126 reduced HIF-1α protein levels in HepG2 cells cultured in either normoxic or hypoxic conditions. In contrast, treatment of cultures with the JNK inhibitor, SP600125, did not alter HIF-1α protein levels. The p38 inhibitor, SB203580, slightly reduced HIF-1α in HepG2 cells in conditions of normoxia but had no effect when the cells were maintained in hypoxic conditions. Taken together, these data are consistent with a predominant role for MKK1/2 in mediating LT blockade on HIF-1α translation in either normoxic or hypoxic conditions.
FIGURE 6.

Blockade of MKK1/2-ERK1/2, mTOR, MNK, and/or RSK signaling pathways have differential effects on HIF-1α protein levels in normoxic versus hypoxic conditions. HepG2 cells were pretreated with the vehicle control (DMSO) or the MAPK inhibitors U0126, SB203580, and SP600125 individually (A); with U0126 (U) plus SB203580 (SB) or the mTOR inhibitors PP242 and rapamycin (Rapa) (B); or with various combinations of an MNK inhibitor (Mi), RSK inhibitor (Ri), and/or PP242 (C) for 3 h. Following pretreatment in these conditions, cells were cultured in normoxic or hypoxic conditions for an additional 4 h. HIF-1α protein levels in treated cells were determined by Western blotting. β-Actin was used as a loading control. The intensity of protein bands in A–C was quantified using Fluorchem Q software; relative amounts are shown below each lane. Data shown are representative of three independent experiments.
We next examined the potential physiological role of mTOR signaling in mediating the LT-dependent inhibition of HIF-1α mRNA translation. Whereas blockade of the mTOR signaling pathway with the specific inhibitor PP242 led to a reduction of HIF-1α protein levels that was even greater than that caused by combined treatment of U0126 and SB203580 in normoxic conditions, it did not cause any decrease of HIF-1α protein levels in HepG2 cells cultured in hypoxic conditions (Fig. 6B). Similarly, treatment with inhibitors of either MNK or RSK alone or together with PP242 led to reduced levels of HIF-1α protein in normoxic but not hypoxic conditions (Fig. 6C). These data suggest that the LT-dependent blockade of cap-dependent HIF-1α translation via its inhibition of the MAPK-MNK/RSK and mTOR signaling cascades is most relevant in normoxic conditions. In contrast, during conditions of hypoxia, LT blocks cap-independent translation of HIF-1α via an MKK1/2-ERK1/2-dependent but mTOR-independent mechanism. These findings are consistent with previous reports that mTOR, a signaling molecule essential for HIF-1α mRNA translation in normoxic conditions, is inactivated under hypoxic conditions (32, 33).
Reduced HIF-1α Levels Correlate with LT-induced Cell Death in Hypoxic Conditions
LT treatment inhibited expression of a variety of direct targets of the HIF-1 transcription factor, including genes encoding proteins that regulate metabolism and cell survival (Fig. 2A). Unfortunately, we were unable to experimentally increase HIF-1α protein levels via an expression vector, due to the LT-dependent blockade in its translation (data not shown). To investigate whether attenuation of HIF-1α translation by LT correlates with a detrimental effect on cell viability, we treated cells with LT either 3 h preceding or at the initiation of a 12-h incubation in normoxic or hypoxic conditions. As hypoxia-induced HIF-1α accumulation occurs very rapidly, a certain amount of HIF-1α can accumulate before extensive inactivation of MKK1/2-ERK1/2 by LT, which occurs 1–2 h following toxin treatment (42). As would be expected, HIF-1α protein levels were higher in HepG2 cells treated with LT at the initiation of hypoxia compared with protein levels in the cells treated with LT 3 h prior to their incubation in hypoxic conditions (Fig. 7A). We next compared viability of HepG2 and Hepa1c1c7 cells treated as described above over a period lasting 24 h. As shown in Fig. 7B, HepG2 and Hep1c1c7 cells treated with LT prior to incubation in hypoxic conditions showed an increase in cell death as compared with those treated with LT at the initiation of hypoxia (Fig. 7B). Therefore, a higher rate of survival was associated with the experimental “trapping” of HIF-1α by delaying LT exposure, thus highlighting a detrimental functional consequence of the LT-induced blockade of HIF-1α translation.
FIGURE 7.
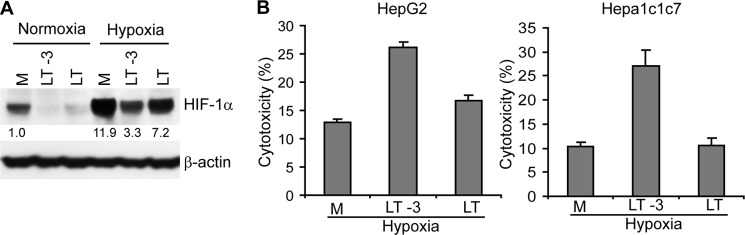
Preinduction of HIF-1α correlates with protection from LT-dependent cytotoxicity during hypoxic stress. A, Western blot assay of HIF-1α protein in HepG2 cells that were treated with LT either 3 h prior to (LT-3) or at the initiation (LT) of the incubation under normoxic or hypoxic conditions for 12 h. Cells grown in medium alone (M) are shown as a control. The intensity of protein bands in A was quantified using Fluorchem Q software; relative amounts are shown below each lane. B, cytotoxicity was assessed in HepG2 (left panel) or Hepa1c1c7 hepatocytes (right panel) treated with LT either 3 h prior to (LT-3) or at the initiation (LT) of the incubation under hypoxia for 24 h. Cells grown in medium alone (M) are shown as a control. Cytotoxicity was determined by the detection of the release of intracellular lactate dehydrogenase into the culture supernatants. Data shown are representative of three independent experiments.
DISCUSSION
Although hypoxia has been reported to be a mediator of LT-induced pathology (14), it was previously unclear whether this mechanism was due entirely to decreased oxygen concentrations in the tissues of LT-treated animals or whether decreased host cell tolerance to hypoxia was also a contributing factor. When host cells sense a decrease in oxygen, they up-regulate the expression of genes encoding proteins critical for angiogenesis, glucose metabolism, cell growth, and survival. Many of these genes are controlled by the master regulator, HIF-1 (18, 19). In this study, we found that LT not only causes a reduction in the basal levels of the HIF-1α subunit in normoxic conditions, but it also inhibits hypoxia-induced HIF-1α accumulation. This blockade in HIF-1α protein accumulation is associated with LT-induced cell death in hypoxic conditions. These findings support a role for LT-induced reduction of HIF-1α protein levels in the mechanism of action for this virulence factor.
The activity of HIF-1 is determined by the cellular level of its O2-labile subunit, HIF-1α, which is regulated at the levels of transcription, translation, and protein turnover. As HIF-1α protein levels are determined primarily by the rate of protein degradation (16, 20), we initially considered this the most likely target for LT. Yet, blockade of protein degradation using various inhibitors fails to restore normal HIF-1α levels in LT-treated cells. Moreover, LT treatment does not affect the turnover rate of HIF-1α protein, further indicating that LT does not act at the level of HIF-1α protein stability. Another possible target for LT activity was at the level of HIF-1α gene transcription. The MAPKs, ERK1/2 and JNK, are required for the LPS-induced up-regulation of HIF-1α mRNA levels in macrophages and hepatocytes, respectively (43). Although LT directly inactivates MAPK signaling pathways, we found that it does not markedly change HIF-1α mRNA levels in LT-treated HepG2 and Hepa1c1c7 cells in normoxic and hypoxic conditions, indicating that the LT-induced reduction of HIF-1α protein is not due to inhibition of its transcription. Instead, LT treatment inhibits MG132-induced HIF-1α protein accumulation by diminishing de novo HIF-1α protein synthesis. Moreover, LT treatment markedly reduces the distribution of HIF-1α mRNA into polysomes with active translation. These findings support the conclusion that LT reduces HIF-1α through inhibiting its translation.
In addition, we have made progress in understanding the molecular mechanisms through which LT acts to inhibit HIF-1α translation. LT treatment disrupts the MKK1/2-ERK1/2 and MKK3/6-p38 signaling pathways and inhibits the phosphorylation of p70S6K and rpS6. Blockade of MKK1/2-ERK1/2 using a selective inhibitor causes a similar reduction of HIF-1α under either normoxic or hypoxic conditions. However, inhibition of mTOR-p70S6K signaling using its specific inhibitor leads to a similar reduction of HIF-1α in normoxic but not hypoxic conditions. These data suggest that although MKK1/2-ERK1/2 signaling is the major target mediating LT's inhibition of HIF-1α translation, LT-dependent disruption of mTOR-p70S6K signaling only plays a role in LT-induced inhibition of HIF-1α translation in normoxic conditions. In contrast, the IRES in the 5UTR of HIF-1α mRNA and the RNA-binding protein PTB have been reported to be important for HIF-1α translation during hypoxia (34, 35). The molecular mechanism linking the ERK1/2 signal transduction pathway and IRES-dependent translation remains enigmatic. Moreover, it is unclear whether the disruptive effect of LT on these critical translation pathways is generalizable to the regulation of other proteins, suggesting the value of surveying the effects of LT on global protein translation.
It is reasonable to speculate that LT-induced disruption of HIF-1α translation would play a role in the pathology of toxemia and infection. The initial linkage of hypoxia to LT-induced pathology was based on its toxic activities in relatively hypoxic tissue environments (e.g. epiphyses of bones and centrilobar regions of the liver) (14). However, very recent data indicate that the cardiovascular system is the major target of LT, which is most relevant to LT-induced mortality in animal models (12). Multiple reports have indicated a critical role for HIF-1α in promoting the survival of cardiomyocytes in hypoxic environments (44, 45), suggesting that inhibition of HIF-1α may contribute to the cardiomyocyte degeneration that has been shown in LT-treated mice (12). The HIF-1α translation blockade could also play a role in establishing and maintaining infection by acting on barrier cells, as HIF-1α is critical for limiting the damage to the mucosal barrier under stress conditions (46, 47). Moreover, the HIF-1α translation blockade could alter host immune responses to infection, as HIF-1α regulates the function of both innate and adaptive immunity (48–52). It is well known that B. anthracis infection thwarts protective immune responses during infection (53). It is interesting to consider the possibility that some of the immunosuppressive activities of LT could be due to its blockade in HIF-1α translation.
In addition, our findings in this study have potential implications for understanding the mechanisms of LT as a protein for cancer therapy. Previous studies report that administration of LT dramatically inhibits tumor growth in vivo, substantially reducing the vascular content of tumors (54–56). The targets of LT, MKKs, have been assumed to promote the vascularization through their direct regulation of VEGF. Attenuation of HIF-1α by LT revealed in this study provides new insights into the mechanism(s) through which LT inhibits vascularization. A recent study demonstrated that a variety of mRNAs containing RNA HREs can form a HIF-2α·RBM4·eIF4E2 complex, which captures the 5′cap and targets mRNAs to polysomes for active translation under hypoxia (57). The proteins encoded by some of the HRE-containing mRNAs are critical for tumor growth. Our preliminary data show that LT treatment also inhibits hypoxia-induced HIF-2α accumulation (data not shown). The effect of LT on translation of HRE-containing mRNAs and tumor development warrants future study.
In conclusion, our results support a model in which LT reduces the level of HIF-1α protein by inhibiting its translation. Attenuation of HIF-1α renders host cells more vulnerable to hypoxia-induced cell death. The effects of anthrax LT both to promote hypoxia and to inhibit homeostatic responses to hypoxia implicate a synergistic effect underlying host pathology. The dysregulation of hypoxic responses via this mechanism may exacerbate the pathology and accelerate the progression of disease associated with the infection of B. anthracis. It may also contribute to the toxin-dependent morbidity and mortality that persist even after bacteremia in patients has been cleared by administration of antibiotics (58).
Acknowledgments
We thank Drs. Lan Xiao and Shan Cao for technical advice regarding the polysome profiling experiment and Drs. Bruce Huang, Milos Dokmanovic, and Kathryn King for thoughtful review of the manuscript.
This work was supported by a Medical Countermeasures Grant from the Food and Drug Administration.
- LF
- lethal factor
- MKK
- mitogen-activated protein kinase kinase
- LT
- lethal toxin
- HIF-1α
- hypoxia-inducible factor 1α
- PA
- protective antigen
- mTOR
- mammalian target of rapamycin
- IRES
- internal ribosome entry site
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- HRE
- hypoxia-responsive element
- CHX
- cycloheximide.
REFERENCES
- 1. Sweeney D. A., Hicks C. W., Cui X., Li Y., Eichacker P. Q. (2011) Anthrax infection. Am. J. Respir. Crit. Care Med. 184, 1333–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mock M., Fouet A. (2001) Anthrax. Annu. Rev. Microbiol. 55, 647–671 [DOI] [PubMed] [Google Scholar]
- 3. Bhatnagar R., Batra S. (2001) Anthrax toxin. Crit. Rev. Microbiol. 27, 167–200 [DOI] [PubMed] [Google Scholar]
- 4. Turk B. E. (2007) Manipulation of host signalling pathways by anthrax toxins. Biochem. J. 402, 405–417 [DOI] [PubMed] [Google Scholar]
- 5. Bromberg-White J. L., Duesbery N. S. (2008) Biological and biochemical characterization of anthrax lethal factor, a proteolytic inhibitor of MEK signaling pathways. Methods Enzymol. 438, 355–365 [DOI] [PubMed] [Google Scholar]
- 6. Frew B. C., Joag V. R., Mogridge J. (2012) Proteolytic processing of Nlrp1b is required for inflammasome activity. PLoS Pathog. 8, e1002659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Levinsohn J. L., Newman Z. L., Hellmich K. A., Fattah R., Getz M. A., Liu S., Sastalla I., Leppla S. H., Moayeri M. (2012) Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog. 8, e1002638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Widmann C., Gibson S., Jarpe M. B., Johnson G. L. (1999) Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol. Rev. 79, 143–180 [DOI] [PubMed] [Google Scholar]
- 9. D'Osualdo A., Reed J. C. (2012) NLRP1, a regulator of innate immunity associated with vitiligo. Pigment Cell Melanoma Res. 25, 5–8 [DOI] [PubMed] [Google Scholar]
- 10. Frankel A. E., Kuo S. R., Dostal D., Watson L., Duesbery N. S., Cheng C. P., Cheng H. J., Leppla S. H. (2009) Pathophysiology of anthrax. Front. Biosci. 14, 4516–4524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Golden H. B., Watson L. E., Lal H., Verma S. K., Foster D. M., Kuo S. R., Sharma A., Frankel A., Dostal D. E. (2009) Anthrax toxin: pathologic effects on the cardiovascular system. Front. Biosci. 14, 2335–2357 [DOI] [PubMed] [Google Scholar]
- 12. Liu S., Zhang Y., Moayeri M., Liu J., Crown D., Fattah R. J., Wein A. N., Yu Z. X., Finkel T., Leppla S. H. (2013) Key tissue targets responsible for anthrax-toxin-induced lethality. Nature 501, 63–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xie T., Auth R. D., Frucht D. M. (2011) The effects of anthrax lethal toxin on host barrier function. Toxins 3, 591–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Moayeri M., Haines D., Young H. A., Leppla S. H. (2003) Bacillus anthracis lethal toxin induces TNF-α-independent hypoxia-mediated toxicity in mice. J. Clin. Invest. 112, 670–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bromberg-White J. L., Boguslawski E., Hekman D., Kort E., Duesbery N. S. (2011) Persistent inhibition of oxygen-induced retinal neovascularization by anthrax lethal toxin. Invest. Ophthalmol. Vis. Sci. 52, 8979–8992 [DOI] [PubMed] [Google Scholar]
- 16. Adams J. M., Difazio L. T., Rolandelli R. H., Luján J. J., Haskó G., Csóka B., Selmeczy Z., Németh Z. H. (2009) HIF-1: a key mediator in hypoxia. Acta Physiol. Hung. 96, 19–28 [DOI] [PubMed] [Google Scholar]
- 17. Semenza G. L. (2007) Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE 2007, cm8 [DOI] [PubMed] [Google Scholar]
- 18. Yeo E. J., Chun Y. S., Park J. W. (2004) New anticancer strategies targeting HIF-1. Biochem. Pharmacol. 68, 1061–1069 [DOI] [PubMed] [Google Scholar]
- 19. Xia Y., Choi H. K., Lee K. (2012) Recent advances in hypoxia-inducible factor (HIF)-1 inhibitors. Eur. J. Med. Chem. 49, 24–40 [DOI] [PubMed] [Google Scholar]
- 20. Yee Koh M., Spivak-Kroizman T. R., Powis G. (2008) HIF-1 regulation: not so easy come, easy go. Trends Biochem. Sci. 33, 526–534 [DOI] [PubMed] [Google Scholar]
- 21. Maxwell P. H., Pugh C. W., Ratcliffe P. J. (2001) The pVHL-hIF-1 system. A key mediator of oxygen homeostasis. Adv. Exp. Med. Biol. 502, 365–376 [PubMed] [Google Scholar]
- 22. Epstein A. C., Gleadle J. M., McNeill L. A., Hewitson K. S., O'Rourke J., Mole D. R., Mukherji M., Metzen E., Wilson M. I., Dhanda A., Tian Y. M., Masson N., Hamilton D. L., Jaakkola P., Barstead R., Hodgkin J., Maxwell P. H., Pugh C. W., Schofield C. J., Ratcliffe P. J. (2001) C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107, 43–54 [DOI] [PubMed] [Google Scholar]
- 23. Maxwell P. H., Wiesener M. S., Chang G. W., Clifford S. C., Vaux E. C., Cockman M. E., Wykoff C. C., Pugh C. W., Maher E. R., Ratcliffe P. J. (1999) The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399, 271–275 [DOI] [PubMed] [Google Scholar]
- 24. Kong X., Alvarez-Castelao B., Lin Z., Castaño J. G., Caro J. (2007) Constitutive/hypoxic degradation of HIF-α proteins by the proteasome is independent of von Hippel Lindau protein ubiquitylation and the transactivation activity of the protein. J. Biol. Chem. 282, 15498–15505 [DOI] [PubMed] [Google Scholar]
- 25. Zhou J., Köhl R., Herr B., Frank R., Brüne B. (2006) Calpain mediates a von Hippel-Lindau protein-independent destruction of hypoxia-inducible factor-1α. Mol. Biol. Cell 17, 1549–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pagé E. L., Robitaille G. A., Pouysségur J., Richard D. E. (2002) Induction of hypoxia-inducible factor-1α by transcriptional and translational mechanisms. J. Biol. Chem. 277, 48403–48409 [DOI] [PubMed] [Google Scholar]
- 27. Chun Y. S., Kim M. S., Park J. W. (2002) Oxygen-dependent and -independent regulation of HIF-1α. J. Korean Med. Sci. 17, 581–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Görlach A. (2009) Regulation of HIF-1α at the transcriptional level. Curr. Pharm. Des. 15, 3844–3852 [DOI] [PubMed] [Google Scholar]
- 29. Bernardi R., Guernah I., Jin D., Grisendi S., Alimonti A., Teruya-Feldstein J., Cordon-Cardo C., Simon M. C., Rafii S., Pandolfi P. P. (2006) PML inhibits HIF-1α translation and neoangiogenesis through repression of mTOR. Nature 442, 779–785 [DOI] [PubMed] [Google Scholar]
- 30. García-Maceira P., Mateo J. (2009) Silibinin inhibits hypoxia-inducible factor-1α and mTOR/p70S6K/4E-BP1 signalling pathway in human cervical and hepatoma cancer cells: implications for anticancer therapy. Oncogene 28, 313–324 [DOI] [PubMed] [Google Scholar]
- 31. Abraham R. T. (2004) mTOR as a positive regulator of tumor cell responses to hypoxia. Curr. Top. Microbiol. Immunol. 279, 299–319 [DOI] [PubMed] [Google Scholar]
- 32. Knaup K. X., Jozefowski K., Schmidt R., Bernhardt W. M., Weidemann A., Juergensen J. S., Warnecke C., Eckardt K. U., Wiesener M. S. (2009) Mutual regulation of hypoxia-inducible factor and mammalian target of rapamycin as a function of oxygen availability. Mol. Cancer Res. 7, 88–98 [DOI] [PubMed] [Google Scholar]
- 33. Cam H., Easton J. B., High A., Houghton P. J. (2010) mTORC1 signaling under hypoxic conditions is controlled by ATM-dependent phosphorylation of HIF-1α. Mol. Cell 40, 509–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lang K. J., Kappel A., Goodall G. J. (2002) Hypoxia-inducible factor-1α mRNA contains an internal ribosome entry site that allows efficient translation during normoxia and hypoxia. Mol. Biol. Cell 13, 1792–1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schepens B., Tinton S. A., Bruynooghe Y., Beyaert R., Cornelis S. (2005) The polypyrimidine tract-binding protein stimulates HIF-1α IRES-mediated translation during hypoxia. Nucleic Acids Res. 33, 6884–6894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xu L., Fang H., Frucht D. M. (2008) Anthrax lethal toxin increases superoxide production in murine neutrophils via differential effects on MAPK signaling pathways. J. Immunol. 180, 4139–4147 [DOI] [PubMed] [Google Scholar]
- 37. Liu L., Rao J. N., Zou T., Xiao L., Wang P. Y., Turner D. J., Gorospe M., Wang J. Y. (2009) Polyamines regulate c-Myc translation through Chk2-dependent HuR phosphorylation. Mol. Biol. Cell 20, 4885–4898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Romeo Y., Zhang X., Roux P. P. (2012) Regulation and function of the RSK family of protein kinases. Biochem. J. 441, 553–569 [DOI] [PubMed] [Google Scholar]
- 39. Ranganathan A., Pearson G. W., Chrestensen C. A., Sturgill T. W., Cobb M. H. (2006) The MAP kinase ERK5 binds to and phosphorylates p90 RSK. Arch. Biochem. Biophys. 449, 8–16 [DOI] [PubMed] [Google Scholar]
- 40. Sonenberg N., Hinnebusch A. G. (2009) Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136, 731–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang J., Cao J., Weng Q., Wu R., Yan Y., Jing H., Zhu H., He Q., Yang B. (2010) Suppression of hypoxia-inducible factor 1α (HIF-1α) by tirapazamine is dependent on eIF2α phosphorylation rather than the mTORC1/4E-BP1 pathway. PLoS One 5, e13910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fang H., Cordoba-Rodriguez R., Lankford C. S., Frucht D. M. (2005) Anthrax lethal toxin blocks MAPK kinase-dependent IL-2 production in CD4+ T cells. J. Immunol. 174, 4966–4971 [DOI] [PubMed] [Google Scholar]
- 43. Vollmer S., Kappler V., Kaczor J., Flügel D., Rolvering C., Kato N., Kietzmann T., Behrmann I., Haan C. (2009) Hypoxia-inducible factor 1α is up-regulated by oncostatin M and participates in oncostatin M signaling. Hepatology 50, 253–260 [DOI] [PubMed] [Google Scholar]
- 44. Loor G., Schumacker P. T. (2008) Role of hypoxia-inducible factor in cell survival during myocardial ischemia-reperfusion. Cell Death Differ. 15, 686–690 [DOI] [PubMed] [Google Scholar]
- 45. Czibik G., Martinov V., Ruusalepp A., Sagave J., Skare Ø., Valen G. (2009) In vivo remote delivery of DNA encoding for hypoxia-inducible factor 1α reduces myocardial infarct size. Clin. Transl. Sci. 2, 33–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Keely S., Campbell E. L., Baird A. W., Hansbro P. M., Shalwitz R. A., Kotsakis A., McNamee E. N., Eltzschig H. K., Kominsky D. J., Colgan S. P. (2014) Contribution of epithelial innate immunity to systemic protection afforded by prolyl hydroxylase inhibition in murine colitis. Mucosal. Immunol. 7, 114–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Karhausen J., Furuta G. T., Tomaszewski J. E., Johnson R. S., Colgan S. P., Haase V. H. (2004) Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J. Clin. Invest. 114, 1098–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Malyshev I., Kruglov S. V., Liamina S. V. (2012) Hypoxia, inflammation and phenotypic plasticity of macrophages: the central role of HIF-1 and NFκB. Patol. Fiziol. Eksp. Ter. 3, 42–50 [PubMed] [Google Scholar]
- 49. Shi L. Z., Wang R., Huang G., Vogel P., Neale G., Green D. R., Chi H. (2011) HIF1α-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 208, 1367–1376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dang E. V., Barbi J., Yang H. Y., Jinasena D., Yu H., Zheng Y., Bordman Z., Fu J., Kim Y., Yen H. R., Luo W., Zeller K., Shimoda L., Topalian S. L., Semenza G. L., Dang C. V., Pardoll D. M., Pan F. (2011) Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell 146, 772–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Olson N., van der Vliet A. (2011) Interactions between nitric oxide and hypoxia-inducible factor signaling pathways in inflammatory disease. Nitric Oxide 25, 125–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rius J., Guma M., Schachtrup C., Akassoglou K., Zinkernagel A. S., Nizet V., Johnson R. S., Haddad G. G., Karin M. (2008) NF-κB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α. Nature 453, 807–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Xu L., Frucht D. M. (2007) Bacillus anthracis: a multi-faceted role for anthrax lethal toxin in thwarting host immune defenses. Int. J. Biochem. Cell Biol. 39, 20–24 [DOI] [PubMed] [Google Scholar]
- 54. Bodart J. F., Chopra A., Liang X., Duesbery N. (2002) Anthrax, MEK and cancer. Cell Cycle 1, 10–15 [PubMed] [Google Scholar]
- 55. Alfano R. W., Leppla S. H., Liu S., Bugge T. H., Ortiz J. M., Lairmore T. C., Duesbery N. S., Mitchell I. C., Nwariaku F., Frankel A. E. (2010) Inhibition of tumor angiogenesis by the matrix metalloproteinase-activated anthrax lethal toxin in an orthotopic model of anaplastic thyroid carcinoma. Mol. Cancer Ther. 9, 190–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Abi-Habib R. J., Singh R., Leppla S. H., Greene J. J., Ding Y., Berghuis B., Duesbery N. S., Frankel A. E. (2006) Systemic anthrax lethal toxin therapy produces regressions of subcutaneous human melanoma tumors in athymic nude mice. Clin. Cancer Res. 12, 7437–7443 [DOI] [PubMed] [Google Scholar]
- 57. Uniacke J., Holterman C. E., Lachance G., Franovic A., Jacob M. D., Fabian M. R., Payette J., Holcik M., Pause A., Lee S. (2012) An oxygen-regulated switch in the protein synthesis machinery. Nature 486, 126–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Artenstein A. W., Opal S. M. (2012) Novel approaches to the treatment of systemic anthrax. Clin. Infect. Dis. 54, 1148–1161 [DOI] [PubMed] [Google Scholar]