

Background: Disruption of the actin cytoskeleton-dependent structural integrity of glomerular epithelial cells (GEC) leads to glomerular dysfunction.
Results: We have identified molecular mechanisms through which the injury-causing complement complex induces cytoskeletal changes in GEC.
Conclusion: Complement-induced activation of the ERK/GEF-H1/RhoA pathway protects GEC from cell death.
Significance: This study furthers our understanding of mechanisms underlying GEC foot process effacement and functional impairment in immune mediated glomerular diseases.
Keywords: Actin, ERK, Guanine Nucleotide Exchange Factor (GEF), Podocytes, Rhoa
Abstract
Visceral glomerular epithelial cells (GEC), also known as podocytes, are vital for the structural and functional integrity of the glomerulus. The actin cytoskeleton plays a central role in maintaining GEC morphology. In a rat model of experimental membranous nephropathy (passive Heymann nephritis (PHN)), complement C5b-9-induced proteinuria was associated with the activation of the actin regulator small GTPase, RhoA. The mechanisms of RhoA activation, however, remained unknown. In this study, we explored the role of the epithelial guanine nucleotide exchange factor, GEF-H1, in complement-induced RhoA activation. Using affinity precipitation to monitor GEF activity, we found that GEF-H1 was activated in glomeruli isolated from rats with PHN. Complement C5b-9 also induced parallel activation of GEF-H1 and RhoA in cultured GEC. In GEC in which GEF-H1 was knocked down, both basal and complement-induced RhoA activity was reduced. On the other hand, GEF-H1 knockdown augmented complement-mediated cytolysis, suggesting a role for GEF-H1 and RhoA in protecting GEC from cell death. The MEK1/2 inhibitor, U0126, and mutation of the ERK-dependent phosphorylation site (T678A) prevented complement-induced GEF-H1 activation, indicating a role for the ERK pathway. Further, complement induced GEF-H1 and microtubule accumulation in the perinuclear region. However, both the perinuclear accumulation and the activation of GEF-H1 were independent of microtubules and myosin-mediated contractility, as shown using drugs that interfere with microtubule dynamics and myosin II activity. In summary, we have identified complement-induced ERK-dependent GEF-H1 activation as the upstream mechanism of RhoA stimulation, and this pathway has a protective role against cell death.
Introduction
The glomerulus is the filtration unit of the kidney where the first step of urine formation occurs. It filters water and small solutes, such as waste products, while retaining large proteins essential for body function, such as albumin. When this barrier function is impaired, protein leakage into the urine (proteinuria) occurs. Proteinuria is not only a marker of glomerular injury but also a prognostic predictor in that heavier proteinuria correlates with a higher risk of kidney failure. Visceral glomerular epithelial cells (GEC),2 commonly known as podocytes, are highly specialized epithelial cells that reside on the outside surface of the glomerular capillary loop and have an important role in maintaining the barrier function of the glomerulus (1). They consist of three morphologically and functionally distinct parts: the large cell body, major processes rich in microtubules, and foot processes maintained by the actin-based cytoskeleton (2). Foot processes from adjacent podocytes form extensive interdigitations and are linked by a modified adherens junction called the “slit diaphragm” (3). These intricate structures are essential for maintaining proper glomerular permselectivity, and loss of foot processes (foot process effacement) is the hallmark of many glomerular diseases accompanied by proteinuria (2). In the last decade, many proteins expressed in podocytes have been identified that regulate the actin cytoskeleton either directly or indirectly (4). Mutations of these proteins disturb the organized structure of the actin cytoskeleton, leading to deranged morphology and function of podocytes (4). Thus, the dynamic regulation of the actin cytoskeleton is crucial for normal physiology of podocytes.
Nephrotic syndrome is a disease characterized by massive proteinuria and hypoalbuminemia and is primarily caused by GEC injury. Membranous nephropathy is one of the most frequent causes of idiopathic nephrotic syndrome in adults and is caused by complement-mediated GEC injury. The rat model of passive Heymann nephritis (PHN) has been used extensively to mimic and study the pathophysiology of membranous nephropathy (5). In PHN, heterologous antibodies bind to their targets on the surface of GEC. The resulting immune complexes activate the complement system, leading to the assembly of the C5b-9 membrane attack complex (6). Nucleated cells require multiple C5b-9 lesions for lysis, but at lower doses, C5b-9 induces sublethal (sublytic) injury and various metabolic effects (7). A previous study reported complement-dependent disruption of the actin microfilaments in cultured GEC (8), which may explain the altered cell-matrix interaction and impaired permselectivity that occur in podocytes in membranous nephropathy (8).
The Rho family of small GTPases consists of more than 22 members, of which RhoA, Rac1, and Cdc42 are the most extensively studied prototypes (9, 10). They are important regulators of actin cytoskeleton dynamics, thus controlling cell morphology and a variety of functions, including motility, adhesion, and malignant transformation (9, 10). Rho proteins act as bimolecular switches cycling between two states: the active (GTP-bound) form, which can bind to effectors and activate downstream pathways, and the inactive (GDP-bound) form. These conformational states are tightly regulated by three families of proteins: guanine nucleotide exchange factors (GEFs), GTPase-activating proteins, and guanine nucleotide dissociation inhibitors.
The GEF family of proteins promotes exchange of GDP for GTP in response to upstream signals (11) and ensures the proper spatio-temporal activation of RhoGTPases (12). To date, more than 80 members of the GEF family have been identified in humans, outnumbering RhoGTPases (12). Although this fact reflects the complex context-specific modes of Rho activation, the mechanisms of GEF regulation remain poorly characterized (12). GEF-H1 (ArhGEF2, also referred to as Lfc in mice) is a GEF that has been implicated in both RhoA and Rac activation induced by various stimuli in epithelia, including renal tubular cells (13, 14). It was initially described as an exchange factor for RhoA and Rac, although its role as a Rho exchange factor is much better characterized (15) (reviewed in Ref. 16). Previous studies have shown an important role of GEF-H1 in the regulation of paracellular permeability in epithelial and endothelial cells (14, 17). GEF-H1 was also shown to mediate RhoA and Rho kinase activation upon Ca2+ removal, leading to the disassembly of the apical junctional complexes (18). In tubular epithelial cells, GEF-H1 is also stimulated by tumor necrosis factor α (TNFα) and mediates RhoA activation and increased paracellular permeability (19). GEF-H1 may thus play a pathological role in the disruption of junctional complexes and epithelial cell barrier.
We have previously reported that complement activates RhoA in GEC in vitro and in vivo (20, 21), but the mechanisms involved remained unknown. The aim of the current study was to identify the upstream signaling mechanisms involved in complement-induced RhoA activation in GEC and to explore a potential role of GEF-H1.
EXPERIMENTAL PROCEDURES
Materials
Tissue culture media and Lipofectamine 2000 were from Invitrogen. Electrophoresis reagents were from Bio-Rad. Enhanced chemiluminescence (ECL) detection reagent and glutathione-Sepharose beads were from Amersham Biosciences. Mini Protease Inhibitor Mixture tablets were from Roche Applied Science. Human C8-deficient serum, purified human C8, and other chemicals were from Sigma-Aldrich. U0126, AG1478, and PP2 were from EMD Biosciences (Mississauga, Canada). The RhoA G-LISATM activation assay colorimetric format was from Cytoskeleton (Denver, CO). Antibodies for GEF-H1, phosphomyosin light chain 2 (Ser-19) and phospho- and total p44/42 MAPK (ERK1/2) were from Cell Signaling (Beverly, MA). Anti-α-tubulin was from Abcam (Cambridge, MA). Anti-RhoA was from EMD Millipore (Billerica, MA). All of the secondary antibodies were from Jackson Immunoresearch (West Grove, PA). Taxol was from Bioshop Canada.
Plasmids
GST-RhoAG17A was from Dr. K. Burridge (University of North Carolina, Chapel Hill, NC). FRET-pRaichu1298x probe was from Dr. M. Matsuda (Osaka University) (22). The plasmids pLKO.1-TRC cloning vector, pMD2.G, psPAX2, and lentiviral shRNA GEF-H1 were from Addgene (Cambridge, MA) (23). The GFP-tagged wild type (WT) GEF-H1 and the point mutant GEF-H1-T678A were gifts from Dr. M. Kohno (24).
Cell Culture and Transfection
Rat GEC culture and characterization were described previously (25, 26). Briefly, a subclone of GEC that grows on plastic was cultured in K1 medium (50% DMEM, 50% Ham/F-12, 10% Nu Serum, hormone supplements), and experiments were carried out between passages 10 and 70. Hormone supplements gave the final concentrations of insulin (5 μg/ml), prostaglandin E1 (25 ng/ml), triiodothyronine (0.325 ng/ml), Na2SeO3 (1.73 ng/ml), apotransferrin (5 μg/ml), and hydrocortisone (18.12 ng/ml). GEC were transiently transfected with Lipofectamine 2000 according to the manufacturer's instructions using 0.5 μg/35-mm plate of plasmids (GFP-GEF-H1-WT or GFP-GEF-H1-T678A) and a 1:2 ratio of the plasmid and Lipofectamine 2000). Conditionally immortalized mouse podocyte culture and characteristics have been described (27, 28). Briefly, undifferentiated mouse podocytes were cultured at 33 °C in RPMI 1640 with 10% fetal bovine serum, 1% penicillin/streptomycin, and 10 units of IFN-γ/ml.
Stimulation with Complements
Complement stimulation of GEC was described previously (25, 26). Briefly, GEC were incubated with anti-GEC antiserum or sheep anti-Fx1A antiserum (5% v/v) for 40 min at room temperature, followed by incubation at 37 °C with normal human serum (NS) to assemble C5b-9 or with decomplemented, heat-inactivated serum (HIS; 56 °C, 1 h) for control for the indicated times. The concentration of NS was 2.5% unless specified otherwise. The anti-GEC antiserum also cross-reacts to mouse podocytes and was used to stimulate mouse podocytes with complement. In some experiments, antibody-sensitized GEC were incubated with C8-deficient serum (C8D; 1.5% (v/v)) with or without reconstitution with purified C8 (2 μg/ml in undiluted serum).
Induction of PHN
PHN was induced in male Sprague-Dawley rats (150–175 g, Charles River Canada, Saint-Constant, Canada) by a single intravenous injection of sheep anti-Fx1A antiserum (400 μl/rat) as described previously (29). Fourteen days postinjection, urine was collected in metabolic cages overnight, and urine protein was quantified using a protein assay kit (Bio-Rad). Rats were then sacrificed, and glomeruli were collected by differential sieving (30). All studies were approved by the McGill University Animal Care Committee.
Immunoblotting
Cultured GEC or isolated glomeruli were washed once and lysed with ice-cold lysis buffer (20 mm HEPES (pH 7.5), 5 mm MgCl2, 150 mm NaCl, 1% Triton X-100, 1 mm PMSF, 1 mm DTT, and a protease inhibitor mixture tablet). After insoluble components were cleared by centrifugation (14,000 rpm, 10 min at 4 °C), protein concentration was determined using protein assay reagent (Bio-Rad). Equal amounts of proteins were separated by SDS-PAGE under reducing conditions and electrophoretically transferred to nitrocellulose membranes. Membranes were blocked with either 5% BSA or skim milk and incubated with primary antibody overnight at 4 °C. After three washes, membranes were incubated with secondary antibodies conjugated with horseradish peroxidase for 1 h at room temperature. Immunoreactive proteins were then detected by ECL. X-ray films were scanned, and densitometric analysis was performed using the ImageJ software. The following antibody dilutions were used: rabbit anti-GEF-H1 (1:1000), mouse anti-tubulin (1:100,000), mouse anti-RhoA (1:1000), mouse anti-pERK (1:2000), mouse anti-ERK1/2 (1:2000), mouse anti-p-MLC (1:1000), anti-mouse and rabbit secondary antibodies (1:2000).
Affinity Precipitation Assay for Active GEF-H1
GST-G17A-RhoA, a mutant with high affinity for activated GEFs, was described previously (31). The assay was performed as described previously (32) with minor modifications. Briefly, treated cells or glomeruli were lysed in lysis buffer. Equal amounts of protein (200–900 μg) were incubated with purified GST-G17A-RhoA (15–20 μg) bound to glutathione-Sepharose beads for 1 h at 4 °C. Beads and protein were washed three times with lysis buffer and were subjected to SDS-PAGE (7.5%) and imunoblotting. X-ray films were scanned, and band intensities were quantified by ImageJ. Active GEF-H1 was normalized to total GEF-H1.
Live Cell Imaging and Fluorescence Resonance Energy Transfer (FRET) Microscopy
GEC were plated on 35-mm glass bottom dishes (MatTek Corp, Ashland, MA) and transfected with 0.5 μg of FRET Raichu1298x probe (pCFP-RBD-pYFP). FRET measurement was carried out 48 h after the transfection following overnight serum starvation using an inverted microscope (IX81, Olympus). After incubation with anti-GEC antiserum at room temperature for 40 min, cells were placed in the microscope chamber. The medium containing the antiserum was removed, and the medium prewarmed at 37 °C was added. After a baseline image was obtained, prewarmed medium containing NS or HIS (for control) was added to the cells to reach a final concentration of 2.5% (v/v), and images were acquired at the indicated times. The biosensors using CFP and YFP as the donor and acceptor, respectively, were excited at a wavelength of 440 nm, and images were taken for CFP and YFP. Corrected FRET (cFRET) intensity was calculated at each pixel using Metamorph Software (Molecular Devices, Sunnyvale, CA) using the following formula: (raw FRET − background)/(CFP − background). cFRET was converted to pseudocolor pixel-to-pixel. The optical filters used for the dual emission imaging were as follows: an XF1071 (440AF21) excitation filter, an XF2034 (455DRLP) dichroic mirror, and two emission filters (XF3075 (480AF30) for CFP and XF3079 (535AF26) for YFP/FRET).
Immunofluorescence Staining
Procedures were carried out at room temperature. Twenty-four hours after transfection, cells were fixed in 4% paraformaldehyde for 15 min, permeabilized with 0.5% Triton X-100 in PBS, and blocked in 3% BSA in PBS for 20 min. α-Tubulin was stained with mouse anti-α-tubulin antibody (1:4000) and rhodamine-conjugated goat anti-mouse antibody (1:500). Nuclei were stained with DAPI (Invitrogen). Images were captured using an AxioObserver-100 microscope (Zeiss). For the subcellular localization study of GEF-H1, images of the cells transfected with GFP-GEF-H1 were analyzed by ImageJ in a blinded manner. A line was drawn from the nuclear membrane to the plasma membrane, and the average GFP fluorescence intensity of the first 5 pixels (perinuclear) was divided by the average of the last 5 pixels (periphery), and the ratio was used as a marker of perinuclear accumulation. Three measurements were averaged per cell, and at least 10 cells were quantified per condition per experiment. Experiments were repeated three times.
Cell Size Quantification
Cross-sectional areas of a cell (cell size) were quantified by the ImageJ software by tracing the cell contour manually.
RhoA Activity Assay
RhoA activity was determined using the RhoA G-LISA activation assay in the colorimetric format (Cytoskeleton, Denver, CO) according to the manufacturer's protocol. Briefly, treated cells were lysed on ice, cleared by centrifugation, and snap-frozen in liquid nitrogen. Prior to the assay, lysates were thawed, and protein concentration was adjusted to 2 μg/μl. Equal amounts of lysates were added to the Rho-GTP affinity plate and incubated for 30 min at 4 °C. After washes, each well was incubated with anti-RhoA antibody for 45 min and then with a secondary antibody conjugated with horseradish peroxidase for 45 min. After washes, buffer containing a substrate was added, and the absorbance at 490 nm was quantified using a microplate spectrophotometer (ELx808IU, BioTek Instruments). The input lysates were analyzed by immunoblotting to ensure that the same amounts of total RhoA were contained in the lysates.
Transduction of GEC with Lentiviral Particles
Lentiviral particles were produced according to the manufacturer's protocol (Addgene). Briefly, low passage HEK293T cells were transfected with pLKO.1-TRC plasmid (control) or GEF-H1 shRNA plasmid, the packaging plasmid psPAX2, and the envelope plasmid pMDG.2 at a ratio of 4:3:1 with Lipofectamine 2000 transfection reagent. The medium was replaced with fresh medium 18 h after transfection, and viral particles contained in the media were collected twice at 24-h intervals. Infection of the target cells with lentiviral particles was used at a multiplicity of infection of 5. GEC-pl cells were exposed to lentivirus encoding GEF-H1 shRNA for 24 h in cell culture medium. Parallel infection with pLKO.1-TRC cloning vector was used as control. All experiments were performed at 48 h after the virus induction was started.
Measurement of Complement-induced Cytotoxicity
Complement-induced cytotoxicity was assessed by measuring the amount of lactate dehydrogenase release as described previously (33). Specific release of lactate dehydrogenase was calculated using the formula, (NS − HIS)/(100 − HIS) × 100, where NS and HIS represent the percentage of lactate dehydrogenase release with NS and HIS, respectively.
Statistics
Data are presented as mean ± S.E. The t statistic was used to determine significant difference between two groups. Two-way analysis of variance was used to determine significant difference in multiple measurements among groups.
RESULTS
Complement C5b-9 Activates GEF-H1 in GEC in Vivo
We reported previously that RhoA is activated in the glomerulus from rats with PHN (20). In PHN, antibody targeted at GEC triggers the assembly of complement C5b-9 on the cell surface, and this leads to GEC injury, morphological changes, and proteinuria (20). To address the mechanisms involved in RhoA activation, we studied whether the Rho exchange factor GEF-H1 is activated in PHN. Active GEF-H1 in glomeruli was detected using affinity precipitation with GST-G17A-RhoA. The G17A mutant of RhoA represents a nucleotide-free form of RhoA that has a high affinity to active GEFs, and therefore the amount of a particular GEF binding to GST-G17A-RhoA reflects the amount of activated form of the GEF (31). Fourteen days after the induction of PHN, rats showed marked proteinuria (384 ± 25 mg/day, n = 5) compared with control rats (5 ± 2 mg/day, n = 4, p < 0.01 versus PHN). Control rat glomeruli showed a small but detectable GEF-H1 activity, which was increased significantly by ∼2.3-fold in PHN (Fig. 1A). Phosphorylation of MLC, a downstream effector of RhoA and Rho-kinase, was also increased significantly (∼2.5-fold) in glomeruli from PHN rats as compared with control (Fig. 1B), consistent with our previous finding that glomerular RhoA activity is increased in PHN (20). Because it is established that GEC is the target of complement-mediated injury in PHN, GEF-H1 activation and MLC phosphorylation are most likely taking place in GEC. Thus, these results suggest that complement C5b-9 activates GEF-H1 in GEC in vivo, leading to the activation of RhoA and its downstream effectors.
FIGURE 1.

GEF-H1 is activated in the glomerulus of rats with PHN. PHN was induced by a single injection of anti-Fx1A antiserum. On day 14, urine was collected, and glomeruli were isolated. A, glomerular lysates were subject to affinity precipitation with GST-RhoG17A to study the amount of active GEF-H1. Precipitates and total lysates were blotted for GEF-H1 (A) or phospho-MLC and tubulin (B). Top, representative blots; bottom, densitometric analysis. **, p < 0.01 versus control. A, n = 4 for control, n = 5 for PHN. B, n = 4 for control, n = 6 for PHN. Error bars, S.E.
Complement C5b-9 Activates GEF-H1 in GEC in Vitro
To further study the mechanisms of GEF-H1 activation by complement, we next studied complement-induced GEF-H1 activation in two podocyte cell lines. Both rat GEC and immortalized mouse podocytes expressed GEF-H1 (Fig. 2, A and B). The RhoG17A pull-down assay demonstrated that GEF-H1 has a small basal activity in unstimulated conditions (i.e. serial incubation with antibody and decomplemented serum; Fig. 2, A and B, HIS). Complement activation was induced by serial incubation with antibody and serum (1–2%) that assemble a sublytic concentration of complement C5b-9 (34). Complement activation increased GEF-H1 activity significantly (∼1.9-fold in rat GEC and ∼4-fold in mouse podocytes with 1% NS), compared with control, without affecting the total amount of GEF-H1 (Fig. 2, A and B, NS). A significant GEF-H1 activation was detected at 15 min after the addition of the serum and continued up to 30 min (Fig. 2C).
FIGURE 2.

Complement activates GEF-H1 in cultured rat GEC. A and B, cultured rat GEC and mouse podocytes (MP) were incubated with rabbit anti-GEC antiserum for 40 min, followed by incubation of NS (to assemble C5b-9) for 30 min. Decomplemented HIS was used as a control. Active GEF-H1 was quantified as in Fig. 1. *, p < 0.05; **, p < 0.01 versus HIS, n = 4 for rat GEC, and n = 6 for MP. C, antibody-sensitized GEC were incubated with complement (NS) for 10, 15, and 30 min, and cell lysates were subject to affinity precipitation for active GEF-H1. Blots with the total cell lysates were also developed for ERK and p-ERK. *, p < 0.05 versus HIS, n = 5. D, antibody-sensitized GEC were incubated with HIS or C8-deficient serum (C8D) with or without reconstitution with purified human C8 for 30 min, and active GEF-H1 was quantified as in Fig. 1. *, p < 0.05 versus HIS and C8D, n = 4. Error bars, S.E.
In order to verify that the complement-induced GEF-H1 activation was dependent on assembly of the full C5b-9 complex, we compared the effects of C8-deficient human serum (C8D), which assembles C5b-7 only, with C8 reconstituted serum, which allows the assembly of C5b-9. When antibody-sensitized GEC were incubated with C8D alone, the amount of active GEF-H1 was not different from control (Fig. 2D, HIS). However, when cells were exposed to the C8D that was reconstituted with purified C8, active GEF-H1 increased significantly ∼1.7-fold) (Fig. 2D), similar to cells stimulated with NS. These results indicate that complement-induced GEF-H1 activation is dependent on C5b-9 assembly.
Complement Activates RhoA in GEC in a Similar Time Course to GEF-H1 Activation
We reported previously that RhoA activity was increased significantly in rat GEC stimulated with complement for 40 min (20). In order to establish the spatio-temporal profile of complement-induced RhoA activation, we utilized a probe that detects RhoA activation through FRET. The FRET probe used (Raichu1298x) contains a yellow fluorescent protein (YFP), a Rho-binding domain, a wild-type RhoA, and a cyan fluorescent protein in tandem (22). Upon activation (GTP binding) RhoA within the probe binds to the Rho-binding domain. This change brings cyan fluorescent protein and YFP to close proximity, allowing energy transfer from cyan fluorescent protein to YFP, resulting in FRET. Thus, the FRET signal intensity correlates with the RhoA activity in a specific site within a cell (Fig. 3A) (35). GEC were transfected with Raichu1298x, and changes in FRET were monitored in live cells. When antibody-sensitized cells were exposed to serum, RhoA activity started to increase at around 15 min and continued to increase up to 30 min (Fig. 3B, NS). RhoA activation was particularly prominent in the perinuclear area of the cells. In contrast, control cells exposed to decomplemented serum did not show RhoA activation (Fig. 3B, HIS). Thus, complement induced localized activation of RhoA in a time course that parallels changes in GEF-H1 activity.
FIGURE 3.

RhoA is activated by complement in GEC. A, schematic representation of the Raichu1298x RhoA FRET probe. RBD, Rho-binding domain of rhotekin; CFP, cyan fluorescent protein. B, GEC were plated onto glass bottom dishes coated with collagen, transfected with the FRET Raichu RhoA probe. Cells were incubated with complement, and FRET images were captured as described under “Experimental Procedures.” The corrected FRET values of each pixel were converted to pseudocolors. Ten cells per condition from three independent experiments were analyzed, and representative cells are shown. Complement (NS) induced RhoA activation starting at ∼15 min. Control cells (HIS) did not show changes in RhoA activity.
GEF-H1 Mediates Complement-induced RhoA Activation in GEC and Protects against Cytotoxicity
Having established that GEF-H1 is expressed and activated by complement in rat GEC in vivo and in vitro, we next studied the functional significance of GEF-H1 activation. Using lentivirus-mediated transduction of an shRNA, we achieved a significant reduction in the expression of GEF-H1 in rat GEC (∼55% knockdown; Fig. 4A). When RhoA activity was quantified by the ELISA-based method (G-LISA), basal as well as complement-stimulated RhoA activity was significantly lower in GEF-H1 knockdown cells as compared with control cells (Fig. 4B). GEF-H1 silencing did not affect RhoA expression. These data indicate that GEF-H1 plays an important role in complement-induced RhoA activation in GEC.
FIGURE 4.

GEF-H1 knockdown inhibits complement-mediated RhoA activation and augments cell injury in GEC. A, GEC were transduced with lentivirus encoding GEF-H1 shRNA or empty vector pLKO.1 (control). Seventy-two hours post-transduction, cell lysates were immunoblotted for GEF-H1. **, p < 0.01 versus pLKO.1, n = 3. B, RhoA activity in GEF-H1 knockdown (shGEF-H1) and control (pLKO.1) cells was assessed by RhoA G-LISA. *, p < 0.05 versus pLKO.1, n = 4. The blot shows the amount of total RhoA in the lysates. C, cytotoxicity was quantified by lactate dehydrogenase release (see “Experimental Procedures”). Control and knockdown cells were stimulated with increasing concentrations of complement (NS, 1, 2.5, 5, and 10%). **, p < 0.01 versus control, n = 4. a.u., arbitrary units; KD, knockdown. Error bars, S.E.
We next studied the functional consequence of GEF-H1 knockdown. We reported previously that a higher concentration of complement C5b-9 induces cytolysis in GEC (lytic concentration) and that RhoA activation protects against complement-induced cytolysis (20). GEC with or without GEF-H1 knockdown were treated with antibody and lytic concentrations of complement (NS, 5 and 10%). Cytolysis was quantified by measuring the release of lactate dehydrogenase in the medium. Complement-induced cytolysis was augmented significantly with GEF-H1 knockdown, as compared with control cells (Fig. 4C). These results suggest that GEF-H1 and subsequent RhoA activation are protective against complement-induced cell death (for discussion, see below).
Complement-mediated GEF-H1 Activation Is Mediated by ERK but Not by the Microtubules
GEF-H1 can be phosphorylated by ERK at Thr-678, and we found that this phosphorylation is necessary for its TNFα-induced activation in tubular cells (19, 24). Consistent with our previous finding, complement C5b-9 activated ERK in rat GEC, and this activation paralleled GEF-H1 activation (Fig. 2C) (36). Moreover, the kinetics of complement-induced ERK and GEF-H1 activation were similar (Fig. 2C). To test whether ERK contributes to complement-induced GEF-H1 activation, we used the pharmacological inhibitor of the ERK pathway (U0126, inhibitor of the ERK activator, MEK1/2, 50 μm). U0126 significantly reduced the activation of GEF-H1 by complement (Fig. 5A). We showed previously that Thr-678 of GEF-H1 can be phosphorylated by ERK (19). When a non-phosphorylatable mutant, T678A, was transfected into GEC, it failed to be activated by complement (Fig. 5B). These results indicate that complement-induced GEF-H1 activation is mediated, at least in part, by the ERK pathway.
FIGURE 5.

Complement-induced GEF-H1 activation is dependent on the ERK pathway. GEC were stimulated with complement in the presence or absence of various inhibitors. The inhibitors were added 30 min prior to the incubation with anti-GEC antiserum and throughout the complement stimulation. After a 30-min incubation with NS (or HIS), cell lysates were subject to affinity precipitation for active GEF-H1. A, U0126 (MEK1/2 inhibitor, 50 μm) inhibited baseline and complement-induced GEF-H1 activity. *, p < 0.05; **, p < 0.01, n = 6 each. B, GEC transfected with WT or the mutant (T678A) GFP-GEF-H1 were stimulated with complement for 30 min. Cell lysates were subjected to affinity precipitation with GST-RhoG17A and immunoblotted for GFP to detect the active amount of the transfected GFP-GEF-H1. WT was activated by complement (NS), whereas the mutant (T678A) was not. **, p < 0.01 versus respective HIS, n = 4 each. C, bisindolylmaleimide I (PKCα inhibitor; 4 μm) partially inhibited complement-induced GEF-H1 activity. *, p < 0.05; **, p < 0.01 versus HIS, n = 4 each. D, AG1478 (EGFR inhibitor) and PP2 (Src inhibitor) (both at 10 μm) did not affect complement-induced GEF-H1 activation. *, p < 0.05; **, p < 0.01 versus respective HIS, n = 3. E, nocodazole (microtubule-disrupting agent) or taxol (microtubule stabilizer) (both at 10 μm) did not affect complement-induced GEF-H1 activation. *, p < 0.05 versus respective HIS, n = 5. Error bars, S.E.
Complement C5b-9 is also known to activate protein kinase C (PKC) via diacylglycerol (37). The PKCα inhibitor, bisindolylmaleimide I (4 μm), significantly but modestly reduced complement-induced GEF-H1, suggesting an additional role of PKCα (Fig. 5C).
In tubular cells, TNFα-induced activation of the ERK/GEF-H1/RhoA pathway was mediated by the receptor tyrosine kinase, epidermal growth factor receptor (EGFR), and Src kinase (38). To test whether a similar mechanism mediates complement-induced GEF-H1 activation, we used inhibitors of these kinases. However, neither the EGFR inhibitor AG1478 nor the Src family inhibitor PP1 affected the basal or complement-stimulated GEF-H1 activities in GEC (Fig. 5D). Thus, complement activates GEF-H1 through signaling pathways that include ERK activation independent of the EGFR and Src.
GEF-H1 is also known to be a microtubule-associated protein (15). It has been shown that GEF-H1 bound to the microtubule is inactive and that microtubule depolymerization liberates GEF-H1, allowing it to activate RhoA (39, 40). Moreover, activation of GEF-H1 by mechanical stimuli was found to be mediated by the microtubules (16, 17, 39). Thus, we next tested the role of the microtubules in complement-induced GEF-H1 activation. Neither the microtubule-disrupting agent, nocodazole, nor the microtubule-stabilizing agent, taxol, had an effect on basal or complement-stimulated GEF-H1 activities (Fig. 5E). These results suggest that in GEC, complement activates GEF-H1 independently of microtubule dynamics (for discussion, see below).
GEF-H1 Accumulates in the Perinuclear Regions upon Stimulation with Complement in GEC
GEF-H1 was found to colocalize with the microtubules in various cultured mammalian cells (15, 16). In order to study the intracellular localization of GEF-H1 in GEC, we transfected GEF-H1 tagged with GFP (GFP-GEF-H1-WT) into cultured rat GEC. In unstimulated cells, GEF-H1 was distributed diffusely in the cytosol, and only a small colocalization with the microtubule filaments was observed (Fig. 6A, HIS). When transfected cells were stimulated with complement (NS), many cells demonstrated a distinct perinuclear distribution of GEF-H1 (Fig. 6A, NS). When we quantified the ratio of the fluorescence intensity adjacent to the nucleus (perinuclear) over that of the cell periphery as a marker of perinuclear accumulation, the ratio increased significantly when cells were stimulated with complement (Fig. 6B). It was noted that complement also altered the structure of the microtubules; in unstimulated cells, microtubules were hairlike filaments projecting from the nucleus to the cell periphery, whereas in complement-stimulated cells, a prominent condensation of the tubulin staining was observed in the perinuclear region (Fig. 6A, HIS versus NS). As a consequence, there was a clear overlap of GEF-H1 and tubulin in the perinuclear region (Fig. 6A, NS). To determine if the perinuclear accumulation of GEH-H1 is dependent on the microtubules, cells were treated with nocodazole or taxol prior to the stimulation with complement. Nocodazole caused the disruption of the microtubule filaments, whereas taxol stabilized and intensified the filamentous staining of the microtubule, demonstrating the efficacy of the two drugs (Fig. 6A). Nonetheless, these two drugs did not affect the perinuclear accumulation of GEF-H1 induced by complement (Fig. 6, A and B). These results indicate that complement-induced perinuclear accumulation of GEF-H1 is independent of the microtubules.
FIGURE 6.
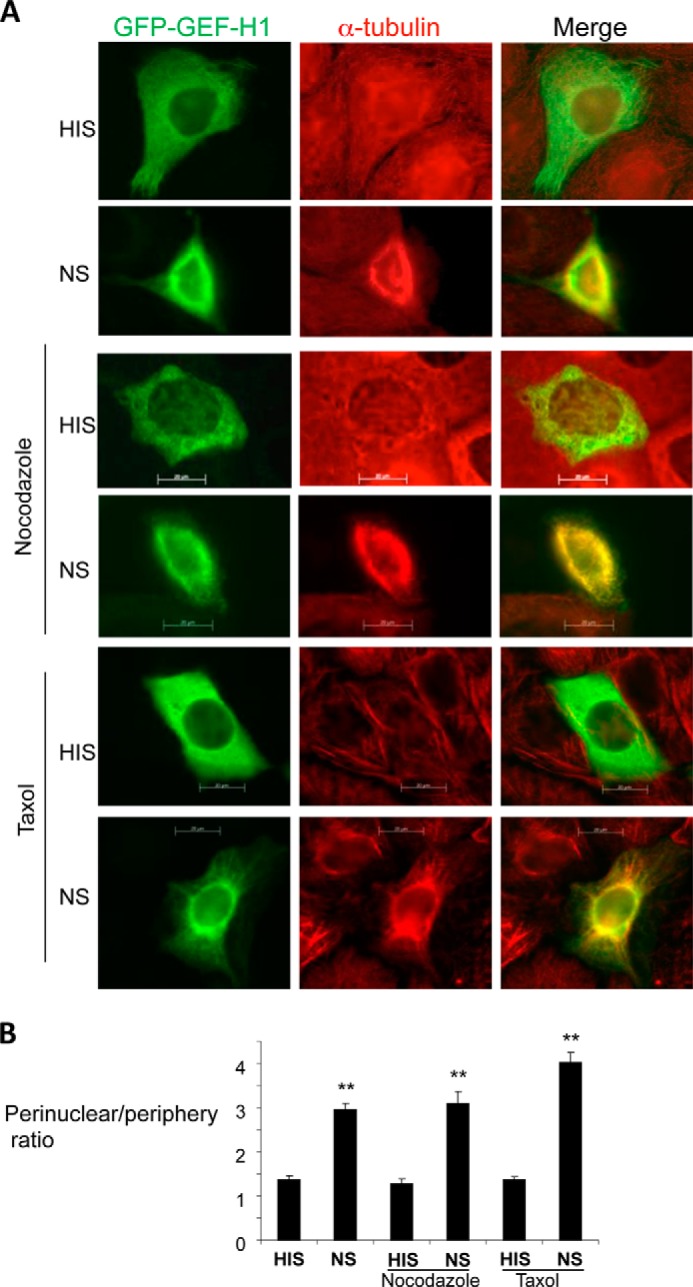
Complement induces perinuclear accumulation of GEF-H1 and alters the microtubule structure in GEC. A, GEC transfected with wild-type GEF-H1 (GFP-GEF-H1) pretreated with vehicle, nocodazole (10 μm), or taxol (10 μm) were stimulated with complement for 30 min. Cells were fixed, permeabilized, and stained for α-tubulin. B, the fluorescence intensity ratio of the perinuclear region over the cell periphery was quantified as described under “Experimental Procedures.” Complement (NS) induced perinuclear accumulation of GEF-H1, and this was not inhibited by nocodazole or taxol. **, p < 0.01 versus respective HIS, n ≥ 30 cells. Error bars, S.E.
We next considered the possibility that the perinuclear accumulation was associated with cell contraction. We tested the effect of a myosin II ATPase inhibitor (blebbistatin; 20 μm) on GEF-H1 activation and translocation. Blebbistatin did not impact the complement-induced GEF-H1 activation as assessed by the pull-down assay (Fig. 7A), nor did it affect the complement-induced perinuclear redistribution of GEF-H1 as assessed by immunofluorescence imaging (Fig. 7B). We also quantified the cross-sectional area as a measure of cell size; complement caused a minor reduction in cell size, which was not significant. In contrast, blebbistatin significantly increased the basal and complement-stimulated cell size, confirming that it was effective (Fig. 7C). Taken together, we concluded that complement induces a small contractile change at the dose used in the current study. Perinuclear relocalization of GEF-H1 induced by complement is not likely to be directly linked to the MLC action because blebbistatin did not affect it.
FIGURE 7.
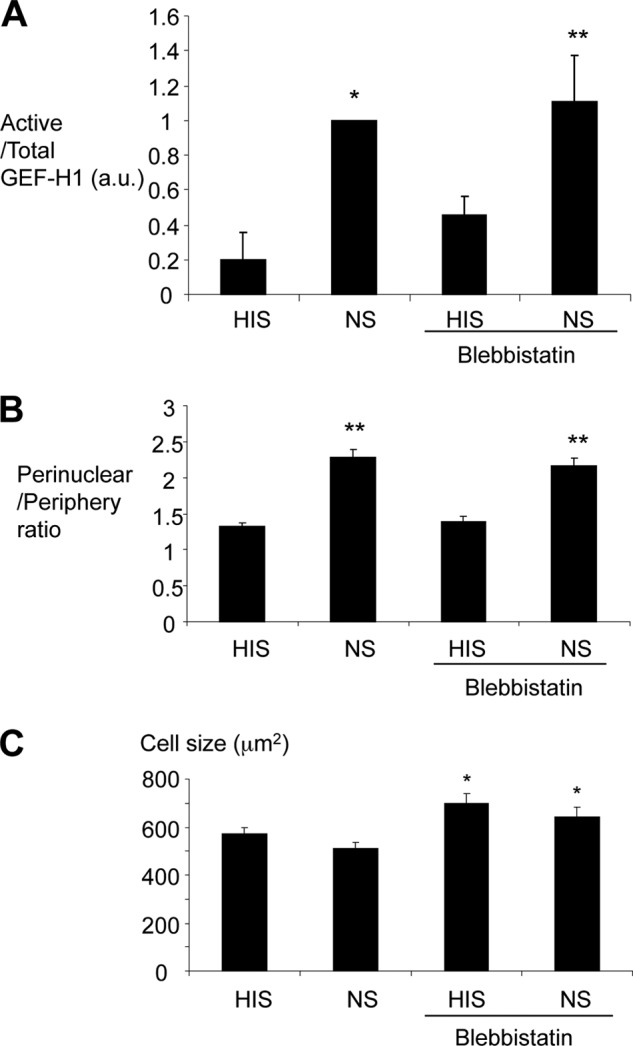
The myosin II ATPase inhibitor, blebbistatin, does not impact complement-induced activation and relocalization of GEF-H1. GEC were stimulated with complement in the presence or absence of blebbistatin (20 μm) for 30 min. A, complement-induced activation of GEF-H1 assessed by pull-down was not affected by blebbistatin. *, p < 0.01; **, p < 0.05 versus HIS, n = 3. B, complement-induced perinuclear relocalization of GEF-H1 assessed as in Fig. 6 was not altered by blebbistatin. **, p < 0.01 versus HIS, n > 40 cells from three experiments. C, complement tended to decrease cell size, but the difference was not significant. Blebbistatin increased cell size significantly. *, p < 0.05 versus no blebbistatin, n > 40 cells from three experiments. (See “Experimental Procedures” for measurement of cell size.) Error bars, S.E.
GEF-H1 Perinuclear Accumulation Is Mediated by ERK
In the previous experiments, we showed that complement-induced GEF-H1 activation was, at least in part, dependent on ERK (Fig. 5). We next studied whether ERK also has a role in the subcellular localization of GEF-H1. Treatment with the MEK1/2 inhibitor, U0126, had no effect on the microtubule structure in unstimulated and complement-stimulated GEC (Fig. 8) but significantly decreased complement-induced perinuclear translocation of GEF-H1 (Fig. 8). In order to test if the effect of the ERK pathway inhibition was directly on GEF-H1, we next utilized the T678A mutant of GEF-H1 (24). Thr-678 of GEF-H1 is the known phosphorylation site by ERK, and the T678A mutant of GEF-H1 was previously shown to lose the ability to be activated by TNFα (19). As shown earlier, this mutant also failed to be activated by complement (Fig. 5B). GFP-GEF-H1-T678A expressed in GEC showed a diffuse cytosolic distribution and a minimal colocalization with the microtubules, similar to the wild-type GEF-H1 (Fig. 8). However, when cells were stimulated with complement, perinuclear accumulation was significantly blunted, similar to the effect of the U0126 (Fig. 8). These results indicate that, in addition to its activation, the change in GEF-H1 intracellular distribution in response to complement is also dependent on the ERK pathway.
FIGURE 8.
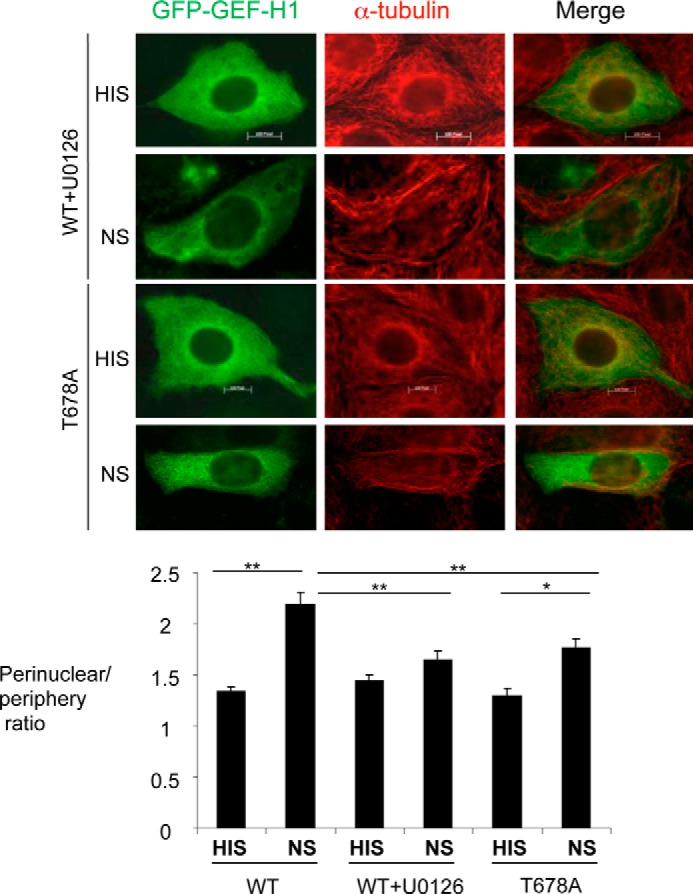
Complement-induced perinuclear accumulation of GEF-H1 is dependent on the ERK pathway. GEC were transfected with WT or the mutant (T678A) GFP-GEF-H1 and were stimulated with complement for 30 min with or without pretreatment with U0126 for 30 min. Cells were fixed, permeabilized, and stained for α-tubulin (red). Both the WT treated with U0126 and the T678A mutant demonstrated reduced perinuclear translocation by complement, as compared with untreated WT. The perinuclear/periphery intensity ratio was quantified as described under “Experimental Procedures.” Complement-induced perinuclear accumulation was inhibited both by the MEK1 inhibitor, U0126, and by using the mutant GEF-H1 (T678A) that is mutated at the ERK phosphorylation site. *, p < 0.05; **, p < 0.01, n ≥ 30 cells.
DISCUSSION
In the present study, we demonstrated a role of GEF-H1 in complement-mediated RhoA activation in GEC. We showed that in the glomerulus from rats with PHN (Fig. 1) and in complement-stimulated cultured GEC (Fig. 2), GEF-H1 activity was increased in a time course that parallels that of the RhoA activation (Fig. 3). GEF-H1 knockdown effectively reduced the basal as well as complement-induced RhoA activity in GEC and augmented complement-induced cytotoxicity (Fig. 4). Complement-induced activation of GEF-H1 was, at least in part, dependent on the ERK pathway but not on the EGFR, Src family kinases, the microtubules (Fig. 5), or myosin-dependent contractility (Fig. 7).
More than 80 human GEFs have been identified, outnumbering the RhoGTPases, suggesting that GEFs have important regulatory effects on Rho-GTPase activities in a cell- and stimulus-dependent manner. However, information on the role of GEFs in podocytes is limited. Our own screen using RT-PCR has shown that in addition to GEF-H1, transcripts of p190 RhoGEF, Net1, and leukemia-associated RhoGEF (LARG) but not p115RhoGEF are also expressed in rat GEC and glomerulus (not shown). LARG (also known as ArhGEF12) was also reported to be enriched in a glomerular expression library prepared from human kidney (41). A recent study showed that the scaffold protein, WT1-interacting protein (Wtip), activated RhoA in cultured mouse podocytes, leading to stress fiber formation. LARG was implicated in Wtip-induced RhoA activation (42). It is likely that the complex and context-dependent effects of GEFs are also mediated by more than one particular Rho family member. For example, podocyte-specific deletion of the atypical aPKCλ/ι in mice caused proteinuria and severe glomerulosclerosis (43), which was attributed to the up-regulation of Def-6, a RacGEF (44–47). In the current study, we focused on RhoA and its regulation because we have previously shown that this small GTPase is activated by complement in GEC (20). Importantly, RhoA activation in podocytes in mice results in proteinuria (21). We chose to explore the role of GEF-H1 in GEC because it has been shown in renal tubular epithelial cells to activate both Rac and RhoA (48), and GEF-H1-mediated RhoA activation was implicated in the regulation of cell-cell junctions. We showed that GEF-H1 knockdown decreased the basal as well as complement-stimulated RhoA activity in GEC (Fig. 4B), verifying a key role for GEF-H1 in both basal and complement-induced RhoA regulation.
The impact of RhoA activation in GEC/podocytes appears to be complex. In cultured mouse podocytes, filamentous actin reorganization by mechanical stretch was dependent on Rho-kinase, a major downstream target of RhoA (49). Pharmacological inhibitors of Rho-kinase have been shown to ameliorate proteinuria and/or kidney functions in a variety of animal models of kidney diseases, including puromycin aminonucleoside nephrosis (50, 51) and hypertensive glomerulosclerosis (52–56). These effects were independent of systemic blood pressure, suggesting that the inhibitors act directly in the kidney, in particular on podocytes (52–56). These findings support the notion that RhoA activation has a negative impact on podocyte morphology and function. On the other hand, RhoA activation was shown to be important for podocyte migration and development (57). Thus, RhoA activation could be beneficial or detrimental in podocyte function, depending on the context. Magnitude, timing, and subcellular localization of RhoA activation might also be an important determinant of the overall outcome. We found that GEF-H1 knockdown cells showed significantly augmented cytolysis by complement (Fig. 4C). These results are consistent with our previous observation that GEC that express a constitutively active mutant of RhoA are more resistant to complement-induced cytolysis (20). These data assign a protective role for GEF-H1 and RhoA. On the other hand, we showed previously that GEC or mouse podocytes transfected with active RhoA lose cellular processes and become contracted with prominent cortical F-actin (20, 21). A similar phenotype was observed in mitotic cells. Thus, the rigid cortex of a rounded cell may protect against external insults (58). Taken together, it is possible that activation of RhoA protects GEC from cell death at the expense of their intricate foot process structures.
Previous studies have demonstrated that GEF-H1 activity is regulated by its association with the microtubules. In HeLa cells, microtubule disruption was shown to release GEF-H1, rendering it active (39). This microtubule-mediated activation of GEF-H1 has also been demonstrated in endothelial cells and dendritic cells (16). Thus, it was surprising that in GEC, GEF-H1 showed limited colocalization with the microtubules and the interfering with microtubule dynamics using nocodazole or taxol did not result in altered GEF-H1 activity. Moreover, complement-induced GEF-H1 activation was also independent of the microtubules (Fig. 5C). It is therefore likely that the contribution of microtubules to GEF-H1 activity is cell type- and stimulus-specific. Indeed, morphology of the podocyte foot processes is predominantly actin-dependent; thus, the influence of the microtubules may be limited there. Further studies are needed to address this hypothesis.
Phosphorylation has also been implicated in GEF-H1 regulation. We found that complement-induced GEF-H1 activation is dependent on ERK. Similar ERK-dependent activation of GEF-H1 toward RhoA induced by TNFα was described in renal tubular epithelial cells (LLC-PK1 and MDCK cells) (19, 24). However, unlike in these cells, complement-induced GEF-H1 activation was independent of the EGF receptor activation (38), suggesting a different mechanism for ERK activation. Other potential candidates that could contribute to complement-induced GEF-H1 activation include increased cytosolic Ca2+ concentration, phospholipase C, PKC, and cytosolic phospholipase A2-α (cPLA2) (25, 34, 36, 37). Several studies showed that PKC signaling is involved in RhoA activation possibly via phosphorylating and activating p115RhoGEF in thrombin-induced endothelial cells, leading to an impaired endothelial barrier (59–61). It was also shown that PKCα activation leads to disruption of renal tubular epithelial (MDCK) apical junctions via a Rho-kinase II-dependent pathway (62). In the current study, a PKCα inhibitor significantly, albeit incompletely, attenuated complement-induced GEF-H1 activation, suggesting that this kinase also contributed to RhoA activation (Fig. 5C). The potential role of other signaling pathways, including other PKC isoforms, in complement-induced GEF-H1 activation requires further investigation.
Activities of some GEFs, such as Net1, Ect2, and Tiam1, depend on their translocation within the cell to the site of RhoGTPase activation (12). Thus, we postulated that the changes in the subcellular localization of GEF-H1 could play a role in its activation. We found that in unstimulated GEC GEF-H1 was diffusely distributed in the cytosol with modest colocalization with the microtubule filaments (Fig. 6A). Upon stimulation with complement, GEF-H1 distribution changed dramatically, and the molecule showed extensive perinuclear accumulation. Curiously, complement also changed the pattern of the microtubules from hairlike filaments projecting from the nucleus to the cell periphery in unstimulated cells to the loss of hairlike filaments and perinuclear condensation (Fig. 6A). However, it is unlikely that microtubule reorganization is the cause of GEF-H1 accumulation because microtubule modulating agents failed to impact complement-induced GEF-H1 accumulation (Fig. 6). Of note, in previous studies, a mutant of GEF-H1 that lacks the carboxyl-terminal region was found to display dense irregular perinuclear aggregates in COS-7 cells (15). It is tempting to speculate that complement induces conformational changes of GEF-H1 mimicking the carboxyl-terminal deletion mutant, thereby causing its redistribution. Of interest, perinuclear accumulation of GEF-H1 appears to correlate with its level of activity, because both were inhibited by the inhibitor of the ERK pathway to a similar degree (Figs. 5A and 8). Furthermore, the T678A mutant of GEF-H1 also demonstrated reduced perinuclear accumulation and activation in response to complement (Figs. 5B and 8). These results are consistent with the hypothesis that complement-induced ERK activation leads to phosphorylation of GEF-H1 at Thr-678, which induces activation and intracellular redistribution of GEF-H1. Indeed, the perinuclear localization of GEF-H1 post-complement stimulation overlaps with the site of RhoA activation observed in live cells (Fig. 3). Further studies are required to address this hypothesis.
In summary, we have identified GEF-H1 as an important regulator that links complement stimulation and RhoA activation in podocytes. Our data support a regulatory model in which the ERK pathway mediates complement-induced RhoA activation by means of GEF-H1 stimulation. The ERK/GEF-H1/RhoA signaling pathway is likely to play a role in the protection of GEC from complement-induced cell death.
Footnotes
- GEC
- glomerular epithelial cells
- NS
- normal human serum
- HIS
- heat-inactivated serum
- PHN
- passive Heymann nephritis
- GEF
- guanine nucleotide exchange factor
- EGFR
- epidermal growth factor receptor.
REFERENCES
- 1. Mundel P., Kriz W. (1995) Structure and function of podocytes. An update. Anat. Embryol. 192, 385–397 [DOI] [PubMed] [Google Scholar]
- 2. Faul C., Asanuma K., Yanagida-Asanuma E., Kim K., Mundel P. (2007) Actin up. Regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol. 17, 428–437 [DOI] [PubMed] [Google Scholar]
- 3. Reiser J., Kriz W., Kretzler M., Mundel P. (2000) The glomerular slit diaphragm is a modified adherens junction. J. Am. Soc. Nephrol. 11, 1–8 [DOI] [PubMed] [Google Scholar]
- 4. Mundel P., Shankland S. J. (2002) Podocyte biology and response to injury. J. Am. Soc. Nephrol. 13, 3005–3015 [DOI] [PubMed] [Google Scholar]
- 5. Greger R., Schlatter E., Hebert S. C. (2001) Milestones in nephrology. Presence of luminal K+, a prerequisite for active NaCl transport in the cortical thick ascending limb of Henle's loop of rabbit kidney. J. Am. Soc. Nephrol. 12, 1788–1793 [DOI] [PubMed] [Google Scholar]
- 6. Jefferson J. A., Pippin J. W., Shankland S. J. (2010) Experimental models of membranous nephropathy. Drug Discov. Today Dis. Models 7, 27–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cybulsky A. V., Quigg R. J., Salant D. J. (2005) Experimental membranous nephropathy redux. Am. J. Physiol. Renal Physiol. 289, F660–F671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Topham P. S., Haydar S. A., Kuphal R., Lightfoot J. D., Salant D. J. (1999) Complement-mediated injury reversibly disrupts glomerular epithelial cell actin microfilaments and focal adhesions. Kidney Int. 55, 1763–1775 [DOI] [PubMed] [Google Scholar]
- 9. Burridge K., Wennerberg K. (2004) Rho and Rac take center stage. Cell 116, 167–179 [DOI] [PubMed] [Google Scholar]
- 10. Jaffe A. B., Hall A. (2005) Rho GTPases. Biochemistry and biology. Annu. Rev. Cell Dev. Biol. 21, 247–269 [DOI] [PubMed] [Google Scholar]
- 11. Quilliam L. A., Khosravi-Far R., Huff S. Y., Der C. J. (1995) Guanine nucleotide exchange factors. Activators of the Ras superfamily of proteins. BioEssays 17, 395–404 [DOI] [PubMed] [Google Scholar]
- 12. Rossman K. L., Der C. J., Sondek J. (2005) GEF means go. Turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 6, 167–180 [DOI] [PubMed] [Google Scholar]
- 13. Terry S. J., Zihni C., Elbediwy A., Vitiello E., Leefa Chong San I. V., Balda M. S., Matter K. (2011) Spatially restricted activation of RhoA signalling at epithelial junctions by p114RhoGEF drives junction formation and morphogenesis. Nat. Cell Biol. 13, 159–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Benais-Pont G., Punn A., Flores-Maldonado C., Eckert J., Raposo G., Fleming T. P., Cereijido M., Balda M. S., Matter K. (2003) Identification of a tight junction-associated guanine nucleotide exchange factor that activates Rho and regulates paracellular permeability. J. Cell Biol. 160, 729–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ren Y., Li R., Zheng Y., Busch H. (1998) Cloning and characterization of GEF-H1, a microtubule-associated guanine nucleotide exchange factor for Rac and Rho GTPases. J. Biol. Chem. 273, 34954–34960 [DOI] [PubMed] [Google Scholar]
- 16. Birkenfeld J., Nalbant P., Yoon S. H., Bokoch G. M. (2008) Cellular functions of GEF-H1, a microtubule-regulated Rho-GEF. Is altered GEF-H1 activity a crucial determinant of disease pathogenesis? Trends Cell Biol. 18, 210–219 [DOI] [PubMed] [Google Scholar]
- 17. Birukova A. A., Adyshev D., Gorshkov B., Bokoch G. M., Birukov K. G., Verin A. D. (2006) GEF-H1 is involved in agonist-induced human pulmonary endothelial barrier dysfunction. Am. J. Physiol. Lung Cell. Mol. Physiol. 290, L540–L548 [DOI] [PubMed] [Google Scholar]
- 18. Samarin S. N., Ivanov A. I., Flatau G., Parkos C. A., Nusrat A. (2007) Rho/Rho-associated kinase-II signaling mediates disassembly of epithelial apical junctions. Mol. Biol. Cell 18, 3429–3439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kakiashvili E., Speight P., Waheed F., Seth R., Lodyga M., Tanimura S., Kohno M., Rotstein O. D., Kapus A., Szászi K. (2009) GEF-H1 mediates tumor necrosis factor-α-induced Rho activation and myosin phosphorylation. Role in the regulation of tubular paracellular permeability. J. Biol. Chem. 284, 11454–11466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang H., Cybulsky A. V., Aoudjit L., Zhu J., Li H., Lamarche-Vane N., Takano T. (2007) Role of Rho-GTPases in complement-mediated glomerular epithelial cell injury. Am. J. Physiol. Renal Physiol. 293, F148–F156 [DOI] [PubMed] [Google Scholar]
- 21. Zhu L., Jiang R., Aoudjit L., Jones N., Takano T. (2011) Activation of RhoA in podocytes induces focal segmental glomerulosclerosis. J. Am. Soc. Nephrol. 22, 1621–1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nakamura T., Kurokawa K., Kiyokawa E., Matsuda M. (2006) Analysis of the spatiotemporal activation of rho GTPases using Raichu probes. Methods Enzymol. 406, 315–332 [DOI] [PubMed] [Google Scholar]
- 23. Kang M. G., Guo Y., Huganir R. L. (2009) AMPA receptor and GEF-H1/Lfc complex regulates dendritic spine development through RhoA signaling cascade. Proc. Natl. Acad. Sci. U.S.A. 106, 3549–3554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fujishiro S. H., Tanimura S., Mure S., Kashimoto Y., Watanabe K., Kohno M. (2008) ERK1/2 phosphorylate GEF-H1 to enhance its guanine nucleotide exchange activity toward RhoA. Biochem. Biophys. Res. Commun. 368, 162–167 [DOI] [PubMed] [Google Scholar]
- 25. Cybulsky A. V., Monge J. C., Papillon J., McTavish A. J. (1995) Complement C5b-9 activates cytosolic phospholipase A2 in glomerular epithelial cells. Am. J. Physiol. 269, F739–F749 [DOI] [PubMed] [Google Scholar]
- 26. Quigg R. J., Cybulsky A. V., Jacobs J. B., Salant D. J. (1988) Anti-Fx1A produces complement-dependent cytotoxicity of glomerular epithelial cells. Kidney Int. 34, 43–52 [DOI] [PubMed] [Google Scholar]
- 27. Mundel P., Kriz W. (1996) Cell culture of podocytes. Exp. Nephrol. 4, 263–266 [PubMed] [Google Scholar]
- 28. Shankland S. J., Pippin J. W., Reiser J., Mundel P. (2007) Podocytes in culture. Past, present, and future. Kidney Int. 72, 26–36 [DOI] [PubMed] [Google Scholar]
- 29. Takano T., Cybulsky A. V., Cupples W. A., Ajikobi D. O., Papillon J., Aoudjit L. (2003) Inhibition of cyclooxygenases reduces complement-induced glomerular epithelial cell injury and proteinuria in passive Heymann nephritis. J. Pharmacol. Exp. Ther. 305, 240–249 [DOI] [PubMed] [Google Scholar]
- 30. Takano T., Cybulsky A. V. (2000) Complement C5b-9-mediated arachidonic acid metabolism in glomerular epithelial cells. Role of cyclooxygenase-1 and -2. Am. J. Pathol. 156, 2091–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. García-Mata R., Wennerberg K., Arthur W. T., Noren N. K., Ellerbroek S. M., Burridge K. (2006) Analysis of activated GAPs and GEFs in cell lysates. Methods Enzymol. 406, 425–437 [DOI] [PubMed] [Google Scholar]
- 32. Waheed F., Speight P., Dan Q., Garcia-Mata R., Szaszi K. (2012) Affinity precipitation of active Rho-GEFs using a GST-tagged mutant Rho protein (GST-RhoA(G17A)) from epithelial cell lysates. J. Vis. Exp. 10.3791/3932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Aoudjit L., Stanciu M., Li H., Lemay S., Takano T. (2003) p38 mitogen-activated protein kinase protects glomerular epithelial cells from complement-mediated cell injury. Am. J. Physiol. Renal Physiol. 285, F765–F774 [DOI] [PubMed] [Google Scholar]
- 34. Cybulsky A. V., Takano T., Papillon J., McTavish A. J. (1999) Complement C5b-9 induces receptor tyrosine kinase transactivation in glomerular epithelial cells. Am. J. Pathol. 155, 1701–1711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Seth A., Otomo T., Yin H. L., Rosen M. K. (2003) Rational design of genetically encoded fluorescence resonance energy transfer-based sensors of cellular Cdc42 signaling. Biochemistry 42, 3997–4008 [DOI] [PubMed] [Google Scholar]
- 36. Cybulsky A. V., Takano T., Papillon J., Bijian K., Guillemette J. (2005) Activation of the extracellular signal-regulated kinase by complement C5b-9. Am. J. Physiol. Renal Physiol. 289, F593–F603 [DOI] [PubMed] [Google Scholar]
- 37. Cybulsky A. V., Papillon J., McTavish A. J. (1998) Complement activates phospholipases and protein kinases in glomerular epithelial cells. Kidney Int. 54, 360–372 [DOI] [PubMed] [Google Scholar]
- 38. Kakiashvili E., Dan Q., Vandermeer M., Zhang Y., Waheed F., Pham M., Szászi K. (2011) The epidermal growth factor receptor mediates tumor necrosis factor-α-induced activation of the ERK/GEF-H1/RhoA pathway in tubular epithelium. J. Biol. Chem. 286, 9268–9279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Krendel M., Zenke F. T., Bokoch G. M. (2002) Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat. Cell Biol. 4, 294–301 [DOI] [PubMed] [Google Scholar]
- 40. Chang Y. C., Nalbant P., Birkenfeld J., Chang Z. F., Bokoch G. M. (2008) GEF-H1 couples nocodazole-induced microtubule disassembly to cell contractility via RhoA. Mol. Biol. Cell 19, 2147–2153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lindenmeyer M. T., Eichinger F., Sen K., Anders H. J., Edenhofer I., Mattinzoli D., Kretzler M., Rastaldi M. P., Cohen C. D. (2010) Systematic analysis of a novel human renal glomerulus-enriched gene expression dataset. PloS One 5, e11545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim J. H., Mukherjee A., Madhavan S. M., Konieczkowski M., Sedor J. R. (2012) WT1-interacting protein (Wtip) regulates podocyte phenotype by cell-cell and cell-matrix contact reorganization. Am. J. Physiol. Renal Physiol. 302, F103–F115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Huber T. B., Hartleben B., Winkelmann K., Schneider L., Becker J. U., Leitges M., Walz G., Haller H., Schiffer M. (2009) Loss of podocyte aPKCλ/ι causes polarity defects and nephrotic syndrome. J. Am. Soc. Nephrol. 20, 798–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Samson T., Will C., Knoblauch A., Sharek L., von der Mark K., Burridge K., Wixler V. (2007) Def-6, a guanine nucleotide exchange factor for Rac1, interacts with the skeletal muscle integrin chain α7A and influences myoblast differentiation. J. Biol. Chem. 282, 15730–15742 [DOI] [PubMed] [Google Scholar]
- 45. Gupta S., Fanzo J. C., Hu C., Cox D., Jang S. Y., Lee A. E., Greenberg S., Pernis A. B. (2003) T cell receptor engagement leads to the recruitment of IBP, a novel guanine nucleotide exchange factor, to the immunological synapse. J. Biol. Chem. 278, 43541–43549 [DOI] [PubMed] [Google Scholar]
- 46. Shinohara M., Terada Y., Iwamatsu A., Shinohara A., Mochizuki N., Higuchi M., Gotoh Y., Ihara S., Nagata S., Itoh H., Fukui Y., Jessberger R. (2002) SWAP-70 is a guanine-nucleotide-exchange factor that mediates signalling of membrane ruffling. Nature 416, 759–763 [DOI] [PubMed] [Google Scholar]
- 47. Worthmann K., M. L., Dittrich-Breiholz O., Kracht M., Haller H., Schiffer M. (2012) Def-6 expression and localization is changed in PKCΛ/I deficient mice and podocytes. Nephrol. Dial. Transplant. 27, ii9–ii10 [Google Scholar]
- 48. Waheed F., Dan Q., Amoozadeh Y., Zhang Y., Tanimura S., Speight P., Kapus A., Szászi K. (2013) Central role of the exchange factor GEF-H1 in TNF-α-induced sequential activation of Rac, ADAM17/TACE, and RhoA in tubular epithelial cells. Mol. Biol. Cell 24, 1068–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Endlich N., Kress K. R., Reiser J., Uttenweiler D., Kriz W., Mundel P., Endlich K. (2001) Podocytes respond to mechanical stress in vitro. J. Am. Soc. Nephrol. 12, 413–422 [DOI] [PubMed] [Google Scholar]
- 50. Wang L., Ellis M. J., Fields T. A., Howell D. N., Spurney R. F. (2008) Beneficial effects of the Rho kinase inhibitor Y27632 in murine puromycin aminonucleoside nephrosis. Kidney Blood Press. Res. 31, 111–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shibata S., Nagase M., Fujita T. (2006) Fluvastatin ameliorates podocyte injury in proteinuric rats via modulation of excessive Rho signaling. J. Am. Soc. Nephrol. 17, 754–764 [DOI] [PubMed] [Google Scholar]
- 52. Kanda T., Wakino S., Hayashi K., Homma K., Ozawa Y., Saruta T. (2003) Effect of fasudil on Rho-kinase and nephropathy in subtotally nephrectomized spontaneously hypertensive rats. Kidney Int. 64, 2009–2019 [DOI] [PubMed] [Google Scholar]
- 53. Nishikimi T., Akimoto K., Wang X., Mori Y., Tadokoro K., Ishikawa Y., Shimokawa H., Ono H., Matsuoka H. (2004) Fasudil, a Rho-kinase inhibitor, attenuates glomerulosclerosis in Dahl salt-sensitive rats. J. Hypertens. 22, 1787–1796 [DOI] [PubMed] [Google Scholar]
- 54. Ishikawa Y., Nishikimi T., Akimoto K., Ishimura K., Ono H., Matsuoka H. (2006) Long-term administration of rho-kinase inhibitor ameliorates renal damage in malignant hypertensive rats. Hypertension 47, 1075–1083 [DOI] [PubMed] [Google Scholar]
- 55. Nishikimi T., Koshikawa S., Ishikawa Y., Akimoto K., Inaba C., Ishimura K., Ono H., Matsuoka H. (2007) Inhibition of Rho-kinase attenuates nephrosclerosis and improves survival in salt-loaded spontaneously hypertensive stroke-prone rats. J. Hypertens. 25, 1053–1063 [DOI] [PubMed] [Google Scholar]
- 56. Shibata S., Mu S., Kawarazaki H., Muraoka K., Ishizawa K., Yoshida S., Kawarazaki W., Takeuchi M., Ayuzawa N., Miyoshi J., Takai Y., Ishikawa A., Shimosawa T., Ando K., Nagase M., Fujita T. (2011) Rac1 GTPase in rodent kidneys is essential for salt-sensitive hypertension via a mineralocorticoid receptor-dependent pathway. J. Clin. Invest. 121, 3233–3243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Asanuma K., Yanagida-Asanuma E., Faul C., Tomino Y., Kim K., Mundel P. (2006) Synaptopodin orchestrates actin organization and cell motility via regulation of RhoA signalling. Nat. Cell Biol. 8, 485–491 [DOI] [PubMed] [Google Scholar]
- 58. Maddox A. S., Burridge K. (2003) RhoA is required for cortical retraction and rigidity during mitotic cell rounding. J. Cell Biol. 160, 255–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Holinstat M., Mehta D., Kozasa T., Minshall R. D., Malik A. B. (2003) Protein kinase Cα-induced p115RhoGEF phosphorylation signals endothelial cytoskeletal rearrangement. J. Biol. Chem. 278, 28793–28798 [DOI] [PubMed] [Google Scholar]
- 60. Pang H., Bitar K. N. (2005) Direct association of RhoA with specific domains of PKC-α. Am. J. Physiol. Cell Physiol. 289, C982–C993 [DOI] [PubMed] [Google Scholar]
- 61. Mehta D., Rahman A., Malik A. B. (2001) Protein kinase C-α signals Rho-guanine nucleotide dissociation inhibitor phosphorylation and Rho activation and regulates the endothelial cell barrier function. J. Biol. Chem. 276, 22614–22620 [DOI] [PubMed] [Google Scholar]
- 62. Ivanov A. I., Samarin S. N., Bachar M., Parkos C. A., Nusrat A. (2009) Protein kinase C activation disrupts epithelial apical junctions via ROCK-II dependent stimulation of actomyosin contractility. BMC Cell Biol. 10, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]