

Background: Malondialdehyde (MDA) released during oxidative stress modifies proteins covalently.
Results: The C terminus of complement regulator factor H binds negative patches on MDA-modified proteins, and aHUS-related mutations affect the interaction.
Conclusion: Defects in factor H binding to MDA-modified surfaces might allow deleterious complement activation on them.
Significance: MDA-modifications are potentially a new factor in aHUS pathogenesis.
Keywords: Complement System, Innate Immunity, Oxidative Stress, Pathogenesis, Protein Chemical Modification, CFH, aHUS, Malondialdehyde, Surface Recognition, Thrombotic Microangiopathy
Abstract
Atypical hemolytic uremic syndrome (aHUS) is a severe thrombotic microangiopathy characterized by uncontrolled complement activation against endothelial and blood cells. Mutations in the C-terminal target recognition domains 19–20 of complement regulator factor H (FH) are strongly associated with aHUS, but the mechanisms triggering disease onset have remained unresolved. Here we report that several aHUS-related mutations alter the binding of FH19–20 to proteins where lysines have reacted with malondialdehyde (MDA). Although FH19–20 did not interact with MDA-modified hexylamine, lysine-containing peptides, or a proteolytically degraded protein, it bound to MDA-modified polylysine. This suggests that FH19–20 recognizes only clustered MDA adducts. Binding of MDA-modified BSA to FH19–20 was ionic by nature, depended on positive residues of FH19–20, and competed with the polyanions heparin and DNA. This could not be explained with the mainly neutral adducts known to form in MDA modification. When positive charges of lysines were eliminated by acetic anhydride instead of MDA, the acetylated BSA started to bind FH19–20. Together, these results indicate that negative charges on the modified proteins dominate the interaction with FH19–20. This is beneficial for the physiological function of FH because by binding to the negative charges of the modified target, FH could prevent excess complement activation initiated by naturally occurring antibodies recognizing MDA epitopes with multiple different structures. We propose that oxidative stress leading to formation of MDA adducts is a common feature for triggers of aHUS and that failure of FH in protecting MDA-modified surfaces from complement activation is involved in the pathogenesis of the disease.
Introduction
Oxidative stress has been associated with the pathogenesis of many human diseases. It involves peroxidation of membrane lipids that leads to the generation of a variety of chemicals, including malondialdehyde (MDA)2 (1). MDA can covalently modify primary amino groups in proteins and lipids, forming adducts with various structures (Fig. 1) (1–3). The presence of MDA epitopes has been shown on apoptotic cells (4), in retinal deposits (5), and in atherosclerotic lesions (6) and low density lipoproteins extracted from them (7).
FIGURE 1.

Schematic structures of the MDA adducts described previously. MDA reacts with primary amino groups of lysines, for example, and various different products have been shown or proposed to be formed in the reactions, including N-2-propenal (A), 2-formyl-3-(alkylamino)butanal (B), MDHDC (C), 1-amino-3-iminopropene (D), and 3,5-diformyl-1,4-dihydropyridine-4-yl-pyridinium derivative (E).
Proteins can also be adducted with MDA in vitro, and, for example in the case of BSA, incubation with MDA yields MDA-BSA. With acetaldehyde added to the modification mixture, the newly formed hybrid structures have been called malondialdehyde-acetaldehyde adducts (MAA) (2, 8). The term MAA has, however, also been used for the single structure MDHDC (Fig. 1C) (9, 10). Although the most prominent one (10), MDHDC is not the only adduct formed in acetaldehyde-enhanced MDA-modification (2). MDHDC can also be formed in the absence of added acetaldehyde (1). Therefore, we have not used the prefix “MAA” in this article. Instead, all MDA-modified targets have been named with the prefix “MDA,” and the specific structure C in Fig. 1 has been designated as MDHDC.
MDA epitopes are potentially dangerous because they can activate immune responses. MDA-modified targets have, for example, been shown to increase the expression of proinflammatory cytokines in monocytes and macrophages (11, 12), most probably by signaling via scavenger receptors (13). In addition, immunoglobulins specific for MDA-modified targets have been detected in healthy and diseased individuals (14, 15). In a large cohort of diabetic patients, analysis of the antibodies against MDA-LDL showed them to be predominantly of types IgG1, IgG3, and IgM (16). These are all known to efficiently activate the complement system, an important arm of innate immunity.
The complement system is a collection of over 30 proteins that act in protecting the body against invading microbes; removing damaged cells, debris, and immune complexes; and alerting and guiding the adaptive immune system (17). Immunoglobulins bound to a surface trigger the classical pathway of complement, whereas the alternative pathway is activated spontaneously on all unprotected surfaces. Both pathways lead to the deposition of the opsonin C3b onto the target surface. Each C3b can form a convertase that generates new C3b molecules and, through this self-amplifying nature, result in prolific local C3b deposition. Propagation of the cascade finally leads to formation of lytic pores to cell membranes.
To prevent attack against host cells, the activation of complement needs to be tightly controlled. The main regulator of the alternative pathway in plasma is factor H (FH), a 155-kDa glycoprotein composed of 20 globular domains (18). The N terminus of FH is responsible for regulatory functions such as the cofactor activity needed for inactivation of C3b. FH, however, inhibits complement activation effectively only on surfaces that contain its ligands (19, 20), and the key in these interactions is the target recognition domains 19–20 (21, 22). The C terminus of FH has been shown to bind mainly anionic molecules, including heparin (23, 24), sialic acids (25), and DNA (26).
Regulation of the complement system is severely disturbed in atypical hemolytic uremic syndrome (aHUS), a rare kidney disease characterized by massive hemolysis and formation of thrombi in the microvasculature (27). Approximately 25% of aHUS patients carry a mutation in the gene encoding FH (28), with the majority of these clustering in domains 19–20 (29–31). A genetic defect in FH, however, only predisposes to aHUS. In addition, a triggering event, such as an infection, drug treatment, or pregnancy, is needed for disease onset (27), but the molecular mechanisms involved are poorly understood.
It has been described recently that FH domains 6–8 and 18–20 bind MDA epitopes (12). FH polymorphism 402H affecting domain 7 reduced the interaction, providing an explanation for the strong association of this variant with age-related macular degeneration. MDA adducts in the context of aHUS have not been studied, but, for example, patients with the disease experience severe damage to the endothelium (27), and endothelial cells injured by induction of apoptosis have been shown to exhibit MDA adducts (4, 32). We hypothesized that dysfunction in binding of FH19–20 to MDA epitopes could be involved in the pathogenesis of aHUS and, for example, explain why triggers associated with oxidative stress may initiate the disease in genetically susceptible individuals. Indeed, we were able to show that many aHUS-related mutations alter the binding of FH19–20 to MDA-modified BSA. Furthermore, although the binding of FH19–20 to MDA epitopes themselves could be detected, the interaction with modified proteins was found to be dominated by the negatively charged side chains exposed by the MDA treatment.
EXPERIMENTAL PROCEDURES
Materials
1,1,3,3-tetramethoxypropane, acetaldehyde, acetic anhydride, malondialdehyde tetrabutylammonium salt (tba-MDA), PMSF, poly-l-lysine (PLL) hydrochloride (molecular weight 15,000–30,000), ubiquitin (from bovine erythrocytes), human serum albumin, human milk lysozyme, ribonuclease A (from bovine pancreas), bovine myelin basic protein, pepsin (from porcine gastric mucosa), and α-chymotrypsin (type II from bovine pancreas) were all from Sigma-Aldrich. Hexylamine was from Merck-Schuchardt. BSA was from multiple suppliers: Sigma-Aldrich, Bovogen Biologicals, and Biowest. FH was purified mainly as described previously (33) or purchased from Complement Technology. All FH fragments were expressed and purified mainly as described previously (24). Production of the mouse mAb 1F83 recognizing the single MDHDC adduct has been documented previously (34). Goat anti-FH antibody was from Calbiochem, and HRP-conjugated anti-mouse, anti-goat, and anti-rabbit antibodies were all from Jackson ImmunoResearch Laboratories. The anti-HA tag antibody was from GeneTex. N-terminally acetylated peptides with sequences SSSSSK, SSSSSKKK, and SSSSSKKKKK were from JPT Peptide Technologies. The microtiter plates used were Nunc MaxiSorp 96 flat-bottom plates. O-phenylenediamine reagent was from Dako. Gel filtrations were done with a HiLoad 16/60, Superdex 200 pg column (GE Healthcare). Heparin beads were from a disintegrated HiTrap Heparin column (GE Healthcare). Agarose was from SeaKem LE Cambrex BioScience Rockland.
Synthesis of MDA and Hexyl-MDHDC
Malondialdehyde sodium salt (sodium-MDA) was synthesized from 1,1,3,3-tetramethoxypropane as described previously (35). 1-hexyl-4-methyl-1,4-dihydropyridine-3,5-dicarbaldehyde (hexyl-MDHDC) was synthesized as described previously (9), except that dichloromethane was used instead of chloroform. Column chromatography (Rf = 0.64, 1:9 methanol:dichloromethane) afforded the product as a yellow solid. 1H NMR analysis of the product (Bruker Avance 400) was performed: δ 9.28 (s, 2H, CHO), 6.67 (s, 2H, C=CH), 3.94 (q, J = 6.5 Hz, 1H, CH-CH3), 3.44 (t, J = 7.0 Hz, 2H, NCH2), 1.74–1.32 (m, 8H, (CH2)4), 1.11 (d, J = 7.5 Hz, 3H, CH3-CH), 0.91 (t, J = 7.0 Hz, 3H, CH3-CH).
Cloning FH6–8 and Tagging FH6–8 and FH19–20
FH domains 6–8 were cloned as described previously for FH19–20 (36) but using primers T ATA GAA CCG CGG CTA AGA TTT AAT GCA CGT GGG TTG AGC TG and ATG TAA TCT GCA GGA ACC TTG AAA CCT TGT GAT TAT CCA GAC. HA tags were added to the N termini of both FH6–8 and FH19–20 in two steps using pPICZαB plasmids containing FH6–8 and FH19–20 sequences as templates. Amplification was first done with primers T ATA GAA CCG CGG CTA AGA TTT AAT GCA CGT GGG TTG AGC TG and TAC GAT GTT CCA GAT TAC GCT ACC TTG AAA CCT TGT GAT TAT CCA GAC (for FH6–8) and TA TAG AAC CGC GGC TAT CTT TTT GCA CAA G and TAC GAT GTT CCA GAT TAC GCT AAA GAT TCT ACA GGA AAA TGT GGG CC (for FH19–20). The products were then reamplified, but with the latter primer in each primer pair changed: AGT AAT TCT GCA GGA TAC CCA TAC GAT GTT CCA GAT TAC GCT ACC (for FH6–8) and AGT AAT TCT GCA GGA TAC CCA TAC GAT GTT CCA GAT TAC GCT AAA GAT TC (for FH19–20).
Protein Measurements
Protein concentrations were determined with a BCA protein assay kit (Novagen). The relative fluorescence of proteins was measured with a Thermo Scientific Fluoroskan Ascent FL microplate reader using an excitation/emission filter pair of 355/460 nm. Absorbances at 492 nm were measured with a Labsystems iEMS Reader MF microplate reader.
Mass Spectrometry
Peptide samples were purified using Millipore C18 Zip Tips (Millipore), dried, and dissolved in 5 μl of 50% acetonitrile containing 0.1% trifluoroacetic acid. Aliquots of 1.0 μl were applied onto the target, and 1.0 μl of saturated α-cyano-4-hydroxycinnamic acid was added, followed by drying in air. The measurements were performed using a MALDI-TOF/TOF Ultraflex instrument (Bruker Daltonics). The parameter settings were optimized for analyses of peptides up to the mass-to-charge ratio (m/z) of 5000. External calibration was done using peptide calibration standard I (Bruker, part no. 206195). The N2 laser (337 nm) was used at a 50-Hz frequency. Results were analyzed with the FlexControl program (Bruker Daltonics).
MDA Modifications
MDA modifications were performed mainly as described previously (35, 37). Briefly, BSA (2 mg/ml) was incubated in 100 mm phosphate (pH 7.2) with 1–85 mm MDA in rotation at 37 °C for 17 h to produce MDA-BSA. In the indicated assays, 20 mm acetaldehyde was added to the reaction mixture. Unreacted aldehydes were removed by changing the buffer to PBS with a concentrator (Amicon Ultra-15, Ultracel 30k, Millipore).
Pepsin, human serum albumin, ubiquitin, lysozyme, ribonuclease A, FH19–20 (each 1 mg/ml) and myelin basic protein (0.5 mg/ml) were modified with 50 mm MDA in 25 mm phosphate (pH 7.3) in rotation at 37 °C for 72 h, followed by storage on ice. Controls were prepared and handled identically, except for omitting MDA or protein from the reaction mixture.
The synthetic peptides (0.9 mg/ml) used were modified with 60 mm sodium-MDA and 20 mm acetaldehyde and PLL (1.6 mg/ml) with 200 mm tba-MDA in 30 mm phosphate (pH 7.2–7.3) at 37 °C for 17 h, followed by storage on ice or at −20 °C. Controls were prepared and handled identically, except for omitting MDA and acetaldehyde, or peptides/PLL, or both from the reaction mixture.
Ubiquitin (4 mg/ml) was modified in 50 mm phosphate buffer (pH 7.4) containing 160 mm tba-MDA at 37 °C for 65 h. Unreacted MDA was removed, and the buffer was exchanged to PBS with a concentrator (3K, Microcon®, Millipore).
Procedures Common for the Microtiter Plate Assays
After coating, microtiter plate wells were blocked with either PBS or TBS containing 0.5% BSA for 1–2 h at room temperature or for 17 h at 4 °C. In binding, PBS or TBS containing 0.1–0.5% BSA was used as a buffer. Bound 1F83 was detected with HRP-conjugated anti-mouse antibody, FH with goat anti-FH antibody followed by HRP-conjugated anti-goat antibody, and anti-HA tag antibody with HRP-conjugated anti-rabbit antibody. The plates were developed with O-phenylenediamine substrate and H2O2 according to the instructions of the manufacturer. Absorbances were measured at 492 nm.
Comparison of MDA Salts
First, the relative fluorescence of 10 μg of BSA modified with sodium-MDA or tba-MDA in the presence of acetaldehyde was measured. Second, ∼2 μg of sodium- or tba-MDA-BSA was run in native PAGE. Third, microtiter plates were coated with 0–24 μg/ml of sodium- or tba-MDA-BSA and incubated with the mAb 1F83 (0.4 μg/ml) or FH (1 μg/ml).
Binding of Mutant FH19–20 to MDA-BSA
Microtiter plates were coated with MDA- and acetaldehyde-modified BSA (10 μg/ml) or native BSA as a control. Wells were incubated with WT or mutant FH19–20 (1.2–1.5 μg/ml).
Effect of NaCl on Binding to MDA-BSA
Microtiter plates were coated with MDA- and acetaldehyde-modified BSA (20 μg/ml) using native BSA as a control. Wells were incubated with FH19–20 (0.5–0.9 μg/ml) and an increasing concentration of NaCl (68.5, 137, 274 or 548 mm).
Heparin Bead Assay
FH19–20 (4 μg) in PBS was mixed with BSA treated with acetaldehyde only (0.18 mg, control) or with both acetaldehyde and malondialdehyde (0.14 mg) in a total volume of 23 μl and incubated at 37 °C for 20 min. 14 μl of this mixture was pipetted onto 7 μl of packed heparin beads in PBS and mixed. Samples of supernatant (FT, 10 μl) and start mixtures (5 μl) were drawn and run in 12.5% SDS-PAGE gel. The gel was stained with Coomassie G-250 and visualized with UV light. Bands were analyzed using ImageJ software (National Institutes of Health). The intensities of FH19–20 bands in FT were compared with the FH19–20 band intensities of the respective start mixtures (percent FT/start).
Gel Shift Assay
Linearized plasmid DNA (0.1 μg) was mixed with FH19–20 (15 μg) and various amounts of MDA- and acetaldehyde-modified BSA (0, 12, 24, 48, and 72 μg) in PBS. Glycerol was added to a final concentration of 10%, and samples run in EtBr-containing 1% agarose gel using TBE as the running buffer. DNA bands were visualized with UV light.
FH19–20 Binding to Acetylated BSA
BSA was acetylated as described previously (38). The acetylated BSA (A-BSA) as well as the control MDA-modified BSA were further purified by gel filtration using PBS as the running buffer. Microtiter plates were coated with native MDA-BSA or A-BSA (10 μg/ml). Wells were incubated with the mAb 1F83 (0.4 μg/ml) or FH19–20 (4 μg/ml).
FH19–20 Binding to Several MDA-modified Proteins with Different pI Values
Microtiter plates were coated with MDA modification mixtures of pepsin, human serum albumin, ubiquitin, ribonuclease A, FH19–20, lysozyme, or myelin basic protein (each 30 μg/ml). Reaction mixtures containing only MDA or native proteins were used as controls. Wells were incubated with the anti-MDHDC mAb 1F83 (0.7 μg/ml) or FH19–20HA (0.15 μg/ml) and anti-HA tag antibody. Isoelectric points of proteins were estimated using the ProtParam Tool (ExPASy, Swiss Institute of Bioinformatics).
Inhibition Assays with Hexyl-MDHDC
Microtiter plate wells were coated with MDA- and acetaldehyde-modified BSA (20 μg/ml) or native BSA as a control. Wells were incubated with FH19–20 (3 μg/ml) or mAb 1F83 (0.2 μg/ml) and increasing concentrations of hexyl-MDHDC (0–115 μg/ml).
Binding to MDA-modified Peptides
Microtiter plates were coated with mixtures containing the MDA-modified peptides (10 μg/ml peptide for the 1F83 and 60 μg/ml for FH19–20HA). Modification mixtures containing only MDA and acetaldehyde or only native peptides served as controls. Wells were incubated with the mAb 1F83 (0.7 μg/ml) or FH19–20HA (0.15 μg/ml) and anti-HA-tag antibody.
Binding to MDA-modified PLL
Microtiter plates were coated with MDA-PLL reaction mixture (0.01 μg/ml for the 1F83 and 10 μg/ml for FH19–20). Reaction mixtures containing MDA only or native PLL only were used as controls. Wells were incubated with the mAb 1F83 (0.7 μg/ml) or FH19–20 (1.2–1.5 μg/ml).
Effect of Proteolysis on Binding of FH19–20 to MDA-modified Protein
Equal amounts of MDA-modified ubiquitin (2.25 mg) were incubated with and without α-chymotrypsin (0.7 mg) in PBS at 37 °C for 2 h. Protease only served as one of the controls. PMSF was added to all three samples to a final concentration of 1.4 mm. The tubes were cooled on ice before adding protease to the non-proteolyzed MDA-ubiquitin control. Samples of these mixtures were incubated for 30 min at 22 °C before adding SDS-PAGE loading dye and running into a gel to verify proteolysis of the digested, but not the undigested, MDA-ubiquitin. The fluorescence of 3 μl of each mixture was measured. Microtiter plates were then coated with MDA- and acetaldehyde-modified BSA (10 μg/ml). Wells were incubated with mAb 1F83 (0.7 μg/ml) or FH19–20 (0.5 μg/ml) and increasing concentrations of proteolyzed or non-proteolyzed MDA-ubiquitin (or protease only as the other control).
Statistical Methods
One-way analysis of variance with Dunnett's multiple comparison post-tests were performed using GraphPad Prism software to assess the statistical significance of observed differences.
Structural Analysis
The location of the residues identified to mediate binding to MDA- and acetaldehyde-modified BSA on the crystal structure of FH19–20 (36) described previously was visualized with PyMOL software (DeLano Scientific). The molecular model of MDHDC was generated with ChemBioDraw Ultra and Chem3D Pro (both from CambridgeSoft). The inter-residue distances and the size of MDHDC were calculated with PyMOL.
Assays with FH6–8
The experiments with FH6–8 were performed as described for FH19–20 but with the following concentrations of FH6–8: heparin bead assay, 3 μg; gel shift assay, 30 μg; MDA-PLL assay, 1 μg/ml; hexyl-MDHDC assay, 1 μg/ml; NaCl effect assay, 0.9 μg/ml; A-BSA assay, 6 μg/ml; and MDA-peptide assay, 0.7 μg/ml.
RESULTS
Commercial tba-MDA Can Also Be Used in MDA Modifications
Because preparation of sodium-MDA commonly used in modifications is somewhat cumbersome, we asked whether the commercially available tba-MDA could be used instead. To answer this, we modified BSA with both salts of MDA and compared the products in three ways. First, the relative fluorescence resulting from MDHDC adducts was observed to be similar for sodium-MDA-BSA and tba-MDA (Fig. 2A). Second, the increase in mobility in native PAGE due to loss of positive charge in reaction with MDA was similar for BSA modified with the different MDA salts (Fig. 2B). Third, binding of the anti-MDHDC mAb 1F83 (Fig. 2C) and FH (D) to sodium- and tba-MDA-BSA was not statistically different at saturating coating. Because BSA modified with sodium- or tba-MDA behaved similarly in all our assays, some modifications were performed using tba-MDA.
FIGURE 2.

Comparison of BSA preparates modified with sodium- and tba-MDA. BSA was modified with sodium- or tba-MDA and acetaldehyde. A, relative fluorescence of 10 μg of native and MDA-modified proteins (excitation/emission, 355/460 nm). Data presented are mean ± S.D. of experiments performed three times in triplicate. B, native, sodium-MDA-modified (Na-MDA), and tba-MDA-modified BSA were run in native PAGE. The gel was stained with Coomassie G-250 and visualized with UV light. C and D, microtiter plates were coated with a dilution series of sodium- and tba-MDA-BSA. Wells were incubated with anti-MDHDC mAb 1F83 (C) or FH (D). Bound 1F83 was detected with HRP-conjugated antibody, and binding of the conjugate in the absence of 1F83 was subtracted from the results. FH was detected with anti-FH and HRP-conjugated anti-goat antibody, and FH binding to native BSA was subtracted from the results. Data presented are mean ± S.D. of experiments performed three times in triplicate.
Positively Charged Residues of FH19–20 Are Needed for Binding to MDA-BSA
Target recognition domains 19–20 of FH, shown recently to interact with MDA epitopes (12), are also a hot spot for mutations in aHUS (29–31). We asked whether the disease-related mutations would affect binding of FH19–20 to MDA-modified targets. Therefore, interactions of 12 different FH19–20 mutants with MDA-BSA were compared with that of the WT FH19–20. Eight mutations, all but one (K1188A) reported in aHUS, resulted in altered binding to MDA-modified BSA (Fig. 3A). The location of these residues on the crystal structure of FH19–20 (36) is shown in Fig. 3B. With the exception of W1183L, a positively charged residue has been mutated to alanine in all the mutants showing decreased binding. Conversely, mutants T1184R and E1198A, interacting more strongly with MDA-BSA, involved a net gain of positive charge. These results suggest that FH19–20 binds MDA-BSA via positively charged residues.
FIGURE 3.

Binding of FH19–20 mutants to MDA-BSA and the effect of NaCl on the interaction. A, microtiter plates coated with MDA-BSA were incubated with WT or mutant FH19–20. Bound proteins were detected with anti-FH antibody, and the respective HRP conjugate. The background signal obtained for FH19–20 binding to native BSA was subtracted from the results. Data presented are mean ± S.D. of experiments performed three times in triplicate. *, p < 0.05. B, previously published structure of FH19–20 (PDB code 2G7I) was used to visualize the location of the residues observed to be involved in MDA-BSA binding. A mesh representation of FH19–20 is shown, with the residues important in the interaction highlighted in dark gray. C, microtiter plates coated with MDA-BSA were incubated with FH19–20 and increasing concentrations of NaCl. Bound protein was detected as in A.
Because of the dependence on positive charges of FH19–20, we next verified the ionic nature of the interaction. Binding of FH19–20 to MDA-BSA was reduced ∼80% by increasing NaCl concentration to twice that of the physiological level (300 mm), indicating that the interaction is salt-dependent (Fig. 3C).
MDA-BSA Competes with Heparin and DNA for Binding to FH19–20
The residues involved in the binding of FH19–20 to MDA-BSA are nearly exactly the same as those responsible for the binding of FH19–20 to heparin (23, 24, 36). The overlap in the recognition sites for these ligands on FH19–20 was verified by the effective inhibition of FH19–20 binding to heparin-coated beads by MDA-BSA (Fig. 4, A and B).
FIGURE 4.

Competitive binding of MDA-BSA with heparin and DNA to FH19–20. For heparin binding assays, FH19–20 was preincubated with BSA modified with MDA and acetaldehyde (MDA-BSA) or with acetaldehyde only (control BSA). These mixtures (start) were added onto heparin beads. A, samples of flow-through (FT) were run in SDS-PAGE, the gels were stained with Coomassie G-250, and FH19–20 bands were visualized with UV light. B, the bands were analyzed using ImageJ software. The intensity of each FT protein band was compared with that of the respective start material (percent), and the means ± S.D. were calculated. The assay was done with three different sets of beads. C, linearized plasmid DNA was mixed with FH19–20 and various amounts of MDA-BSA (0, 12, 24, 48, and 72 μg). Samples were run in agarose gel electrophoresis, and DNA migration was visualized with UV light. An image of one representative gel of three is shown. The identity of each band is indicated for clarity.
It has been shown previously that FH19–20 also binds to DNA (26), but the residues responsible for the interaction are unknown. In a gel shift assay, MDA-BSA, but not native BSA, prevented the binding of FH19–20 to linearized plasmid DNA (Fig. 4C), indicating an overlap between these recognition sites as well.
Removal of Positive Charges of BSA Enhances FH19–20 Binding
Dependence on positively charged residues of FH19–20 and overlap of the recognition site with those for the polyanionic ligands suggested that the target on MDA-BSA was negatively charged. This was surprising because none of the described structures generated by MDA modification contain negative charges (Fig. 1). To test whether the removal of positive charges of lysines of BSA due to MDA modification is responsible for FH19–20 binding, we used acetic anhydride to replace the amino groups of lysines with neutral structures different from the MDA adducts. The net negative charges of the prepared acetylated and MDA-modified BSA were nearly equal, according to very similar mobilities in native PAGE (Fig. 5A), whereas only the MDA-modified BSA was recognized by the anti-MDHDC mAb 1F83 (B). Both acetylation and MDA modification resulted in enhanced binding of FH19–20 (Fig. 5C), although binding to MDA-BSA was higher. These results indicate that removal of positive charges of lysines, i.e. the increase in the net negative charge due to MDA modification of BSA, is at least partially responsible for the interaction with FH19–20.
FIGURE 5.

FH19–20 binding to acetylated BSA and several MDA-modified proteins with different pI values. A, native BSA, MDA-BSA, and A-BSA were run in native PAGE. The gel was stained with Coomassie G-250 and visualized with UV light. B and C, native, MDA-BSA, and A-BSA were immobilized on a microtiter plate, and the wells were incubated with the anti-MDHDC mAb 1F83 (B) or FH19–20 (C). Bound 1F83 was detected with an anti-mouse HRP conjugate and FH19–20 with goat anti-FH antibody and the respective HRP-conjugate. Background signals obtained with detection antibodies only were subtracted from the results. Data presented are mean ± S.D. of experiments performed three times in triplicate. *, p < 0.05. D and E, microtiter plates were coated with MDA modification mixtures of pepsin (pI 3.2), human serum albumin (pI 5.9), ubiquitin (pI 6.6), ribonuclease A (pI 8.6), FH19–20 (pI 9.1), lysozyme (pI 9.3), and myelin basic protein (pI 11.3). Buffer containing only native proteins or MDA were used as controls. Wells were incubated with anti-MDHDC mAb 1F83 (D) or FH19–20HA and anti-HA-tag antibody (E). Bound 1F83 and anti-HA antibodies were detected with HRP-conjugated anti-mouse and anti-rabbit antibody, respectively. Background signals obtained with detection antibodies and buffer only were subtracted from the results. Data presented are mean ± S.D. of experiments performed three times in triplicate.
Modification of Proteins with Decreasing Isoelectric Points Results in Increasing FH19–20 Binding
To get more support for the role of negative charges in binding of FH19–20 to MDA-modified targets, seven proteins with different pIs were modified with MDA. Binding of FH19–20 showed an increasing trend with decreasing pI of the target protein (Fig. 5E). The observed effect was not related to the MDA adducts per se because binding of anti-MDHDC mAb 1F83 was not increased with a decreasing pI (Fig. 5D). It is noteworthy that unmodified pepsin with a pI of only 3 and MDA-BSA bound approximately equally to FH19–20 (Fig. 5E). It should also be noted that pepsin has a very low content of modifiable lysines (only a single residue or 0.3%) when compared with the other proteins used (4–10%). Taken together, these results give support for FH19–20 interacting with negative charges on modified proteins.
FH19–20 Interacts Only with an Abundance of MDA Adducts
Because the binding of FH19–20 was weaker to acetylated than to MDA-modified BSA, we asked whether FH19–20 would interact with the MDA adducts themselves. To test the binding of FH19–20 to a single adduct, we synthesized hexyl-MDHDC. This compound resembles the MDHDC adduct in a side chain of lysine (9) and has been shown earlier to inhibit the binding of immunoglobulins to MDA-modified albumin (10, 39). Also, in our hands, hexyl-MDHDC dose-dependently prevented the binding of the anti-MDHDC mAb to MDA-BSA, with approximately equimolar concentrations needed to reduce the binding by 50% (Fig. 6A). Hexyl-MDHDC did not, however, inhibit the binding of FH19–20 to MDA-BSA, not even when present at 250-fold molar excess (Fig. 6A). This indicates that single MDHDC adducts on MDA-BSA are not responsible for the binding of FH19–20.
FIGURE 6.

Binding of FH19–20 to hexyl-MDHDC and MDA-modified peptides. A, MDA-BSA was immobilized on a microtiter plate, and wells were incubated with anti-MDHDC mAb 1F83 or FH19–20 and increasing concentrations of hexyl-MDHDC. Bound 1F83 was detected with an HRP-conjugate, and background binding in the absence of 1F83 was subtracted from the results. FH19–20 was detected with anti-FH antibody and the respective HRP-conjugate, and binding to the BSA background was subtracted from the results. Data presented are mean ± S.D. of experiments performed three times in triplicate. B, peptides with sequences SSSSSK (K1), SSSSSKKK (K3), SSSSSKKKKK (K5) were modified with MDA and acetaldehyde and then subjected to analysis with MALDI-TOF/TOF. Detected ion masses were compared with those of unmodified peptides. Resulting changes in mass-to-charge ratios (Δm/z) were tested against different combinations of possible adducts known to be formed in MDA modifications, exemplified by the MDHDC structure (mass increase of 134.04). Data for the peaks with the highest intensities for peptides containing three and five lysines (MDA-K3 and MDA-K5) is given. C and D, microtiter plates were coated with the peptide modification mixtures or controls (buffer containing only native peptides or MDA and acetaldehyde). Wells were incubated with the anti-MDHDC mAb 1F83 (C) or FH19–20HA and anti-HA tag antibody (D). Bound 1F83 and anti-HA antibody were detected with HRP-conjugated anti-mouse and anti-rabbit antibodies, respectively. Background signals obtained with detection antibodies only were subtracted from the results. Data presented are mean ± S.D. of experiments performed three times in triplicate.*, p < 0.05; ns, not significant.
Instead of a single adduct, the binding of FH19–20 could require multiple adducts in a row. To test this, we chose to modify peptides containing one, three, or five lysines with MDA. The formation of MDHDC adducts was verified by showing the binding of the anti-MDHDC mAb 1F83 (Fig. 6C), and the multiplicity of these structures within the peptides containing three or five lysines was confirmed with mass spectrometry (MALDI-TOF/TOF) (B). No interaction of FH19–20 with MDA-modified peptides was observed (Fig. 6E). These results indicate that FH19–20 does not recognize up to five neighboring MDA adducts.
Because MDA adducts might also appear in large clusters on modified proteins, we next tested whether a surface with several MDA-modified lysines would bind to FH19–20. To avoid the contribution of the negatively charged residues to the interaction, we used MDA-modified poly-l-lysine (MDA-PLL). The presence of MDA adducts in modified PLL was confirmed by showing the binding of the anti-MDHDC mAb 1F83 (Fig. 7A). Also, FH19–20 interacted with the modified, but not with the native, PLL (Fig. 7B). This indicates that FH19–20 interacts with MDA adducts when these are present in abundance on a surface, even in the absence of negatively charged side chains of modified proteins.
FIGURE 7.
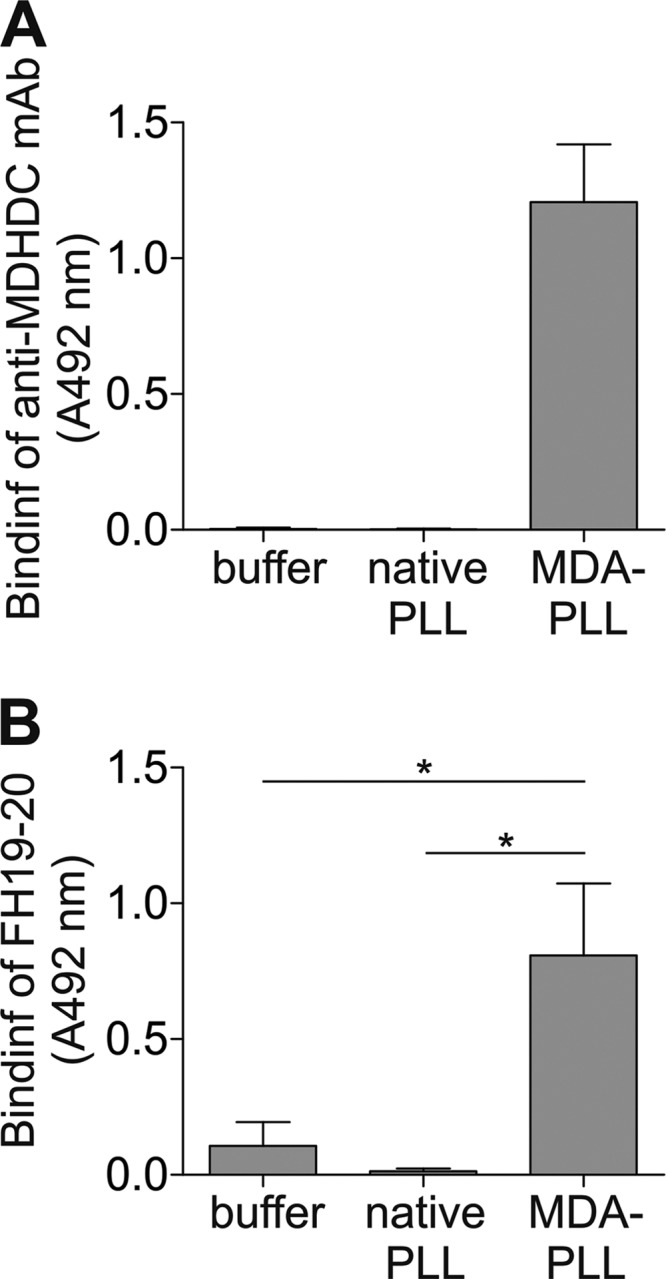
Binding of FH19–20 to MDA-modified PLL. Microtiter plates were coated with PLL modification mixture containing MDA or controls (native PLL or buffer with MDA only). Wells were incubated with the anti-MDHDC mAb 1F83 (A) or FH19–20 (B). Bound 1F83 was detected with HRP-conjugated antibody. FH19–20 was detected with anti-FH antibody and an HRP conjugate. Background signals obtained with detection antibodies only were subtracted from the results. Data presented are mean ± S.D. of experiments performed three times in triplicate. *, p < 0.05.
Integrity of the MDA-modified Protein Is Essential for FH19–20 Binding
Because FH19–20 bound only to a surface with multiple MDA adducts, we asked whether the interactions of FH19–20 with MDA-modified proteins would also require large binding sites with several closely located structures. We first used the crystal structure of FH19–20 (36) to visualize the location of residues whose mutation had an effect on binding to MDA-BSA. Compared with the size of a modeled MDHDC adduct (∼7 Å), the binding site on FH19–20 was found to be very large, with inter-residue distances of up to 20 Å (Fig. 8).
FIGURE 8.

Location of the residues involved in binding to MDA-BSA in the crystal structure of FH19–20. The structure of FH19–20 published previously (PDB code 2G7I) was used to visualize the location of the residues observed to be involved in MDA-BSA binding (Fig. 3A). A mesh representation of FH19–20 is shown, with the residues important in the interaction with MDA-BSA marked with dark gray. Distances between the tips of the side chains are indicated in ångströms next to the dashed lines. A modeled structure of an MDHDC adduct is shown on the right for size comparison.
To test whether the structures on MDA-modified proteins responsible for binding to FH19–20 also need to be present as clusters, we chose to analyze the effect of proteolysis on interactions of MDA-ubiquitin with FH19–20. Digestion of modified ubiquitin into peptides had no effect on the MDA adducts as such because binding of anti-MDHDC mAb 1F83 and fluorescence resulting from the MDHDC adducts were unaltered (Fig. 9, A–C). Although the intact MDA-ubiquitin was able to prevent binding of FH19–20 to MDA-modified BSA, protease treatment abolished the inhibition (Fig. 9D). These results indicate that the FH19–20 interaction needs the integrity of the modified protein and suggest that the sites recognized on the MDA-modified proteins are surfaces consisting of several closely spaced structures.
FIGURE 9.

Effect of proteolysis on the ability of MDA-ubiquitin to inhibit FH19–20 binding to MDA-BSA. A, MDA-ubiquitin was first incubated with α-chymotrypsin, and then the proteolysis was stopped with PMSF. Unproteolyzed MDA-ubiquitin with PMSF-inhibited α-chymotrypsin and the protease with inhibitor only served as controls. Samples were run in SDS-PAGE, and the gel was stained with Coomassie G-250 and visualized with UV light. B, relative fluorescence values of proteolysis mixtures of MDA-ubiquitin were measured using an excitation/emission pair of 355/460 nm. C and D, MDA-BSA was immobilized on microtiter plates. The wells were incubated with the anti-MDHDC mAb 1F83 (C) or FH19–20 (D) and increasing concentrations of proteolyzed or unproteolyzed MDA-ubiquitin. Reaction mixtures containing the protease and PMSF only or native ubiquitin served as controls. Bound 1F83 was detected with an HRP-conjugate, and background binding in the absence of 1F83 was subtracted from the results. Binding of FH19–20 was detected with anti-FH antibody and the respective HRP conjugate. FH19–20 binding to the BSA background was subtracted from the results. Data presented are mean ± S.D. of experiments performed three times in triplicate.
FH6–8 Behaves Differently from FH19–20 in Binding to MDA-modified Targets
Because domains 6–8 of FH have also been shown to interact with MDA-modified BSA (12), we repeated most of the assays using FH6–8. Similar to FH19–20, interactions of FH6–8 with both heparin (Fig. 10A) and DNA (B) could be inhibited with MDA-BSA. Binding to MDA-PLL was observed also for FH6–8 (Fig. 10C), and, as with FH19–20, hexyl-MDHDC was unable to inhibit the interaction of FH6–8 with MDA-BSA (D). Unlike FH19–20, the binding of FH6–8 to MDA-BSA was only partially inhibited with NaCl (Fig. 10E), and no interaction with acetylated BSA was detected (F). Further, dissimilar to FH19–20, the binding of FH6–8 to MDA-modified peptides was observed (Fig. 10G). Taken together, FH domains 6–8 and 19–20 behave partially differently with MDA-modified targets.
FIGURE 10.

Interactions of FH6–8 with modified targets. A, FH6–8 was preincubated with native or MDA-modified BSA. Samples of these mixtures (start) and FT after incubation of those mixtures with heparin beads were run in SDS-PAGE and analyzed as the data shown in Fig. 4. The assay was done with three different sets of beads, and data from a representative experiment are shown. B, a linearized plasmid DNA (0.1 μg) was mixed with FH6–8 and various amounts of MDA-BSA (0, 27, 54, and 81 μg) in 30 μl. Samples were run in agarose gel electrophoresis, and DNA migration was visualized with UV light. An image of one representative gel of three is shown. C–G, microtiter plate assays of FH6–8 binding to modified proteins and peptides. C, wells were coated with PLL modification mixture containing MDA or controls (native PLL or buffer with MDA only) and then incubated with FH6–8. D, wells were immobilized with MDA-BSA following incubation with FH6–8 and increasing concentrations of hexyl-MDHDC. E, wells were coated with MDA-BSA and then incubated with FH6–8 and increasing concentrations of NaCl. F, wells were immobilized with native BSA, MDA-BSA, or A-BSA, followed by incubation with FH6–8. G, wells were coated with the peptide modification mixtures or controls (buffer containing only native peptides or MDA and acetaldehyde) and then incubated with FH6–8HA and anti-HA tag antibody. C–F, the bound FH6–8 was detected with anti-FH antibody and an HRP-conjugate. G, the anti-HA antibody was detected with an HRP conjugate. The background signal obtained with detection antibodies only was subtracted from the results. Data presented are mean ± S.D. of experiments performed three times in triplicate. *, p < 0.05; ns, not significant.
DISCUSSION
Factor H has been shown recently to interact with MDA epitopes via domains 6–8 and 18–20 (12). In this study, we show that FH19–20 binds only to clustered MDA adducts, and, even in that case, the negative charges exposed on the protein by the MDA modification dominate the interaction. Further, several aHUS-related mutations in FH19–20 were found to alter binding to MDA-modified targets, proposing that defects in FH-MDA interactions could be involved in the pathogenesis of aHUS.
FH domains 19–20, important for target discrimination, have been shown to bind various anionic ligands, including glycosaminoglycans represented by heparin (23, 24), DNA (26), and sialic acids (25). Taking into account the high electropositivity of FH domains 19–20 (theoretical pI ≈9), our observation of the binding target on MDA-modified BSA being negatively charged is not surprising. None of the reported MDA adducts themselves, however, are negatively charged (Fig. 1) (1–3). BSA, on the other hand, is a rather anionic protein (pI 5) (40), and modification of its abundant lysines (10%) is expected to turn it very negative overall. In other words, masking the positive charges of lysines presumably makes the negatively charged side chains of BSA more exposed and, therefore, available for interactions. To support this, FH19–20 was shown to interact with acetylated BSA in which the positive charges of lysines had been removed without introduction of MDA adducts (Fig. 5C). Because acetyl groups themselves have not been found to affect FH-mediated complement regulation (41, 42), the carboxylic acid groups of amino acid side chains are likely to be responsible for the interaction. Further, when multiple different proteins were modified with MDA, binding of FH19–20 was shown to increase with decreasing pI of the target protein (Fig. 5E). On the basis of these aspects, it appears plausible that the interaction of FH19–20 with MDA-modified BSA is at least partially mediated by negatively charged residues of the latter.
Binding of FH19–20 to MDA-polylysine devoid of negatively charged residues indicates that FH19–20 also interacts with MDA adducts per se (Fig. 7B). Like all our MDA-modified targets, MDA-polylysine was shown to contain MDHDC adducts using the specific anti-MDHDC antibody 1F83. Because MDA modification can result in the formation of other adducts besides MDHDC (1, 2), and because the presence of those could not be excluded in our samples, we cannot conclude which of the MDA adducts FH19–20 interacts with. Nevertheless, negative charges on MDA-modified proteins probably dominate the interactions with FH19–20. Being neutral structures (Fig. 1), the MDA adducts cannot explain either the ionic nature of the interaction (Fig. 3C) or its dependence on the widely spread positively charged residues of FH19–20 involved in the interaction (A). Therefore, the surfaces with multiple, closely spaced structures essential for FH19–20 binding, as indicated by the effect of proteolysis on the interaction (Fig. 9D), are best interpreted as clusters of negatively charged amino acid side chains.
The ability of FH to recognize MDA-modified surfaces on the basis of their negative charge without a requirement for certain adducts like MDHDC could actually be beneficial. When proteins are treated with MDA, N-propenal (Fig. 1) is the major adduct with very little complex structures formed (37, 43, 44). If MDHDC is desired in abundance, extra acetaldehyde is needed in modifications (10). Although MDHDC has been shown in atherosclerotic aortas (34), concentrations of both MDA and acetaldehyde in serum are generally very low (45, 46). Therefore, in vivo complex adducts are likely to be very sparse, and simple structures presumably dominate. The complement-activating anti-MDA immunoglobulins of human serum (16), on the other hand, supposedly recognize a multitude of different adduct structures. Thus, by recognizing modified targets mainly on the basis of their negative charges, FH would be able to regulate complement activation on any surface, regardless of the complexity of the MDA adducts formed.
A clear situation when complement inhibition on surfaces with MDA modifications could be needed is aHUS. In this disease, there is severe damage to the endothelium (27), and MDA adducts have been shown on endothelial cells injured by induction of apoptosis (4, 32). Another characteristic of aHUS is the formation of thrombi in the microvasculature (27). During platelet activation, MDA is formed in equimolar amounts compared with the synthetized thromboxane A2 (47). Further, the predominating acidic pH in the inflammatory environment in the thrombotic vessels favors the more reactive form of MDA (1), leading to the enhancement of modifications. In addition to endothelial damage and thrombosis, patients with aHUS also experience intravascular hemolysis (27), leading to liberation of hemoglobin and iron into plasma. “Free” redox-active iron cannot only initiate but particularly efficiently catalyze lipid peroxidation (48, 49), a source of MDA (1). Taken together, it is evident that MDA is formed in aHUS.
Supporting a role for oxidative modifications like MDA adducts in complement regulation, the binding of FH to human endothelial cells has been shown to increase after their exposure to purified heme (50). In this context, our finding of altered binding of FH19–20 to MDA-modified BSA because of several aHUS-related mutations (Fig. 3A) becomes highly interesting. Mutations in FH19–20, however, affect its interactions with other ligands as well (23, 24). Of the two binding sites for the C3d part of C3b (51, 52), the one in domain 19 that is close to but does not overlap with the MDA-BSA site, can be used simultaneously with the heparin binding site in domain 20 (52). The heparin binding site is, however, overlapping with that for MDA-BSA (Fig. 4A). Because treatment with glycosaminoglycan-degrading enzymes does not affect FH binding to apoptotic cells (26) and because it only partially abolishes FH19–20 interaction with kidney tissue (53), some other ligand in addition to heparin or glycosaminoglycans must be present on self surfaces. Therefore, MDA adducts and defects in FH-mediated protection of surfaces containing them are brought forward as an additional possible factor in the pathogenesis of aHUS.
Mutations in the C terminus of FH, however, only predispose to the syndrome (27). Although one individual falls ill as a newborn baby, another with the same mutation encounters the disease only in adulthood. This is likely because, in addition to a genetic defect, a triggering event such as an infection, a drug, or pregnancy (27) is needed for disease onset. Interestingly, many of the agents implicated as triggers of aHUS have also been associated with oxidative stress. For example, the immunosuppressant drug cyclosporin A has been shown to result mainly in increased levels of MDA when injected into rats (54, 55). Further, the concentration of MDA in the plasma of both patients infected with HIV (56, 57) and women with a normal pregnancy (58, 59) has been found to be elevated. Hence, should a trigger lead to formation of MDA adducts on surfaces exposed to serum, anti-MDA immunoglobulins might initiate complement activation. If mutations in FH19–20 limit the proper interaction of FH with the modified targets on the cells, the regulation of the complement cascade is lost, with deleterious consequences. In other words, malfunction of the C terminus of FH in recognizing MDA-modified surfaces could explain why a trigger associated with oxidative stress could initiate the disease in individuals with a mutation in FH19–20.
Although not within the focus of this study, interactions of FH6–8 with MDA-modified targets were also studied. Interestingly, FH6–8 was shown to behave differently from FH19–20 in some binding assays. Further studies are needed to confirm whether dissimilar MDA modifications are formed in age-related macular degeneration and aHUS. If this is the case, a difference in the recognition of the variable MDA adducts by the two sites of FH could explain the association of polymorphism in domain 7 with age-related macular degeneration and mutations in domains 19–20 with aHUS.
In conclusion, the data presented in this study provide a detailed picture of the binding of FH19–20 to MDA-modified targets. Although FH19–20 was found to bind MDA adducts as such, the results indicate that the negatively charged side chains exposed during the modification of proteins presumably dominate the interaction. This is clearly beneficial, considering the physiological role of the C terminus of FH in protecting self cells from complement attack. By interacting with the negatively charged residues of MDA-modified proteins, FH could prevent excess complement activation on surfaces containing adducts with multiple different structures. Finally, and clinically most importantly, we showed the interaction of MDA-modified BSA with FH19–20 to be altered by many aHUS-related mutations, proposing that defects in complement regulation on surfaces containing MDA modifications are involved in the pathogenesis of the disease.
Acknowledgments
We thank Heli Flink for assistance with purification and 1H NMR analysis of hexyl-MDHDC; Sini Miettinen for MS analyses; and Marjatta Ahonen, Pirkko Kokkonen, and Kirsti Widing for general technical assistance.
This work was supported by research grants from the Sigrid Jusélius Foundation and by Academy of Finland Grants 128646, 255922, and 259793.
- MDA
- malondialdehyde
- MAA
- malondialdehyde-acetaldehyde adduct
- MDHDC
- 4-methyl-1,4-dihydropyridine-3,5-dicarbaldehyde
- FH
- factor H
- aHUS
- atypical hemolytic uremic syndrome
- tba-MDA
- tetrabutylammonium salt of malondialdehyde
- PLL
- poly-l-lysine
- A-BSA
- BSA modified with acetic anhydride
- FT
- flow-through.
REFERENCES
- 1. Esterbauer H., Schaur R. J., Zollner H. (1991) Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic. Biol. Med. 11, 81–128 [DOI] [PubMed] [Google Scholar]
- 2. Kearley M. L., Patel A., Chien J., Tuma D. J. (1999) Observation of a new nonfluorescent malondialdehyde-acetaldehyde-protein adduct by 13C NMR spectroscopy. Chem. Res. Toxicol. 12, 100–105 [DOI] [PubMed] [Google Scholar]
- 3. Itakura K., Uchida K., Osawa T. (1996) A novel fluorescent malondialdehyde-lysine adduct. Chem. Phys. Lipids 84, 75–79 [Google Scholar]
- 4. Chang M. K., Bergmark C., Laurila A., Hörkkö S., Han K. H., Friedman P., Dennis E. A., Witztum J. L. (1999) Monoclonal antibodies against oxidized low-density lipoprotein bind to apoptotic cells and inhibit their phagocytosis by elicited macrophages. Evidence that oxidation-specific epitopes mediate macrophage recognition. Proc. Natl. Acad. Sci. U.S.A. 96, 6353–6358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schutt F., Bergmann M., Holz F. G., Kopitz J. (2003) Proteins modified by malondialdehyde, 4-hydroxynonenal, or advanced glycation end products in lipofuscin of human retinal pigment epithelium. Invest. Ophthalmol. Vis. Sci. 44, 3663–3668 [DOI] [PubMed] [Google Scholar]
- 6. Haberland M. E., Fong D., Cheng L. (1988) Malondialdehyde-altered protein occurs in atheroma of Watanabe heritable hyperlipidemic rabbits. Science 241, 215–218 [DOI] [PubMed] [Google Scholar]
- 7. Ylä-Herttuala S., Palinski W., Rosenfeld M. E., Parthasarathy S., Carew T. E., Butler S., Witztum J. L., Steinberg D. (1989) Evidence for the presence of oxidatively modified low density lipoprotein in atherosclerotic lesions of rabbit and man. J. Clin. Invest. 84, 1086–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tuma D. J., Thiele G. M., Xu D., Klassen L. W., Sorrell M. F. (1996) Acetaldehyde and malondialdehyde react together to generate distinct protein adducts in the liver during long-term ethanol administration. Hepatology 23, 872–880 [DOI] [PubMed] [Google Scholar]
- 9. Xu D., Thiele G. M., Kearley M. L., Haugen M. D., Klassen L. W., Sorrell M. F., Tuma D. J. (1997) Epitope characterization of malondialdehyde-acetaldehyde adducts using an enzyme-linked immunosorbent assay. Chem. Res. Toxicol. 10, 978–986 [DOI] [PubMed] [Google Scholar]
- 10. Duryee M. J., Klassen L. W., Schaffert C. S., Tuma D. J., Hunter C. D., Garvin R. P., Anderson D. R., Thiele G. M. (2010) Malondialdehyde-acetaldehyde adduct is the dominant epitope after MDA modification of proteins in atherosclerosis. Free Radic. Biol. Med. 49, 1480–1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shanmugam N., Figarola J. L., Li Y., Swiderski P. M., Rahbar S., Natarajan R. (2008) Proinflammatory effects of advanced lipoxidation end products in monocytes. Diabetes 57, 879–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weismann D., Hartvigsen K., Lauer N., Bennett K. L., Scholl H. P., Charbel Issa P., Cano M., Brandstätter H., Tsimikas S., Skerka C., Superti-Furga G., Handa J. T., Zipfel P. F., Witztum J. L., Binder C. J. (2011) Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature 478, 76–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shechter I., Fogelman A. M., Haberland M. E., Seager J., Hokom M., Edwards P. A. (1981) The metabolism of native and malondialdehyde-altered low density lipoproteins by human monocyte-macrophages. J. Lipid Res. 22, 63–71 [PubMed] [Google Scholar]
- 14. Salonen J. T., Ylä-Herttuala S., Yamamoto R., Butler S., Korpela H., Salonen R., Nyyssönen K., Palinski W., Witztum J. L. (1992) Autoantibody against oxidised LDL and progression of carotid atherosclerosis. Lancet 339, 883–887 [DOI] [PubMed] [Google Scholar]
- 15. Maggi E., Bellazzi R., Gazo A., Seccia M., Bellomo G. (1994) Autoantibodies against oxidatively-modified LDL in uremic patients undergoing dialysis. Kidney Int. 46, 869–876 [DOI] [PubMed] [Google Scholar]
- 16. Virella G., Koskinen S., Krings G., Onorato J. M., Thorpe S. R., Lopes-Virella M. (2000) Immunochemical characterization of purified human oxidized low-density lipoprotein antibodies. Clin. Immunol. 95, 135–144 [DOI] [PubMed] [Google Scholar]
- 17. Ricklin D., Hajishengallis G., Yang K., Lambris J. D. (2010) Complement. A key system for immune surveillance and homeostasis. Nat. Immunol. 11, 785–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Makou E., Herbert A. P., Barlow P. N. (2013) Functional anatomy of complement factor H. Biochemistry 52, 3949–3962 [DOI] [PubMed] [Google Scholar]
- 19. Fearon D. T. (1978) Regulation by membrane sialic acid of β1H-dependent decay-dissociation of amplification C3 convertase of the alternative complement pathway. Proc. Natl. Acad. Sci. U.S.A. 75, 1971–1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pangburn M. K., Müller-Eberhard H. J. (1978) Complement C3 convertase. Cell surface restriction of β1H control and generation of restriction on neuraminidase-treated cells. Proc. Natl. Acad. Sci. U.S.A. 75, 2416–2420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ferreira V. P., Herbert A. P., Hocking H. G., Barlow P. N., Pangburn M. K. (2006) Critical role of the C-terminal domains of factor H in regulating complement activation at cell surfaces. J. Immunol. 177, 6308–6316 [DOI] [PubMed] [Google Scholar]
- 22. Józsi M., Oppermann M., Lambris J. D., Zipfel P. F. (2007) The C-terminus of complement factor H is essential for host cell protection. Mol. Immunol. 44, 2697–2706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ferreira V. P., Herbert A. P., Cortés C., McKee K. A., Blaum B. S., Esswein S. T., Uhrín D., Barlow P. N., Pangburn M. K., Kavanagh D. (2009) The binding of factor H to a complex of physiological polyanions and C3b on cells is impaired in atypical hemolytic uremic syndrome. J. Immunol. 182, 7009–7018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lehtinen M. J., Rops A. L., Isenman D. E., van der Vlag J., Jokiranta T. S. (2009) Mutations of factor H impair regulation of surface-bound C3b by three mechanisms in atypical hemolytic uremic syndrome. J. Biol. Chem. 284, 15650–15658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ram S., Sharma A. K., Simpson S. D., Gulati S., McQuillen D. P., Pangburn M. K., Rice P. A. (1998) A novel sialic acid binding site on factor H mediates serum resistance of sialylated Neisseria gonorrhoeae. J. Exp. Med. 187, 743–752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Leffler J., Herbert A. P., Norström E., Schmidt C. Q., Barlow P. N., Blom A. M., Martin M. (2010) Annexin-II, DNA, and histones serve as factor H ligands on the surface of apoptotic cells. J. Biol. Chem. 285, 3766–3776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Noris M., Remuzzi G. (2009) Atypical hemolytic-uremic syndrome. N. Engl. J. Med. 361, 1676–1687 [DOI] [PubMed] [Google Scholar]
- 28. Loirat C., Frémeaux-Bacchi V. (2011) Atypical hemolytic uremic syndrome. Orphanet. J. Rare Dis. 6, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Caprioli J., Bettinaglio P., Zipfel P. F., Amadei B., Daina E., Gamba S., Skerka C., Marziliano N., Remuzzi G., Noris M., Italian Registry of Familial and Recurrent HUS/TTP (2001) The molecular basis of familial hemolytic uremic syndrome. Mutation analysis of factor H gene reveals a hot spot in short consensus repeat 20. J. Am. Soc. Nephrol. 12, 297–307 [DOI] [PubMed] [Google Scholar]
- 30. Pérez-Caballero D., González-Rubio C., Gallardo M. E., Vera M., López-Trascasa M., Rodríguez de Córdoba S., Sánchez-Corral P. (2001) Clustering of missense mutations in the C-terminal region of factor H in atypical hemolytic uremic syndrome. Am. J. Hum. Genet. 68, 478–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Richards A., Buddles M. R., Donne R. L., Kaplan B. S., Kirk E., Venning M. C., Tielemans C. L., Goodship J. A., Goodship T. H. (2001) Factor H mutations in hemolytic uremic syndrome cluster in exons 18–20, a domain important for host cell recognition. Am. J. Hum. Genet. 68, 485–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang C., Turunen S. P., Kummu O., Veneskoski M., Lehtimäki J., Nissinen A. E., Hörkkö S. (2013) Natural antibodies of newborns recognize oxidative stress-related malondialdehyde acetaldehyde adducts on apoptotic cells and atherosclerotic plaques. Int. Immunol. 25, 575–587 [DOI] [PubMed] [Google Scholar]
- 33. Koistinen V., Wessberg S., Leikola J. (1989) Common binding region of complement factors B, H and CR1 on C3b revealed by monoclonal anti-C3d. Complement Inflamm. 6, 270–280 [DOI] [PubMed] [Google Scholar]
- 34. Yamada S., Kumazawa S., Ishii T., Nakayama T., Itakura K., Shibata N., Kobayashi M., Sakai K., Osawa T., Uchida K. (2001) Immunochemical detection of a lipofuscin-like fluorophore derived from malondialdehyde and lysine. J. Lipid Res. 42, 1187–1196 [PubMed] [Google Scholar]
- 35. Thiele G. M., Klassen L. W., Tuma D. J. (2008) Formation and immunological properties of aldehyde-derived protein adducts following alcohol consumption. Methods Mol. Biol. 447, 235–257 [DOI] [PubMed] [Google Scholar]
- 36. Jokiranta T. S., Jaakola V. P., Lehtinen M. J., Pärepalo M., Meri S., Goldman A. (2006) Structure of complement factor H carboxyl-terminus reveals molecular basis of atypical haemolytic uremic syndrome. EMBO J. 25, 1784–1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ishii T., Kumazawa S., Sakurai T., Nakayama T., Uchida K. (2006) Mass spectroscopic characterization of protein modification by malondialdehyde. Chem. Res. Toxicol. 19, 122–129 [DOI] [PubMed] [Google Scholar]
- 38. Tabachnick M., Sobotka H. (1960) Azoproteins. II. A spectrophotometric study of the coupling of diazotized arsanilic acid with proteins. J. Biol. Chem. 235, 1051–1054 [PubMed] [Google Scholar]
- 39. Rolla R., Vay D., Mottaran E., Parodi M., Traverso N., Aricó S., Sartori M., Bellomo G., Klassen L. W., Thiele G. M., Tuma D. J., Albano E. (2000) Detection of circulating antibodies against malondialdehyde-acetaldehyde adducts in patients with alcohol-induced liver disease. Hepatology 31, 878–884 [DOI] [PubMed] [Google Scholar]
- 40. Salis A., Boström M., Medda L., Cugia F., Barse B., Parsons D. F., Ninham B. W., Monduzzi M. (2011) Measurements and theoretical interpretation of points of zero charge/potential of BSA protein. Langmuir 27, 11597–11604 [DOI] [PubMed] [Google Scholar]
- 41. Michalek M. T., Bremer E. G., Mold C. (1988) Effect of gangliosides on activation of the alternative pathway of human complement. J. Immunol. 140, 1581–1587 [PubMed] [Google Scholar]
- 42. Michalek M. T., Mold C., Bremer E. G. (1988) Inhibition of the alternative pathway of human complement by structural analogues of sialic acid. J. Immunol. 140, 1588–1594 [PubMed] [Google Scholar]
- 43. Ishii T., Ito S., Kumazawa S., Sakurai T., Yamaguchi S., Mori T., Nakayama T., Uchida K. (2008) Site-specific modification of positively-charged surfaces on human serum albumin by malondialdehyde. Biochem. Biophys. Res. Commun. 371, 28–32 [DOI] [PubMed] [Google Scholar]
- 44. Shao B., Pennathur S., Pagani I., Oda M. N., Witztum J. L., Oram J. F., Heinecke J. W. (2010) Modifying apolipoprotein A-I by malondialdehyde, but not by an array of other reactive carbonyls, blocks cholesterol efflux by the ABCA1 pathway. J. Biol. Chem. 285, 18473–18484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Giustarini D., Dalle-Donne I., Tsikas D., Rossi R. (2009) Oxidative stress and human diseases. Origin, link, measurement, mechanisms, and biomarkers. Crit. Rev. Clin. Lab. Sci. 46, 241–281 [DOI] [PubMed] [Google Scholar]
- 46. Halvorson M. R., Noffsinger J. K., Peterson C. M. (1993) Studies of whole blood-associated acetaldehyde levels in teetotalers. Alcohol 10, 409–413 [DOI] [PubMed] [Google Scholar]
- 47. Hecker M., Haurand M., Ullrich V., Diczfalusy U., Hammarström S. (1987) Products, kinetics, and substrate specificity of homogeneous thromboxane synthase from human platelets. Development of a novel enzyme assay. Arch. Biochem. Biophys. 254, 124–135 [DOI] [PubMed] [Google Scholar]
- 48. Balla J., Vercellotti G. M., Jeney V., Yachie A., Varga Z., Jacob H. S., Eaton J. W., Balla G. (2007) Heme, heme oxygenase, and ferritin. How the vascular endothelium survives (and dies) in an iron-rich environment. Antioxid. Redox Signal 9, 2119–2137 [DOI] [PubMed] [Google Scholar]
- 49. Cheng Z., Li Y. (2007) What is responsible for the initiating chemistry of iron-mediated lipid peroxidation. An update. Chem. Rev. 107, 748–766 [DOI] [PubMed] [Google Scholar]
- 50. Frimat M., Tabarin F., Dimitrov J. D., Poitou C., Halbwachs-Mecarelli L., Fremeaux-Bacchi V., Roumenina L. T. (2013) Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood 122, 282–292 [DOI] [PubMed] [Google Scholar]
- 51. Kajander T., Lehtinen M. J., Hyvärinen S., Bhattacharjee A., Leung E., Isenman D. E., Meri S., Goldman A., Jokiranta T. S. (2011) Dual interaction of factor H with C3d and glycosaminoglycans in host-nonhost discrimination by complement. Proc. Natl. Acad. Sci. U.S.A. 108, 2897–2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Morgan H. P., Schmidt C. Q., Guariento M., Blaum B. S., Gillespie D., Herbert A. P., Kavanagh D., Mertens H. D., Svergun D. I., Johansson C. M., Uhrín D., Barlow P. N., Hannan J. P. (2011) Structural basis for engagement by complement factor H of C3b on a self surface. Nat. Struct. Mol. Biol. 18, 463–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Clark S. J., Ridge L. A., Herbert A. P., Hakobyan S., Mulloy B., Lennon R., Würzner R., Morgan B. P., Uhrín D., Bishop P. N., Day A. J. (2013) Tissue-specific host recognition by complement factor H is mediated by differential activities of its glycosaminoglycan-binding regions. J. Immunol. 190, 2049–2057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wang C., Salahudeen A. K. (1995) Lipid peroxidation accompanies cyclosporine nephrotoxicity. Effects of vitamin E. Kidney Int. 47, 927–934 [DOI] [PubMed] [Google Scholar]
- 55. Parra T., de Arriba G., Arribas I., Perez de Lema G., Rodriguez-Puyol D., Rodriguez-Puyol M. (1998) Cyclosporine A nephrotoxicity: role of thromboxane and reactive oxygen species. J. Lab. Clin. Med. 131, 63–70 [DOI] [PubMed] [Google Scholar]
- 56. Jareño E. J., Bosch-Morell F., Fernández-Delgado R., Donat J., Romero F. J. (1998) Serum malondialdehyde in HIV seropositive children. Free Radic. Biol. Med. 24, 503–506 [DOI] [PubMed] [Google Scholar]
- 57. Suresh D. R., Annam V., Pratibha K., Prasad B. V. (2009) Total antioxidant capacity. A novel early bio-chemical marker of oxidative stress in HIV infected individuals. J Biomed. Sci. 16, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ilhan N., Ilhan N., Simsek M. (2002) The changes of trace elements, malondialdehyde levels and superoxide dismutase activities in pregnancy with or without preeclampsia. Clin. Biochem. 35, 393–397 [DOI] [PubMed] [Google Scholar]
- 59. Morris J. M., Gopaul N. K., Endresen M. J., Knight M., Linton E. A., Dhir S., Anggård E. E., Redman C. W. (1998) Circulating markers of oxidative stress are raised in normal pregnancy and pre-eclampsia. Br. J. Obstet. Gynaecol. 105, 1195–1199 [DOI] [PubMed] [Google Scholar]