

Background: MitoNEET is a target of the type II diabetes drug pioglitazone and contains a [2Fe-2S] cluster.
Results: The mitoNEET [2Fe-2S] cluster can be reduced by biological thiols and reversibly oxidized by hydrogen peroxide.
Conclusion: The redox state of mitoNEET [2Fe-2S] clusters can be regulated by thiols and oxidative signals.
Significance: MitoNEET may act as a redox sensor to modulate mitochondrial functions.
Keywords: Diabetes, Hydrogen Peroxide, Iron-Sulfur Protein, Redox Regulation, Thioredoxin
Abstract
The human mitochondrial outer membrane protein mitoNEET is a novel target of the type II diabetes drug pioglitazone. The C-terminal cytosolic domain of mitoNEET hosts a redox-active [2Fe-2S] cluster via an unusual ligand arrangement of three cysteine residues and one histidine residue. Here we report that human mitoNEET [2Fe-2S] clusters are fully reduced when expressed in Escherichia coli cells. In vitro studies show that purified mitoNEET [2Fe-2S] clusters can be partially reduced by monothiols such as reduced glutathione, l-cysteine or N-acetyl-l-cysteine and fully reduced by dithiothreitol or the E. coli thioredoxin/thioredoxin reductase system under anaerobic conditions. Importantly, thiol-reduced mitoNEET [2Fe-2S] clusters can be reversibly oxidized by hydrogen peroxide without disruption of the clusters in vitro and in E. coli cells, indicating that mitoNEET may act as a sensor of oxidative signals to regulate mitochondrial functions via its [2Fe-2S] clusters. Furthermore, the binding of the type II diabetes drug pioglitazone in mitoNEET effectively inhibits the thiol-mediated reduction of [2Fe-2S] clusters, suggesting that pioglitazone may modulate the function of mitoNEET by blocking the thiol-mediated reduction of [2Fe-2S] clusters in the protein.
Introduction
The human mitochondrial protein mitoNEET is a novel target of the type II diabetes drugs thiazolidinediones, such as pioglitazone (1). Genetic studies have shown that the deletion of mitoNEET in mice results in a reduced oxidative phosphorylation capacity in mitochondria (2). Although an increased expression of mitoNEET in adipocytes in mice enhances lipid uptake and storage and inhibits mitochondrial iron transport into the matrix, depletion of mitoNEET in adipocytes leads to less weight gain in mice (3), suggesting that mitoNEET may regulate the energy metabolism in mitochondria (1). Recent studies further indicated that mitoNEET may have a central role in neurodegenerative diseases (4, 5) and breast cancer proliferation (6) by maintaining mitochondrial homeostasis in cells.
MitoNEET localizes on mitochondrial outer membranes (2) via the N-terminal transmembrane α-helix (residues 14–32) (1). Expression of the soluble C-terminal domain (residues 33–108) of mitoNEET in Escherichia coli cells produced a protein that contains a [2Fe-2S] cluster (7). Crystallographic studies revealed that mitoNEET exists as a homodimer, with each monomer hosting a [2Fe-2S] cluster via an unusual ligand arrangement of three cysteine residues (Cys-72, Cys-74, and Cys-83) and one histidine residue (His-87) (8–10). The [2Fe-2S] clusters in mitoNEET are redox-active, with a midpoint redox potential at pH 7 (Em7) of ∼0 mV (11, 12). Substitution of the unique His-87 with cysteine in mitoNEET shifts the Em7 value of the [2Fe-2S] clusters from 0 to −320 mV (11) and increases the stability of the cluster in the protein (13). The redox property and stability of the [2Fe-2S] clusters in mitoNEET are also modulated by the type II diabetes drug pioglitazone (11), excess zinc (14), resveratrol-3-sulfate (15), NADPH (16), the interdomain interactions (17, 18), and the hydrogen bond network in the protein (19), suggesting that mitoNEET may act as a sensor of multiple signals to regulate mitochondrial functions (19). Interestingly, purified mitoNEET can also transfer the [2Fe-2S] clusters to apo-protein with a 50% completion time of about 120 min at room temperature (20, 21). Because mitochondria are the primary sites for iron-sulfur cluster biogenesis (22), it is appealing to consider that mitoNEET may participate in the iron-sulfur cluster biogenesis process in human cells (20, 21). Nevertheless, the iron-sulfur cluster transfer occurs only when the [2Fe-2S] clusters in mitoNEET are oxidized (20) and the cluster transfer process is inhibited by NADPH (21), suggesting that the mitoNEET-mediated iron-sulfur cluster transfer could be regulated by the redox state of the [2Fe-2S] clusters and by intracellular NADPH.
Because the cytosolic redox potential in eukaryotic cells is estimated to be around −325 mV (pH 7.0) (23), it is expected that the mitoNEET [2Fe-2S] clusters (Em7 = 0 mV) (11, 12) would be in a reduced state in cells under physiological conditions. Nevertheless, the redox state of the mitoNEET [2Fe-2S] clusters in cells has not been demonstrated, and specific cellular components that may reduce the mitoNEET [2Fe-2S] clusters have not been identified. Here we report that human mitoNEET [2Fe-2S] clusters are in the fully reduced state when expressed in E. coli cells under normal growth conditions and that purified mitoNEET [2Fe-2S] clusters can be partially reduced by monothiols such as reduced glutathione, l-cysteine, and N-acetyl-l-cysteine and fully reduced by dithiothreitol and the E. coli thioredoxin-1 reduced by thioredoxin reductase and NADPH. Importantly, the reduced mitoNEET [2Fe-2S] clusters can be reversibly oxidized by hydrogen peroxide without disruption of the clusters, indicating that the redox state of the mitoNEET [2Fe-2S] clusters can be modulated by biological thiols and oxidative signals. Furthermore, we find that the type II diabetes drug pioglitazone can effectively inhibit the thiol-mediated reduction of the mitoNEET [2Fe-2S] clusters in vitro, suggesting that pioglitazone may modulate the function of mitoNEET by blocking the thiol-mediated reduction of the [2Fe-2S] clusters in the protein.
EXPERIMENTAL PROCEDURES
Protein Purification
The gene encoding human mitoNEET33–108 (containing amino acid residues 33–108) was cloned previously from the human cDNA library as described in Ref. 14. The mitoNEET mutant in which histidine 87 was substituted with cysteine (H87C) was constructed using the QuikChange site-directed mutagenesis kit (Stratagene). The constructed mutation was confirmed by direct sequencing (Operon Co.). Human mitoNEET and the mitoNEET mutant proteins were prepared following procedures described previously (14). E. coli thioredoxin-1 (TrxA) and thioredoxin reductase (TrxB) were produced from E. coli cells using the expression vectors pDL59 (24) and pTrR301 (25), respectively, and purified as described in Ref. 26. Both E. coli thioredoxin and thioredoxin reductase were purified as the native form. The purity of purified proteins was greater than 95%, as judged by electrophoresis analysis on a 15% polyacrylamide gel containing SDS, followed by staining with Coomassie Blue. The protein concentration of purified mitoNEET or the mitoNEET mutant H87C was measured at 280 nm using an extinction coefficient of 8.6 mm−1cm−1. The protein concentration of thioredoxin-1 and thioredoxin reductase was determined at 280 nm using extinction coefficients of 14.2 and 17.7 mm−1cm−1, respectively. A Beckman DU640 UV-visible spectrometer equipped with a temperature control was used for measuring absorption spectra.
Other Chemicals
Pioglitazone, l-cysteine, N-acetyl-l-cysteine, reduced glutathione, and other chemicals were purchased from Sigma. Isopropyl β-d-1-thiogalactopyranoside, NADPH, kanamycin, ampicillin, and dithiothreitol were from Research Product International Co. Pioglitazone was dissolved in dimethyl sulfoxide as a stock solution containing 25 mm pioglitazone. An equal amount of dimethyl sulfoxide was added to the samples as a control.
EPR Measurements
For whole-cell EPR measurements, E. coli cells were grown for 2 h before the recombinant protein expression was induced. After 2 h of protein expression, cells were harvested, washed once with phosphate buffer (pH 7.5), and resuspended in phosphate buffer or fresh LB medium. An aliquot (350 μl) of the E. coli cells was then transferred to the EPR tube and frozen immediately in liquid nitrogen. For the reduced samples, freshly prepared sodium dithionite was added to the E. coli cells before the EPR samples were prepared. For protein sample preparation, purified mitoNEET [2Fe-2S] clusters dissolved in buffer containing 20 mm Tris (pH 8.0) and 500 mm NaCl were purged with pure argon gas in sealed vials for 15 min before reductant was added under anaerobic conditions. After incubation at 37 °C for 20 min, samples were transferred to the EPR tubes and frozen immediately in liquid nitrogen. The X-band EPR spectra were recorded using a Bruker model ESR-300 spectrometer equipped with an Oxford Instruments 910 continuous flow cryostat. Routine EPR conditions were as follows: microwave frequency, 9.47 GHz; microwave power, 10.0 milliwatt; modulation frequency, 100 kHz; modulation amplitude, 1.2 millitesla; temperature, 20 K; receive gain, 105.
RESULTS
Human MitoNEET [2Fe-2S] Clusters Expressed in E. coli Cells Are Fully Reduced
Purified human mitoNEET [2Fe-2S] clusters are in an oxidized state and have no EPR signals (Fig. 1A). When purified mitoNEET [2Fe-2S]] clusters are reduced with sodium dithionite (Em7 = -600 mV (27)), a rhombic EPR spectrum with gx= 1.895, gy=1.937, and gz = 2.005 appears (Fig. 1A), as reported previously (7, 19, 28, 29). The gx, gy, and gz values represent the anisotropic g-factors of a typical [2Fe-2S] cluster in proteins. In parallel, we also prepared the mitoNEET mutant H87C, in which the unique ligand His-87 of the [2Fe-2S] cluster is replaced with Cys. Although purified mitoNEET H87C has no EPR signal, addition of sodium dithionite produces a new rhombic EPR spectrum of the [2Fe-2S] cluster, with gx = 1.896, gy = 1.970, and gz = 1.992, that is distinct from that of the wild-type mitoNEET (Fig. 1A).
FIGURE 1.
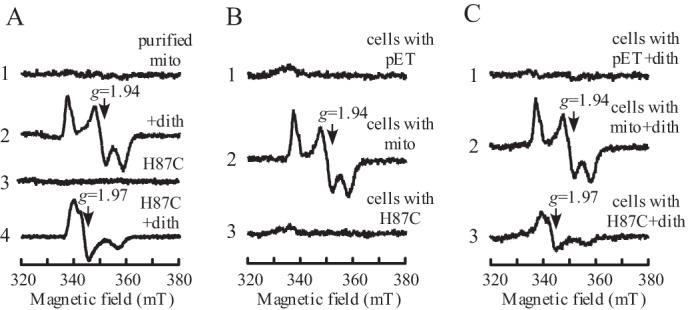
MitoNEET [2Fe-2S] clusters are fully reduced in E. coli cells. A, EPR spectra of purified mitoNEET (mito) [2Fe-2S] clusters. Purified mitoNEET and mitoNEET mutant H87C proteins (20 μm [2Fe-2S] clusters) were reduced with sodium dithionite (dith) (2 mm). Spectrum 1 shows wild-type mitoNEET [2Fe-2S] clusters. Spectrum 2 shows wild-type mitoNEET [2Fe-2S] clusters reduced with dithionite. Spectrum 3 shows mitoNEET mutant H87C [2Fe-2S] clusters. Spectrum 4 shows mitoNEET mutant H87C [2Fe-2S] clusters reduced with dithionite. mT, millitesla. B, EPR spectra of E. coli cells with or without recombinant mitoNEET. Recombinant proteins were expressed in E. coli cells for 2 h at room temperature, and then cells were concentrated to an A600 nm of 15. Spectrum 1 shows E. coli cells containing the expression plasmid pET28b+ (pET) only. Spectrum 2 shows E. coli cells expressing wild-type mitoNEET. Spectrum 3 shows E. coli cells expressing the mitoNEET mutant H87C. C, EPR spectra of E. coli cells after reduction with sodium dithionite (2 mm). Spectrum 1 shows E. coli cells containing the expression plasmid pET28b+ only. Spectrum 2 shows E. coli cells expressing wild-type mitoNEET. Spectrum 3 shows E. coli cells expressing the mitoNEET mutant H87C. The results are representative of three independent experiments.
The EPR measurement provides an efficient and non-intrusive approach to determine the redox state of iron-sulfur clusters in proteins in solutions or in whole cells (30, 31). Here, we took the advantage of EPR to explore the redox state of the mitoNEET [2Fe-2S] clusters expressed in E. coli cells. Fig. 1B shows that E. coli cells without any recombinant proteins have very little or no EPR signals because of low concentrations of endogenous iron-sulfur proteins in cells (30, 31). Under the same experimental conditions, E. coli cells expressing human mitoNEET show a rhombic EPR signal at g = 1.94, which is identical to that of the reduced mitoNEET [2Fe-2S] clusters (Fig. 1A). In contrast, E. coli cells expressing the mitoNEET mutant H87C do not have any EPR signals (Fig. 1B), although both mitoNEET and the mitoNEET mutant H87C are expressed similarly in E. coli cells on the basis of SDS-PAGE gel analysis (data not shown). This is likely because the mutation of His-87 to Cys shifts the Em7 value of the mitoNEET [2Fe-2S] clusters from 0 to −320 mV (11). Because the cytosolic redox potential of E. coli cells is about −260 mV (32), the mitoNEET mutant H87C [2Fe-2S] clusters would be mostly in an oxidized (EPR-silent) state in E. coli cells.
To further examine the redox state of the [2Fe-2S] clusters in wild-type mitoNEET and the mitoNEET mutant H87C expressed in E. coli cells, excess sodium dithionite is added to the cells to reduce all iron-sulfur proteins. Fig. 1C shows that addition of sodium dithionite to E. coli cells without recombinant proteins does not produce any new EPR signals. On the other hand, addition of sodium dithionite to E. coli cells expressing the mitoNEET mutant H87C generates an EPR signal at g = 1.97, which is the same as that of the reduced mitoNEET mutant H87C [2Fe-2S] clusters (Fig. 1A). Thus, the oxidized [2Fe-2S] clusters in the mitoNEET mutant H87C in E. coli cells can be reduced by sodium dithionite. In contrast, the EPR signal at g = 1.94 of the E. coli cells expressing the wild-type mitoNEET is not changed by addition of sodium dithionite, suggesting that the wild-type mitoNEET [2Fe-2S] clusters are fully reduced in E. coli cells even before the addition of sodium dithionite. Because the cytosolic redox potential in eukaryotic cells (-325 mV (23)) is about 65 mV lower than that in E. coli cells (-260 mV (32)), we postulate that the mitoNEET [2Fe-2S] clusters are most likely in a reduced state in human cells under physiological conditions.
Human MitoNEET [2Fe-2S] Clusters Can Be Reduced by Biological Thiols
Although it has been shown that the mitoNEET [2Fe-2S] clusters are redox-active (11, 12), specific biological molecules that can reduce or oxidize the mitoNEET [2Fe-2S] clusters are unknown. Because the mitoNEET [2Fe-2S] clusters are fully reduced in E. coli cells, we reason that the mitoNEET [2Fe-2S] clusters may be reduced by cellular reducing components that are common in both human and E. coli cells. However, NADPH is not one of them because NADPH fails to reduce the mitoNEET [2Fe-2S] clusters in vitro (21). Among other cellular reducing components, biological thiols are abundant. More importantly, biological thiols are able to form relatively stable intermediate thiol free radicals (33, 34) that can potentially provide electrons for the single-electron reduction of the oxidized [2Fe-2S] clusters in mitoNEET.
To test this idea, purified mitoNEET was incubated with thiols under anaerobic conditions. After incubation, the samples were subjected to EPR and UV-visible absorption measurements. The reduced mitoNEET [2Fe-2S] clusters were observed from the EPR signal at g = 1.94, and the UV-visible absorption peaks at 420 and 550 nm. As shown in Fig. 2, all biological thiols tested (reduced glutathione, l-cysteine, and N-acetyl-l-cysteine) are able to partially reduce the mitoNEET [2Fe-2S] clusters. Reduced glutathione appears to have the least ability to reduce the mitoNEET [2Fe-2S] clusters. l-cysteine and N-acetyl-l-cysteine are able to reduce about 50% of the mitoNEET [2Fe-2S] clusters in solution under anaerobic conditions, in comparison with the fully reduced mitoNEET [2Fe-2S] clusters by sodium dithionite. A further increase of l-cysteine concentration (up to 50-fold excess over the mitoNEET [2Fe-2S] clusters) or incubation time (up to 60 min) does not increase the amount of the reduced mitoNEET [2Fe-2S] clusters in the incubation solution (data not shown), suggesting that l-cysteine could not fully reduce the mitoNEET [2Fe-2S] clusters under the experimental conditions. Interestingly, incubation with dithiothreitol, a dithiol that has an Em7 value of −323 mV (35), almost fully reduced the mitoNEET [2Fe-2S] clusters under the same experimental conditions (Fig. 2).
FIGURE 2.
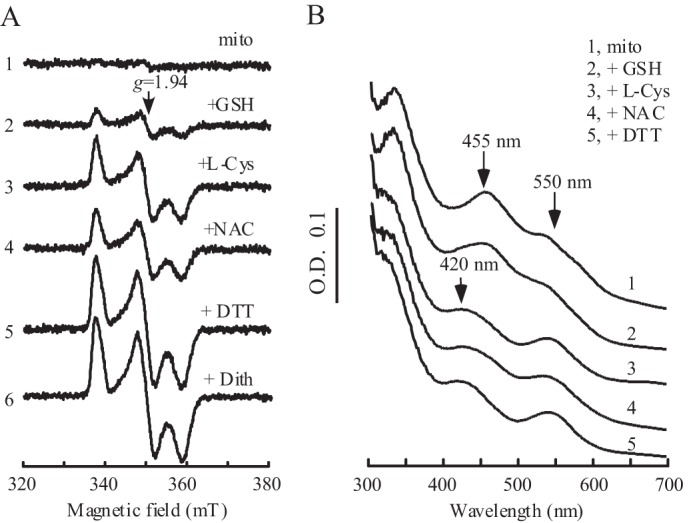
MitoNEET [2Fe-2S] clusters can be reduced by biological thiols in vitro. A, EPR spectra of the mitoNEET [2Fe-2S] clusters after reduction with different thiols. Purified mitoNEET [2Fe-2S] clusters (14 μm) (spectrum 1) were incubated with 2 mm of GSH (spectrum 2), l-cysteine (l-Cys) (spectrum 3), N-acetyl-l-cysteine (NAC) (spectrum 4), or DTT (dith) (spectrum 5) at 37 °C for 20 min under anaerobic conditions. The EPR spectrum of fully reduced mitoNEET [2Fe-2S] clusters (spectrum 6) was obtained by adding sodium dithionite (2 mm). mT, millitesla. B, UV-visible spectra of mitoNEET [2Fe-2S] clusters after incubation with biological thiols. Purified mitoNEET [2Fe-2S] clusters (14 μm) (spectrum 1) was incubated with 2 mm of GSH (spectrum 2), l-cysteine (spectrum 3), N-acetyl-l-cysteine (spectrum 4), or DTT (spectrum 5) at 37 °C for 20 min under anaerobic conditions. The absorption peaks at 455 nm and 550 nm represent the oxidized mitoNEET [2Fe-2S] clusters, and the new absorption peak at 420 nm represents the reduced mitoNEET [2Fe-2S] clusters.
Human MitoNEET [2Fe-2S] Clusters Can Be Fully Reduced by the E. coli Thioredoxin/Thioredoxin Reductase System
The biological equivalent of dithiothreitol is the ubiquitous thioredoxin (36). The Em7 value of E. coli thioredoxin-1 is about −270 mV (37), which is close to that of dithiothreitol (35). To test whether the oxidized mitoNEET [2Fe-2S] clusters can be reduced by thioredoxin-1, we prepared E. coli thioredoxin-1 and thioredoxin reductase as described previously (26). When purified mitoNEET [2Fe-2S] clusters are incubated with equimolar concentrations of the E. coli thioredoxin/thioredoxin reductase or an excess amount of NADPH under anaerobic conditions, the mitoNEET [2Fe-2S] clusters remained in an oxidized state (Fig. 3). This could be because purified thioredoxin-1 is mostly oxidized during the protein purification process. Indeed, when purified mitoNEET [2Fe-2S] clusters were incubated with thioredoxin-1 prereduced with thioredoxin reductase and NADPH, the mitoNEET [2Fe-2S] clusters were fully reduced (Fig. 3). On the other hand, the mitoNEET mutant H87C [2Fe-2S] clusters that have an Em7 of −320 mV (11) cannot be reduced by the E. coli thioredoxin/thioredoxin reductase system under the same experimental conditions (Fig. 3). This is consistent with the observation that the mitoNEET mutant H87C [2Fe-2S] clusters are in an oxidized state when expressed in E. coli cells (Fig. 1B). Taken together, the results suggest that the wild-type mitoNEET [2Fe-2S] clusters can be fully reduced by the E. coli thioredoxin/thioredoxin reductase system in vitro.
FIGURE 3.
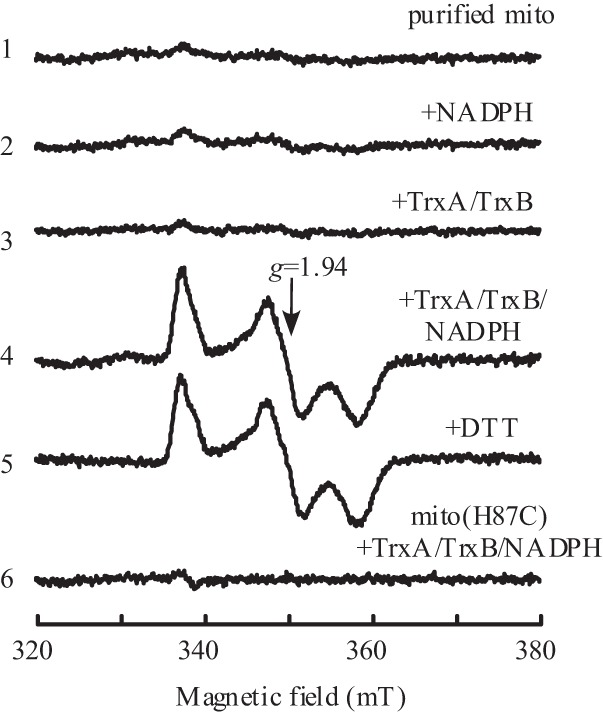
Reduction of mitoNEET [2Fe-2S] clusters by the thioredoxin reductase system in vitro. Purified mitoNEET (mito) [2Fe-2S] clusters (10 μm) (spectrum 1) were incubated with NADPH (500 μm) (spectrum 2); E. coli thioredoxin (TrxA, 10 μm) and thioredoxin reductase (TrxB, 10 μm) (spectrum 3); or E. coli thioredoxin (10 μm), thioredoxin reductase (10 μm), and NADPH (500 μm) (spectrum 4) at 37 °C for 20 min under anaerobic conditions. Spectrum 5 shows purified mitoNEET [2Fe-2S] clusters reduced with dithiothreitol (2 mm). Spectrum 6 shows the mitoNEET mutant H87C [2Fe-2S] clusters (10 μm) incubated with E. coli thioredoxin-1 (10 μm), thioredoxin reductase (10 μm), and NADPH (500 μm) at 37 °C for 20 min under anaerobic conditions. After incubation, the samples were transferred to EPR tubes for EPR measurements. The results are representative of three independent experiments. mT, millitesla.
Reduced mitoNEET [2Fe-2S] Clusters Can Be Reversibly Oxidized by Hydrogen Peroxide
Previous studies have shown that iron-sulfur clusters in regulatory proteins can undergo redox transition in response to oxidative signals without disruption of the clusters (38–41). To explore whether the mitoNEET [2Fe-2S] clusters may also act as a redox sensor to oxidative signals, we examined the stability of the mitoNEET [2Fe-2S] clusters under oxidative stress conditions. Fig. 4A shows that, when purified mitoNEET [2Fe-2S] clusters were incubated with increasing concentrations of hydrogen peroxide (up to 50-fold excess), the UV-visible absorption spectrum of the mitoNEET [2Fe-2S] clusters remains essentially unchanged, indicating that the mitoNEET [2Fe-2S] clusters are stable in the presence of hydrogen peroxide.
FIGURE 4.

Reversible oxidation of mitoNEET [2Fe-2S] clusters by hydrogen peroxide in vitro. A, stability of mitoNEET (mito) [2Fe-2S] clusters in the presence of hydrogen peroxide. UV-visible absorption spectra of mitoNEET [2Fe-2S] clusters were taken after purified mitoNEET [2Fe-2S] clusters (20 μm) were incubated with hydrogen peroxide (0 to 1.0 mm). B, reversible oxidation of mitoNEET [2Fe-2S] clusters by hydrogen peroxide. UV-visible absorption spectra were taken when purified mitoNEET (20 μm) (spectrum 1) was incubated with excess dithiothreitol (10 mm) in an anaerobic cuvette at 37 °C for 20 min (spectrum 2), followed by addition of hydrogen peroxide (0.5 mm) (spectrum 3) and 14 min of additional incubation (spectrum 4). C, time course of rereduction of oxidized mitoNEET [2Fe-2S] clusters by dithiothreitol. The absorption peak at 455 nm of the oxidized mitoNEET [2Fe-2S] clusters was plotted as a function of incubation time after addition of hydrogen peroxide. D, EPR spectra of mitoNEET [2Fe-2S] clusters. Purified mitoNEET [2Fe-2S] clusters (20 μm) (spectrum 1) were reduced with dithiothreitol (10 mm) (spectrum 2), oxidized by hydrogen peroxide (0.5 mm) (spectrum 3), and rereduced after additional incubation with dithiothreitol for 14 min (spectrum 4). mT, millitesla.
We then asked whether the prereduced mitoNEET [2Fe-2S] clusters can be oxidized by hydrogen peroxide. To avoid possible effects of NADPH on the stability of the mitoNEET [2Fe-2S] clusters (16), we prereduced the mitoNEET [2Fe-2S] clusters with excess dithiothreitol under anaerobic conditions. As shown in Fig. 4B, the prereduced mitoNEET [2Fe-2S] clusters were quickly oxidized upon addition of hydrogen peroxide. Further incubation with dithiothreitol after hydrogen peroxide treatment led to a full rereduction of the oxidized mitoNEET [2Fe-2S] clusters within about 14 min (Fig. 4, B and C). The EPR measurements of the protein samples taken from the reaction solution further confirm that the amount of the mitoNEET [2Fe-2S] clusters remain the same before and after hydrogen peroxide treatment (Fig. 4D), suggesting that the mitoNEET [2Fe-2S] clusters can undergo redox transition in response to hydrogen peroxide without disruption of the cluster.
To further explore whether the mitoNEET [2Fe-2S] clusters could undergo redox transition in vivo in response to hydrogen peroxide, E. coli cells expressing mitoNEET [2Fe-2S] clusters are treated with hydrogen peroxide. As shown in Fig. 5, addition of hydrogen peroxide (1 mm) to the E. coli cells largely eliminates the EPR signal at g = 1.94 of the reduced mitoNEET [2Fe-2S] clusters. Reincubation of the E. coli cells in LB medium after hydrogen peroxide treatment quickly restores the EPR signal at g = 1.94 of the reduced mitoNEET [2Fe-2S] clusters, demonstrating that the mitoNEET [2Fe-2S] clusters can undergo redox transition in response to hydrogen peroxide in E. coli cells.
FIGURE 5.
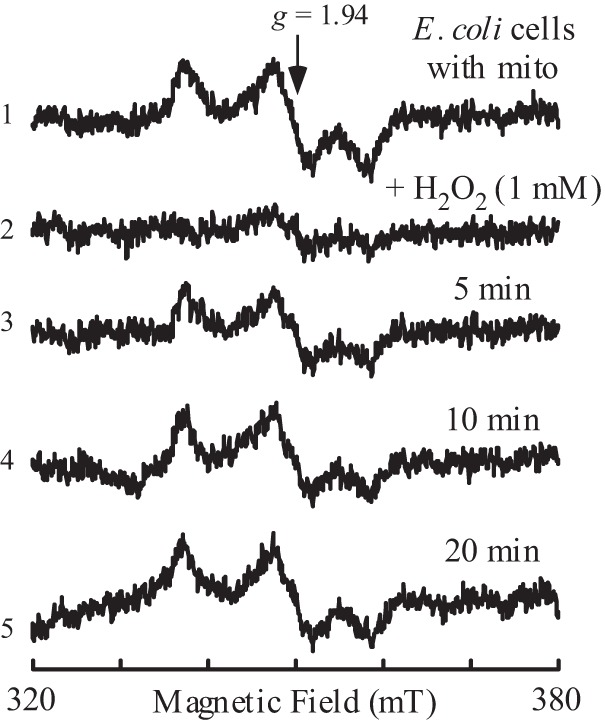
Reversible oxidation of mitoNEET [2Fe-2S] clusters by hydrogen peroxide in E. coli cells. E. coli cells containing recombinant human mitoNEET (mito) [2Fe-2S] clusters were resuspended in fresh LB medium to an A600 nm of 4.0. The cells (spectrum 1) were then treated with hydrogen peroxide (1 mm) (spectrum 2), followed by reincubation in LB medium with aeration at 37 °C for 5 min (spectrum 3), 10 min (spectrum 4), and 20 min (spectrum 5). The EPR signal at g = 1.94 indicates the reduced mitoNEET [2Fe-2S] clusters in E. coli cells. The results are representative of three independent experiments. mT, millitesla.
The Type II Diabetes Drug Pioglitazone Inhibits the Thiol-mediated Reduction of the mitoNEET [2Fe-2S] Clusters
It has been reported that binding of the type II diabetes drug pioglitazone shifts the Em7 value of the mitoNEET [2Fe-2S] clusters by −100 mV (11). The negative shift of the Em7 value would make it more difficult to reduce the mitoNEET [2Fe-2S] clusters. Nevertheless, how the redox state of the mitoNEET [2Fe-2S] clusters may be modulated by pioglitazone is unknown. With the finding that the mitoNEET [2Fe-2S] clusters can be reduced by biological thiols, we sought to investigate the effect of pioglitazone on the thiol-mediated reduction of the mitoNEET [2Fe-2S] clusters. Purified mitoNEET is preincubated with pioglitazone, followed by reduction with thiols under anaerobic conditions. Fig. 6A shows that binding of pioglitazone had little or no effect on the dithionite-mediated reduction of the mitoNEET [2Fe-2S] clusters because dithionite is a strong reductant (Em7 = −660 mV (27)). On the other hand, binding of pioglitazone largely inhibits the dithiothreitol-mediated reduction of the mitoNEET [2Fe-2S] clusters (Fig. 6B). Titration experiments showed that, as the concentration of pioglitazone increased gradually in the incubation solutions (from 0 to 2.5 mm), the amount of the dithiothreitol-reduced mitoNEET [2Fe-2S] clusters was decreased progressively (Fig. 6C). We also tested whether pioglitazone affected the thioredoxin-mediated reduction of the mitoNEET [2Fe-2S] clusters in vitro. As shown in Fig. 6D, binding of pioglitazone greatly inhibited the thioredoxin-mediated reduction of the mitoNEET [2Fe-2S] clusters, suggesting that pioglitazone can effectively block the biological thiol-mediated reduction of the mitoNEET [2Fe-2S] clusters in cells. Nevertheless, attempts to modulate the redox state of the mitoNEET [2Fe-2S] clusters expressed in E. coli cells with pioglitazone were not successful. This is likely because pioglitazone may not easily penetrate bacterial cell walls and/or cell membranes because addition of pioglitazone to the cell extracts prepared from E. coli cells effectively inhibits the thiol-mediated reduction of the mitoNEET [2Fe-2S] clusters in the cell extracts (data not shown).
FIGURE 6.
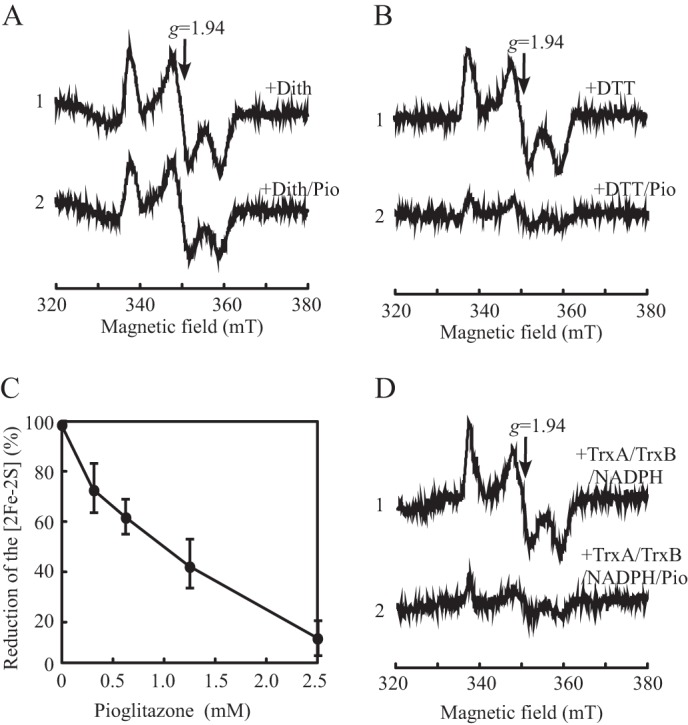
Pioglitazone inhibits the thiol-mediated reduction of mitoNEET [2Fe-2S] clusters. A, effect of pioglitazone (Pio) on the dithionite-mediated reduction of mitoNEET [2Fe-2S] clusters. Purified mitoNEET [2Fe-2S] clusters (10 μm) were preincubated with 0 (spectrum 1) or 2.5 mm (spectrum 2) pioglitazone for 15 min at 37 °C, followed by addition of sodium dithionite (Dith) (2 mm) and incubation for 15 min under anaerobic conditions. mT, millitesla. B, purified mitoNEET [2Fe-2S] clusters (10 μm) were preincubated with 0 (spectrum 1) or 2.5 mm (spectrum 2) pioglitazone for 15 min at 37 °C, followed by addition of dithiothreitol (2 mm) and incubation for 15 min under anaerobic conditions. C, titration of pioglitazone. Purified mitoNEET [2Fe-2S] clusters (10 μm) were preincubated with the indicated concentrations of pioglitazone. The amounts of the reduced mitoNEET [2Fe-2S] clusters were measured from the amplitudes of the EPR signal at g = 1.94 and plotted as a function of pioglitazone concentration after incubation with dithiothreitol (2 mm) under anaerobic conditions. D, purified mitoNEET [2Fe-2S] clusters (10 μm) were preincubated with 0 (spectrum 1) or 2.5 mm (spectrum 2) pioglitazone for 15 min at 37 °C, followed by incubation with E. coli thioredoxin (TrxA)/thioredoxin reductase (TrxB) system (10 μm E. coli thioredoxin, 10 μm thioredoxin reductase, and 500 μm NADPH) at 37 °C for 15 min under anaerobic conditions.
DISCUSSION
In this study, we report that human mitoNEET [2Fe-2S] clusters are fully reduced when expressed in E. coli cells under normal growth conditions and that the mitoNEET [2Fe-2S] clusters can be partially reduced by monothiols and fully reduced by dithiothreitol and the E. coli thioredoxin-1 reduced by thioredoxin reductase and NADPH in vitro. Importantly, the thiol-reduced mitoNEET [2Fe-2S] clusters can be reversibly oxidized by hydrogen peroxide without disruption of the clusters. Furthermore, binding of the type II diabetes drug pioglitazone effectively blocks the thiol-mediated reduction of the mitoNEET [2Fe-2S] clusters. The results suggest that the mitoNEET [2Fe-2S] cluster may act as a sensor of oxidative signals to regulate mitochondrial function and that the type II diabetes drug pioglitazone may modulate the function of mitoNEET by blocking the thiol-mediated reduction of the [2Fe-2S] clusters in cells. The possible interplay among biological thiols, hydrogen peroxide, and the type II diabetes drug pioglitazone in regulating the redox state of the mitoNEET [2Fe-2S] clusters is shown in Fig. 7.
FIGURE 7.
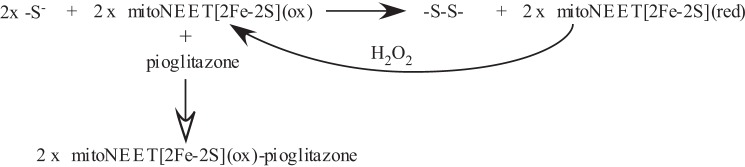
Proposed model for redox regulation of mitoNEET [2Fe-2S] clusters in response to biological thiols, hydrogen peroxide, and the type II diabetes drug pioglitazone. Two thiols reduce two mitoNEET [2Fe-2S] clusters via the single-electron reduction process to produce two thiol free radicals that may form a disulfide. Reduced [2Fe-2S] clusters in mitoNEET are reversibly oxidized by hydrogen peroxide. Binding of the type II diabetes drug pioglitazone to mitoNEET shifts the Em7 value of the [2Fe-2S] clusters by −100 mV and blocks the thiol-mediated reduction of the clusters in the protein.
The interaction between thioredoxin and iron-sulfur clusters in proteins has been well established in ferredoxin-thioredoxin reductase, where the reduced iron-sulfur clusters provide electrons for reduction of the oxidized thioredoxin (42, 43). However, to the best of our knowledge, the reduction of iron-sulfur clusters in proteins by biological thiols has not been reported previously. The unique ligand arrangement (Cys-72, Cys-74, Cys-83, and His-87) (8–10) and relatively high Em7 value (0 mV) (11, 12) may facilitate the reduction of the mitoNEET [2Fe-2S] clusters by biological thiols because a single mutation (His-87 to Cys) in mitoNEET shifts the Em7 value of the [2Fe-2S] clusters from 0 to −320 mV (11) and effectively prevents the reduction of the clusters by biological thiols (Fig. 3). Unlike NADPH, which fails to reduce the mitoNEET [2Fe-2S] clusters in vitro (21), biological thiols have a relatively stable intermediate state of thiol free radical (33, 34) that can provide electrons for the single-electron reduction of the [2Fe-2S] clusters in mitoNEET. Interestingly, monothiols, such as reduced glutathione, l-cysteine, and N-acetyl-l-cysteine, can only partially reduce the mitoNEET [2Fe-2S] clusters, even at high concentrations and with extended incubation time. On the other hand, dithiothreitol (Fig. 2) and E. coli thioredoxin prereduced with thioredoxin reductase and NADPH (Fig. 3) can fully reduce the mitoNEET [2Fe-2S] clusters. The redox reactions underlying the reduction of the mitoNEET [2Fe-2S] clusters by biological thiols remain to be further investigated. Nevertheless, the finding that the reduced thioredoxin can reduce the mitoNEET [2Fe-2S] clusters is highly significant. Thioredoxin is highly conserved from bacteria to humans (36). In human cells, there are two major thioredoxins: thioredoxin-1, which is located in the cytosol and nucleus, and thioredoxin 2, which is located in mitochondria (44). The Em7 value of human cytosolic thioredoxin-1 is about −230 mV (pH 7.0) (45), which is close to that of E. coli thioredoxin-1 (-270 mV) (37). Therefore, human thioredoxin-1 could be responsible for reducing the mitoNEET [2Fe-2S] clusters. Because human thioredoxin 1 has an essential role in the regulation of complex diseases, including cancer (46), cardiovascular disease, type II diabetes, and aging (47), the proposed role of human thioredoxin-1 in reducing the mitoNEET [2Fe-2S] clusters may provide fresh clues for the links between thioredoxin and various human diseases. However, it should be pointed out that other biological thiols, such as glutathione, l-cysteine, and glutaredoxins (48), may also reduce the mitoNEET [2Fe-2S] clusters in cells.
As a novel target of the type II diabetes drug pioglitazone, mitoNEET has a specific interaction with pioglitazone (1). It has been speculated that pioglitazone may modulate the energy metabolism in mitochondria via direct interaction with mitoNEET (49). Previous studies indicated that binding of pioglitazone stabilizes the [2Fe-2S] clusters in mitoNEET (10) and shifts the Em7 value of the [2Fe-2S] clusters from 0 to −100 mV (11). Here, we find that binding of pioglitazone effectively inhibits the thiol-mediated reduction of the mitoNEET [2Fe-2S] clusters, likely because of the decreased Em7 value of the [2Fe-2S] clusters in the protein. Thus, pioglitazone may alter the function of mitoNEET by blocking the thiol-mediated reduction of the [2Fe-2S] clusters in cells. A set of novel ligands have been identified recently that may target mitochondrial mitoNEET (50, 51). It would be of interest to explore the effect of these ligands on the thiol-mediated reduction of the mitoNEET [2Fe-2S] clusters.
The finding that the mitoNEET [2Fe-2S] clusters can be reversibly oxidized by hydrogen peroxide without disruption of the clusters strongly suggests that the mitoNEET [2Fe-2S] cluster may act as a redox sensor to oxidative signals. The redox state of the [2Fe-2S] clusters in mitoNEET has already been attributed to regulating the iron-sulfur cluster transfer from mitoNEET to target proteins (20, 21). It would also be possible that the redox state of the [2Fe-2S] clusters may directly modulate the energy metabolism in mitochondria via specific protein-protein interactions between mitoNEET and mitochondrial proteins, such as pyruvate dehydrogenase, in mitochondria (49). In crystal structural models, mitoNEET exists as a homodimer with two mitoNEET monomers tightly associated via the “β cap” structure (8–10). The closest distance between the two [2Fe-2S] clusters in the mitoNEET dimer is about 14 Å. We envision that, when the reduced [2Fe-2S] clusters are oxidized, two positive charges are introduced to the mitoNEET dimer. The electrostatic repulsion between the two [2Fe-2S] clusters in the mitoNEET dimer could lead to subtle conformation change and, consequently, modulate mitochondrial functions. A similar redox regulation of iron-sulfur clusters has been proposed for a number of proteins (40). For example, in the redox transcription factor SoxR, oxidation of the [2Fe-2S] clusters switches on its transcription activity (38, 41). In the bacterial DNA damage-inducible DNA helicase DinG, the enzyme is active only when the reduced [4Fe-4S] cluster is oxidized (39). In this context, we propose that mitoNEET may represent a new member of this redox signaling protein family in human cells.
A genome-wide search revealed that humans have at least two mitoNEET related proteins, Miner1 (52) and Miner2 (2), that share the conserved CDGSH domain with mitoNEET (53). Miner1 was initially localized in the endoplasmic reticulum (54), but a recent study indicated that Miner1 is mostly associated with the mitochondrial outer membrane (55). Mutations in the Miner1 gene have been attributed to Wolfram syndrome 2, a disease characterized by juvenile-onset diabetes mellitus and optic atrophy (54). Biochemical studies have shown that Miner1 interacts with BCL2 (B-cell lymphoma 2) (56) and regulates the sulfhydryl redox status, the unfolded protein response, and Ca2+ homeostasis in mitochondria via an unknown mechanism (57). Like mitoNEET, Miner1 hosts a [2Fe-2S] cluster via an unusual three cysteine residues and one histidine residue in its C-terminal domain (52). Although the function of Miner2 remains unknown, Miner2 is also located on the mitochondrial outer membrane and, most likely, contains a [2Fe-2S] cluster hosted by three cysteine residues and one histidine residue (2). The existence of [2Fe-2S] clusters in mitoNEET (7), Miner1 (52), and, most likely, in Miner2 (2) strongly suggests that the [2Fe-2S] clusters in this group of mitochondrial outer membrane proteins (53) may share similar and crucial regulatory functions. If the [2Fe-2S] clusters in Miner1 and Miner2, like that in mitoNEET, can be reduced by biological thiols and reversibly oxidized by hydrogen peroxide, Miner1 and Miner2 may also act as redox sensors to modulate specific functions in response to oxidative signals in human cells.
Acknowledgments
We thank Dr. Christine Winterbourn (Christchurch School of Medicine, Christchurch, New Zealand) for advice regarding the proposed mechanisms of thiol-mediated reduction of mitoNEET [2Fe-2S] clusters.
Footnotes
This work was supported, in whole or in part, by NCI, National Institutes of Health Grant R01CA107494. This work was also supported by American Heart Association Grant 13GRNT16890014 and by a Louisiana Board of Regents graduate scholarship.
REFERENCES
- 1. Colca J. R., McDonald W. G., Waldon D. J., Leone J. W., Lull J. M., Bannow C. A., Lund E. T., Mathews W. R. (2004) Identification of a novel mitochondrial protein (“mitoNEET”) cross-linked specifically by a thiazolidinedione photoprobe. Am. J. Physiol. Endocrinol. Metab. 286, E252–E260 [DOI] [PubMed] [Google Scholar]
- 2. Wiley S. E., Murphy A. N., Ross S. A., van der Geer P., Dixon J. E. (2007) MitoNEET is an iron-containing outer mitochondrial membrane protein that regulates oxidative capacity. Proc. Natl. Acad. Sci. U.S.A. 104, 5318–5323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kusminski C. M., Holland W. L., Sun K., Park J., Spurgin S. B., Lin Y., Askew G. R., Simcox J. A., McClain D. A., Li C., Scherer P. E. (2012) MitoNEET-driven alterations in adipocyte mitochondrial activity reveal a crucial adaptive process that preserves insulin sensitivity in obesity. Nat. Med. 18, 1539–1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yonutas H. M., Sullivan P. G. (2013) Targeting PPAR isoforms following CNS injury. Curr. Drug Targets 14, 733–742 [DOI] [PubMed] [Google Scholar]
- 5. Geldenhuys W. J., Van der Schyf C. J. (2013) Rationally designed multi-targeted agents against neurodegenerative diseases. Curr. Med. Chem. 20, 1662–1672 [DOI] [PubMed] [Google Scholar]
- 6. Sohn Y. S., Tamir S., Song L., Michaeli D., Matouk I., Conlan A. R., Harir Y., Holt S. H., Shulaev V., Paddock M. L., Hochberg A., Cabanchick I. Z., Onuchic J. N., Jennings P. A., Nechushtai R., Mittler R. (2013) NAF-1 and mitoNEET are central to human breast cancer proliferation by maintaining mitochondrial homeostasis and promoting tumor growth. Proc. Natl. Acad. Sci. U.S.A. 110, 14676–14681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wiley S. E., Paddock M. L., Abresch E. C., Gross L., van der Geer P., Nechushtai R., Murphy A. N., Jennings P. A., Dixon J. E. (2007) The outer mitochondrial membrane protein mitoNEET contains a novel redox-active 2Fe-2S cluster. J. Biol. Chem. 282, 23745–23749 [DOI] [PubMed] [Google Scholar]
- 8. Hou X., Liu R., Ross S., Smart E. J., Zhu H., Gong W. (2007) Crystallographic studies of human MitoNEET. J. Biol. Chem. 282, 33242–33246 [DOI] [PubMed] [Google Scholar]
- 9. Lin J., Zhou T., Ye K., Wang J. (2007) Crystal structure of human mitoNEET reveals distinct groups of iron-sulfur proteins. Proc. Natl. Acad. Sci. U.S.A. 104, 14640–14645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Paddock M. L., Wiley S. E., Axelrod H. L., Cohen A. E., Roy M., Abresch E. C., Capraro D., Murphy A. N., Nechushtai R., Dixon J. E., Jennings P. A. (2007) MitoNEET is a uniquely folded 2Fe 2S outer mitochondrial membrane protein stabilized by pioglitazone. Proc. Natl. Acad. Sci. U.S.A. 104, 14342–14347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bak D. W., Zuris J. A., Paddock M. L., Jennings P. A., Elliott S. J. (2009) Redox characterization of the FeS protein MitoNEET and impact of thiazolidinedione drug binding. Biochemistry 48, 10193–10195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tirrell T. F., Paddock M. L., Conlan A. R., Smoll E. J., Jr., Nechushtai R., Jennings P. A., Kim J. E. (2009) Resonance Raman studies of the (His)(Cys)(3) 2Fe-2S cluster of MitoNEET. Comparison to the (Cys)(4) mutant and implications of the effects of pH on the labile metal center. Biochemistry 48, 4747–4752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Conlan A. R., Paddock M. L., Homer C., Axelrod H. L., Cohen A. E., Abresch E. C., Zuris J. A., Nechushtai R., Jennings P. A. (2011) Mutation of the His ligand in mitoNEET stabilizes the 2Fe-2S cluster despite conformational heterogeneity in the ligand environment. Acta Crystallogr. D. Biol. Crystallogr. 67, 516–523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tan G., Landry A. P., Dai R., Wang L., Lu J., Ding H. (2012) Competition of zinc ion for the [2Fe-2S] cluster binding site in the diabetes drug target protein mitoNEET. Biometals 25, 1177–1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arif W., Xu S., Isailovic D., Geldenhuys W. J., Carroll R. T., Funk M. O. (2011) Complexes of the outer mitochondrial membrane protein mitoNEET with resveratrol-3-sulfate. Biochemistry 50, 5806–5811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhou T., Lin J., Feng Y., Wang J. (2010) Binding of reduced nicotinamide adenine dinucleotide phosphate destabilizes the iron-sulfur clusters of human mitoNEET. Biochemistry 49, 9604–9612 [DOI] [PubMed] [Google Scholar]
- 17. Baxter E. L., Jennings P. A., Onuchic J. N. (2011) Interdomain communication revealed in the diabetes drug target mitoNEET. Proc. Natl. Acad. Sci. U.S.A. 108, 5266–5271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Baxter E. L., Jennings P. A., Onuchic J. N. (2012) Strand swapping regulates the iron-sulfur cluster in the diabetes drug target mitoNEET. Proc. Natl. Acad. Sci. U.S.A. 109, 1955–1960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bak D. W., Elliott S. J. (2013) Conserved hydrogen bonding networks of MitoNEET Tune Fe-S cluster binding and structural stability. Biochemistry 52, 4687–4696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zuris J. A., Harir Y., Conlan A. R., Shvartsman M., Michaeli D., Tamir S., Paddock M. L., Onuchic J. N., Mittler R., Cabantchik Z. I., Jennings P. A., Nechushtai R. (2011) Facile transfer of [2Fe-2S] clusters from the diabetes drug target mitoNEET to an apo-acceptor protein. Proc. Natl. Acad. Sci. U.S.A. 108, 13047–13052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zuris J. A., Ali S. S., Yeh H., Nguyen T. A., Nechushtai R., Paddock M. L., Jennings P. A. (2012) NADPH inhibits [2Fe-2S] cluster protein transfer from diabetes drug target MitoNEET to an apo-acceptor protein. J. Biol. Chem. 287, 11649–11655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lill R. (2009) Function and biogenesis of iron-sulphur proteins. Nature 460, 831–838 [DOI] [PubMed] [Google Scholar]
- 23. Dooley C. T., Dore T. M., Hanson G. T., Jackson W. C., Remington S. J., Tsien R. Y. (2004) Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J. Biol. Chem. 279, 22284–22293 [DOI] [PubMed] [Google Scholar]
- 24. Veine D. M., Mulrooney S. B., Wang P. F., Williams C. H., Jr. (1998) Formation and properties of mixed disulfides between thioredoxin reductase from Escherichia coli and thioredoxin. Evidence that cysteine-138 functions to initiate dithiol-disulfide interchange and to accept the reducing equivalent from reduced flavin. Protein Sci. 7, 1441–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mulrooney S. B. (1997) Application of a single-plasmid vector for mutagenesis and high-level expression of thioredoxin reductase and its use to examine flavin cofactor incorporation. Protein Expr. Purif. 9, 372–378 [DOI] [PubMed] [Google Scholar]
- 26. Ding H., Harrison K., Lu J. (2005) Thioredoxin reductase system mediates iron binding in IscA and iron delivery for the iron-sulfur cluster assembly in IscU. J. Biol. Chem. 280, 30432–30437 [DOI] [PubMed] [Google Scholar]
- 27. Mayhew S. G. (1978) The redox potential of dithionite and SO-2 from equilibrium reactions with flavodoxins, methyl viologen and hydrogen plus hydrogenase. Eur. J. Biochem. 85, 535–547 [DOI] [PubMed] [Google Scholar]
- 28. Dicus M. M., Conlan A., Nechushtai R., Jennings P. A., Paddock M. L., Britt R. D., Stoll S. (2010) Binding of histidine in the (Cys)(3)(His)(1)-coordinated [2Fe-2S] cluster of human mitoNEET. J. Am. Chem. Soc. 132, 2037–2049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Iwasaki T., Samoilova R. I., Kounosu A., Ohmori D., Dikanov S. A. (2009) Continuous-wave and pulsed EPR characterization of the [2Fe-2S](Cys)3(His)1 cluster in rat MitoNEET. J. Am. Chem. Soc. 131, 13659–13667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ding H., Demple B. (1997) In vivo kinetics of a redox-regulated transcriptional switch. Proc. Natl. Acad. Sci. U.S.A. 94, 8445–8449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Djaman O., Outten F. W., Imlay J. A. (2004) Repair of oxidized iron-sulfur clusters in Escherichia coli. J. Biol. Chem. 279, 44590–44599 [DOI] [PubMed] [Google Scholar]
- 32. Gaudu P., Weiss B. (1996) SoxR, a [2Fe-2S] transcription factor, is active only in its oxidized form. Proc. Natl. Acad. Sci. U.S.A. 93, 10094–10098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Quijano C., Alvarez B., Gatti R. M., Augusto O., Radi R. (1997) Pathways of peroxynitrite oxidation of thiol groups. Biochem. J. 322, 167–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Eaton P. (2006) Protein thiol oxidation in health and disease. Techniques for measuring disulfides and related modifications in complex protein mixtures. Free Radic. Biol. Med. 40, 1889–1899 [DOI] [PubMed] [Google Scholar]
- 35. Szajewski R. P., Whitesides G. M. (1980) Rate constants and equilibrium constants for thiol-disulfide interchange reactions involving oxidized glutathione. J. Am. Chem. Soc. 102, 2011–2026 [Google Scholar]
- 36. Arnér E. S., Holmgren A. (2000) Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 267, 6102–6109 [DOI] [PubMed] [Google Scholar]
- 37. Krause G., Lundström J., Barea J. L., Pueyo de la Cuesta C., Holmgren A. (1991) Mimicking the active site of protein disulfide-isomerase by substitution of proline 34 in Escherichia coli thioredoxin. J. Biol. Chem. 266, 9494–9500 [PubMed] [Google Scholar]
- 38. Ding H., Hidalgo E., Demple B. (1996) The redox state of the [2Fe-2S] clusters in SoxR protein regulates its activity as a transcription factor. J. Biol. Chem. 271, 33173–33175 [DOI] [PubMed] [Google Scholar]
- 39. Ren B., Duan X., Ding H. (2009) Redox control of the DNA damage-inducible protein DinG helicase activity via its iron-sulfur cluster. J. Biol. Chem. 284, 4829–4835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Crack J. C., Green J., Thomson A. J., Le Brun N. E. (2012) Iron-sulfur cluster sensor-regulators. Curr. Opin. Chem. Biol. 16, 35–44 [DOI] [PubMed] [Google Scholar]
- 41. Gu M., Imlay J. A. (2011) The SoxRS response of Escherichia coli is directly activated by redox-cycling drugs rather than by superoxide. Mol. Microbiol. 79, 1136–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dai S., Schwendtmayer C., Schürmann P., Ramaswamy S., Eklund H. (2000) Redox signaling in chloroplasts. Cleavage of disulfides by an iron-sulfur cluster. Science 287, 655–658 [DOI] [PubMed] [Google Scholar]
- 43. Walters E. M., Garcia-Serres R., Naik S. G., Bourquin F., Glauser D. A., Schürmann P., Huynh B. H., Johnson M. K. (2009) Role of histidine-86 in the catalytic mechanism of ferredoxin:thioredoxin reductase. Biochemistry 48, 1016–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Collet J. F., Messens J. (2010) Structure, function, and mechanism of thioredoxin proteins. Antioxid. Redox Signal. 13, 1205–1216 [DOI] [PubMed] [Google Scholar]
- 45. Watson W. H., Pohl J., Montfort W. R., Stuchlik O., Reed M. S., Powis G., Jones D. P. (2003) Redox potential of human thioredoxin 1 and identification of a second dithiol/disulfide motif. J. Biol. Chem. 278, 33408–33415 [DOI] [PubMed] [Google Scholar]
- 46. Wondrak G. T. (2009) Redox-directed cancer therapeutics. Molecular mechanisms and opportunities. Antioxid. Redox Signal. 11, 3013–3069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zschauer T. C., Matsushima S., Altschmied J., Shao D., Sadoshima J., Haendeler J. (2013) Interacting with thioredoxin-1. Disease or no disease? Antioxid. Redox Signal. 18, 1053–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hanschmann E. M., Godoy J. R., Berndt C., Hudemann C., Lillig C. H. (2013) Thioredoxins, glutaredoxins, and peroxiredoxins-molecular mechanisms and health significance. From cofactors to antioxidants to redox signaling. Antioxid. Redox Signal. 19, 1539–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Feinstein D. L., Spagnolo A., Akar C., Weinberg G., Murphy P., Gavrilyuk V., Dello Russo C. (2005) Receptor-independent actions of PPAR thiazolidinedione agonists. Is mitochondrial function the key? Biochem. Pharmacol. 70, 177–188 [DOI] [PubMed] [Google Scholar]
- 50. Geldenhuys W. J., Funk M. O., Awale P. S., Lin L., Carroll R. T. (2011) A novel binding assay identifies high affinity ligands to the rosiglitazone binding site of mitoNEET. Bioorg. Med. Chem. Lett. 21, 5498–5501 [DOI] [PubMed] [Google Scholar]
- 51. Bieganski R. M., Yarmush M. L. (2011) Novel ligands that target the mitochondrial membrane protein mitoNEET. J. Mol. Graph. Model. 29, 965–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Conlan A. R., Axelrod H. L., Cohen A. E., Abresch E. C., Zuris J., Yee D., Nechushtai R., Jennings P. A., Paddock M. L. (2009) Crystal structure of Miner1. The redox-active 2Fe-2S protein causative in Wolfram syndrome 2. J. Mol. Biol. 392, 143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lin J., Zhang L., Lai S., Ye K. (2011) Structure and molecular evolution of CDGSH iron-sulfur domains. PLoS ONE 6, e24790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Amr S., Heisey C., Zhang M., Xia X. J., Shows K. H., Ajlouni K., Pandya A., Satin L. S., El-Shanti H., Shiang R. (2007) A homozygous mutation in a novel zinc-finger protein, ERIS, is responsible for Wolfram syndrome 2. Am. J. Hum. Genet. 81, 673–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chen Y. F., Kao C. H., Chen Y. T., Wang C. H., Wu C. Y., Tsai C. Y., Liu F. C., Yang C. W., Wei Y. H., Hsu M. T., Tsai S. F., Tsai T. F. (2009) Cisd2 deficiency drives premature aging and causes mitochondria-mediated defects in mice. Genes Dev. 23, 1183–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chang N. C., Nguyen M., Shore G. C. (2012) BCL2-CISD2. An ER complex at the nexus of autophagy and calcium homeostasis? Autophagy 8, 856–857 [DOI] [PubMed] [Google Scholar]
- 57. Wiley S. E., Andreyev A. Y., Divakaruni A. S., Karisch R., Perkins G., Wall E. A., van der Geer P., Chen Y. F., Tsai T. F., Simon M. I., Neel B. G., Dixon J. E., Murphy A. N. (2013) Wolfram syndrome protein, Miner1, regulates sulphydryl redox status, the unfolded protein response, and Ca homeostasis. EMBO Mol. Med. 5, 904–918 [DOI] [PMC free article] [PubMed] [Google Scholar]