

Background: γ-Secretase complexes generate amyloid-β (Aβ) in Alzheimer disease.
Results: Aβ profiles of the four γ-secretase complexes expressed in humans show that PSEN regulates total peptide levels and the Aβ38 pathway, whereas APH1 affects mainly the efficiency of the carboxypeptidase-like activity.
Conclusion: γ-Secretase subunit composition regulates Aβ generation.
Significance: These intrinsic differences could be used to advance AD therapeutic development.
Keywords: Alzheimer Disease, Amyloid Precursor Protein, Enzyme Mechanisms, Neurobiology, Secretases, Amyloid Beta, Gamma-Secretase
Abstract
γ-Secretase complexes are involved in the generation of amyloid-β (Aβ) in the brain. Therefore, γ-secretase has been proposed as a potential therapeutic target in Alzheimer disease (AD). Targeting γ-secretase activity in AD requires the pharmacological dissociation of the processing of physiological relevant substrates and the generation of “toxic” Aβ. Previous reports suggest the differential targeting of γ-secretase complexes, based on their subunit composition, as a valid strategy. However, little is known about the biochemical properties of the different complexes, and key questions regarding their Aβ product profiles should be first addressed. Here, we expressed, purified, and analyzed, under the same conditions, the endopeptidase and carboxypeptidase-like activities of the four γ-secretase complexes present in humans. We find that the nature of the catalytic subunit in the complex affects both activities. Interestingly, PSEN2 complexes discriminate between the Aβ40 and Aβ38 production lines, indicating that Aβ generation in one or the other pathway can be dissociated. In contrast, the APH1 subunit mainly affects the carboxypeptidase-like activity, with APH1B complexes favoring the generation of longer Aβ peptides. In addition, we determined that expression of a single human γ-secretase complex in cell lines retains the intrinsic attributes of the protease while present in the membrane, providing validation for the in vitro studies. In conclusion, our data show that each γ-secretase complex produces a characteristic Aβ signature. The qualitative and quantitative differences between different γ-secretase complexes could be used to advance drug development in AD and other disorders.
Introduction
The γ-secretase complexes are membrane-associated aspartyl proteases involved in the pathogenesis of Alzheimer disease (AD).5 γ-Secretase complexes consist of four essential subunits: nicastrin (NCT), PSEN (presenilin), APH1 (anterior pharynx defective 1), and PEN-2 (presenilin enhancer 2) (1–3). Two different PSEN subunits (PSEN1 and PSEN2) and two different APH1 subunits (APH1A and APH1B) are encoded by separate genes in humans, which results in four different protease complexes (see Fig. 1A). Furthermore, the mRNA for the PSEN and APH1 subunits can be alternatively spliced (4), contributing to the structural diversity of the γ-secretase population.
FIGURE 1.

Purification of γ-secretase complexes. A, schematic representation of the subunit composition and structural heterogeneity of the γ-secretase complex. A color code, indicated on top, is applied to the PSEN and APH1 subunits to show local homology between PSEN1 and PSEN2 or APH1A and APH1B subunits, respectively (based on ClustalW using Blosum matrix). PSEN1 and PSEN2 show 65% homology, whereas APH1A and APH1B show 56% homology at the amino acid level. Stars on presenilins TMD6 and TMD7 denote catalytic aspartate residues Asp-257 and Asp-385, respectively (46). Stars on the ectodomain of NCT indicate complex glycosylation. B, schematic representation of the purification methodology. Baculovirus expressed γ-secretase complex is purified using the GFP tag linked to the cytoplasmic domain of NCT. The complex is eluted with Prescission protease. C, Coomassie staining of purified γ-secretase complexes. Next to the γ-secretase subunits, the Prescission protease remains present in all purified samples. An unidentified band is also visible in all purifications (asterisk). D, Western blot of purified γ-secretase complexes. PEN-2 subunit immunoreactivity was used to estimate and normalize γ-secretase complex levels used in further in vitro activity assays.
Cleavage of the amyloid precursor protein (APP) by β-secretase results in the release of the APP ectodomain into the extracellular environment and the generation of a 99-amino acid-long APP C-terminal fragment (APP-C99) within the membrane (5). APP-C99 is then sequentially processed by the γ-secretase complexes to release C-terminal heterogeneous amyloid-β (Aβ) peptides. Aβ40 is the main product of the γ-secretase (6), but the more aggregating-prone and neurotoxic Aβ42 and Aβ43 are also generated in the brain. Aβ42 and Aβ43 enhance (seed) the formation of neurotoxic oligomers in the brain leading to neuronal dysfunction in AD patients (7, 8).
According to the current model for the function of the γ-secretase complex, APP-C99 is sequentially processed along two production lines. The first endoproteolytic (ϵ-) cleavage occurs either between residues 50 and 51 or between 49 and 50 of APP-C99 and results in the release of an APP intracellular domain fragment called AICD50–99 and the corresponding N-terminal fragment Aβ49 or AICD49–99 and the Aβ48 peptide, respectively. Aβ49/Aβ48 are subsequently shortened by consecutive carboxypeptidase-like γ-cleavages, which progressively decrease their hydrophobicity and increase the probability of their release into the extracellular environment (see Fig. 2) (9, 10). Whereas the efficiency of the endopeptidase cleavage of the γ-secretase defines AICD and total Aβ levels, the carboxypeptidase-like functionality defines the type of Aβ peptides that are generated (Aβ product profiles). The latter is strongly affected by mutations causing AD (11, 12), indicating that changes in the profiles of Aβ peptides affect seeding and toxicity of amyloid oligomers (13–15).
FIGURE 2.
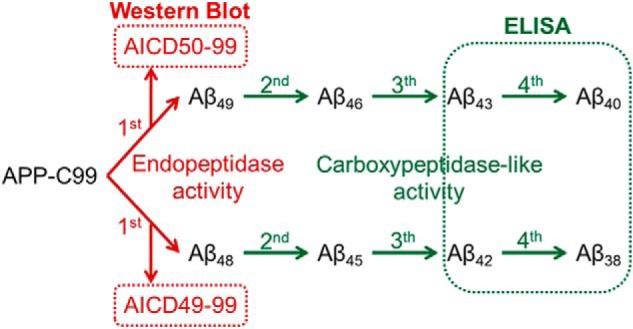
Schematic overview of the current model for γ-secretase activity. APP-C99 is first cleaved by an endopeptidase activity to release the intracellular domain and a long Aβ peptide (Aβ48 or Aβ49). These longer Aβ peptides are then further processed by a consecutive carboxypeptidase-like activity at every third amino acid, except for the processing of Aβ42 into Aβ38, where four amino acids are released. Methods used for the detection and quantification of AICD and Aβ products are indicated.
In addition to APP, γ-secretase complexes cleave many other type I transmembrane proteins, such as Notch1–4, N-cadherin, ErbB4, and neuregulin (16). These substrates are involved in various physiological processes, implying that the pathological and clinical significance of γ-secretase goes beyond AD (17, 18). In fact, the broad substrate specificity of the γ-secretase complexes likely explains the serious side effects (mostly related to Notch toxicity) observed in clinical trials that targeted Aβ production by general inhibition of the γ-secretase complexes (19, 20). For instance, in the phase III clinical trial with the broad spectrum inhibitor semagacestat (20), Aβ levels were not altered in the cerebrospinal fluid, but toxic effects related to the inhibition of the processing of Notch were observed. This negative outcome of the clinical trial is actually not surprising as semagacestat has a stronger IC50 for Notch processing than for APP processing (12). Importantly and unfortunately, this trial did not address whether lowering Aβ production would be beneficial in AD but actually confirmed that general inhibition of the γ-secretase complexes is not a therapeutic option in AD.
The associated toxicity with γ-secretase inhibition could be circumvented if the γ-secretase activity required for the processing of physiological relevant substrates and the generation of toxic Aβ can be pharmacologically dissociated in the brain. In this scenario, the selective targeting of the γ-secretase complex(es) involved in the production of toxic Aβ in the brain could be applied in the clinic.
Gene targeting studies have indeed indicated that the structural heterogeneity of γ-secretase is important in physiology and pathology. Although Psen1 knock-out and Aph1A knock-out mice die during embryogenesis due to impaired Notch signaling (21–24), Psen2 null mice do not exhibit embryonic lethal phenotypes (21, 25–27). However, PSEN1 type γ-secretase complexes seem to produce most of the Aβ in the CNS (28), whereas PSEN2 complexes have a minor contribution (29). But, interestingly, PSEN2 containing γ-secretase complexes appear to carry out an important part of the physiological processing of Notch in peripheral organs. The differential activity profiles of PSEN1/2 complexes were actually exploited in a study conducted by Borgegård et al. (30) in which in vivo inhibition of APP processing by a PSEN2-sparing γ-secretase inhibitor (MRK-560) circumvented to a large extent Notch-related side effects. These results demonstrate that PSEN1 and PSEN2 γ-secretase complexes can be targeted specifically and provide the first preclinical proof of concept that differential targeting of γ-secretase complexes is a worthwhile strategy in therapy development for AD.
Also, the heterogeneity at the APH1 subunit of the γ-secretase complex might provide alternative ways to develop drugs acting specifically on some types of complexes and therefore inhibiting or modulating their physiological substrates. Genetic ablation of the APH1B subunit in mice causes no major phenotypes during development and adulthood. In addition and importantly, it reduced significantly amyloid pathology and restored memory in a murine AD model, while leaving unaffected the processing of the Notch receptor (31). Although more in-depth analysis revealed that the APH1B containing γ-secretase complexes are involved in the processing of neuregulin in the brain (32). Thus, this work evidenced that APH1B containing γ-secretase complexes play a major role in the production of toxic Aβ (amyloid pathology) without contributing significantly to the biology of other substrates and pointed at the selective inhibition of APH1B containing γ-secretase complexes as a potential approach to tackle amyloid pathology in the AD brain. The underlying reason for the biological or pathological activity of the different protease complexes is not fully understood. Although differential expression and co-localization of substrates and enzyme complexes at the tissue, cellular, or subcellular level needs certainly to be considered, distinctive intrinsic enzymatic properties may account for differential processing of substrates by the different γ-secretase complexes (33, 34). In fact, the two possibilities are non-mutually exclusive.
In this study, we focus on the processing of APP by the different γ-secretase complexes and investigate how the structural heterogeneity of the protease affects its endopeptidase and carboxypeptidase-like activities. We show that the PSEN subunit regulates mainly the endopeptidase activity levels whereas the APH1 subunit regulates the functionality of the carboxypeptidase-like activity, leading to important changes in the Aβ product profiles. Most importantly, our data demonstrate that each protease complex has different intrinsic biochemical properties that result in distinctive Aβ product profiles.
EXPERIMENTAL PROCEDURES
Antibodies
Rabbit polyclonal antibodies against APH1A (B80.3), APH1B (B78.2), PEN-2 (B126.2), and APP C terminus (B63.3) and the mouse monoclonal antibody 9C3 against nicastrin have been described (47, 48). Rabbit monoclonal neo-epitope AICD was a gift from Eli Lilly and Co. Commercially available antibodies were as follows: anti-PSEN1 loop (MAB5232) from Chemicon, anti-PSEN2-CTF (D30G3) from Cell Signaling, anti-FLAG M2 from Sigma, goat anti-mouse IRDye800 from Rockland, goat anti-rabbit Alexa Fluor 680 from Invitrogen, 82E1 from Demeditec Diagnostics, biotinylated anti-mouse IgG from Vector Laboratories, and streptavidin-HRP from GE Healthcare. ELISA-capturing antibodies were as follows: JRF AB038 for Aβ38, JRF/cAb40/28 for Aβ40, and JRF/cAb42/26 for Aβ42 from Janssen Pharmaceutica (Beerse, Belgium). 9C4 for Aβ43 from Signet Labs, Inc. Detection antibody huAB25-HRPO was obtained from Janssen Pharmaceutica (Zhou et al. (49)). The anti-GFP nanobody used for the purification of the γ-secretase complexes was obtained from the VIB Nanobody Service Facility (35).
Purification of γ-Secretase Complexes
The human coding sequences of PSEN1 or 2, NCT, APH1AL or B, and PEN-2 were cloned into a pAcAB4 transfer vector (BD Biosciences). Co-transfection of a transfer vector (containing the heterologous genes) and BaculoGoldTM DNA into Sf9 cells allowed homologous recombination and the production of recombinant viruses (36). The GFP was cloned at the C-terminal site of NCT. γ-Secretase complexes were purified using agarose beads (NHS-activated beads, GE Healthcare) coupled with anti-GFP nanobodies. A PreScission cleavage site was included between NCT and GFP and used to elute untagged γ-secretase complexes (see Fig. 1B). Partial removal of the PreScission protease was carried out by concentrating the sample trough a 100 kDa cut-off Amicon Ultra centrifugal filter (Millipore).
Cell Culture and Generation of Stable Cell Lines
Conditional Psen1/2 double knock-out mice were crossed with conditional Aph1ABC triple knock-out mice. At embryonic day 7.5, embryos were dissected and dissociated, and cells were plated in the presence of DMEM/f12 + 50% FCS (Invitrogen). Primary mouse embryonic fibroblasts (MEFs) were immortalized by transduction with the LargeT antigen. Psen1/2 double knock-out/Aph1ABC triple knock-out MEFs were generated by transduction with a Cre-GFP expressing adenoviral vector and GFP-positive MEFs were sorted by FACS analysis. Psen1/2 Aph1ABC-deficient MEFs were maintained in DMEM/f12 + 10% FCS. To rescue γ-secretase expression, Psen1/2 double knock-out/Aph1ABC triple knock-out MEFs were transduced using pMSCV viral vectors (Clontech) containing the human coding sequences of the different PSEN and APH1 homologues and the zeocin selection marker. An IRES sequence was cloned between the coding sequences for PSEN and APH1 to ensure co-expression of both proteins. Stable transfected cell lines were selected using 500 μg/ml zeocin (Invitrogen). Four different combinations were made: PSEN1 and APH1AL, PSEN1 and APH1B, PSEN2 and APH1AL, and PSEN2 and APH1B. These cell lines were transduced with pMSCV viral vectors (Clontech) expressing APP-C99-GFP-puromycin. After puromycin selection (5 μg/ml), GFP-positive cells were selected through FACS sorting.
In Vitro Activity Assays Using Purified γ-Secretase
The γ-secretase in vitro activity assay was performed as described previously (37) with minor modifications. Briefly, in vitro reactions with 30 ng/μl purified γ-secretase and 1.75 μm APP-C99–3×FLAG were performed in 50 mm citric acid, pH 6.7, 0.25 m sucrose, 1 mm EGTA, 1× EDTA-free complete proteinase inhibitors (Roche Applied Science), 2.5% dimethyl sulfoxide, and 0.1% phosphatidylcholine. Twenty μl reactions were incubated for 3 h at 37 °C. Lipids and substrates were extracted by adding 1 volume chloroform/methanol (2:1, v/v). Then, the aqueous fraction (ICD Products) was taken and subjected to SDS-PAGE and quantitative Western immunoblot. Known amounts of APP-C99–3×FLAG (0.1–0.5 pmol), were included as standards for AICD-3×FLAG quantifications. AICD-3×FLAG and standards were determined with the anti-FLAG M2 (Sigma) and goat anti-mouse IR800 (Pierce) antibodies, whereas the AICD50–99 product was determined with a neo-epitope mAb and a goat anti-rabbit Alexa Fluor 680 secondary antibody (Rockland). Infrared signals were detected using the Odyssey Infrared Imaging System. Specific endopeptidase activities (pm/min) in reactions containing equal enzyme levels were calculated by dividing AICD product levels by incubation time.
Quantification of Soluble Aβ Peptides Using Sandwich ELISA
Ninety-six-well plates (Nunc) were coated with 1.5 μg/ml anti-Aβ capture antibodies, except for the anti-Aβ43 antibody that was coated at 7.5 μg/ml, all in a final volume of 50 μl of 10 mm Tris-HCl, 10 mm NaCl, 10 mm NaN3, pH 8.5. After overnight incubation at 4 °C, the plates were rinsed with PBS + 0.05% Tween 20 and blocked with 100 μl per well of casein buffer (1× phosphate-buffered saline with 1% casein, pH 7.4) for 4 h at room temperature. Samples and standards (synthetic human Aβ1–38, Aβ1–40, Aβ1–42, or Aβ1–43 peptides) were diluted in casein buffer. After overnight incubation at 4 °C, plates were rinsed and developed using 50 μl per well of 100 mm NaOAc, pH 4.9, 0.83 mm 3,3′,5,5′-tetramethylbenzidine (Sigma), 0.03% H2O2 (v/v). Reactions were stopped with 50 μl per well of 2 n H2SO4 and read on a Perkin-Elmer Life Science Envision 2103 multilabel reader at 450 nm. Aβ43 peptides in the cell-based assays were quantified by the Aβ1–43 ELISA kit from IBL, according to manufacturer's instructions.
Urea Gels
Aβ peptides were analyzed by a modified version of the urea-based SDS-PAGE (11% T/5% C instead of 12% T/5% C polyacrylamide and 0.075 m instead of 0.1 m H2SO4 in the separation gel) (38). Western immunoblot was performed using 82E1 antibody, biotinylated anti-mouse IgG, and streptavidin-HRP. Signals were detected using ECL chemiluminescence with a Fujifilm LAS-3000 Imager.
Statistics
In vitro experiments were repeated 12 times. Cell-based experiments were repeated four times. Statistical significance of the data was tested with one-way analysis of variance and Dunnett's post test. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001.
RESULTS
Reconstitution and Purification of γ-Secretase Complexes
The baculovirus expression system was used to produce independently the following γ-secretase complexes: NCT·PSEN1·APH1A·PEN-2, NCT·PSEN1·APH1B·PEN-2, NCT·PSEN2·APH1A·PEN-2, and NCT·PSEN2·APH1B·PEN-2. To express the different complexes, we generated four recombinant baculoviruses, each of them expressing the four essential components of the γ-secretase complexes mentioned above. Thus, infection of insect cells with any of these recombinant baculoviruses ensures co-expression of the four essential components of the respective γ-secretase complex in every infected cell, facilitating the reconstitution of a functional enzyme. For purification purposes, the NCT subunit was expressed as a fusion protein with the GFP separated by a PreScission protease cleavage site. Hi5 insect cells infected with each of the recombinant baculoviruses were collected at 72 h post infection and crude cellular extracts were prepared in 1% CHAPSO. After removal of insoluble material by ultracentrifugation, immunoaffinity purification of the respective γ-secretase complexes was performed using a high affinity anti-GFP nanobody coupled to agarose beads. GFP was then cleaved from NCT by specific proteolysis to elute untagged γ-secretase (Fig. 1B) (39). All of the γ-secretase subunits (NCT, APH1, PSEN-CTF, PSEN-NTF, and PEN-2) were present in the purified samples, and purity of the different γ-secretase complexes was high as evaluated by SDS-PAGE followed by Coomassie staining (Fig. 1C). Because NCT and PEN-2 are the only common subunits among the different complexes and we purified through a GFP tag linked to the NCT subunit, we used the intensity of the PEN-2 immunostaining (Fig. 1D) instead of total protein to normalize for γ-secretase complex levels among different samples and purifications. We made all our estimations relative to the NCT·PSEN1·APH1A·PEN-2 complex, which was considered as the reference γ-secretase complex in this study.
PSEN2 Containing γ-Secretase Complexes Display Lower Endopeptidase Activities Than the Corresponding PSEN1 Complexes
To investigate the functional relevance of the subunit composition of the γ-secretase complex, we characterized the intrinsic kinetic properties of the four different γ-secretase complexes. First, we evaluated their endopeptidase activity by measuring the de novo generation of AICD in an in vitro γ-secretase activity assay (12, 37), in which equivalent amounts of purified enzymes were tested against purified APP-C99-FLAG substrate at saturating concentrations. AICD product was quantified by SDS-PAGE and Western blotting against the FLAG tag. Addition of 10 μm γ-secretase inhibitor X (L-685,458; InhX, Merck) blocked the production of AICD demonstrating the specificity of the proteolytic assay (see Fig. 3A). APH1A containing γ-secretase complexes produce similar AICD levels compared with the corresponding APH1B containing complexes. In contrast, the endopeptidase activity was decreased by ∼35% in PSEN2 containing γ-secretase complexes (see Fig. 3B). Furthermore, we asked whether the subunit composition of the γ-secretase has an effect on the position of the (ϵ-) endopeptidase cleavage and therefore on the product line preference of the enzyme (Fig. 2). To address this question, we used an antibody that recognizes specifically the AICD50–99-FLAG product (12) and the M2 FLAG antibody for the detection of total AICD (AICD50–99-FLAG and AICD49–99-FLAG). The AICD50–99/total AICD ratio showed that all γ-secretase complexes cleave the APP-C99 substrate in a similar way with regard to the position of the endoproteolytic cleavage (Fig. 3C).
FIGURE 3.
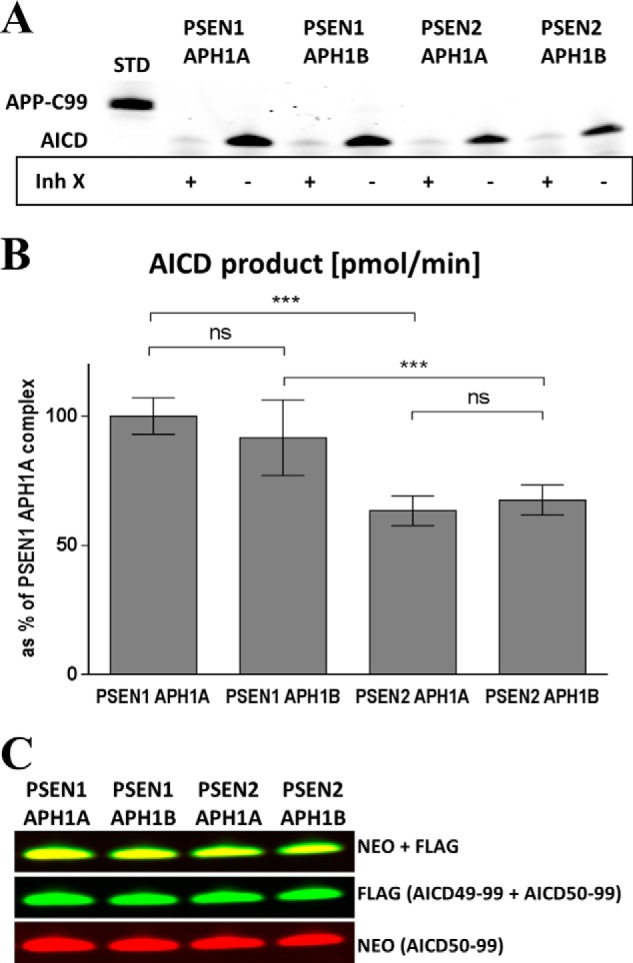
PSEN2 containing γ-secretase complexes display lower endopeptidase activity (ϵ-cleavage), relative to PSEN1 complexes. A, total AICD generated by the different γ-secretase complexes in the presence or absence of 10 μm inhibitor X (Inh X; L-685,458, Merck). B, total AICD levels plotted as % of AICD generated by the PSEN1·APH1A γ-secretase complex (mean ± 95% CI, n = 12). C, detection of AICD50–99 and total AICD using a neo epitope (Neo) antibody and the FLAG-M2 antibody, respectively. Relative staining is not different. ns, not significant.
Subunit Composition of the γ-Secretase Complex Affects the Functionality of the Carboxypeptidase-like Activity
To evaluate the carboxypeptidase-like activity of the different γ-secretase complexes, we quantified Aβ38, Aβ40, Aβ42, and Aβ43 by ELISA. These Aβ peptides represent substrates (Aβ43 and Aβ42) and products (Aβ40 and Aβ38) of the fourth enzymatic turnover in each of the product lines of the APP-C99 processing by the γ-secretase (Fig. 2). Because the endopeptidase activities of the γ-secretase complexes differ, Aβ peptides were normalized relative to total AICD, which reflects total Aβ substrate (Aβ49 + Aβ48) available for the carboxypeptidase-like activity. Clearly, the four γ-secretase complexes appear to generate distinct Aβ product profiles (Fig. 4, A and C). For instance, the production of the short Aβ38 and Aβ40 peptides are decreased in the APH1B-containing complexes, compared with the corresponding APH1A complexes (Fig. 4A and Table 1 for statistical significance).
FIGURE 4.

γ-Secretase complexes generate distinctive Aβ product profiles. A, Aβ products were measured using ELISA techniques and normalized to initial AICD generation. Results are plotted as % of the Aβ products generated by the PSEN1·APH1A γ-secretase complex (mean ± 95% CI, n = 12). Aβ38 peptides were undetectable in reactions with the PSEN2·APH1B complex (ND). B, the sum of Aβ38, Aβ40, Aβ42, and Aβ43 was plotted as % of the PSEN1·APH1A complex (mean ± 95% CI, n = 12). Lower total Aβ levels suggest the accumulation of longer Aβ (>43) peptides. C, Aβ profiles analyzed in urea-based gels corroborate the accumulation of longer Aβ peptides by these complexes. Aβ42 and Aβ43 are running as one band, the asterisk denotes an aspecific band (between Aβ40 and Aβ38).
TABLE 1.
p values corresponding to pairwise comparisons of the Aβ product profiles in Fig. 4A
p values for the pairwise comparison evaluated by one-way analysis of variance and Dunnett's post test are given for each Aβ peptide. ND, not determined. (Aβ peptides were undetectable in reactions with the PSEN2·APH1B complex.)
| Aβ38 | Aβ40 | Aβ42 | Aβ43 | |
|---|---|---|---|---|
| 1A vs. 1B | <0.0001 | <0.0001 | <0.0001 | 0.3296 |
| 1A vs. 2A | <0.0001 | 0.0534 | <0.0001 | 0.6509 |
| 1B vs. 2B | ND | <0.0001 | 0.0019 | 0.1292 |
| 2A vs. 2B | ND | <0.0001 | 0.9801 | 0.3087 |
We took the sum of Aβ38, Aβ40, Aβ42, and Aβ43 peptides (third + fourth Aβ products) as a measure for the overall functionality of the carboxypeptidase-like activity of the different complexes (i.e. how efficient are the enzymes in hydrolyzing the long Aβ48 and Aβ49 peptides initially generated upon endoproteolysis). The significant reductions observed in these Aβ products for the PSEN1·APH1B and the two PSEN2 containing γ-secretase complexes, when compared with PSEN1·APH1A (Fig. 4B), implies the concomitant accumulation of longer Aβ precursors (Aβ>43). This was confirmed by analyzing total Aβ products in urea based gels (Fig. 4C). Clearly, the PSEN1·APH1B complex (Fig. 4C, lane 3) or PSEN2 containing γ-secretase complexes (Fig. 4C, lanes 4 and 5) maintain proportionally longer Aβ peptide species than the reference complex (PSEN1·APH1A) (Fig. 4C, lane 2).
APH1B Containing γ-Secretase Complexes Are Less Functional Carboxypeptidases Than Their APH1A Counterparts
The Aβ38/Aβ42 and Aβ40/Aβ43 product/substrate ratios provide information on the efficiency of the fourth enzymatic turnover. These ratios reveal that in the PSEN1·APH1B complex the fourth turnover in both product lines is decreased by ∼70%, relative to the reference complex (Fig. 5). Similarly, the PSEN2·APH1B complex lowers the Aβ40/Aβ43 ratio by ∼50% relative to the PSEN2·APH1A complex (Fig. 5, gray bars). These decrements indicate that the APH1 subunit is involved in the mechanism(s) that regulate(s) the efficiency of the fourth catalytic turnover of the γ-secretase complex and likely the functionality of the carboxypeptidase.
FIGURE 5.

The APH1 subunit affects the fourth cleavage in vitro. Aβ product/substrate ratios for the fourth turnover were plotted as % of the ratio generated by the PSEN1/APH1A complex (mean ± 95% CI, n = 12). Aβ38/Aβ42 ratios could not be calculated for the PSEN2/APH1B complex because the levels of Aβ38 were undetectable (ND). APH1B containing γ-secretase complexes lower the efficiency of the fourth cycle of γ-secretase activity, when compared with the corresponding APH1A containing complexes.
PSEN2 Subunit Affects Differentially the Aβ40 and Aβ38 Production Pathways
Aβ40/Aβ43 ratios indicate that PSEN1·APH1A versus PSEN2·APH1A and PSEN1·APH1B versus PSEN2·APH1B complexes are equally efficient at the processing of Aβ43 to Aβ40 peptide (Fig. 5, gray bars). However, the Aβ38/Aβ42 ratios suggest that the processing of Aβ42 by the PSEN2-catalytic subunit occurs in a less efficient manner than in the PSEN1 active site (Fig. 5, white bars). In addition, lower Aβ42 levels are observed in the PSEN2 reactions, relative to the corresponding PSEN1 assays (Fig. 4A and Table 1). Because Aβ peptides are normalized toward total AICD (Aβ49 + Aβ48) produced in each reaction, lower Aβ42 levels likely result from lower turnover of the Aβ precursors (Aβ45 and/or Aβ48) in the PSEN2 (relative to PSEN1) reactions. In agreement, Aβ profiles in urea-based gels show higher levels of the longer Aβ peptides in the reactions for PSEN2 complexes than in those for PSEN1, when the APH1 subunit is constant (i.e. PSEN1·APH1A versus PSEN2·APH1A and PSEN1·APH1B versus PSEN2·APH1B) (Fig. 4C) (band refer as > Aβ46, may contain peptides longer than 46 amino acids). Thus, although all the different complexes do not change the product line preference (position of ϵ-cleavage, Fig. 3C), PSEN2-containing complexes seem to process Aβ peptides less efficiently in the Aβ38 pathway.
The Subunit Composition of the γ-Secretase Has a Significant Effect on Aβ Product Profiles in Cells
We then asked whether the changes in the Aβ product profiles observed in vitro could also be observed in a cell-based assay. To evaluate the biochemical properties of the different γ-secretase complexes in cells, we generated MEFs deficient for the Psen and Aph1 genes (Psen1/2−/−, Aph1ABC−/−) and rescued them with either human PSEN1 or PSEN2 and with either human APH1A or APH1B to generate four independent cell lines, with each expressing exclusively one type of γ-secretase complex, i.e. the different combinations for PSEN1·PSEN2 and APH1A·APH1B. SDS-PAGE and Western blotting against the different γ-secretase subunits validate this claim (Fig. 6A). Because the endogeneous APP in MEF cells is predominantly processed to APP-C83, all cell lines expressed stably human APP-C99-GFP, the direct substrate of γ-secretase and precursor of Aβ peptides. Human APP-C99-GFP protein levels are comparable and in the same range as the endogeneous APP and APP-C83 (Fig. 6B). Importantly, the reconstitution of active γ-secretase complexes could rescue the accumulation of endogeneous APP-C83 substrate, which could be inhibited by a γ-secretase inhibitor (Fig. 6B). This inhibitor also prevented the processing of the human APP-C99 into AICD favoring its processing into APP-C83 instead (Fig. 6B). Secreted Aβ products (Aβ38, Aβ40, Aβ42, and Aβ43) were quantified from the extracellular medium by ELISA techniques.
FIGURE 6.

Cell-based assays confirm the effects of APH1 and PSEN subunit composition on γ-secretase activities. A, expression of γ-secretase subunits in MEFs deficient for the two endogenous PSEN and the three endogenous APH1 subunits and rescued with either human PSEN1 or PSEN2 and with either human APH1A or APH1B. B, expression of endogeneous APP and APP-C83 and the introduced human APP-C99-GFP and its products in the presence or absence of 10 μm semagacestat (Janssen Pharmaceutica). Pictures come from the same blot developed at one exposure, so protein levels can be mutually compared. C, plotted as % of the ratio generated by the MEF cell line expressing PSEN1 and APH1A (mean ± 95% CI, n = 5).
We calculated the product/substrate ratios (Aβ40/Aβ43 and Aβ38/Aβ42) for the fourth enzymatic turnover of the γ-secretase complex. With regard to the APH1 subunit, the Aβ40/Aβ43 and Aβ38/Aβ42 ratios indicate that PSEN1·APH1B complexes are less functional than their PSEN1·APH1A counterparts in the cell (Fig. 6C), which supports the observations in vitro. Additionally, the Aβ40/Aβ43 ratio for the PSEN2·APH1B complex evidences a less functional turnover of Aβ43, with respect to the PSEN2·APH1A complex, but this difference does not reach statistical significance on the other product line (Aβ38/Aβ42 ratio). However, PSEN2 complexes show an increased turnover of Aβ43 but decrease the conversion of Aβ42 into Aβ38 with respect to their PSEN1 counterparts. Thus, as expected from our in vitro observations, the effect of the PSEN2 subunit on the processing of APP depends on the product line (Fig. 6C).
DISCUSSION
We demonstrate that the structural heterogeneity of the γ-secretase has an effect on the intrinsic enzymatic properties of the complex by analyzing the processing of APP-C99 towards Aβ peptides. Using purified γ-secretase complexes expressed in insect cells (Fig. 1, B–D), we found that the endopeptidase activity levels are regulated by the nature of the catalytic subunit of the complex. The PSEN2 type complexes appear to be the less active endopeptidases (Fig. 3B). These results expand previous observations that show lower activity levels in samples containing PSEN2 type γ-secretase complexes, relative to those with PSEN1 as catalytic subunit (solubilized microsomal membranes from MEFs, blastocyst derived cell cultures or mouse brain lysates) (12, 33, 34, 40). Our results differ from data published by Yonemura et al. in 2011 (42), who used a yeast reconstitution system to express the different γ-secretase complexes (41). They found that solubilized microsomes prepared from cells expressing the PSEN1·APH1A γ-secretase complex produced ∼24-fold more Aβ than those containing PSEN2·APH1A complexes (42), but γ-secretase levels were ∼28-fold higher in microsomes containing PSEN1 than in those with PSEN2. Thus, apparent specific activities for the PSEN1·APH1A and PSEN2·APH1A γ-secretase complexes in their experiments seemed similar. However, the use of microsomes complicates considerably the interpretation of the data. Questions such as to what extent substrate and enzyme interact or to what extent the interaction occurs under similar conditions in the different preparations are critical issues that cannot be controlled nor addressed under these conditions. Our approach using purified enzymes and purified substrate allows control of critical variables such as substrate and enzyme concentrations or lipids present in the assay. Our work further differentiates from previous publications because we evaluated the specificity of the PSEN1 and PSEN2 endopeptidase cleavage by quantifying the position of the initial (ϵ-) cleavage in the APP sequence. Our results show that PSEN2 and PSEN1 complexes differ at their endopeptidase activity levels but display similar endopeptidase cleavage specificities against the APP-C99 substrate (Fig. 3C), which results in lower Aβ48 and Aβ49 product levels in the PSEN2 reactions, but similar Aβ48/Aβ49 ratios, compared with PSEN1 reactions.
We investigated the effect of subunit composition on the functionality of the carboxypeptidase-like activity of the γ-secretase complex. Importantly, Aβ peptides were normalized relative to total AICD produced in the assay, i.e. for the initial endopeptidase activity of the complexes (Fig. 4A and Table 1). PSEN2 type carboxypeptidases appeared to be less functional than the PSEN1 counterparts, as indicated by the Aβ38, Aβ40, Aβ42, and Aβ43 product levels (Fig. 4B) and the accumulation of longer and more hydrophobic Aβ peptides in the reactions. Aβ45 is relatively increased in the PSEN2/APH1B profile compared with the PSEN1/APH1B (Fig. 4C, lane 5 versus 3); whereas the >Aβ46 band suggests a higher relative abundance of longer Aβ peptides in the PSEN2/APH1A profile versus PSEN1/APH1A (Fig. 4C, lane 4 versus 2). However, and interestingly, our Aβ product profiles show that although Aβ38 and Aβ42 levels are decreased in PSEN2 versus corresponding PSEN1 reactions, Aβ40 and Aβ43 products are produced to similar levels (Fig. 4A). This results in decreased Aβ38/Aβ42 but similar Aβ40/Aβ43 ratios in PSEN2 versus PSEN1 counterpart reactions (PSEN2·APH1A versus PSEN1·APH1A and PSEN2·APH1B versus PSEN1·APH1B) (Fig. 5). Our results thus indicate that PSEN2-containing complexes affect differentially the Aβ product lines, being less functional at the Aβ38 product line. Furthermore, the fact that PSEN2 type complexes produce lower Aβ42 than the corresponding PSEN1 complexes suggests that the product line is actually affected at the second and/or third enzymatic cycles of the Aβ38 product line (Aβ48 to Aβ45 and/or Aβ45 to Aβ42). Recently, Okochi et al. (43) reported that 40% of Aβ38 is actually derived from Aβ43 in HEK cells. We cannot truly evaluate the extent to which this observation influences our results, as it is unknown what type of γ-secretase complexes are present in HEK cells and the proportion in which they are expressed. In addition, it is unclear whether this observation applies to all the different complexes. In case this observation is valid for all, then the Aβ38/Aβ42 ratio actually underestimates the magnitude of the effects.
Taken together, the nature of the catalytic subunit in the γ-secretase complex is determinant for both endo- and carboxypeptidase-like activities, but surprisingly, it mainly affects the carboxypeptidase functionality in one production line. The mechanism(s) of this observation needs further functional and structural understanding of the γ-secretase complex, probably requiring a deep comparison of PSEN1 versus PSEN2 complexes once the structure of the γ-secretase complex is available at atomic resolution.
In contrast, structural heterogeneity at the APH1 subunit affects mainly the carboxypeptidase-like activity of the complex (Fig. 4A). Both APH1A and APH1B type γ-secretase complexes display similar endopeptidase activities (Fig. 3B). However, APH1B containing complexes are less efficient carboxypeptidases compared with the corresponding APH1A complexes (Figs. 4 and 5). This reinforces the idea that endo- and carboxypeptidase-like activities of γ-secretase are uncoupled (12, 44) and also confirms our previous work (31), demonstrating that APH1B complexes generate longer Aβ peptides. Interestingly, the impact of APH1B on the fourth enzymatic turnover of the γ-secretase results in relative more Aβ42 or Aβ43 (Figs. 4 and 5) than in the APH1A product profiles; an effect that has also been observed with γ-secretase complexes containing familial AD-linked PSEN mutants. We have previously suggested that this could reflect a “premature” release of intermediary Aβ products, i.e. a less efficient retaining of longer Aβ by the γ-secretase complex containing the APH1B subunit similar to what is seen with familial AD-linked PSEN mutants (12). These effects (APH1A versus APH1B complexes or WT versus familial AD-linked complexes) correlate with a change in the conformation of the γ-secretase complexes as indirectly measured by fluorescence lifetime imaging microscopy technology (31, 45). Taken together, this suggests that the APH1 subunit stabilizes PSEN in particular conformations, referred to as open/closed (31), and the conformational status of PSEN has an effect on the efficiency of the carboxypeptidase-like activity. Interestingly, these data indicate that APH1 may act as an allosteric subunit that activates/inhibits the production of short Aβ peptides but does not regulate the generation of APP intracellular domain (endopeptidase activity). Theoretically, this opens the possibility that molecules that bind to APH1 could affect Aβ generation.
Finally, the results obtained in the in vitro enzymatic characterization are well supported by the results obtained in cell lines expressing only one γ-secretase complex. Thus, APH1B complexes present a lower Aβ40/Aβ43 ratio than their APH1A counterparts and PSEN2 affects considerably the efficiency of the carboxypeptidase-like activity in the Aβ38 production line compared with PSEN1 (Fig. 6C). We conclude that the functionality and product line preference of the carboxypeptidase-like activity are intrinsic attributes of the enzyme complexes and that those properties are well maintained in solubilized conditions, something that was also observed when characterizing the familial AD linked PSEN mutations in vitro and in cell culture (12).
In conclusion, this is the first time that both endo- and carboxypeptidase-like activities of human γ-secretase complexes are compared under the same conditions. Our results show that the nature of the PSEN catalytic subunit present in the complex affects both γ-secretase activities. Interestingly, PSEN2 complexes discriminate between the Aβ40 and Aβ38 production lines, indicating that Aβ40 and Aβ42 generation can be dissociated at the level of the carboxypeptidase-like activity. In contrast, the APH1 subunit mainly affects the carboxypeptidase-like activity, with APH1B complexes favoring the generation of longer Aβ peptides. Clearly, each γ-secretase complex produces a characteristic Aβ signature. Such differences could be used to advance AD therapeutic development.
Acknowledgments
We thank Dr. Veerle Vulsteke, Heidi Lauridsen, and Dr. An Herreman for technical support; Dr. Philip Skezeres and Dr. Pat May (Eli-Lilly, Indianapolis, IN) for AICD antibody; and Dr. Marc Mercken (Janssen Pharmaceutica) for the Aβ antibodies.
This work was supported by the Fonds voor Wetenschappelijk Onderzoek, the University of Leuven and the Flemish Institute for Biology, Stichting Alzheimer Onderzoek Grant SAO-FRA P-12007, the Interuniversity Attraction Poles Program IUAP P6P7/1658 of the Belgian Federal Science Policy Office, a Methusalem grant of the Flemish government, the University of Leuven (to B. D. S.), the Arthur Bax-Anna Vanluffelen chair for Alzheimer research (to. B. D. S.), and an anonymous foundation.
- AD
- Alzheimer disease
- Aβ
- amyloid β
- APP
- amyloid precursor protein
- APP-C99
- 99-amino acid-long APP C-terminal fragment
- AICD
- APP intracellular domain
- PEN-2
- presenilin enhancer 2
- PSEN
- presenilin
- APH1
- anterior pharynx defective 1
- CI
- confidence interval
- MEF
- mouse embryonic fibroblast
- NCT
- nicastrin.
REFERENCES
- 1. Edbauer D., Winkler E., Regula J. T., Pesold B., Steiner H., Haass C. (2003) Reconstitution of γ-secretase activity. Nat. Cell Biol. 5, 486–488 [DOI] [PubMed] [Google Scholar]
- 2. Tolia A., De Strooper B. (2009) Structure and function of γ-secretase. Semin. Cell Dev. Biol. 20, 211–218 [DOI] [PubMed] [Google Scholar]
- 3. De Strooper B. (2003) Aph-1, Pen-2, and Nicastrin with Presenilin Generate an Active γ-Secretase Complex. Neuron 38, 9–12 [DOI] [PubMed] [Google Scholar]
- 4. Gu Y., Chen F., Sanjo N., Kawarai T., Hasegawa H., Duthie M., Li W., Ruan X., Luthra A., Mount H. T., Tandon A., Fraser P. E., St George-Hyslop P. (2003) APH-1 interacts with mature and immature forms of presenilins and nicastrin and may play a role in maturation of presenilin.nicastrin complexes. J. Biol. Chem. 278, 7374–7380 [DOI] [PubMed] [Google Scholar]
- 5. Vassar R., Bennett B. D., Babu-Khan S., Kahn S., Mendiaz E. A., Denis P., Teplow D. B., Ross S., Amarante P., Loeloff R., Luo Y., Fisher S., Fuller J., Edenson S., Lile J., Jarosinski M. A., Biere A. L., Curran E., Burgess T., Louis J. C., Collins F., Treanor J., Rogers G., Citron M. (1999) β-Secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286, 735–741 [DOI] [PubMed] [Google Scholar]
- 6. Mori H., Takio K., Ogawara M., Selkoe D. J. (1992) Mass spectrometry of purified amyloid β protein in Alzheimer's disease. J. Biol. Chem. 267, 17082–17086 [PubMed] [Google Scholar]
- 7. Welander H., Frånberg J., Graff C., Sundström E., Winblad B., Tjernberg L. O. (2009) Aβ43 is more frequent than Aβ40 in amyloid plaque cores from Alzheimer disease brains. J. Neurochem. 110, 697–706 [DOI] [PubMed] [Google Scholar]
- 8. Benilova I., Karran E., De Strooper B. (2012) The toxic A[β] oligomer and Alzheimer's disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357 [DOI] [PubMed] [Google Scholar]
- 9. Takami M., Nagashima Y., Sano Y., Ishihara S., Morishima-Kawashima M., Funamoto S., Ihara Y. (2009) γ-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of β-carboxyl terminal fragment. J. Neurosci. 29, 13042–13052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Qi-Takahara Y., Morishima-Kawashima M., Tanimura Y., Dolios G., Hirotani N., Horikoshi Y., Kametani F., Maeda M., Saido T. C., Wang R., Ihara Y. (2005) Longer forms of amyloid β protein: implications for the mechanism of intramembrane cleavage by γ-secretase. J. Neurosci. 25, 436–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Funamoto S., Morishima-Kawashima M., Tanimura Y., Hirotani N., Saido T. C., Ihara Y. (2004) Truncated Carboxyl-Terminal Fragments of β-Amyloid Precursor Protein Are Processed to Amyloid β-Proteins 40 and 42†. Biochemistry 43, 13532–13540 [DOI] [PubMed] [Google Scholar]
- 12. Chávez-Gutiérrez L., Bammens L., Benilova I., Vandersteen A., Benurwar M., Borgers M., Lismont S., Zhou L., Van Cleynenbreugel S., Esselmann H., Wiltfang J., Serneels L., Karran E., Gijsen H., Schymkowitz J., Rousseau F., Broersen K., De Strooper B. (2012) The mechanism of [γ]-Secretase dysfunction in familial Alzheimer disease. EMBO J. 31, 2261–2274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kuperstein I., Broersen K., Benilova I., Rozenski J., Jonckheere W., Debulpaep M., Vandersteen A., Segers-Nolten I., Van Der Werf K., Subramaniam V., Braeken D., Callewaert G., Bartic C., D'Hooge R., Martins I. C., Rousseau F., Schymkowitz J., De Strooper B. (2010) Neurotoxicity of Alzheimer's disease A[β] peptides is induced by small changes in the A[β]42 to A[β]40 ratio. EMBO J. 29, 3408–3420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yoshiike Y., Chui D. H., Akagi T., Tanaka N., Takashima A. (2003) Specific compositions of amyloid-β peptides as the determinant of toxic β-aggregation. J. Biol. Chem. 278, 23648–23655 [DOI] [PubMed] [Google Scholar]
- 15. Pauwels K., Williams T. L., Morris K. L., Jonckheere W., Vandersteen A., Kelly G., Schymkowitz J., Rousseau F., Pastore A., Serpell L. C., Broersen K. (2012) The structural basis for increased toxicity of pathological Aβ42:Aβ40 ratios in Alzheimer's disease. J. Biol. Chem. 287, 5650–5660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wakabayashi T., De Strooper B. (2008) Presenilins: members of the γ-secretase quartets, but part-time soloists too. Physiology 23, 194–204 [DOI] [PubMed] [Google Scholar]
- 17. McCarthy J. V., Twomey C., Wujek P. (2009) Presenilin-dependent regulated intramembrane proteolysis and γ-secretase activity. Cell Mol. Life Sci. 66, 1534–1555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. De Strooper B., Annaert W. (2010) Novel research horizons for presenilins and γ-secretases in cell biology and disease. Annu. Rev. Cell Dev. Biol. 26, 235–260 [DOI] [PubMed] [Google Scholar]
- 19. Wong G. T., Manfra D., Poulet F. M., Zhang Q., Josien H., Bara T., Engstrom L., Pinzon-Ortiz M., Fine J. S., Lee H. J., Zhang L., Higgins G. A., Parker E. M. (2004) Chronic treatment with the γ-secretase inhibitor LY-411,575 inhibits β-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J. Biol. Chem. 279, 12876–12882 [DOI] [PubMed] [Google Scholar]
- 20. Doody R. S., Raman R., Farlow M., Iwatsubo T., Vellas B., Joffe S., Kieburtz K., He F., Sun X., Thomas R. G., Aisen P. S., Alzheimer's Disease Cooperative Study Steering Committee, Siemers E., Sethuraman G., Mohs R. (2013) A phase 3 trial of semagacestat for treatment of Alzheimer's disease. New Engl. J. Med. 369, 341–350 [DOI] [PubMed] [Google Scholar]
- 21. Shen J., Bronson R. T., Chen D. F., Xia W., Selkoe D. J., Tonegawa S. (1997) Skeletal and CNS defects in presenilin-1-deficient mice. Cell 89, 629–639 [DOI] [PubMed] [Google Scholar]
- 22. Serneels L., Dejaegere T., Craessaerts K., Horré K., Jorissen E., Tousseyn T., Hébert S., Coolen M., Martens G., Zwijsen A., Annaert W., Hartmann D., De Strooper B. (2005) Differential contribution of the three Aph1 genes to γ-secretase activity in vivo. Proc. Natl. Acad. Sci. U.S.A. 102, 1719–1724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. De Strooper B., Annaert W., Cupers P., Saftig P., Craessaerts K., Mumm J. S., Schroeter E. H., Schrijvers V., Wolfe M. S., Ray W. J., Goate A., Kopan R. (1999) A presenilin-1-dependent [γ]-secretase-like protease mediates release of Notch intracellular domain. Nature 398, 518–522 [DOI] [PubMed] [Google Scholar]
- 24. Struhl G., Greenwald I. (1999) Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature 398, 522–525 [DOI] [PubMed] [Google Scholar]
- 25. Herreman A., Hartmann D., Annaert W., Saftig P., Craessaerts K., Serneels L., Umans L., Schrijvers V., Checler F., Vanderstichele H., Baekelandt V., Dressel R., Cupers P., Huylebroeck D., Zwijsen A., Van Leuven F., De Strooper B. (1999) Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc. Natl. Acad. Sci. U.S.A. 96, 11872–11877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Steiner H., Duff K., Capell A., Romig H., Grim M. G., Lincoln S., Hardy J., Yu X., Picciano M., Fechteler K., Citron M., Kopan R., Pesold B., Keck S., Baader M., Tomita T., Iwatsubo T., Baumeister R., Haass C. (1999) A loss of function mutation of presenilin-2 interferes with amyloid β-peptide production and Notch signaling. J. Biol. Chem. 274, 28669–28673 [DOI] [PubMed] [Google Scholar]
- 27. Donoviel D. B., Hadjantonakis A. K., Ikeda M., Zheng H., Hyslop P. S., Bernstein A. (1999) Mice lacking both presenilin genes exhibit early embryonic patterning defects. Genes Dev. 13, 2801–2810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. De Strooper B., Saftig P., Craessaerts K., Vanderstichele H., Guhde G., Annaert W., Von Figura K., Van Leuven F. (1998) Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 391, 387–390 [DOI] [PubMed] [Google Scholar]
- 29. Frånberg J., Svensson A. I., Winblad B., Karlström H., Frykman S. (2011) Minor contribution of presenilin 2 for γ-secretase activity in mouse embryonic fibroblasts and adult mouse brain. Biochem. Biophys. Res. Commun. 404, 564–568 [DOI] [PubMed] [Google Scholar]
- 30. Borgegård T., Gustavsson S., Nilsson C., Parpal S., Klintenberg R., Berg A. L., Rosqvist S., Serneels L., Svensson S., Olsson F., Jin S., Yan H., Wanngren J., Jureus A., Ridderstad-Wollberg A., Wollberg P., Stockling K., Karlström H., Malmberg A., Lund J., Arvidsson P. I., De Strooper B., Lendahl U., Lundkvist J. (2012) Alzheimer's disease: presenilin 2-sparing γ-secretase inhibition is a tolerable Aβ peptide-lowering strategy. J. Neurosci. 32, 17297–17305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Serneels L., Van Biervliet J., Craessaerts K., Dejaegere T., Horré K., Van Houtvin T., Esselmann H., Paul S., Schäfer M. K., Berezovska O., Hyman B. T., Sprangers B., Sciot R., Moons L., Jucker M., Yang Z., May P. C., Karran E., Wiltfang J., D'Hooge R., De Strooper B. (2009) γ-Secretase heterogeneity in the Aph1 subunit: relevance for Alzheimer's disease. Science 324, 639–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dejaegere T., Serneels L., Schäfer M. K., Van Biervliet J., Horré K., Depboylu C., Alvarez-Fischer D., Herreman A., Willem M., Haass C., Höglinger G. U., D'Hooge R., De Strooper B. (2008) Deficiency of Aph1B/C-γ-secretase disturbs Nrg1 cleavage and sensorimotor gating that can be reversed with antipsychotic treatment. Proc. Natl. Acad. Sci. U.S.A. 105, 9775–9780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lai M. T., Chen E., Crouthamel M. C., DiMuzio-Mower J., Xu M., Huang Q., Price E., Register R. B., Shi X. P., Donoviel D. B., Bernstein A., Hazuda D., Gardell S. J., Li Y. M. (2003) Presenilin-1 and presenilin-2 exhibit distinct yet overlapping γ-secretase activities. J. Biol. Chem. 278, 22475–22481 [DOI] [PubMed] [Google Scholar]
- 34. Mastrangelo P., Mathews P. M., Chishti M. A., Schmidt S. D., Gu Y., Yang J., Mazzella M. J., Coomaraswamy J., Horne P., Strome B., Pelly H., Levesque G., Ebeling C., Jiang Y., Nixon R. A., Rozmahel R., Fraser P. E., St George-Hyslop P., Carlson G. A., Westaway D. (2005) Dissociated phenotypes in presenilin transgenic mice define functionally distinct γ-secretases. Proc. Natl. Acad. Sci. U.S.A. 102, 8972–8977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rothbauer U., Zolghadr K., Muyldermans S., Schepers A., Cardoso M. C., Leonhardt H. (2008) A versatile nanotrap for biochemical and functional studies with fluorescent fusion proteins. Mol. Cell. Proteomics 7, 282–289 [DOI] [PubMed] [Google Scholar]
- 36. Belyaev A. S., Roy P. (1993) Development of baculovirus triple and quadruple expression vectors: co-expression of three or four bluetongue virus proteins and the synthesis of bluetongue virus-like particles in insect cells. Nucleic Acids Res. 21, 1219–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kakuda N., Funamoto S., Yagishita S., Takami M., Osawa S., Dohmae N., Ihara Y. (2006) Equimolar production of amyloid β-protein and amyloid precursor protein intracellular domain from β-carboxyl-terminal fragment by γ-secretase. J. Biol. Chem. 281, 14776–14786 [DOI] [PubMed] [Google Scholar]
- 38. Wiltfang J., Esselmann H., Bibl M., Smirnov A., Otto M., Paul S., Schmidt B., Klafki H. W., Maler M., Dyrks T., Bienert M., Beyermann M., Rüther E., Kornhuber J. (2002) Highly conserved and disease-specific patterns of carboxyterminally truncated Aβ peptides 1–37/38/39 in addition to 1–40/42 in Alzheimer's disease and in patients with chronic neuroinflammation. J. Neurochem. 81, 481–496 [DOI] [PubMed] [Google Scholar]
- 39. Walker P. A., Leong L. E., Ng P. W., Tan S. H., Waller S., Murphy D., Porter A. G. (1994) Efficient and rapid affinity purification of proteins using recombinant fusion proteases. Bio/technology 12, 601–605 [DOI] [PubMed] [Google Scholar]
- 40. Bentahir M., Nyabi O., Verhamme J., Tolia A., Horré K., Wiltfang J., Esselmann H., De Strooper B. (2006) Presenilin clinical mutations can affect γ-secretase activity by different mechanisms. J. Neurochem. 96, 732–742 [DOI] [PubMed] [Google Scholar]
- 41. Yagishita S., Futai E., Ishiura S. (2008) In vitro reconstitution of γ-secretase activity using yeast microsomes. Biochem. Biophys. Res. Commun. 377, 141–145 [DOI] [PubMed] [Google Scholar]
- 42. Yonemura Y., Futai E., Yagishita S., Suo S., Tomita T., Iwatsubo T., Ishiura S. (2011) Comparison of presenilin 1 and presenilin 2 γ-secretase activities using a yeast reconstitution system. J. Biol. Chem. 286, 44569–44575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Okochi M., Tagami S., Yanagida K., Takami M., Kodama T. S., Mori K., Nakayama T., Ihara Y., Takeda M. (2013) γ-Secretase modulators and presenilin 1 mutants act differently on presenilin/γ-secretase function to cleave Aβ42 and Aβ43. Cell Rep. 3, 42–51 [DOI] [PubMed] [Google Scholar]
- 44. Quintero-Monzon O., Martin M. M., Fernandez M. A., Cappello C. A., Krzysiak A. J., Osenkowski P., Wolfe M. S. (2011) Dissociation between the processivity and total activity of γ-secretase: implications for the mechanism of Alzheimer's disease-causing presenilin mutations. Biochemistry 50, 9023–9035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Berezovska O., Lleo A., Herl L. D., Frosch M. P., Stern E. A., Bacskai B. J., Hyman B. T. (2005) Familial Alzheimer's disease presenilin 1 mutations cause alterations in the conformation of presenilin and interactions with amyloid precursor protein. J. Neurosci. 25, 3009–3017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wolfe M. S., Xia W., Ostaszewski B. L., Diehl T. S., Kimberly W. T., Selkoe D. J. (1999) Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and [γ]-secretase activity. Nature 398, 513–517 [DOI] [PubMed] [Google Scholar]
- 47. Annaert W. G., Esselens C., Baert V., Boeve C., Snellings G., Cupers P., Craessaerts K., De Strooper B. (2001) Interaction with telencephalin and the amyloid precursor protein predicts a ring structure for presenilins. Neuron 32, 579–589 [DOI] [PubMed] [Google Scholar]
- 48. Esselens C., Oorschot V., Baert V., Raemakers T., Spittaels K., Serneels I., Zheng H., Saftig P., De Strooper B., Klumperman J., Annaert W. (2004) Presenilin 1 mediates the turnover of telencephalin in hippocampal neurons via an autophagic degradative pathway. J. Cell Biol. 166, 1041–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]