

Background: Programmed cell death protein 5 (PDCD5) has been proposed to act as a pro-apoptotic factor with tumor suppressor capabilities.
Results: PDCD5 forms a complex with the cytosolic chaperonin CCT and inhibits β-tubulin folding.
Conclusion: PDCD5 functions as a modulator of CCT to regulate β-tubulin folding.
Significance: PDCD5 may exert its pro-apoptotic function by blocking β-tubulin folding.
Keywords: Apoptosis, Chaperone Chaperonin, Electron Microscopy (EM), Protein Folding, Tubulin, Programmed Cell Death Protein 5
Abstract
Programmed cell death protein 5 (PDCD5) has been proposed to act as a pro-apoptotic factor and tumor suppressor. However, the mechanisms underlying its apoptotic function are largely unknown. A proteomics search for binding partners of phosducin-like protein, a co-chaperone for the cytosolic chaperonin containing tailless complex polypeptide 1 (CCT), revealed a robust interaction between PDCD5 and CCT. PDCD5 formed a complex with CCT and β-tubulin, a key CCT-folding substrate, and specifically inhibited β-tubulin folding. Cryo-electron microscopy studies of the PDCD5·CCT complex suggested a possible mechanism of inhibition of β-tubulin folding. PDCD5 bound the apical domain of the CCTβ subunit, projecting above the folding cavity without entering it. Like PDCD5, β-tubulin also interacts with the CCTβ apical domain, but a second site is found at the sensor loop deep within the folding cavity. These orientations of PDCD5 and β-tubulin suggest that PDCD5 sterically interferes with β-tubulin binding to the CCTβ apical domain and inhibits β-tubulin folding. Given the importance of tubulins in cell division and proliferation, PDCD5 might exert its apoptotic function at least in part through inhibition of β-tubulin folding.
Introduction
A fundamental question in biology is how proteins, which are synthesized by the ribosome as a linear sequence of amino acids, fold into their native functional state. It is now clear that many proteins require the assistance of molecular chaperones to maneuver through the folding process. Molecular chaperones are themselves proteins that protect newly synthesized or unfolded proteins from aggregation and help them reach their native state in the very concentrated protein environment of the cell (1). One important class of molecular chaperones is the chaperonins, which are large multisubunit complexes that form stacked double-ring structures with a central cavity in each ring. These cavities provide an isolated environment for client proteins to bind and fold (2, 3). Each subunit consists of three domains as follows: an equatorial domain that binds and hydrolyzes ATP, an apical domain that traps substrates, and an intermediate domain that connects the two other domains and facilitates interdomain communication (3). There are two types of chaperonins. Group I chaperonins are found in bacteria (i.e. GroEL from Escherichia coli), mitochondria, and chloroplasts (Hsp60). Their ring structures are composed of seven identical subunits that bind and hydrolyze ATP. Binding of ATP is coordinated with encapsulation of substrates within the folding cavity by a co-chaperone called GroES in E. coli and Hsp10 in eukaryotes (1, 2). The group II chaperonins are found in archaebacteria (named thermosomes) and in the eukaryotic cytosol (CCT,3 cytosolic chaperonin containing tailless complex polypeptide 1, also called TRiC). CCT is the most complex of all the chaperonins with each of the two rings composed of eight paralogous subunits that orchestrate the folding of many proteins, with the most abundant substrates being actins and tubulins (3). CCT substrates tend to have complex domain topologies and range up to ∼70 kDa in size (4, 5). Nascent polypeptides or denatured proteins bind inside the folding cavity to regions of both the equatorial and apical domains of the CCT subunits (6). The process of ATP binding and hydrolysis induces dramatic conformational changes in the apical domains that result in closure of the folding cavity by finger-like helical protrusions found at the tip of the apical domains (7–12). The substrate then folds in this sequestered environment. After phosphate release from the nucleotide-binding pocket, the apical domains reopen and the folded protein dissociates from CCT. If the protein has not yet reached its native structure, it can reassociate for another round of ATP binding and hydrolysis (7, 8, 10–12).
CCT substrates often require additional proteins called co-chaperones for efficient delivery or release from CCT. For example, the co-chaperone prefoldin is required for transfer of nascent actin or tubulin to CCT (13, 14). The unfolded actin or tubulin binds to hydrophobic residues at the tips of the tentacle-like extensions of prefoldin (13). The complex then binds and transfers the nascent substrate to CCT to continue folding (13, 15, 16). Another CCT co-chaperone is Hsp70 that, besides acting as a chaperone on its own, can transfer different substrates to the eukaryotic chaperonin for more efficient folding (17). A contrasting CCT co-chaperone function is found with phosducin-like protein 1 (PhLP1), which is involved in the final folding and release of the β-subunit of the guanine nucleotide-binding protein (Gβ) from CCT (18–22). PhLP1 stabilizes the β-propeller fold of Gβ so that it can release from CCT and interact with its obligate binding partners Gγ, in the case of Gβ1–4, or regulator of G protein signaling (RGS) proteins in the case of Gβ5 (23–25). These examples show how co-chaperones enhance the repertoire of CCT substrates to include a variety of proteins with essential cellular functions.
Gβ belongs to the large family of WD40 repeat proteins with multiple β-sheets that form β-propeller structures, many of which are folded by CCT (26–28). In seeking to determine whether PhLP1 were involved in the folding of other CCT substrates, particularly other β-propeller proteins, we identified a novel interaction of CCT with programmed cell death protein 5 (PDCD5). PDCD5 was originally described as an up-regulated gene from cells undergoing apoptosis (29). Cellular expression of PDCD5 is decreased in many cancer cell lines and tumors (30, 31). Moreover, overexpression of PDCD5 accelerates apoptosis in tumor cells (32), suggesting that PDCD5 plays a role in stemming uncontrolled cell proliferation by triggering apoptosis. It has been proposed that apoptotic stimuli cause translocation of PDCD5 to the nucleus, where it interacts with the histone acetyltransferase Tip60 and the transcription factor p53 to promote programmed cell death (33, 34). In contrast to these observations, our data show that PDCD5 interacts with CCT in the cytosol as a regulatory co-chaperone that specifically inhibits β-tubulin folding. Based on these findings, we propose that the apoptotic activity of PDCD5 may result at least in part by impairing CCT-mediated β-tubulin folding.
EXPERIMENTAL PROCEDURES
Cell Culture
HEK-293T and U2OS cells were cultured in 1:1 DMEM/F-12 growth media containing 2.5 mm l-glutamine and 15 mm HEPES supplemented with 10% fetal bovine serum. The cells were subcultured regularly to maintain growth but were not used beyond 20 passages.
Preparation of cDNA Constructs
Human PDCD5 (N-terminal FLAG-tagged, N-terminal FLAG-TEV-tagged, or C-terminal FLAG-tagged) and tubulin-binding protein co-factors A and B with C-terminal FLAG tags (Open Biosystems) were cloned in the pcDNA3.1/Myc-His B vector (Invitrogen) using PCR. C-terminally c-Myc-tagged human PhLP1 in pcDNA3.1/Myc-His B vector was described previously (21). For recombinant protein purification, His6-PDCD5-FLAG was cloned into the first multiple cloning site of the bacterial expression vector pETDuet, and PhLP1-Myc-His was cloned into the bacterial expression vector pET15b (Novagen) using PCR. The integrity of all constructs was confirmed by sequence analysis. The N-terminal hemagglutinin (HA)-tagged Gγ2 and FLAG epitope-tagged Gβ1 cDNAs also in the pcDNA3.1 vector were obtained from the UMR cDNA Resource Center. BzF tRNA and synthetase cDNAs were a generous gift from Thomas Sakmar (Rockefeller University).
Transient Transfections
HEK-293T and U2OS cells were grown in 6-well plates, 60-mm dishes, or 100-mm dishes to 80–90% confluency at which point they were transfected with 1 μg (6-well plate), 2–5 μg (60-mm dishes), or 6 μg (100-mm dishes) each of the indicated vectors using Lipofectamine 2000 according to the manufacturer's protocol (Invitrogen). After 48 h, the cells were harvested for immunoprecipitation, mass spectrometry, or radiolabel pulse-chase experiments.
Protein Expression and Purification
E. coli DE3 cells were transformed with human PhLP1 in the pET15b vector or PDCD5 in the pETDuet vector. The recombinant proteins were then purified using Co2+ affinity chromatography as described previously for Ni2+-chelate chromatography (35). The purified proteins were concentrated and exchanged into 20 mm HEPES, pH 7.2, 150 mm NaCl by ultrafiltration and were stored in 40% glycerol at −20 °C. Protein concentrations were determined using BCA protein assay reagent (Pierce). CCT was purified from bovine testis as described previously (13).
Immunoprecipitation Experiments
Transfected or untreated HEK-293T or U2OS cells were washed with phosphate-buffered saline (PBS) and solubilized in one of the following immunoprecipitation (IP) buffers depending on the experiment: standard IP buffer (PBS, pH 7.4, 1% Nonidet P-40 (Sigma)), β-tubulin IP buffer (50 mm Na2HPO4, pH 7.5, 150 mm NaCl, 0.1% Tween 20, 1 mm GTP), β-actin IP buffer (20 mm HEPES, pH 7.4, 50 mm KCl, 1 mm DTT, 0.2 mm CaCl2, 0.5% Nonidet P-40, 4 μm cycloheximide, 40 mm glucose), or ATP-depletion buffer (PBS, pH 7.4, 1% Nonidet P-40, 100 mm deoxyglucose, 1 mm azide, 5 mm EDTA). All were supplemented with 0.6 mm PMSF and 6 μl/ml protease inhibitor mixture (Sigma P8340). The lysates were passed through a 25-gauge needle 10 times and centrifuged at maximum speed for 10 min at 4 °C in an Eppendorf microcentrifuge. The protein concentration for each sample was determined using the DC Protein Assay Kit II (Bio-Rad), and equal amounts of protein were used in the subsequent immunoprecipitations. The clarified lysates were incubated for 30 min at 4 °C with one of the following antibodies as indicated: 3–5 μg of anti-Myc (clone 9E10, Enzo Life Sciences), 3–8 μg of anti-CCTϵ antibody (clone PK/29/23/8d, AbD Serotec), 3–6 μg of anti-FLAG (clone M2, Sigma), or 0.4 μg of anti-HA (clone 3F10, Roche Applied Science). Next, 30 μl of protein A/G Plus-agarose slurry (Santa Cruz Biotechnology) was added, and the mixture was incubated for 20–30 min at 4 °C. In β-actin precipitations, DNase I-agarose beads were used to precipitate folded actin as described previously (36). Immunoprecipitated proteins and lysates were resolved on 10 or 14% Tris-glycine/SDS gels or 16.5% Tricine/SDS gels. The proteins were transferred to nitrocellulose and immunoblotted using the following antibodies as indicated: anti-CCTα, -β, -δ, -ϵ, -η, and -θ (AbD Serotec); anti-CCTζ (Santa Cruz Biotechnology); anti-CCTγ, α-tubulin, β-tubulin, and PDCD5 (Abcam); anti-FLAG or anti-Myc antibodies. Immunoblots were incubated with the appropriate anti-mouse, anti-rat, anti-rabbit, or anti-goat secondary antibody conjugated to an infrared dye (Li-Cor Biosciences). Blots were scanned using an Odyssey Infrared Imaging System (Li-Cor Biosciences), and protein band intensities were quantified using the Odyssey software. In all cases, the ratio of the co-immunoprecipitated protein to the immunoprecipitated protein was calculated and then normalized to the control.
RNA Interference Experiments
HEK-293T cells were grown in 12- or 6-well plates to 40–50% confluency at which point they were transfected with CCTζ (Dharmacon), PDCD5 (Ambion), or negative control 1 (Ambion) siRNA using Oligofectamine (Invitrogen) reagent as described previously (21) or RNAiMAX reagent (Invitrogen) according to the manufacturer's protocol. In some cases, cells were transfected 24 h later with 0.5 μg (12-well) or 1.0 μg (6-well) of the indicated cDNAs using Lipofectamine 2000 according to the manufacturer's protocol (Invitrogen). Cells were harvested for subsequent immunoprecipitation experiments 4 days after the knockdown. A total of 10 μg of cell lysates were immunoblotted with anti-CCTζ or anti-PDCD5 antibodies to assess the percent knockdown.
Mass Spectrometry
PhLP1 and phosducin (Pdc) binding partners were determined by transfecting their cDNAs with C-terminal tags containing a tobacco etch virus (TEV) cleavage site followed by a Myc epitope site (PhLP1-TEV-Myc). PDCD5-binding partners were identified by transfecting U2OS cells with an N-terminally tagged FLAG-TEV-PDCD5. Empty vector transfected cells served as a control. After 48 h, cells were harvested, and the lysates (1 mg of total protein) were immunoprecipitated with the indicated antibodies. The proteins were released from the antibody·bead complex via TEV protease cleavage according to the manufacturer's protocol (Promega). The released co-immunoprecipitates were reduced with DTT, alkylated with iodoacetamide, and digested with trypsin as described previously (37). Proteins in the co-immunoprecipitates were identified by tandem mass spectrometry (MS/MS). When different, details for the PhLP1 and Pdc MS/MS analysis are indicated first followed by those from the PDCD5 analysis. MS/MS was performed using an LTQ-Orbitrap mass spectrometer interfaced with a Waters nanoAcquity UPLC and outfitted with a BEH C18 reversed phase column (25 cm × 75 μm inner diameter, 1.7 μm, 100 or 130 Å, Waters). Peptide mixtures were separated by acetonitrile gradients for 90 or 150 min at flow rates of 300 or 325 nl/min. MS/MS were collected with m/z window = 475–1600 or 300–2000 Da enabling monoisotopic precursor and charge selection settings. Ions with unassigned charge state or charge state of 1 were excluded. For each MS scan, the 5 or 6 most intense ions were targeted with a dynamic exclusion of 30 s, a 20 or 10 ppm exclusion width, and a repeat count of 2 or 1. The maximum injection time for Orbitrap parent scans was 500 ms with 1 microscan and automatic gain control of 1 × 106. The maximum injection time for the LTQ MS/MS was 250 ms with 1 microscan and automatic gain control of 1 × 104 or 3 × 104. The normalized collision energy was 35%, with activation Q of 0.25 for 30 ms. Raw files were searched against the UniprotKB human database (including variants) with Sequest, SequestHT, and Mascot (version 2.3) using Proteome Discoverer 1.4. Database search engine parameters were as follows: trypsin digestion, two missed cleavages, b and y ion series, precursor mass tolerance of 10 ppm, fragment mass tolerance of 0.8 Da (both assuming monoisotopic peaks), and variable cysteine carbamidomethylation and methionine oxidation. Additional processing was performed using mspire version 0.8.6.2–1-g85f741e (38). Data were transformed to a raw list of spectral counts and filtered to accept only those with a Q value of less than 0.01 (false discovery rate of less than 1%), and protein sequences were inferred using peptide_hit_qvalues_ to_spectral_ counts_table.rb from mspire which uses QSpec (version 2) (39) with the normalized flag, which normalizes total spectral counts per sample. Table 2 shows average QSpec normalized spectral counts, and Table 1 shows the sum from two biological replicates without QSpec normalization. A separate PhLP1 versus control analysis was performed using QSpec so that a reasonable significance value could be provided in Table 1.
TABLE 2.
PDCD5 interacts with β-tubulin
U2OS cells were transfected with FLAG-TEV-PDCD5 or empty vector. PDCD5 immunoprecipitates were analyzed for binding partners by tandem mass spectrometry. This table displays significant hits found in the proteomics screen. The values in the first 2 columns indicate the normalized spectral counts for each sample. Values in the 3rd column indicate the significance of the peptide hits (Decibans were calculated from Bayes factors).
| Control | PDCD5 | Decibans | Gene | UniProt | Description |
|---|---|---|---|---|---|
| 1.5 | 225 | 183.9 | CCT8 | P50990 | T-complex protein 1 subunit θ |
| 0 | 217.7 | 201.6 | TCP1 | P17987 | T-complex protein 1 subunit α |
| 0 | 169 | 172.4 | CCT2 | P78371 | T-complex protein 1 subunit β |
| 0 | 108.5 | 155.2 | CCT5 | P48643 | T-complex protein 1 subunit ϵ |
| 0 | 99.3 | 150.6 | PDCD5 | O14737 | Programmed cell death protein 5 |
| 0 | 98.7 | 149.7 | CCT7 | Q99832 | T-complex protein 1 subunit η |
| 0 | 88.5 | 135.8 | CCT6A | P40227 | T-complex protein 1 subunit ζ |
| 0 | 85.5 | 142.3 | CCT4 | P50991 | T-complex protein 1 subunit δ |
| 5.2 | 32.9 | 64.9 | TUBB | P07437 | Tubulin β-5 chain |
| 5.9 | 32.8 | 63.4 | TUBB2C | P68371 | Tubulin β-2C chain |
| 5.9 | 32.7 | 60.8 | TUBB4 | P04350 | Tubulin β-4 chain |
| 0 | 31.8 | 90.8 | CCT3 | P49368 | T-complex protein 1 subunit γ |
| 4.5 | 26.7 | 57.7 | TUBB2A | Q13885 | Tubulin β-2A chain |
| 1.4 | 15.7 | 53.4 | TUBB6 | Q9BUF5 | Tubulin β-6 chain |
| 0 | 3.3 | 7.4 | FLG2 | Q5D862 | Filaggrin-2 |
| 0 | 2.9 | 5.8 | GNB2L1 | P63244 | G protein subunit β-2-like 1 |
| 0 | 2 | 6.8 | PIP | P12273 | Prolactin-inducible protein |
| 0 | 1.7 | 4.2 | TUBA4B | Q9H853 | Putative tubulin-like protein α-4B |
TABLE 1.
PDCD5 interacts with PhLP1
HEK-293T cells were transfected with PhLP-TEV-myc or Pdc-TEV-myc for comparison. An empty vector transfection served as a negative control. Myc immunoprecipitates were analyzed for binding partners by tandem mass spectrometry. This table displays significant hits found in the proteomics screen. The numbers in the first 3 columns indicate the normalized spectral counts for each sample. Values in the 4th column indicate the significance of the peptide hits.
| Control | Pdc | PhLP1 | Decibansa | Gene | Uniprot | Description |
|---|---|---|---|---|---|---|
| 0 | 0 | 120 | 52.5 | CCT4 | P50991 | T-complex protein 1 subunit δ |
| 0 | 0 | 69.5 | 30.7 | PDCL | Q13371 | Phosducin-like protein |
| 0.5 | 0 | 104 | 164.3 | CCT7 | Q99832 | T-complex protein 1 subunit η |
| 0 | 0 | 68.5 | 29.8 | HSD17B4 | P51659 | Peroxisomal multifunctional enzyme type 2 |
| 1.5 | 0 | 140 | 209.5 | CCT2 | P78371 | T-complex protein 1 subunit β |
| 5 | 0 | 273 | 381.1 | TCP1 | P17987 | T-complex protein 1 subunit α |
| 0 | 0 | 38 | 17.3 | PDCD5 | O14737 | Programmed cell death protein 5 |
| 0.6 | 0 | 52 | 75.1 | CCT3 | P49368 | T-complex protein 1 subunit γ |
| 9 | 0 | 288 | 359.0 | CCT8 | P50990 | T-complex protein 1 subunit θ |
| 1 | 0.6 | 59.6 | 83.6 | CCT5 | P48643 | T-complex protein 1 subunit ϵ |
| 1 | 0 | 34.75 | 15.7 | CCT6A | P40227 | T-complex protein 1 subunit ζ |
| 0 | 28.3 | 13 | 3.6 | GNB2 | P62879 | G protein subunit β-2 |
| 0 | 25.3 | 13 | 3.6 | B2R6K4 | P62873 | Transducin β chain 1 |
| 0 | 31 | 8 | GNB4 | Q9HAV0 | G protein subunit β-4 | |
| 0 | 1.5 | 5 | 4.7 | HNRNPA2B1 | P22626 | Heterogeneous nuclear ribonucleoproteins A2/B1 |
a Decibans calculated from Bayes factors were taken from separately run Qspec analysis of PhLP1 versus Control.
Radiolabeled Pulse-Chase Assays
For the rate of CCT association, HEK-293T cells in 6-well plates were transfected with PDCD5-FLAG. After 48 h, cells were washed and incubated in methionine-free DMEM (Mediatech Inc.) supplemented with 4 mm l-glutamine (Sigma), 0.063 g/liter l-cystine dihydrochloride (United States Biochemical Corp.), and 10% dialyzed fetal bovine serum (Invitrogen) for 1 h. The media were discarded, and the cells were pulsed with new media supplemented with 200 μCi/ml radiolabeled l-[35S]methionine (PerkinElmer Life Sciences) for 10 min. The cells were washed and incubated in DMEM/F-12 growth media supplemented with an extra 10 mm l-methionine (Sigma) to stop [35S]methionine incorporation. At increasing times, CCT was immunoprecipitated, and the proteins were resolved on 10% Tris-glycine/SDS or 16.5% Tricine/SDS gels. The radiolabeled gels were dried, exposed on a phosphor screen (GE Healthcare), and imaged on a Storm 860 phosphorimager. The band intensities were quantified using ImageQuant software (GE Healthcare) and corrected for the number of methionine residues found in each protein. The molar ratios of nascent proteins to CCTϵ were then calculated. The rate data were fit to a first-order rate equation to determine the rate constant (k), and the t½ for assembly or dissociation was calculated as t½ = ln 2/k.
For protein folding experiments, HEK-293T cells in 12-well plates were treated with PDCD5 siRNA and then transfected 24 h later with FLAG-cofactor B (for α-tubulin folding), FLAG-cofactor A (for β-tubulin folding), FLAG-Gβ1 and HA-Gγ2 (for Gβ folding), or nothing (for β-actin folding). At 96 h, cells were treated with [35S]methionine as described above and chased for 60 min (α- and β-tubulin), 30 min (Gβ), or 15 min (β-actin). Folded protein was then determined by the amount of nascent, labeled protein co-immunoprecipitating with its binding partner. Binding partners, tubulin-binding protein co-factor B for α-tubulin, co-factor A (TBCA) for β-tubulin, Gγ for Gβ, and DNase I beads for β-actin, associate with their targets immediately after folding by CCT. Radioactive bands were detected, and the ratios of the folded protein to its binding partner were calculated and normalized to the control siRNA.
EM and Image Processing
PDCD5·CCT complexes were prepared by mixing purified components in a 10:1 PDCD5/CCT ratio. For the two-dimensional EM analysis, aliquots of the different samples (CCT, PDCD5·CCT complex, or the immunocomplex between PDCD5, CCT, and a monoclonal antibody against CCTδ (PK/9/86b from GenWay)) were applied onto carbon-coated copper grids and stained with 2% uranyl acetate. Micrographs were taken under minimal dose conditions in a JEOL JEM1200EXII microscope operated at 100 kV and digitized in a Zeiss SCAI scanner with a sampling window corresponding to 3.5 Å/pixel. Individual particles were manually selected using XMIPP (40). Image classification was performed using a free pattern, maximum likelihood, multiple reference refinement (ML2D) (41). Homogeneous populations were obtained and averaged for a final two-dimensional characterization.
For the three-dimensional reconstruction of the PDCD5·CCT complex, aliquots of the solution were applied to Quantifoil 2-μm holey carbon grids for 1 min, blotted for 3 s, and frozen rapidly in liquid ethane at −180 °C. Images were acquired with a defocus range of 2–3.5 μm at 1.75 Å per pixel sampling rate on an 4K × 4K Eagle CCD camera (Gatan Inc.) mounted on an FEI Tecnai G2 FEG200 electron microscope at 200 kV with a Gatan side-entry cryo-holder. A total of 13,100 particles (down-sampled to 3.5 Å per pixel) were selected, normalized, and CTF-corrected using standard XMIPP procedures (40). Images were classified using ML2D, and the most representative classes were used to generate an initial three-dimensional model using the startcsym program from the EMAN package (42). The resulting model was subsequently refined without imposing any symmetry. The model was filtered at 50 Å and used for three-dimensional maximum likelihood classification (ML3D) with internal correction for normalization errors (43) to separate PDCD5-bound CCT particles from the PDCD5-free ones. The selected particles with bound PDCD5 were refined with EMAN to obtain the final model. The resolution of the reconstructions was determined to be 25 Å by the Fourier shell correlation coefficient (FSC) 0.5 criterion between two independent reconstructions (Fig. 5). The density maps and atomic structures were visualized with UCSF Chimera (44). The atomic structures were manually fitted into the three-dimensional reconstructions.
FIGURE 5.

Three-dimensional reconstruction of the PDCD5·CCT complex. A, representative area of a micrograph of a vitrified sample of PDCD5·CCT complexes. Green circles show end-on views of the complex, and red rectangles mark side views. B, gallery of selected images of PDCD5·CCT complexes. C, average images corresponding to different classes used for the generation of the first volume. D, selected projection views (top panel) and the corresponding class averages (bottom panel) of the final model. E, Fourier shell correlation (FSC) plot of the PDCD5-CCT reconstruction showing 25 Å resolution at a 0.5 Fourier shell correlation value.
Amino Acid Incorporation and Photo-cross-linking
Amber codon suppression technology was used to incorporate the photo-cross-linking unnatural amino acid BzF into residues at the C terminus of PDCD5 as described previously (45). PDCD5 variants were engineered with amber codons inserted at residues 111, 113, 115, 117, or 119 of the PDCD5 C terminus. Five μg of each variant cDNA were transfected into HEK-293T cells plated on 60-mm dishes using Lipofectamine 2000, along with 5 μg of BzF tRNA and 0.5 μg of BzF tRNA synthetase cDNA. Three hours post-transfection, the cell media were supplemented with fresh media containing BzF, bringing the final concentration to 1 mm. Cells were then harvested in IP buffer at 40–48 h post-transfection, and clarified lysates were exposed to UV light for 4 min at 4 °C using a 600-watt UV lamp (150 milliwatt/cm2, Integrated Dispensing Solutions) set to half-intensity at a distance of 27 cm. FLAG-PDCD5 variants or CCTϵ were immunoprecipitated, and immunoprecipitates and lysates were analyzed by immunoblotting. To test the effects of BzF alone on cross-linking, similar experiments were performed, but without transfecting the BzF tRNA or synthetase. BzF (1 mm) was added to the cell culture media as indicated.
RESULTS
PDCD5 Forms a Ternary Complex with PhLP1 and CCT
To investigate possible contributions of PhLP1 to the folding of other proteins besides Gβ, we performed an extensive proteomic analysis of PhLP1-binding partners (Table 1). Immunoprecipitates of PhLP1 were analyzed for potential interactors by tryptic digestion and mass spectrometric identification of the resulting peptides. Multiple peptides from all of the CCT subunits were found in the PhLP1 sample along with Gβ1, Gβ2, and Gβ4 as expected. A Pdc immunoprecipitate was also analyzed for comparison. The Pdc sample contained peptides from the same Gβ subunits but none of the CCT subunits. These results are consistent with previous findings that showed an interaction between PhLP1 and CCT that was not shared with Pdc (36). Interestingly, several peptides from PDCD5 and the peroxisomal multifunctional enzyme type 2 (HSD17B4) were also found in the PhLP1 sample, suggesting an interaction between PhLP1 and these proteins. The apparent interaction between PhLP1 and PDCD5 was intriguing given the proposed tumor suppressor role of PDCD5 (32), so we decided to explore it further. To determine the specificity of the interaction, several PhLP1 isoforms were co-expressed along with PDCD5 in HEK-293T cells, and interactions were assessed by co-immunoprecipitation and immunoblotting. PDCD5 associated with PhLP1 but not with Pdc, PhLP2A, or PhLP3, confirming that a specific interaction between PhLP1 and PDCD5 was occurring (Fig. 1A).
FIGURE 1.
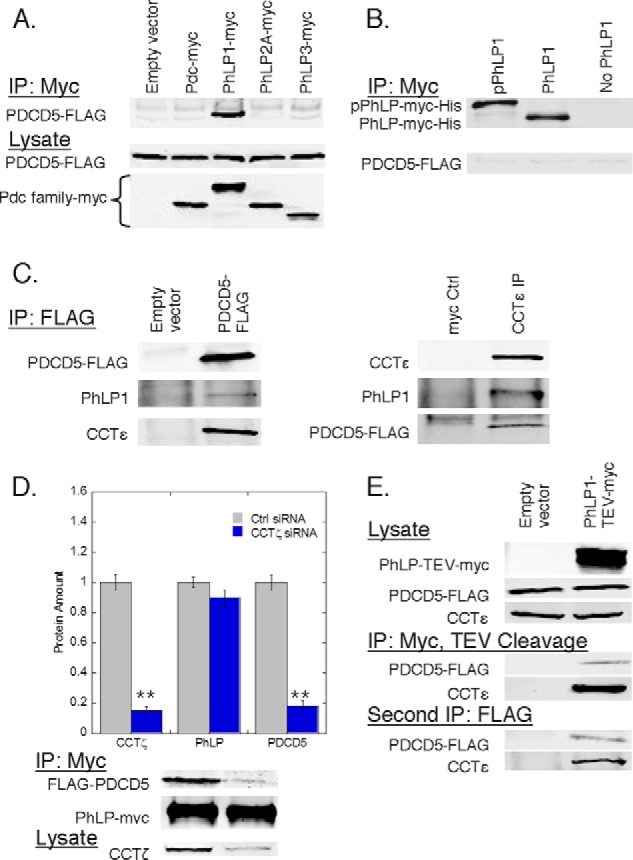
PDCD5 forms a complex with PhLP1 and CCT. A, binding of PDCD5 to phosducin family members was measured by co-immunoprecipitation from HEK-293T cells transfected with PDCD5-FLAG along with Myc-tagged phosducin family members as indicated. After 48 h, cells were lysed, immunoprecipitated with an anti-Myc antibody, and immunoblotted for PDCD5-FLAG. B, binding of purified PDCD5 to PhLP1 or CK2-phosphorylated PhLP1 was assessed by co-immunoprecipitation in vitro. Phosphorylated PhLP1, unphosphorylated PhLP1, or no PhLP1 was incubated with PDCD5, immunoprecipitated with an Myc antibody, and blotted as indicated. C, simultaneous binding of PDCD5 and PhLP1 was measured by co-immunoprecipitation. HEK-293T cells were transfected with PDCD5-FLAG or empty vector, immunoprecipitated with FLAG, and blotted for endogenous PhLP1 and CCTϵ (left panel). Endogenous CCTϵ was also immunoprecipitated and blotted for endogenous PhLP1 and PDCD5-FLAG (right panel). A nontargeting Myc antibody served as a negative control. D, effect of CCT knockdown on PDCD5 binding to PhLP1 was measured by co-immunoprecipitation of PhLP1-Myc from HEK-293T cells treated with CCTζ siRNA or a control siRNA and later transfected with FLAG-PDCD5 and PhLP1-Myc. The ratio of the PDCD5 band to the PhLP1 band was calculated and normalized to the control. Bars represent the average ± S.E. of the mean from at least three experiments. Representative blots are shown below the graphs. E, formation of a PhLP1·PDCD5·CCT complex was demonstrated in double immunoprecipitation experiments from HEK-293T cells transfected with PDCD5-FLAG along with PhLP1-TEV-Myc or empty vector.
To determine whether the co-immunoprecipitation of PDCD5 with PhLP1 resulted from a direct interaction or from indirect interactions through other proteins in a common complex, the binding of recombinant purified PhLP1 and PDCD5 was tested in vitro by co-immunoprecipitation. Surprisingly, no PDCD5 was found in the PhLP1 immunoprecipitate whether PhLP1 was in its CK2-phosphorylated form (20) or not (Fig. 1B). These findings suggest that PhLP1 and PDCD5 do not interact directly but instead associate indirectly as components of the same complex.
Knowing that PhLP1 binds CCT, we investigated whether PDCD5 and PhLP1 were part of the same CCT complex. We first looked at the ability of PDCD5 to bind CCT by co-immunoprecipitation. PDCD5 immunoprecipitates contained a significant amount of endogenous CCTϵ, even more than PhLP1 (Fig. 1C). In a reciprocal experiment, both PDCD5 and PhLP1 were found in a CCT immunoprecipitate (Fig. 1C). These results suggest that PhLP1 and PDCD5 might be interacting through CCT. To investigate this possibility, we measured the effect of siRNA-mediated depletion of CCTζ on the ability of PhLP1 to co-immunoprecipitate PDCD5. We found that an 80% depletion of CCTζ caused a corresponding decrease in the association of PDCD5 with PhLP1 (Fig. 1D). This finding is consistent with the idea that PhLP1 and PDCD5 interact indirectly through the CCT complex. To confirm this result, we performed a double co-immunoprecipitation experiment. PhLP1 with a C-terminal tag consisting of a TEV cleavage site and a Myc epitope was overexpressed along with FLAG-tagged PDCD5 in HEK-293T cells. PhLP1 was immunoprecipitated and released from the antibody and beads with TEV protease. The resulting supernatant was then subject to a second immunoprecipitation using a FLAG antibody. CCT was found in both the first and the second immunoprecipitates, demonstrating that the same CCT complexes that were associated with PhLP1 were also bound to PDCD5 (Fig. 1E). Collectively, these data show that PDCD5 does not bind PhLP1 directly but that they interact indirectly through a ternary complex with CCT.
Functional Analysis of the PDCD5/CCT Interaction
The formation of a ternary complex between PhLP1, PDCD5, and CCT suggests that PDCD5 and PhLP1 may be functionally linked. To begin to test this possibility, we measured the effects of overexpression and siRNA-mediated depletion of PhLP1 or PDCD5 on the binding of the other to CCT. Surprisingly, neither overexpression nor depletion of PDCD5 had any effect on the interaction of PhLP1 with CCT in co-immunoprecipitation experiments (Fig. 2, A and B). Likewise, overexpression or depletion of PhLP1 had no effect on PDCD5 binding to CCT (Fig. 2, C and D). These results indicate that PhLP1 and PDCD5 interact with CCT independently of each other, raising questions about the functional significance of PDCD5 binding to CCT. It does not appear that PDCD5 requires CCT and PhLP1 for folding because PDCD5 does not have the structure of a typical CCT substrate. It is a small 125-amino acid protein with a simple three-helical bundle fold without multiple domains or a complex folding pattern that is common among CCT substrates (46). Furthermore, PhLP1 has profound effects on the binding of its known substrate, Gβ, to CCT (20, 25), but this was not observed with PDCD5 (Fig. 2). These observations suggest that PDCD5 is not a CCT substrate but interacts with CCT for another reason. We tested this idea further by measuring the binding of nascent PDCD5 to CCT in a pulse-chase experimental format. Normally, nascent proteins that are CCT substrates rapidly bind to CCT upon synthesis and are released more slowly after folding (5, 20). In contrast, nascent subunits of the CCT complex or co-chaperones like PhLP1 accumulate as part of CCT complexes over time. In a pulse-chase experiment, the CCT complex was immunoprecipitated with an antibody to CCTϵ at increasing chase times after a [35S]methionine radiolabeling pulse, and the binding of nascent interacting partners was tracked over time (Fig. 3A). Newly synthesized PDCD5 accumulated with the CCT complex, as did nascent CCT subunits, such as CCTα and -γ. In contrast, nascent tubulin, a known CCT substrate (47), was released over time. These results show that PDCD5 is not a CCT-folding substrate but an interacting partner that could act as a co-chaperone or another type of regulator of CCT function.
FIGURE 2.
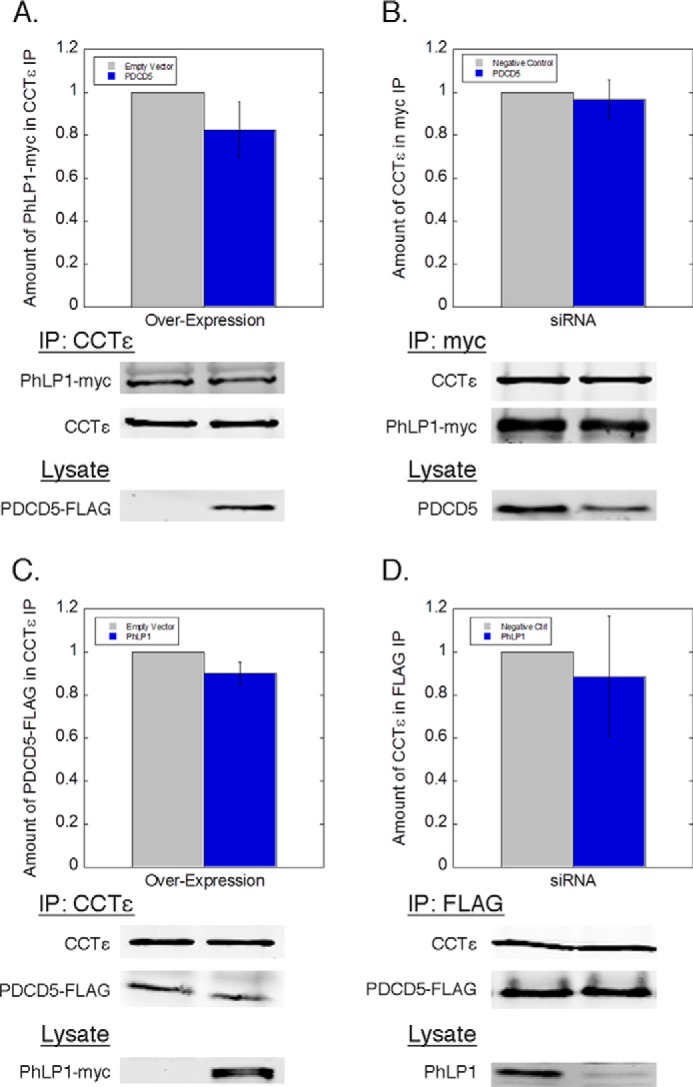
PhLP1 and PDCD5 bind CCT independently of each other. PDCD5 was either overexpressed (A) or knocked down (B), along with PhLP1-Myc overexpression in HEK-293T cells. Cell lysates were immunoprecipitated with anti-CCTϵ (A) or anti-Myc (B) and blotted as indicated. PhLP1 was either overexpressed (C) or knocked down (D), along with PDCD5-FLAG overexpression in HEK-293T cells. Cell lysates were immunoprecipitated with anti-CCTϵ (C) or anti-FLAG (D) and blotted as indicated. Bars represent the average ± S.E. from at least three experiments. Cell lysates were blotted for PDCD5-FLAG, endogenous PDCD5, PhLP1-Myc, or endogenous PhLP1 as indicated to verify the overexpression and knockdowns. Representative blots are shown below the graphs.
FIGURE 3.

PDCD5 inhibits β-tubulin folding. A, rate of association or dissociation from CCT complexes was measured by pulse-chase immunoprecipitations of CCTϵ from HEK-293T cells transfected with PDCD5-FLAG. The rate of association of CCTα and -γ subunits (black, t½ = 112 ± 18 min) and PDCD5 (red, t½= 44 ± 2 min) was calculated along with the rate of dissociation for tubulin (blue, t½ = 39 ± 1 min). B, binding of β-tubulin to PDCD5 was measured by co-immunoprecipitation from HEK-293T cells transfected with FLAG-PDCD5 or empty vector. C, effect of CCT knockdown on β-tubulin binding to PDCD5 was measured by co-immunoprecipitation from HEK-293T cells treated with CCTζ siRNA or a control siRNA and later transfected with FLAG-PDCD5. The ratio of the β-tubulin band to the PDCD5 band was calculated and normalized to the control. D, folding of the indicated proteins by CCT was measured by pulse-chase co-immunoprecipitations from HEK-293T cells treated with PDCD5 siRNA or negative control as indicated (see “Experimental Procedures”). E and F, effect of PDCD5 knockdown (E) or overexpression (F) on β-tubulin binding to CCT was measured by co-immunoprecipitation with CCTϵ and immunoblotting as indicated. The ratio of the β-tubulin band to the CCTϵ band was calculated and normalized to the control. In all experiments, bars represent the average ± S.E. from at least three experiments. Representative gels or blots are shown below each graph. PDCD5 knockdown averaged between 65 and 80% as measured by immunoblotting.
To explore a possible co-chaperone function of PDCD5, we performed an analysis of PDCD5-binding partners. PDCD5 was immunoprecipitated in an ATP depletion buffer to trap CCT substrates on the complex, and potential interactors were identified by mass spectrometry (Table 2). Each of the CCT subunits was found, as were several isoforms of β-tubulin. The interaction between PDCD5 and β-tubulin was confirmed by co-immunoprecipitation. β-Tubulin was clearly identified in PDCD5 immunoprecipitates, although there was no specific interaction with α-tubulin (Fig. 3B). The fact that both PDCD5 and β-tubulin interact with CCT suggests that they may form a co-complex on CCT. To test this possibility, we measured the effect of siRNA-mediated CCT depletion on the PDCD5/β-tubulin interaction (Fig. 3C). An 80% reduction in CCTζ resulted in a 50% reduction in β-tubulin binding to PDCD5, suggesting that the PDCD5/β-tubulin interaction occurs at least in part through a co-complex with CCT. This interaction points to a possible role of PDCD5 in β-tubulin folding. To test this possibility, we measured the effect of PDCD5 knockdown on β-tubulin folding as well as several other known CCT substrates. To perform this measurement, we developed a new approach to assess tubulin folding by determining the rate of association of nascent α- and β-tubulin with co-factor A (for β-tubulin) and co-factor B (for α-tubulin) in a pulse-chase experimental format. These co-factors are the first to interact with their respective tubulins after they are folded by CCT in the process of tubulin dimer formation (48). For comparison, we also measured the effects of PDCD5 knockdown on β-actin and Gβ folding using previously established methods (21, 36). PDCD5 knockdown increased the rate of β-tubulin folding by more than 50%, although it had no effect on α-tubulin, β-actin, or Gβ folding (Fig. 3D). These findings are consistent with the binding of PDCD5 and β-tubulin to CCT and indicate that PDCD5 interacts with CCT to specifically down-regulate β-tubulin folding.
To further investigate the mechanism by which PDCD5 inhibited β-tubulin folding, we measured the effects of PDCD5 knockdown and overexpression on the binding of β-tubulin to CCT. An 80% siRNA reduction of PDCD5 increased the binding of β-tubulin to CCT by 35% (Fig. 3E), whereas PDCD5 overexpression decreased the binding of β-tubulin to CCT by 40% (Fig. 3F). In contrast, the binding of α-tubulin to CCT was unaffected by these changes in PDCD5 expression. Together, these findings indicate that PDCD5 specifically inhibits β-tubulin folding by disrupting the interaction of β-tubulin with CCT. This result was unexpected in light of the data from Fig. 3, B and C, showing that PDCD5 and β-tubulin form a complex with CCT. However, these observations can be reconciled if PDCD5 only partially inhibits β-tubulin binding to CCT (see under “Discussion”).
Structural Analysis of the PDCD5/CCT Interaction
To begin to understand how PDCD5 might disrupt the interaction of β-tubulin with CCT, we performed a structural analysis of the PDCD5·CCT complex. An excess of purified PDCD5 was combined with purified CCT in vitro in the absence of nucleotide. The resulting complex was subjected to native gel electrophoresis after which the high molecular weight band corresponding to the CCT complex was excised and resolved on a denaturing gel. The denaturing gel showed the bands corresponding to the eight CCT subunits and a band corresponding to PDCD5, indicating the formation of a stable PDCD5·CCT complex (Fig. 4A). The existence of the PDCD5·CCT complex was confirmed by electron microscopy (EM). Negatively stained EM images showed the typical doughnut-shaped structure corresponding to end-on views of CCT (Fig. 4B). Most of the particles revealed a small elongated mass protruding into the CCT cavity. This mass was clearly observed upon processing and averaging 1354 of these particles (Fig. 4B), but it was not present in the average image of 1128 PDCD5-free CCT particles (Fig. 4C); thus, the mass is attributable to PDCD5. The PDCD5 protruded from only one of the eight CCT subunits. To identify this CCT subunit, we employed an immunomicroscopy approach. A monoclonal antibody against CCTδ labeled the CCT subunit very near the PDCD5 mass, suggesting that PDCD5 binds CCTδ or an adjacent subunit (Fig. 4D).
FIGURE 4.
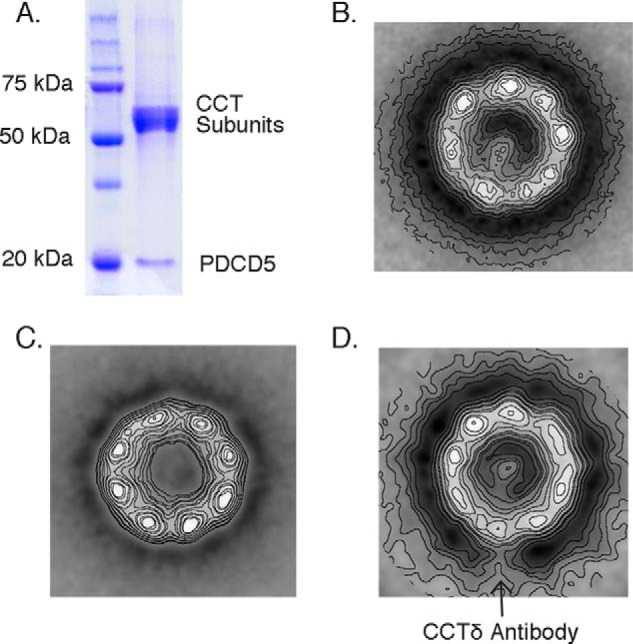
PDCD5 binds CCT near the δ subunit. A, gel analysis of the PDCD5·CCT complex. PDCD5 was mixed with CCT at a 10:1 molar ratio in the absence of nucleotide, and the mixture was resolved on a native polyacrylamide gel. The CCT oligomer (∼960 kDa), which runs with a mobility clearly distinct from that of PDCD5 (∼14 kDa), was excised and run on a denaturing acrylamide gel revealing bands corresponding to the eight CCT subunits and a band corresponding to PDCD5. B–D, average electron microscopy images obtained from negatively stained particles. B, PDCD5-CCT; C, apo-CCT; D, anti-CCTδ-PDCD5. Images were averaged from 1356, 1128, and 1018 particles, respectively.
To determine its three-dimensional structure, the PDCD5·CCT complex was frozen-hydrated and subjected to cryo-EM. A three-dimensional reconstruction carried out with 13,000 particles to a 25-Å resolution (Fig. 5) revealed the typical barrel-shape structure built by the two octameric CCT rings in an open substrate-receptive conformation, as is the case with CCT in the absence of nucleotide (Fig. 6). The reconstruction clearly shows a small mass, attributable to PDCD5, protruding from one of the CCT subunits. Although the PDCD5 mass points toward the interior of CCT, it does not enter into the folding cavity but rather extends above it in a position not observed with CCT substrates actin and tubulin. A docking analysis using the 5.5-Å crystal structure of CCT in its open conformation (6) and an NMR solution structure of a PDCD5 fragment containing all but the last 12 residues of the C terminus (46) gave a good fit into the cryo-EM three-dimensional reconstruction (Fig. 6B). The facts that the mass attributed to PDCD5 accommodates the native atomic structure of the protein and that this mass is positioned on top of the folding cavity reinforce the idea that PDCD5 is not a substrate of CCT but rather may have a regulatory function.
FIGURE 6.
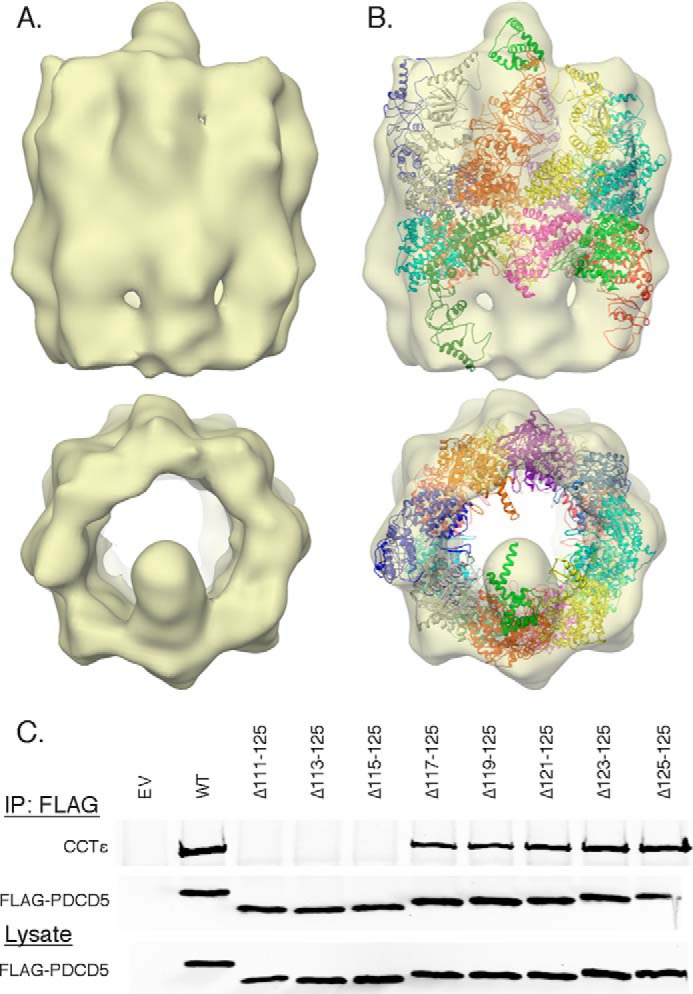
PDCD5 binds the apical domain of one CCT subunit. A, two orthogonal views of the three-dimensional reconstruction of the PDCD5·CCT complex carried out with 13,000 particles at 25 Å resolution. B, same two views showing the docking of the crystal structure of the open form of CCT colored by subunit (Protein Data Bank code 2XSM) and the atomic structure of PDCD5 in green (Protein Data Bank code 2K6B). C, binding of various C-terminal truncations of PDCD5 to CCT was measured by co-immunoprecipitation and immunoblotting.
The docking of the PDCD5 atomic structure was best when the C terminus was oriented toward the CCT subunit (Fig. 6B). To determine whether the C terminus of PDCD5 was involved in the interaction, the effects of several C-terminal truncations on PDCD5 binding to CCT were measured (Fig. 6C). Near normal binding was observed for truncations up to the last nine residues (PDCD5 Δ117–125). However, further truncation (PDCD5 Δ115–125) resulted in a complete loss of binding. Thus, it is clear that the PDCD5 C terminus participates in contacts with CCT, supporting the docking orientation shown in Fig. 6B.
Chemical Cross-linking of PDCD5 to CCT
The immuno-EM showed that PDCD5 was associated with CCTδ or an adjacent subunit. We sought to identify precisely which CCT subunit was involved by employing amber codon suppression technology to incorporate the photo-crosslinking unnatural amino acid BzF into residues at the C terminus of PDCD5 and specifically cross-link the CCT subunit in close proximity to these residues (45). We engineered amber codons (TAG) at positions 115, 117, or 119 of the PDCD5 cDNA and transfected these variants into HEK-293 cells along with the BzF tRNA and BzF tRNA synthetase. Cells were subsequently incubated with BzF, and cell extracts were cross-linked with ultraviolet (UV) light (Fig. 7A). PDCD5 was immunoprecipitated and immunoblotted to detect potential PDCD5 cross-links. Unfortunately, incorporation of BzF into PDCD5 was unusually low, less than 10% as judged by the intensity of the full-length BzF-incorporated PDCD5 band in immunoblots of cell lysates compared with the truncated PDCD5 band resulting from failed incorporation (Fig. 7B). In contract, ∼75% BzF incorporation was common for Gβ using this same method (Fig. 7B). Despite the low degree of BzF incorporation, UV-induced PDCD5 cross-links were still observed but not as expected (Fig. 8A). Surprisingly, we could detect a strong UV-dependent PDCD5 cross-link at ∼75 kDa in the control sample, transfected with wild-type PDCD5 that had no amber codon incorporation site. This cross-link was also seen in the variants with amber codons at residues 117 and 119 producing the Δ117–125 and Δ119–125 truncations that retained binding to CCT, but the cross-link was greatly reduced in the residue 115 variant producing the Δ115–125 truncation that had lost binding to CCT. This unexpected result indicates that UV irradiation induced a PDCD5 cross-link independent of incorporation of BzF into the amber codon site.
FIGURE 7.
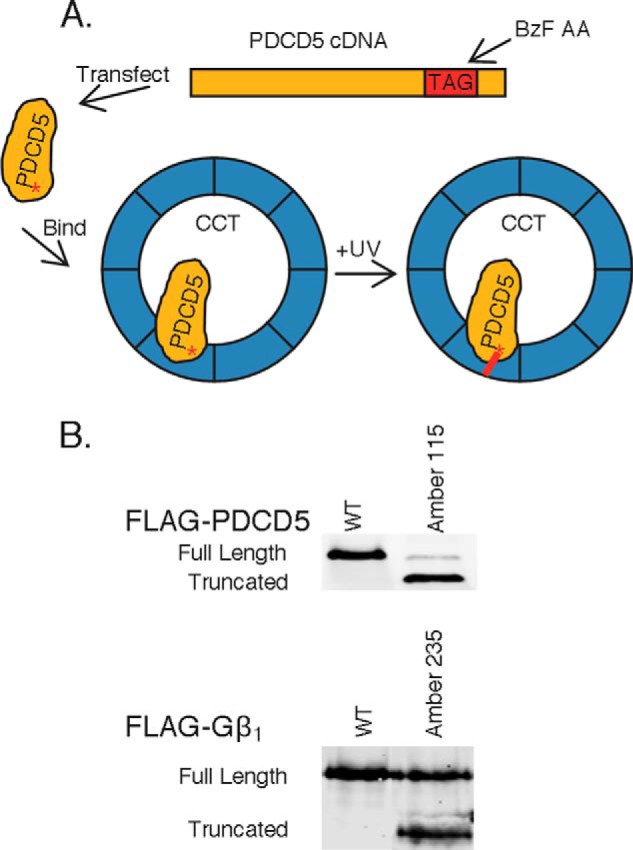
Unnatural amino acid incorporation into PDCD5. A, scheme of unnatural amino acid UV cross-linking using BzF. B, BzF incorporation into Amber codon variants of PDCD5 and the G protein β-subunit. HEK-293T cells were transfected with the indicated FLAG-PDCD5 constructs, BzF tRNA and BzF amino acid synthetase, and treated with 1 mm BzF 3 h post-transfection. Lysates were immunoblotted to determine the level of expression of the variants. The percent incorporation was measured as the ratio of the truncated protein band, resulting from a lack of BzF incorporation into the Amber codon, to the full-length protein band, resulting from BzF incorporation into the Amber codon. PDCD5 averaged 6% incorporation compared with 75% for Gβ1.
FIGURE 8.

PDCD5 interacts directly with CCTβ. A, cross-linking of PDCD5 to CCT. HEK-293T cells were transfected with the indicated FLAG-PDCD5 constructs, BzF tRNA, and BzF amino acid synthetase and treated with 1 mm BzF 3 h post-transfection. Lysates were exposed ± UV light prior to immunoprecipitation and blotting for FLAG-PDCD5. The asterisk indicates a PDCD5 cross-linked band. HC, heavy chain. B, cross-linking of PDCD5 to the CCT subunits was measured by co-immunoprecipitation from HEK-293T cells treated as in A and exposed to UV light prior to immunoprecipitation with a CCTϵ antibody and blotting for each of the CCT subunits. The asterisk indicates a CCTβ cross-linked band. The arrows mark the position of the CCT subunits. NS, nonspecific band. C, PDCD5 cross-linking to CCTβ depends on an interaction with the PDCD5 C terminus. HEK-293T cells were treated as in A and lysates were exposed ± UV light prior to immunoprecipitation with a CCTϵ antibody and blotting for CCTβ. The asterisk indicates a CCTβ cross-linked band. D, BzF catalyzes PDCD5 cross-linking to CCT without incorporation into Amber codon sites. HEK-293T cells were transfected with FLAG-PDCD5 (WT) in the absence of BzF tRNA and BzF AA synthetase. Cells were treated ± 1 mm BzF 3 h post-transfection. Lysates were exposed ± UV light prior to immunoprecipitation with CCTϵ and immunoblotting for CCTβ. The asterisk indicates a CCTβ cross-linked band.
Two observations suggested that the observed PDCD5 cross-link was to a CCT subunit. First, the cross-linking efficiency corresponded closely to the ability of the wild-type or truncated PDCD5 variants to bind CCT (compare Figs. 6C and 8A). Second, the ∼75-kDa size of the cross-link was approximately equal to the sum of the ∼60-kDa mass of a CCT subunit and the 16-kDa mass of FLAG-tagged PDCD5. To further explore this possibility and to determine which CCT subunit might be involved, the cross-linked PDCD5 immunoprecipitates were immunoblotted for all eight CCT subunits. The ∼75-kDa cross-link in the WT PDCD5 sample was detected only in the CCTβ immunoblot (Fig. 8B). Furthermore, the CCTβ cross-link was also found only in immunoprecipitates of the WT, Δ117–125, and Δ119–125 PDCD5 variants that bound CCT and not with the Δ115–125 variants that did not bind CCT (Fig. 8C). These results clearly show that PDCD5 was specifically cross-linked to CCTβ. Given that UV-induced cross-links are short lived and thus occur over distances of ∼3 Å (45), PDCD5 must be in close proximity to CCTβ. This finding is consistent with the immuno-EM images showing that PDCD5 bound near CCTδ because, in the recently revised orientation of the subunits within the CCT complex, CCTβ is adjacent to CCTδ (49, 50).
From these results, it is not obvious how the UV-induced cross-linking of PDCD5 to CCTβ occurs, but the cross-linking was dependent on BzF and was independent of the BzF tRNA or synthetase. In cells not transfected with the BzF tRNA or synthetase, a robust PDCD5-CCTβ cross-link was observed in the presence of BzF, but not in its absence (Fig. 8D). Thus, the free BzF in the cell extract must be catalyzing the cross-linking reaction. An examination of the free radical chemistry of BzF suggests possible ways that free BzF could cross-link two proteins. UV light initiates a BzF diradical intermediate that then removes a hydrogen from an adjacent protein, creating an alkyl radical on the protein and a ketyl radical on the BzF. Normally, the two radicals then recombine to yield a benzhydrol modification of the protein (51). However, in this case, a sufficient amount of the alkyl radical formed on either PDCD5 or CCTβ must react with its binding partner to form a detectable inter-protein cross-link. This unusual radical chemistry confirms a close binding interaction between PDCD5 and CCTβ.
DISCUSSION
Our search for novel PhLP1-binding partners has led to the serendipitous finding that the pro-apoptotic protein PDCD5 interacts with CCT. Investigation of the physiological role of the PDCD5/CCT interaction suggests that PDCD5 acts as a regulator of CCT function. The significant increase in β-tubulin folding upon PDCD5 depletion points to an inhibitory role for PDCD5 in β-tubulin folding (Fig. 3D). This inhibition was specific for β-tubulin and was not shared with other CCT substrates α-tubulin, β-actin, and Gβ. Accordingly, PDCD5 interfered with the binding of β-tubulin to CCT but not α-tubulin, indicating that PDCD5 specifically blocked β-tubulin folding by disrupting its interaction with CCT (Fig. 3, E and F). This disruption could result from a number of factors, but steric overlap of the binding sites of PDCD5 and β-tubulin on CCTβ seems likely. The cryo-EM reconstruction and cross-linking identifies an interaction of PDCD5 with the CCTβ helical protrusion at the tip of the apical domain (Figs. 4–8), and the crystal structure of the β-tubulin·CCT complex shows contacts between β-tubulin and the helical protrusion of CCTβ (6). A second β-tubulin contact site was also observed deeper within the CCT folding cavity, contacting the sensor loop of the equatorial domain (6). Thus, PDCD5 could interfere with β-tubulin binding in the helical protrusion without affecting its interaction with the sensor loop. Such partial inhibition is consistent with the observed effects of PDCD5 on β-tubulin binding to CCT and on β-tubulin folding (Fig. 3).
The ability of PDCD5 to discriminate between α- and β-tubulin was unexpected. The two proteins are structurally comparable, and both interact similarly with CCT (6, 9), yet PDCD5 only binds the β-tubulin·CCT complex and only inhibits β-tubulin folding (Fig. 3). It appears that when α-tubulin is bound to CCT, PDCD5 is excluded and has no effect on α-tubulin folding. The source of this specificity is unknown, but it could result from PDCD5 disrupting CCT contacts specific to β-tubulin or from a direct interaction between PDCD5 and β-tubulin. The residual binding of β-tubulin to PDCD5 upon CCT knockdown suggests that direct interactions between PDCD5 and β-tubulin do exist (Fig. 3C).
PDCD5 has been proposed to act as a pro-apoptotic factor with tumor suppressor capabilities (32). However, the molecular mechanisms underlying its apoptotic function are largely unknown. Interactions between PDCD5 and two pro-apoptotic proteins, the histone acetyltransferase Tip60 (33) and the transcription factor p53 (34), have been proposed to contribute to its apoptotic role. A key question arising from our studies is what contribution PDCD5 binding to CCT and inhibition of β-tubulin folding contributes to PDCD5-mediated apoptosis. Interestingly, the C-terminal truncations of PDCD5 that were found to disrupt its interaction with CCT (Fig. 6C) have been reported previously to be less effective in inducing cellular apoptosis (46). This correlation suggests that CCT binding and inhibition of β-tubulin folding could contribute to the apoptotic function of PDCD5. Tubulin dimer formation is a complex process, involving multiple co-factors that bring α- and β-tubulin together after their release from CCT (48). The process is vital for cell function in providing the building blocks for the microtubules that make the mitotic spindle and other important cellular structures. If mitotic spindle formation is disrupted, cells cannot divide and will eventually die and thus the large number of anti-cancer drugs that disrupt microtubule dynamics (52). PDCD5-mediated inhibition of β-tubulin folding could disrupt tubulin dimer formation and microtubule assembly and thus contribute to apoptosis.
The image emerging from this work and other previous studies shows CCT decorated with co-chaperones and regulators that modulate its protein folding function. For example, prefoldin delivers actin and tubulin to CCT for folding (16). PhLP1 allows release of Gβ from CCT to associate with Gγ and form the Gβγ dimer (21), and now PDCD5 binds CCT to slow β-tubulin folding and possibly disrupt microtubule formation. By employing co-chaperones in this manner, CCT is able to expand and fine-tune its already versatile protein folding capacity.
This work was supported, in whole or in part, by National Institutes of Health Grant GM078550 (to B. M. W.). This work was also supported by MINECO Grant BFU2010-15703 (to J. M. V.) and Madrid Regional Government Grant S2009MAT-1507 (to J. M. V.).
This article was selected as a Paper of the Week.
- CCT
- cytosolic chaperonin containing tailless complex polypeptide 1
- PDCD5
- programmed cell death protein 5
- Pdc
- phosducin
- PhLP1
- phosducin-like protein 1
- Gβ
- GTP-binding protein β subunit
- Gγ
- GTP-binding protein γ subunit
- BzF
- p-benzoyl-l-phenylalanine
- TEV
- tobacco etch virus
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- IP
- immunoprecipitation.
REFERENCES
- 1. Hartl F. U., Bracher A., Hayer-Hartl M. (2011) Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332 [DOI] [PubMed] [Google Scholar]
- 2. Saibil H. R., Fenton W. A., Clare D. K., Horwich A. L. (2013) Structure and allostery of the chaperonin GroEL. J. Mol. Biol. 425, 1476–1487 [DOI] [PubMed] [Google Scholar]
- 3. Yébenes H., Mesa P., Muñoz I. G., Montoya G., Valpuesta J. M. (2011) Chaperonins: two rings for folding. Trends Biochem. Sci. 36, 424–432 [DOI] [PubMed] [Google Scholar]
- 4. Rüssmann F., Stemp M. J., Mönkemeyer L., Etchells S. A., Bracher A., Hartl F. U. (2012) Folding of large multidomain proteins by partial encapsulation in the chaperonin TRiC/CCT. Proc. Natl. Acad. Sci. U.S.A. 109, 21208–21215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yam A. Y., Xia Y., Lin H. T., Burlingame A., Gerstein M., Frydman J. (2008) Defining the TRiC/CCT interactome links chaperonin function to stabilization of newly made proteins with complex topologies. Nat. Struct. Mol. Biol. 15, 1255–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Muñoz I. G., Yébenes H., Zhou M., Mesa P., Serna M., Park A. Y., Bragado-Nilsson E., Beloso A., de Cárcer G., Malumbres M., Robinson C. V., Valpuesta J. M., Montoya G. (2011) Crystal structure of the open conformation of the mammalian chaperonin CCT in complex with tubulin. Nat. Struct. Mol. Biol. 18, 14–19 [DOI] [PubMed] [Google Scholar]
- 7. Booth C. R., Meyer A. S., Cong Y., Topf M., Sali A., Ludtke S. J., Chiu W., Frydman J. (2008) Mechanism of lid closure in the eukaryotic chaperonin TRiC/CCT. Nat. Struct. Mol. Biol. 15, 746–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Douglas N. R., Reissmann S., Zhang J., Chen B., Jakana J., Kumar R., Chiu W., Frydman J. (2011) Dual action of ATP hydrolysis couples lid closure to substrate release into the group II chaperonin chamber. Cell 144, 240–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Llorca O., Martín-Benito J., Ritco-Vonsovici M., Grantham J., Hynes G. M., Willison K. R., Carrascosa J. L., Valpuesta J. M. (2000) Eukaryotic chaperonin CCT stabilizes actin and tubulin folding intermediates in open quasi-native conformations. EMBO J. 19, 5971–5979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Meyer A. S., Gillespie J. R., Walther D., Millet I. S., Doniach S., Frydman J. (2003) Closing the folding chamber of the eukaryotic chaperonin requires the transition state of ATP hydrolysis. Cell 113, 369–381 [DOI] [PubMed] [Google Scholar]
- 11. Zhang J., Baker M. L., Schröder G. F., Douglas N. R., Reissmann S., Jakana J., Dougherty M., Fu C. J., Levitt M., Ludtke S. J., Frydman J., Chiu W. (2010) Mechanism of folding chamber closure in a group II chaperonin. Nature 463, 379–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang J., Ma B., DiMaio F., Douglas N. R., Joachimiak L. A., Baker D., Frydman J., Levitt M., Chiu W. (2011) Cryo-EM structure of a group II chaperonin in the prehydrolysis ATP-bound state leading to lid closure. Structure 19, 633–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Martín-Benito J., Boskovic J., Gómez-Puertas P., Carrascosa J. L., Simons C. T., Lewis S. A., Bartolini F., Cowan N. J., Valpuesta J. M. (2002) Structure of eukaryotic prefoldin and of its complexes with unfolded actin and the cytosolic chaperonin CCT. EMBO J. 21, 6377–6386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Siegers K., Waldmann T., Leroux M. R., Grein K., Shevchenko A., Schiebel E., Hartl F. U. (1999) Compartmentation of protein folding in vivo: sequestration of non-native polypeptide by the chaperonin-GimC system. EMBO J. 18, 75–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hansen W. J., Cowan N. J., Welch W. J. (1999) Prefoldin-nascent chain complexes in the folding of cytoskeletal proteins. J. Cell Biol. 145, 265–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vainberg I. E., Lewis S. A., Rommelaere H., Ampe C., Vandekerckhove J., Klein H. L., Cowan N. J. (1998) Prefoldin, a chaperone that delivers unfolded proteins to cytosolic chaperonin. Cell 93, 863–873 [DOI] [PubMed] [Google Scholar]
- 17. Cuéllar J., Martín-Benito J., Scheres S. H., Sousa R., Moro F., López-Viñas E., Gómez-Puertas P., Muga A., Carrascosa J. L., Valpuesta J. M. (2008) The structure of CCT-Hsc70 NBD suggests a mechanism for Hsp70 delivery of substrates to the chaperonin. Nat. Struct. Mol. Biol. 15, 858–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Humrich J., Bermel C., Bünemann M., Härmark L., Frost R., Quitterer U., Lohse M. J. (2005) Phosducin-like protein regulates G-protein βγ folding by interaction with TCP-1α. J. Biol. Chem. 280, 20042–20050 [DOI] [PubMed] [Google Scholar]
- 19. Knol J. C., Engel R., Blaauw M., Visser A. J., van Haastert P. J. (2005) The phosducin-like protein PhLP1 is essential for Gβγ dimer formation in Dictyostelium discoideum. Mol. Cell. Biol. 25, 8393–8400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lukov G. L., Baker C. M., Ludtke P. J., Hu T., Carter M. D., Hackett R. A., Thulin C. D., Willardson B. M. (2006) Mechanism of assembly of G protein βγ subunits by protein kinase CK2-phosphorylated phosducin-like protein and the cytosolic chaperonin complex. J. Biol. Chem. 281, 22261–22274 [DOI] [PubMed] [Google Scholar]
- 21. Lukov G. L., Hu T., McLaughlin J. N., Hamm H. E., Willardson B. M. (2005) Phosducin-like protein acts as a molecular chaperone for G protein βγ dimer assembly. EMBO J. 24, 1965–1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wells C. A., Dingus J., Hildebrandt J. D. (2006) Role of the chaperonin CCT/TRiC complex in G protein βγ-dimer assembly. J. Biol. Chem. 281, 20221–20232 [DOI] [PubMed] [Google Scholar]
- 23. Lai C. W., Kolesnikov A. V., Frederick J. M., Blake D. R., Jiang L., Stewart J. S., Chen C. K., Barrow J. R., Baehr W., Kefalov V. J., Willardson B. M. (2013) Phosducin-like protein 1 is essential for G-protein assembly and signaling in retinal rod photoreceptors. J. Neurosci. 33, 7941–7951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Willardson B. M., Howlett A. C. (2007) Function of phosducin-like proteins in G protein signaling and chaperone-assisted protein folding. Cell. Signal. 19, 2417–2427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Howlett A. C., Gray A. J., Hunter J. M., Willardson B. M. (2009) Role of molecular chaperones in G protein β5/regulator of G protein signaling dimer assembly and G protein βγ dimer specificity. J. Biol. Chem. 284, 16386–16399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Camasses A., Bogdanova A., Shevchenko A., Zachariae W. (2003) The CCT chaperonin promotes activation of the anaphase-promoting complex through the generation of functional Cdc20. Mol. Cell 12, 87–100 [DOI] [PubMed] [Google Scholar]
- 27. Sondek J., Bohm A., Lambright D. G., Hamm H. E., Sigler P. B. (1996) Crystal structure of a G-protein βγ dimer at 2.1Å resolution. Nature 379, 369–374 [DOI] [PubMed] [Google Scholar]
- 28. Valpuesta J. M., Martín-Benito J., Gómez-Puertas P., Carrascosa J. L., Willison K. R. (2002) Structure and function of a protein folding machine: the eukaryotic cytosolic chaperonin CCT. FEBS Lett. 529, 11–16 [DOI] [PubMed] [Google Scholar]
- 29. Liu H., Wang Y., Zhang Y., Song Q., Di C., Chen G., Tang J., Ma D. (1999) TFAR19, a novel apoptosis-related gene cloned from human leukemia cell line TF-1, could enhance apoptosis of some tumor cells induced by growth factor withdrawal. Biochem. Biophys. Res. Commun. 254, 203–210 [DOI] [PubMed] [Google Scholar]
- 30. Li H., Wang Q., Gao F., Zhu F., Wang X., Zhou C., Liu C., Chen Y., Ma C., Sun W., Zhang L. (2008) Reduced expression of PDCD5 is associated with high-grade astrocytic gliomas. Oncol. Rep. 20, 573–579 [PubMed] [Google Scholar]
- 31. Spinola M., Meyer P., Kammerer S., Falvella F. S., Boettger M. B., Hoyal C. R., Pignatiello C., Fischer R., Roth R. B., Pastorino U., Haeussinger K., Nelson M. R., Dierkesmann R., Dragani T. A., Braun A. (2006) Association of the PDCD5 locus with lung cancer risk and prognosis in smokers. J. Clin. Oncol. 24, 1672–1678 [DOI] [PubMed] [Google Scholar]
- 32. Han X. R., Sun Y., Bai X. Z. (2012) The anti-tumor role and mechanism of integrated and truncated PDCD5 proteins in osteosarcoma cells. Cell. Signal. 24, 1713–1721 [DOI] [PubMed] [Google Scholar]
- 33. Xu L., Chen Y., Song Q., Xu D., Wang Y., Ma D. (2009) PDCD5 interacts with Tip60 and functions as a cooperator in acetyltransferase activity and DNA damage-induced apoptosis. Neoplasia 11, 345–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Xu L., Hu J., Zhao Y., Hu J., Xiao J., Wang Y., Ma D., Chen Y. (2012) PDCD5 interacts with p53 and functions as a positive regulator in the p53 pathway. Apoptosis 17, 1235–1245 [DOI] [PubMed] [Google Scholar]
- 35. Savage J. R., McLaughlin J. N., Skiba N. P., Hamm H. E., Willardson B. M. (2000) Functional roles of the two domains of phosducin and phosducin-like protein. J. Biol. Chem. 275, 30399–30407 [DOI] [PubMed] [Google Scholar]
- 36. McLaughlin J. N., Thulin C. D., Hart S. J., Resing K. A., Ahn N. G., Willardson B. M. (2002) Regulatory interaction of phosducin-like protein with the cytosolic chaperonin complex. Proc. Natl. Acad. Sci. U.S.A. 99, 7962–7967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shen P. S., Domek M. J., Sanz-García E., Makaju A., Taylor R. M., Hoggan R., Culumber M. D., Oberg C. J., Breakwell D. P., Prince J. T., Belnap D. M. (2012) Sequence and structural characterization of great salt lake bacteriophage CW02, a member of the T7-like supergroup. J. Virol. 86, 7907–7917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Prince J. T., Marcotte E. M. (2008) mspire: mass spectrometry proteomics in Ruby. Bioinformatics 24, 2796–2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Choi H., Fermin D., Nesvizhskii A. I. (2008) Significance analysis of spectral count data in label-free shotgun proteomics. Mol. Cell. Proteomics 7, 2373–2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sorzano C. O., Marabini R., Velázquez-Muriel J., Bilbao-Castro J. R., Scheres S. H., Carazo J. M., Pascual-Montano A. (2004) XMIPP: a new generation of an open-source image processing package for electron microscopy. J. Struct. Biol. 148, 194–204 [DOI] [PubMed] [Google Scholar]
- 41. Scheres S. H., Valle M., Nuñez R., Sorzano C. O., Marabini R., Herman G. T., Carazo J.-M. (2005) Maximum-likelihood multi-reference refinement for electron microscopy images. J. Mol. Biol. 348, 139–149 [DOI] [PubMed] [Google Scholar]
- 42. Ludtke S. J., Baldwin P. R., Chiu W. (1999) EMAN: Semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 128, 82–97 [DOI] [PubMed] [Google Scholar]
- 43. Scheres S. H., Valle M., Grob P., Nogales E., Carazo J.-M. (2009) Maximum likelihood refinement of electron microscopy data with normalization errors. J. Struct. Biol. 166, 234–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004) UCSF Chimera—A visualization system for exploratory research and analysis. J. Comp. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 45. Grunbeck A., Huber T., Abrol R., Trzaskowski B., Goddard W. A., 3rd, Sakmar T. P. (2012) Genetically encoded photo-cross-linkers map the binding site of an allosteric drug on a G protein-coupled receptor. ACS Chem. Biol. 7, 967–972 [DOI] [PubMed] [Google Scholar]
- 46. Yao H., Xu L., Feng Y., Liu D., Chen Y., Wang J. (2009) Structure-function correlation of human programmed cell death 5 protein. Arch. Biochem. Biophys. 486, 141–149 [DOI] [PubMed] [Google Scholar]
- 47. Yaffe M. B., Farr G. W., Miklos D., Horwich A. L., Sternlicht M. L., Sternlicht H. (1992) TCP1 complex is a molecular chaperone in tubulin biogenesis. Nature 358, 245–248 [DOI] [PubMed] [Google Scholar]
- 48. Lopez-Fanarraga M., Avila J., Guasch A., Coll M., Zabala J. C. (2001) Review: postchaperonin tubulin folding cofactors and their role in microtubule dynamics. J. Struct. Biol. 135, 219–229 [DOI] [PubMed] [Google Scholar]
- 49. Kalisman N., Adams C. M., Levitt M. (2012) Subunit order of eukaryotic TRiC/CCT chaperonin by cross-linking, mass spectrometry, and combinatorial homology modeling. Proc. Natl. Acad. Sci. U.S.A. 109, 2884–2889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Leitner A., Joachimiak L. A., Bracher A., Mönkemeyer L., Walzthoeni T., Chen B., Pechmann S., Holmes S., Cong Y., Ma B., Ludtke S., Chiu W., Hartl F. U., Aebersold R., Frydman J. (2012) The molecular architecture of the eukaryotic chaperonin TRiC/CCT. Structure 20, 814–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dormán G., Prestwich G. D. (1994) Benzophenone photophores in biochemistry. Biochemistry 33, 5661–5673 [DOI] [PubMed] [Google Scholar]
- 52. Matson D. R., Stukenberg P. T. (2011) Spindle poisons and cell fate: a tale of two pathways. Mol. Interv. 11, 141–150 [DOI] [PMC free article] [PubMed] [Google Scholar]