

Abstract
Epithelial to mesenchymal transition (EMT) is an important and complex phenomenon that determines the aggressiveness of cancer cells. The morphological transformation of cancerous cells is accompanied by various cellular processes such as alterations in cell-cell adhesion, cell matrix degradation, down regulation of epithelial marker E-cadherin and upregulation of mesenchymal markers N-cadherin and Vimentin. Besides these markers several other important tumor antigens/mucins are also involved in the EMT process. Mainly high molecular weight glycoproteins such as mucin molecules (MUC1, MUC4 and MUC16) play a major role in the cellular transformation and signaling alteration in EMT process. In addition to these factors, EMT may be an essential process triggering the emergence or expansion of the CSC population, which slowly results in the initiation of tumor at metastatic sites. Furthermore, mucins have been demonstrated to be involved in the EMT process and also in the enrichment of cancer stem cell population. Mucin mediated EMT is very complex since the key components of tumor microenvironment are also regulating mucin molecules. In this review, we have discussed all the aforementioned factors and their mechanistic involvement for EMT process.
Keywords: EMT transcription factors, EMT signaling, Mucins, MUC1, MUC4, MUC16, Cancer stem cells, Tumor microenvironment
INTRODUCTION
Epithelial to mesenchymal transition (EMT) plays an important role to understand the basis and potential mechanisms for tumor progression and metastasis. During this process, cancer cells attain unique characteristic features by altering cell-cell interaction and cell- matrix interaction to facilitate the process of invasion and motility thereby enhancing the risk of developing metastasis process. In the recent years, several important transcription factors or transformation modulators were found to be associated with the altered physiological changes in the cellular phenotype during EMT. Certain EMT activating, mediating and inducing transcription factors such as TWIST, SNAIL, SLUG, ZEB-1 and ZEB-2 and important conventional signaling molecules such as TGFβ, WNT, Notch and Hedgehog were arising during the EMT signaling alterations.
Though this transition process was mediated and orchestrated by various transcriptional modulators and signaling intermediates stated above, mucins play a significant role in cellular differentiation process and is associated with the aggressive behavior of metastatic tumor cells. Mucins are high molecular weight glycoproteins, heavily glycosylated with O-linked oligosaccharides produced majorly in the secretory epithelium and rarely in endothelial cells. Mucins are expressed in various organs of epithelial cell and are involved in the protective function under normal physiological functions. The function of mucins has been implicated for the process of epithelial cell differentiation, growth regulation, modulation of cell-cell contact and cell matrix interactions [1]. Apart from their protective function, certain mucins are differentially expressed in several pathological diseases such as cancer, cystic fibrosis, severe asthma, acute and chronic bronchitis [2]. Moreover, aberrant expression of mucins has been observed in various cancers and has also been reported to be associated with proliferation, altered cellular adhesion, invasion and metastasis properties of cancer cells. Among the various membrane bound mucins, MUC1, MUC4 and MUC16 are so far identified to be associated with the EMT process (Fig. 1). The mucin mediated EMT process is very complex and specifically in this section we have focused on the predominant signaling pathways up to now elucidated for all the mucins stated above. Additionally, recent evidences demonstrate that a subgroup of cells in the tumor core regarded as cancer stem cells are the important determinants for the initiation and progression of cancers, enhanced genomic instability and acquisition of drug resistance leading to aggressive behavior of tumor along with the initiation of EMT process. In our recent studies we have demonstrated that mucins may also play an important role in the maintenance of CSCs [3]. The study by Bafna et al., showed that MUC4 is instrumental in imparting gemcitabine resistance in pancreatic cancer cells and MUC4 down-regulation reverses chemoresistance of pancreatic cancer stem/progenitor cells thereby preventing tumor relapse [4, 5]. Furthermore, we also demonstrated that the siRNA mediated knockdown of MUC4 decreases the number of CSC population in pancreatic cancer cells [5]. Interestingly, in our recent study we have shown that exogenous MUC4 overexpressed ovarian cancer cells enriches the ovarian cancer stem cells (OCSCs) [3] suggesting MUC4 directed CSCs may also be involved in the EMT process. MUC1, another membrane-bound mucin is also shown to be involved in increasing the proportion of stem/progenitor cells in breast cancer [6].
Fig. 1. Aberrant expression of mucins induce EMT phenotype.

(i) Normal epithelial cells carry many epithelial markers and junctional proteins that anchor epithelial cells to the basement membrane. (ii) Many of the cancerous cells apparently express mucin molecules and upregulates the EMT markers. (iii) Mucin molecules involved in the induction of mesenchymal cell type. (iv) Metastatic process initiated by mesenchymal cell types in relation to mucin expression.
Finally, it is becoming evident that the heterotypic cellular interactions in tumor microenvironment by several factors such as growth factors, cytokines released by neighboring cells, inflammation, hypoxia stimulation and tissue necrosis are implicated in EMT and metastasis of different cancers [7–10]. In a tumor, the niche is mainly modified by certain non-cancerous cells like macrophages, monocytes, mast cells, neutrophils, endothelial cells and stromal fibroblast cells etc. favoring tumor cell proliferation, growth, survival and metastasis. Overall, in this review we have discussed the correlation between EMT related transcription factors and its signaling alterations, role of mucins in EMT process, cancer stem cells as an important player of EMT and the functional relationship between tumor microenvironment and EMT initiation process.
ESSENTIAL ROLE OF TRANSCRIPTIONAL REGULATORS AND MICRORNA IN EMT PROCESS
EMT is based on the potency of different transcription factors which are involved in the cellular transformation by repressing and activating the EMT regulatory network proteins. Several studies have demonstrated that SNAIL 1 directly interacts with the E-Box sequence present in the proximal promoter region of E-cadherin, to repress its expression in epithelial cells [11, 12] and many other transcription factors such as SNAIL2 [13], ZEB1, ZEB2 [14, 15], KLF8 [16], E47 [17] and Brachyury (a nuclear transcription factor essential for mesoderm layer and notochord formation) [18] are also involved in repressing of EMT markers and further leading to repression of junctional proteins (Claudins and Desmosomes) [19–21]. Altogether, in cancerous cells decreased expression of epithelial markers (E-cadherin and CK18) and increased expression of mesenchymal markers (Vimentin and N-cadherin) are correlated with endogenous expression of above mentioned transcription factors. Furthermore, numerous transcription factors were demonstrated to be involved indirectly through a control mechanism over the EMT process such as TWIST [22], SIX1 [23], FOXC2 [24], E2-2 [25] and PRRX1 [26]. Majority of transcription factors are involved in the cellular transformation to mesenchymal phenotype which emphasize the idea of suppression of epithelial phenotype [27]. Emerging evidences provide a strong link between microRNA regulation and EMT associated phenotypic alterations that triggers the metastatic potential of cancer cells. Notably, miRNA-200 has been demonstrated to be a direct target of ZEB1/2 [28], an important transcription factor for the repression of E-cadherin molecule. In a similar fashion, Smad3 and TGF-β pathways were also controlled by miRNA-200 via E-cadherin in cancer cells [29]. Consistently, in another study the expression of miRNA-27 was correlated with EMT phenotype by increasing the expression of EMT associated transcription factors SNAIL and ZEB1/2, mesenchymal marker Vimentin and diminished expression of epithelial marker E-cadherin in human gastric cancer cells [30]. These studies suggest that EMT transcription factors and miRNAs are involved in the activation of EMT based signaling pathways and trigger the metastatic potential of various cancers.
MULTIPLE SIGNALING PATHWAYS IN EMT PROCESS
In cancer progression several molecular hallmarks and signaling pathways are activated thereby initiating the EMT process. A brief description of the major signaling pathways involved in EMT process such as TGF-β, Wnt, Notch and Hedgehog is given below.
TGFβ Signaling
TGF-β is a pro-EMT signaling stimulus which exerts the potential of cell proliferation, apoptosis, migration and invasion of human cancer cells [31]. TGF-β signaling is controlled by many transcription factors such as AP1, ETS, zinc finger transcription factors, p300/CBP and helix-loop-helix family proteins [32]. TGF-β is involved in two different pathways such as canonical TGF-β signaling and downstream activation of other canonical pathways, including Hedgehog, Notch and Wnt. TGF-β signaling is an important master regulator in both physiological and pathological EMT progression [33] by the activation of specific transcription factors such as TWIST, SNAIL, SLUG and ZEB [34, 35]. This signaling also triggers the cells towards disassembling cell-cell interactions and actin-cytoskeleton rearrangement in cancer cells [36]. It was also demonstrated by the treatment of TGF-β which was found to increase the cell invasion and spindle shape phenotype in cholangiocarcinoma cells leading to increased expression of N-cadherin and decreased expression of E-cadherin in cholangiocarcinoma [37]. These studies suggest that TGF-β signaling is a master switch of the EMT process and initiation of metastatic potential in many types of cancers.
Wnt Signaling
Wnt signaling is an important canonical pathway in the cell fate determination during embryonic development and cancer progression. Basically, Wnt binds with Frizzled receptor to activate the canonical pathway, which further causing accumulation of β-catenin and its translocation to the nucleus for the transcriptional activation of oncogenes such as c-Myc, Cyclin D1, MMP-7, CD44, SOX-9 and Runx2, elements essential for cellular proliferation, cell cycle control, DNA replication and altered mitosis. The accumulation of β-catenin in cytoplasm will not occur without Wnt signaling since the GSK3β complex degrades β-catenin by targeting it for ubiquitination. Wnt signaling also activates Axin2 and GSK-3β and regulates SNAIL family of transcription factors leading to initiation of EMT and invasion process in breast cancer cells [38]. Secreted Frizzled-related protein 1 (SFRP1) is one of the antagonists of Wnt receptor and silencing SFRP1 leads to altered EMT phenotype thereby increasing the expression of ZEB1 in breast cancer cells [39]. Another Wnt activator serum response factor (SRF) also plays a vital role in EMT by up-regulating Vimentin, N-cadherin, and RhoA in HeLa cells, thereby leading to increased cellular migration and invasion [40]. Studies have demonstrated that the β-catenin signaling and its importance in altering the expression of SNAIL, TWIST and SLUG to repress E-cadherin expression are associated with poor prognosis of various cancers [41, 42]. Overall, Wnt signaling is one of the important activator for the EMT process and cancer metastasis by regulating different downstream effectors.
Notch Signaling
Notch signaling is also one of the key mechanism implicated in embryogenesis and cancer progression [43]. In recent studies, Notch signaling was shown to be involved in the EMT process and maintenance of cancer stem cells [44, 45]. The mutants of Notch pathway are involved in altering the expression of SNAIL and TGFβ2 molecules, which are the important factors for EMT development [46]. A recent study demonstrates that Notch expression leads to EMT induction through down regulation of miRNA-200 [47]. The interactions of ZEB1 and miRNA 200 with Notch lead to the cellular transformations in breast and pancreatic cancer cells [48]. Further, the clinical implications of Notch 1 expression were also correlated with the EMT markers such as, E-cadherin and Vimentin in the primary and metastatic specimens of prostate cancer [49]. These recent findings suggest that Notch signaling is the most pivotal pathway for EMT and metastasis of cancer progression.
Hedgehog Signaling
Hedgehog pathway was originally identified during the various stages of embryonic development and later it was also implicated in the development of cancer progression. Three known hedgehog pathways are established in cancer progression such as, sonic hedgehog, indian hedgehog and desert hedgehog. The downstream activator of Smo and Gli for hedgehog are playing major role in EMT process by up-regulating SNAIL transcription factor [50]. TGF-β treatment in lung cancer cells induces sonic hedgehog which further leads to the induction of EMT by altering the cellular expression of E-cadherin and ZEB1 proteins [51]. A recent study has also demonstrated that overexpression of sonic hedgehog leads to metastasis in gastric cancer through P13K/Akt pathway and the inhibition of sonic hedgehog also reduced the EMT markers [52]. The sonic hedgehog activates Gli1 which results in EMT changes by altering the expression of SNAIL, SLUG and E-cadherin [53]. These studies suggest that hedgehog is an important determinant for the cell type dependent EMT mechanism in cancer progression.
MUCINS IN EPITHELIAL TO MESENCHYMAL TRANSITION
Apart from above mentioned EMT pathways, many other molecules like mucins are also found to be involved in the process of EMT in various cancers (Fig. 1). Our research group and other groups have recently established the emerging role of mucin molecules in different cancers [1, 2, 54–59]. Furthermore, in this review we have specifically discussed the importance of mucins in EMT mechanism.
Role of MUC1 in EMT Initiation and Acquisition of Multistep Metastatic Process
The membrane bound mucin MUC1 is aberrantly expressed in several epithelial cancers such as breast, ovarian and pancreatic cancer [60–63]. Among cell adhesion mucin molecules MUC1 was initially shown to be transcriptionally repressed when transfected with SNAIL transcription factor in epithelial cells. Earlier experiments with in vitro model of mouse mammary tumor cells (DA3) have demonstrated that stable overexpression of truncated form of human MUC1 leads to EMT with altered cellular signaling. Furthermore, EMT was shown to be associated with ERK dependent activation of Fibronectin for cell invasion and PI3K dependent activation (Fig. 2). This process is essential for cellular proliferation in DA3/TRUNC-MUC1 trasfected mammary cells as compared to epithelia like DA3 cells [64]. Later, the study by Roy et al., was the first evidence elucidating the direct association between MUC1 mucin aberrant expression and initiation of EMT phenotype in cancer cells. Through stable overexpression of MUC1 in pancreatic cancer cells, they have provided the direct association of EMT related transcription factors such as SNAIL and SLUG and their involvement in the initiation of EMT, further enhancing the invasiveness and acquisition of metastatic potential of pancreatic cancer. Additionally, by mutating the tyrosine residue in the cytoplasmic region of MUC1 in pancreatic cancer cells, they demonstrated the functional significance of phosphorylation status of MUC1 and EMT phenotype initiation. The genetically engineered mouse model system also reflects the in vitro data demonstrating the knockdown effect of Muc1 leading to significant reduction of EMT and metastasis process [63]. It was also demonstrated that the interaction between the androgen receptor (AR) and carboxyl-terminal part of MUC1 mucin is critical in driving the EMT- associated phenotypical changes in prostate cancer cells. Furthermore, carboxy-terminal part of MUC1-C terminal interacts with the DNA binding domain of androgen receptor enhancing the invasiveness of prostate cancer. Additionally, the MUC1-C also suppresses the AR expression, resulting in more aggressive phenotypical variations that are sensitive when suppressed with MUC1-C specific inhibitor G0-203 in prostate cancer cells [62, 65] (Fig. 2).
Fig. 2. Mechanism of MUC4, MUC1 and MUC5AC mucins in EMT process.

In cancer cells, MUC4 interacts with HER2 and subsequently activates FAK/MKK7/JNK and c-Jun to regulate the N-cadherin expression. MUC4 dependent up-regulation of N-cadherin involved in the cellular alteration of EMT in pancreatic, ovarian and breast cancer cells. MUC4 induced EMT increase the expression of mesenchymal markers N-cadherin and Vimentin as well as decrease the expression of epithelial markers E-cadherin, CK-18 and Occludin. MUC1 activates ERK1/2 and leads to the regulation of Fibronectin for EMT process. Furthermore, the cytoplasmic tail (CT) of MUC1 interacts with the DNA binding domain of androgen receptor (AR) and resulted in the activation of EMT process and invasiveness of cancer cells. MUC5AC, a secretory mucin involved in differential localization of E-cadherin and ERK1/2 activation for the EMT process.
MUC4 Mucin Expression Regulates EMT Process
Our previous studies have illustrated the role of MUC4 in the malignant transformation, involvement in metastasis and altered cellular signaling in the tissues of pancreatic, ovarian, endometrial and breast cancer [55, 57, 58]. Studies pertaining to the expression of MUC4 in tissues have demonstrated the malignant and invasive potential of MUC4 in initiating the multistep process of metastasis. Ectopic overexpression of MUC4 mucin in SKOV3 ovarian cancer cells have established the altered morphological characteristics associated with the EMT process (Fig. 2). Further, Ponnusamy et al., has reported that MUC4 induced changes in the cellular phenotype is accompanied by altered expression of epithelial marker (E-cadherin and CK-18) followed by mesenchymal marker (N-cadherin and Vimentin) which in turn leads to the ovarian cancer cells to acquire enhanced invasive property accompanied by cellular, phenotypical and morphological changes leading to more malignant phenotype. In addition, two EMT related pro-transcription factors such as TWIST and SNAIL were found to be significantly upregulated in the MUC4 overexpressed ovarian cancer cells. Finally, it was identified that the central mechanism of EMT in ovarian cancer cells were through the activation of focal adhesion kinase (FAK) pathway and upregulation of N-cadherin by MUC4 mucin (Fig. 2). Our previous studies have also shown MUC4 to be a modulator of cellular signaling pathway by interacting with epidermal growth factor receptor family protein (HER2). Through various interaction studies it was demonstrated that MUC4 interacts and stabilizes HER2 both at membranous and cytoplasmic region in pancreatic and ovarian cancer cells [55, 57]. Additionally, stable silencing of MUC4 had led to decreased expression of phosphorylated form of HER2 at tyrosine residue 1248 further leading to down regulation of FAK thus providing a mechanistic insight of MUC4 regulated cellular signaling in cancer cells [55, 57, 58]. Although the loss of E-cadherin is regarded as the hallmark of EMT process, stable silencing of transmembrane mucin MUC4 leads to down regulation of N-cadherin and its interacting partner fibroblast growth factor receptor 1 (FGFR1) in pancreatic cancer cells [59]. Rachagani et al., have demonstrated the precise molecular mechanism associated with the MUC4 mediated EMT initiation in pancreatic cancer cells. Interestingly, downregulation of FGFR1 in MUC4 knockdown pancreatic cancer cells led to changes in the cellular morphology, downregulation of mesenchymal markers such as Vimentin and N-cadherin along with EMT regulators TWIST, SLUG and ZEB1 and upregulation of epithelial markers E-cadherin, Occludin, and Cytokeratin-18 [59]. Thus, MUC4 suppression in pancreatic cancer cells is associated with enhanced expression of epithelial markers and absence of mesenchymal marker resulting in altered EMT phenotype, which was further believed to be involved in diminishing cellular migration property. Subsequently, MUC4 mucin was also shown to enhance the invasive and migratory potential of triple negative breast cancer cells through upregulation of EGFR family proteins. Another study from our group have demonstrated that knockdown of MUC4 in breast cancer cells leads to molecular biochemical alterations necessary for mesenchymal to epithelial transition. Furthermore, reduced expression of mesenchymal markers such as Vimentin and Vitronectin and increased expression of epithelial marker cytokeratin-18 in MUC4 knockdown cells suggests an important role of MUC4 in acquisition of EMT phenotype, thereby transforming to more migratory and aggressive phenotype of breast cancer cells [56]. In contrast, the recent publication by Majhi et al., shows that MUC4 expression in lung cancer cells could abrogate migration and invasion through altering the phosphorylation of FAK. Subsequently, the MUC4 expression leads to significant changes in the expression of mesenchymal and epithelial markers of lung cancer cells. Moreover, the association of EMT phenotype and differential expression of mesenchymal marker N-cadherin were also correlated with overexpression and knockdown of MUC4 in A549 lung cancer cells [66]. These studies suggest that MUC4 acts as an intermediate protein for the EMT development in different cancer cells.
MUC16/CA125 in Epithelial to Mesenchymal Transition
In contrast to other membrane bound mucins, MUC16 was shown to be involved in the EMT process by altering cell surface proteins as well as intracellular downstream signaling molecules. A recent study describes that silencing of cell surface expression of MUC16 by single-chain variable fragment (ScFV) resulted in cellular morphological changes [67]. Loss of cell surface expression of MUC16 induces mesenchymal features such as longer and fibroblast like structures [cobblestone-like monolayer, round boundary and intact cell-cell contact] [67] and these changes have been well correlated with MUC16 negative Ovarian Surface Epithelium (OSE) cells, which have mesenchymal–type signature. In the same study they have also observed increased invasion and motility in MUC16 deficient cells due to enhanced expression of mesenchymal marker N-cadherin and Vimentin [67]. Furthermore, silencing of cell surface MUC16 induced the internalization of E-cadherin; this invariably enhances the N-cadherin and Vimentin expression. Knockdown of MUC16 also activates the EGFR signaling that target Akt, ERK, MMP-2 and MMP-9 to promote the motility of ovarian cancer cells. These studies suggest that expression of MUC16 provides strong cell-cell contact by interaction with N-cadherin that leads to inhibit invasive process in cancer cells [67]. Another study of the same group also demonstrated that overexpression of cytoplasmic tail domain region of MUC16 (MUC16 CTD) affects the increased motility and invasive properties, suggesting that cytoplasmic region of MUC16 is required for motility and invasion of ovarian cancer cells [68]. Additionally, overexpression of cytoplasmic portion of MUC16 is associated with invasive properties of ovarian cancer cells by repressing the E-cadherin expression and gain of mesenchymal characteristic features. Based on the available evidences, cell surface localization and cytoplasmic tail region of MUC16 performs two different mechanisms for epithelial to mesenchymal transition process by altering E-cadherin expression [68].
Several line of studies have demonstrated that MUC16/CA125 is overexpressed in ovarian cancer and it has been used as a biomarker for progression of ovarian cancer following chemotherapy [69]. Cell binding assays demonstrated that N-linked glycan of MUC16 is necessary for mesothelin interaction, both at membranous and cytoplasmic levels. Overexpression of MUC16 and mesothelin in ovarian tumor cells recruits other tumor cells at the sites of metastasis [70]. Heterotypic interaction of MUC16 and mesothelin initiates primary mechanism for peritoneal metastasis of ovarian cancer. In a nut shell, inhibiting MUC16 and mesothelin would be a better approach to prevent the ovarian cancer metastasis.
Role of Secretory Mucin in EMT
The gel forming mucin MUC5AC appears to be expressed in many cancers such as pancreas, colon and lung etc. A study demonstrated that MUC5AC is not expressed in normal pancreas but overexpressed in pancreatic cancer. The overexpression of MUC5AC is significantly correlated with proliferation, invasion and motility of cancer cells [71–73]. Recent study have suggested that GLI1 and GLI2 can regulate MUC5AC gene expression by binding with CACCC-box like cis-regulatory elements of MUC5AC promoter region [74]. GLI1 mediated upregulation of MUC5AC was expressed in cultured pancreatic ductal adenocarcinoma cells and alter the membrane localization of E-cadherin resulted to increased migration and invasion of pancreatic ductal adenocarcinoma cells [74] (Fig. 2). Overexpression of MUC5AC induced the invasive and motile properties of pancreatic cancer cells by altering the vascular endothelial growth factor receptor-1(VEGFR-1). In addition, MUC5AC also induces expression of integrins, MMP-3 and ERK pathway during the invasive process of pancreatic cancer cells [75] (Fig. 2). MUC5AC appears to be expressed in early stage of colon carcino- genesis and it has been suggested that expression of MUC5AC is essential for invasive behavior of colon cancer cells. To demonstrate the invasive potential of colon cancer cells a set of sub-population of HT-29 5M21 clones, derived from HT-29 parental cell line was examined for morphological studies. The sub- clones appeared to grow as a monolayer of polarized cells with aberrant expression of MUC5AC mucins, whereas the parental HT-29 displayed a round structural morphology with rare expression of MUC1 and MUC5AC [76]. Overexpression of these mucins correlated with high potential invasive behavior of HT-29 5M21, which was also associated with diminished expression of E-cadherin than parental cell line, HT-29. These findings were also confirmed by using anti-MUC1 and MUC5AC antibody which resulted in abrogation of cellular invasiveness due to restoration of E-cadherin function [76]. These findings suggested that co-operation between MUC1 and MUC5AC affects the E-cadherin downregulation for invasive process during metastasis [76]. Another gel forming mucin such as MUC5B was also shown to be involved in tumor cell invasion and metastasis in luminal type of breast cancer cells by affecting cell-cell and cell-matrix interaction [77].
Like MUC16, MUC6 was also shown to inhibit the invasion of pancreatic cancer cells through altering basement membrane organization [78]. Overexpression of different portions of MUC6 such as N- and C-terminal region inhibits cell adhesion and cell matrix associated proteins such as collagen I, collagen IV, fibronectin and laminin intervening the process of invasion [78].
EMERGING EVIDENCES OF CANCER STEM CELLS IN EMT
Cancer stem cells also known as tumor initiating cells are multi-potent progenitor cells. These cells resemble stem cells in their self-renewal property. CSCs have the ability to divide asymmetrically and are highly resistant to chemotherapy and radiotherapy. They originate from either stem cells or progenitor cells or from terminally differentiated cells, which have acquired oncogenic lesions thereby, resulting in dedifferentiation to a stem cell like state [79]. In addition to the above mentioned features, CSCs also display EMT-like phenotype through loss of E-cadherin [80] (Fig. 3).
Fig. 3. Role of CSCs in EMT process.
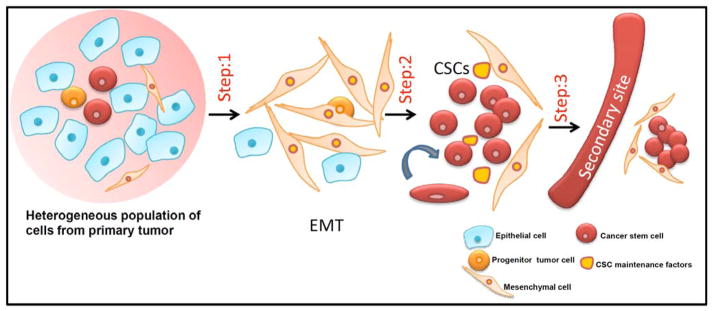
Primary tumor consists of a heterogeneous population of cells, including epithelial cells, progenitor tumor cells, mesenchymal cells and cancer stem cells. Step 1: Epithelial cells from primary tumor undergo EMT to give rise to mesenchymal cells. Step 2: On undergoing an insult such as conventional chemotherapy and radiation it enriches the cancer stem cell population. These CSCs maintain their self-renewal property in the presence of CSC maintenance factors. Step 3: CSCs evade to the secondary sites and colonize at distant metastatic sites.
Although, CSCs initiate tumors and helps in the maintenance and recurrence of tumors, it is also essential in mediating tumor invasion and metastasis. Once the cells disseminate from the primary site and evade to the secondary sites, it could be argued that these cells behave like CSCs by carrying out the tumor initiating property and thereby seeding new colonies in the secondary sites. Tang et al., showed that from a tumor bearing a highly heterogeneous pool of tumorigenic cells, a very small population of CSCs bearing specific surface markers, such as CD90+ has been responsible for the aggressive and metastatic phenotype of the esophageal squamous cell carcinoma (ESCC) [76].
Essential Players Linking CSCs and EMT
Wellner et al., showed that ZEB1 (zinc finger E-box binding homeobox 1), an EMT activator is an important promoter of metastasis and ZEB1 expression facilitates inhibition of microRNA 200 (miR200) family has been shown to be involved in epithelial differentiation induction. They clearly showed that ZEB1 is a major player in inducing EMT by inhibiting the miRNAs and promoting migratory CSCs [81]. In Non-Small-Cell Lung Carcinoma, TGF-β1 causes increase in motility in CD133+ cells (a subpopulation of CSCs isolated from A549 cell line) whereas there is no difference in the motility behavior of CD133− cells [82].
CXC chemokine receptor 4 (CXCR4) is another essential marker which is expressed on the surface of CSCs in Renal Cell Carcinoma (RCC) and its expression clearly distinguishes between the high tumorigenic cell line (having higher CXCR4 expression) and less tumorigenic cell line. The CXCR4 positive cells were able to form spheres faster than that of the CXCR4 non-expressing cells. Also, these cells were highly resistant to tyrosine kinase inhibitors such as sorafenib, pazopanib and sorafenib [83]. This study have suggested that the presence of high CXCR4+ CSCs in RCC facilitates invasion and cancer cell dissemination. As soon as the cells metastasize, there is very little influence of these mutated stem cells or CSCs on disease progression [83].
CD44 is an essential marker of stem cells and it has been found to play an important role in tumorigenicity, invasion, cell migration and bone metastases This molecule has been found to be highly expressed in various cancer cells such as MDA-MB231, PC-3, A375 and HARA-B and also have which finally metastasize to bone when injected into nude mice [84–86]. CSCs rich in CD44 expression has been shown to be involved in bone metastasis through interaction with hyaluronan, an essential part of the CSC niche. However, an interesting study in head and neck cancer by Giudice et al., have shown that inhibition of histone deacetylase (HDAC) disrupts the CSC population and induces EMT phenotype of head and neck cancer cells. All these studies suggests that by modulating the CSC population in a therapeutic aspect, will help targeting cancer cells having the metastatic phenotype and not perturbing the CSC populations which are the major contributors to the recurrence of several cancers [87].
Is EMT a Consequence of CSCs or CSCs a Consequence of EMT
Occurrence of EMT due to prevailing CSC population or whether EMT confers the stem cell properties to the existing cancer cells in a tumor has been a major debatable issue. Mani et al., has addressed this issue, by inducing EMT (cells transformed with HER2/neu oncogene along with a vector expressing tamoxifen-activatable form of TWIST or SNAIL transcription factors) in an immortalized human mammary epithelial cell, which resulted in harboring mesenchymal and stem cell traits [80]. They suspect that EMT could trigger and drive the generation of CSCs phenotype from differentiated neoplastic cells [80] as evidenced by the results, where EMT induced cells formed a 10-fold higher tumorspheres as well as 10-fold more colonies as opposed to the control cells. In addition, there were elevated levels of CD44high/CD24low cells in the EMT induced cells. Constant EMT inducing signals has been shown to play a vital role for the maintenance of cells in its stem cell state [80]. These studies have shown that there could be a possible crosstalk between the EMT process and CSC phenotype.
IMPORTANCE OF TUMOR MICROENVIRONMENT IN EMT
Crosstalk between tumor cells and tumor microenvironment (TME) has been acknowledged to play an essential role in tumor progression. Tumor microenvironment comprises of stroma and non-cancerous cells. There is a significant involvement of TME through its key components such as immune cells, non-immune cells, hypoxia and extracellular matrix in driving EMT process (Fig. 4). Diverse populations of cells in stromal compartment are essential for inducing EMT phenotype. A number of studies have shown the participation of these microenvironment elements in EMT process. Among the immune cells, tumor associated macrophages (TAMs) stimulate tumor growth and progression through secretion of various growth factors and proteases. Jing et al., using co-culture studies have shown that macrophages enhance EMT of tumor cells through secretion of TNFα. Also mesenchymal stem cells (MSCs), multipotent stromal cells are actively recruited to the microenvironment of hepatocellular and breast carcinoma and have been found to induce EMT phenotype [7]. More importantly, pancreatic stellate cells, a major source for contributing to pancreatic desmoplasia also promotes EMT in pancreatic cancer cells [9]. Furthermore, another important stromal fraction such as cancer associated fibroblasts (CAFs) also communicates with cancer cells through secretion of various secretory factors. In contrast to normal fibroblasts which play a role in epithelial homeostasis, CAFs have been found to promote tumor growth [10] through secretion of MMP-2 and MMP-9. CAFs can induce EMT in various cancer models [8] and can also activate EMT by engaging transcription factors such as NFκB and HIF-1 and transcriptional regulation of an inducible enzyme COX-2 in various cancer cells [88].
Fig. 4. Diverse signaling from tumor microenvironment inducing EMT.

Key components of tumor microenvironments such as stellate cells, cancer associated fibroblasts, macrophages, hypoxia, cytokines, growth factors, MMPs and extracellular matrix is majorly driving EMT process.
Apart from the cellular population, various cytokines which can be autocrine or paracrine in origin comprise tumor microenvironment. Some of these cytokines like IL-6, TGF-β and TNFα in the inflammatory tumor microenvironment are also involved in facilitating EMT. IL-6, an inflammatory cytokine was shown to induce epithelial to mesenchymal transition in breast cancer cells [89]. TGF-β is an important cytokine produced by myeloid, mesenchymal and other cells which have also been shown to be an important player in inducing EMT in embryonic development as well as in tumor progression [90]. Also TNFα, a cytokine involved in systemic inflammation is secreted primarily by macrophages but can also be produced by variety of other cell types like fibroblasts, smooth muscle cells and tumor cells. Various studies have demonstrated the role of TNFα in EMT initiation. One of the studies have shown that TNFα can enhance EMT by increasing transcriptional repressors such as Slug and ZEB1 which further down-regulates E-cadherin and promotes Vimentin expression [91]. TNFα has also been shown to accelerate TGF-β induced EMT in human colorectal cancer cells [91]. Nevertheless, hypoxia is another pathological condition that arises in tumor microenvironment and is linked to EMT through notch signaling pathway [92]. The surrounding extracellular matrix components like MMPs and other proteins play crucial roles in inducing EMT phenotype. Stromelysin-1/MMP-3 also plays a major role in EMT by inducing expression of Rac1b, an activated splice isoform which causes an increase in cellular reactive oxygen species which stimulate cell invasion by altering gene expression [93]. Increased level of Fibronectin, stromal extracellular matrix protein has been found to induce EMT in mammary tumors through interaction with epithelial surface integrins [94]. Interestingly, endotrophin, processing product of COL6 released by stromal adipocytes in mammary tumor microenvironment is also found to stimulate TGF-β dependent EMT initiation [94]. Hence most of the cellular and non-cellular components of tumor microenvironment interact with tumor cells thereby triggering EMT associated molecular and morphological changes.
CONCLUSION AND PERSPECTIVES
The mucin glycoproteins, secreted by epithelial carcinoma cells are the initiators and key modulators of various proliferative signals including the signaling pathways activated for the EMT process. In general, an epithelial cell secretes mucin and other extracellular matrix proteins for their protective function against injurious insult. However, in case of cancer initiation or tumor progression these mucins and their associated proteins are abnormally expressed, to an extent that they can also influence the epithelial tissue integrity by disassembling and remodeling the epithelial junction during the EMT process. MUC1 influences EMT pathway through SNAIL, SLUG and ZEB-1 transcription factors whereas, MUC4 regulates FAK and c-JUN pathways. Among various signaling pathways and transcription factors that decide the EMT and MET process, the exact mechanism behind the activation of two processes in normal and diseased condition is elusive. Recent reports have demonstrated that mucin targeting approaches through MUC1 aptamer and miRNA mediated effect on mucin expression such as miR-125b, miRNA-145, miR200C and miR-1226 have been detained in the reviews [60, 62, 95]. Taken together, all the research approaches discussed were predominantly based on the studies in pancreatic, breast and ovarian cancer. It would be interesting to observe if these results on MUC1 and MUC4 mucin might have the same effect on other cancers, upon which we can focus the FAK pathway for targeting MUC4, and SNAIL, SLUG and ZEB1 for MUC1 to identify a novel targeted therapeutic interventional approach for mucins in cancer progression and EMT initiation. On the other hand, using an in vitro ovarian cancer cell model system, MUC16 mucin was also shown to be associated with EMT process. However, MUC16 promoter sequence and mechanism of activation is not fully understood like other mucins. It is believed that MUC16 promoter characterization will enable us to predict the EMT related regulation associated with MUC16. Thus targeting signaling molecules activated by MUC16 would be an efficient strategy to abrogate EMT phenotype and cancer progression.
The molecular mechanisms that regulate EMT transcription factors and the biological features that they exhibit over CSCs should be explored for designing novel therapeutic strategies. It can predominantly target the regulatory mechanism that controls the EMT process which in turn will control the CSC generation. Thus, a better understanding of the behavior of CSCs that underlie metastasis will further have profound implications in treatment of cancer. Expression of tumor specific mucin antigen may play an important role in CSCs and in the future to devise novel approaches for therapeutic intervention and a better management of this lethal disease. Several factors from tumor microenvironment such as macrophages, stellate cells, hypoxia, CAFs, cytokines/growth factors and ECM/MMP molecules plays a major role in the epithelial to mesenchymal cellular transformation. Understanding further mechanisms by which different components of tumor microenvironment mediate EMT will help us in designing efficacious therapeutics against various cancers. Overall, how transcription factors related to mucins and other downstream signaling activation is still not well understood. Opening the black box of mucin associated EMT initiation will reveal the specific signatures associated with various cancers, as mucins are cancer specific tumor antigen. The best examples are as follows: as MUC1 is expressed majorly in breast cancer, MUC4 in pancreatic, MUC5AC in lung cancer and MUC16 in ovarian cancer. These efforts to explore the mucin associated EMT phenotype transformation will fragment the EMT and MET process towards mucin based therapeutic interventions.
Acknowledgments
The authors on this article were, in part, supported by grants from the National Institutes of Health (EDRN UO1 CA111294, SPORE P50 CA127297, TMEN U54 CA163120, RO1 CA133774 and RO3 CA176342) and the Elsa U. Pardee Foundation (2013). The authors acknowledge the invaluable technical support from Ms. Kavita Mallya.
ABBREVIATIONS
- EMT
Epithelial to mesenchymal transition
- CSC
Cancer stem cells
- TME
tumor microenvironment
- OCSC
Ovarian cancer stem cells
- SFRP1
Secreted Frizzled-related protein 1
- FAK
focal adhesion kinase
- FGFR1
fibroblast growth factor receptor 1
- SRF
Serum response factor
- OSE
Ovarian Surface Epithelium
- ESCC
esophageal squamous cell carcinoma
- MSCs
mesenchymal stem cells
- TAMs
tumor associated macrophages
- CAFs
cancer associated fibroblasts
Footnotes
CONFLICT OF INTEREST
The author(s) confirm that this article content has no conflict of interest.
References
- 1.Hollingsworth MA, Swanson BJ. Mucins in cancer: protection and control of the cell surface. Nat Rev Cancer. 2004;4:45–60. doi: 10.1038/nrc1251. [DOI] [PubMed] [Google Scholar]
- 2.Chaturvedi P, Singh AP, Batra SK. Structure, evolution, and biology of the MUC4 mucin. FASEB J. 2008;22:966–981. doi: 10.1096/fj.07-9673rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ponnusamy MP, Seshacharyulu P, Vaz A, Dey P, Batra SK. MUC4 stabilizes HER2 expression and maintains the cancer stem cell population in ovarian cancer cells. J Ovarian Res. 2011;4:7. doi: 10.1186/1757-2215-4-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bafna S, Kaur S, Momi N, Batra SK. Pancreatic cancer cells resistance to gemcitabine: the role of MUC4 mucin. Br J Cancer. 2009;101:1155–1161. doi: 10.1038/sj.bjc.6605285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mimeault M, Johansson SL, Senapati S, Momi N, Chakraborty S, Batra SK. MUC4 down-regulation reverses chemoresistance of pancreatic cancer stem/progenitor cells and their progenies. Cancer Lett. 2010;295:69–84. doi: 10.1016/j.canlet.2010.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Engelmann K, Shen H, Finn OJ. MCF7 side population cells with characteristics of cancer stem/progenitor cells express the tumor antigen MUC1. Cancer Res. 2008;68:2419–2426. doi: 10.1158/0008-5472.CAN-07-2249. [DOI] [PubMed] [Google Scholar]
- 7.Jing Y, Han Z, Liu Y, Sun K, Zhang S, Jiang G, Li R, Gao L, Zhao X, Wu D, Cai X, Wu M, Wei L. Mesenchymal stem cells in inflammation microenvironment accelerates hepatocellular carcinoma metastasis by inducing epithelial-mesenchymal transition. PLoS One. 2012;7:e43272. doi: 10.1371/journal.pone.0043272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401. doi: 10.1038/nrc1877. [DOI] [PubMed] [Google Scholar]
- 9.Kikuta K, Masamune A, Watanabe T, Ariga H, Itoh H, Hamada S, Satoh K, Egawa S, Unno M, Shimosegawa T. Pancreatic stellate cells promote epithelial-mesenchymal transition in pancreatic cancer cells. Biochem Biophys Res Commun. 2010;403:380–384. doi: 10.1016/j.bbrc.2010.11.040. [DOI] [PubMed] [Google Scholar]
- 10.Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–348. doi: 10.1016/j.cell.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 11.Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, Garcia De HA. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- 12.Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, Portillo F, Nieto MA. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- 13.Hajra KM, Chen DY, Fearon ER. The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res. 2002;62:1613–1618. [PubMed] [Google Scholar]
- 14.Comijn J, Berx G, Vermassen P, Verschueren K, van GL, Bruyneel E, Mareel M, Huylebroeck D, van RF. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol Cell. 2001;7:1267–1278. doi: 10.1016/s1097-2765(01)00260-x. [DOI] [PubMed] [Google Scholar]
- 15.Eger A, Aigner K, Sonderegger S, Dampier B, Oehler S, Schreiber M, Berx G, Cano A, Beug H, Foisner R. DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene. 2005;24(14):2375–2385. doi: 10.1038/sj.onc.1208429. [DOI] [PubMed] [Google Scholar]
- 16.Wang X, Zheng M, Liu G, Xia W, McKeown-Longo PJ, Hung MC, Zhao J. Kruppel-like factor 8 induces epithelial to mesenchymal transition and epithelial cell invasion. Cancer Res. 2007;67:7184–7193. doi: 10.1158/0008-5472.CAN-06-4729. [DOI] [PubMed] [Google Scholar]
- 17.Perez-Moreno MA, Locascio A, Rodrigo I, Dhondt G, Portillo F, Nieto MA, Cano A. A new role for E12/E47 in the repression of E-cadherin expression and epithelial-mesenchymal transitions. J Biol Chem. 2001;276:27424–27431. doi: 10.1074/jbc.M100827200. [DOI] [PubMed] [Google Scholar]
- 18.Fernando RI, Litzinger M, Trono P, Hamilton DH, Schlom J, Palena C. The T-box transcription factor Brachyury promotes epithelial-mesenchymal transition in human tumor cells. J Clin Invest. 2010;120:533–544. doi: 10.1172/JCI38379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De CB, Gilbert B, Stove C, Bruyneel E, van RF, Berx G. The transcription factor snail induces tumor cell invasion through modulation of the epithelial cell differentiation program. Cancer Res. 2005;65:6237–6244. doi: 10.1158/0008-5472.CAN-04-3545. [DOI] [PubMed] [Google Scholar]
- 20.Moreno-Bueno G, Cubillo E, Sarrio D, Peinado H, Rodriguez-Pinilla SM, Villa S, Bolos V, Jorda M, Fabra A, Portillo F, Palacios J, Cano A. Genetic profiling of epithelial cells expressing E-cadherin repressors reveals a distinct role for Snail, Slug, and E47 factors in epithelial-mesenchymal transition. Cancer Res. 2006;66:9543–9556. doi: 10.1158/0008-5472.CAN-06-0479. [DOI] [PubMed] [Google Scholar]
- 21.Vandewalle C, Comijn J, De CB, Vermassen P, Bruyneel E, Andersen H, Tulchinsky E, van RF, Berx G. SIP1/ZEB2 induces EMT by repressing genes of different epithelial cell-cell junctions. Nucleic Acids Res. 2005;33:6566–6578. doi: 10.1093/nar/gki965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A, Weinberg RA. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 23.McCoy EL, Iwanaga R, Jedlicka P, Abbey NS, Chodosh LA, Heichman KA, Welm AL, Ford HL. Six1 expands the mouse mammary epithelial stem/progenitor cell pool and induces mammary tumors that undergo epithelial-mesenchymal transition. J Clin Invest. 2009;119:2663–2677. doi: 10.1172/JCI37691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mani SA, Yang J, Brooks M, Schwaninger G, Zhou A, Miura N, Kutok JL, Hartwell K, Richardson AL, Weinberg RA. Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc Natl Acad Sci USA. 2007;104:10069–10074. doi: 10.1073/pnas.0703900104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sobrado VR, Moreno-Bueno G, Cubillo E, Holt LJ, Nieto MA, Portillo F, Cano A. The class I bHLH factors E2-2A and E2-2B regulate EMT. J Cell Sci. 2009;122:1014–1024. doi: 10.1242/jcs.028241. [DOI] [PubMed] [Google Scholar]
- 26.Ocana OH, Corcoles R, Fabra A, Moreno-Bueno G, Acloque H, Vega S, Barrallo-Gimeno A, Cano A, Nieto MA. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell. 2012;22:709–724. doi: 10.1016/j.ccr.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 27.Frisch SM. The epithelial cell default-phenotype hypothesis and its implications for cancer. Bioessays. 1997;19:705–709. doi: 10.1002/bies.950190811. [DOI] [PubMed] [Google Scholar]
- 28.Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283:14910–14914. doi: 10.1074/jbc.C800074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahn SM, Cha JY, Kim J, Kim D, Trang HT, Kim YM, Cho YH, Park D, Hong S. Smad3 regulates E-cadherin via miRNA-200 pathway. Oncogene. 2012;31:3051–3059. doi: 10.1038/onc.2011.484. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Z, Liu S, Shi R, Zhao G. miR-27 promotes human gastric cancer cell metastasis by inducing epithelial-to-mesenchymal transition. Cancer Genet. 2011;204:486–491. doi: 10.1016/j.cancergen.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 31.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 32.Koinuma D, Tsutsumi S, Kamimura N, Imamura T, Aburatani H, Miyazono K. Promoter-wide analysis of Smad4 binding sites in human epithelial cells. Cancer Sci. 2009;100:2133–2142. doi: 10.1111/j.1349-7006.2009.01299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tian M, Neil JR, Schiemann WP. Transforming growth factor-beta and the hallmarks of cancer. Cell Signal. 2011;23:951–962. doi: 10.1016/j.cellsig.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leptin M. TWIST and SNAIL as positive and negative regulators during Drosophila mesoderm development. Genes Dev. 1991;5:1568–1576. doi: 10.1101/gad.5.9.1568. [DOI] [PubMed] [Google Scholar]
- 35.Wendt MK, Tian M, Schiemann WP. Deconstructing the mechanisms and consequences of TGF-beta-induced EMT during cancer progression. Cell Tissue Res. 2012;347:85–101. doi: 10.1007/s00441-011-1199-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.D’Inzeo S, Nicolussi A, Donini CF, Zani M, Mancini P, Nardi F, Coppa A. A novel human Smad4 mutation is involved in papillary thyroid carcinoma progression. Endocr Relat Cancer. 2012;19:39–55. doi: 10.1530/ERC-11-0233. [DOI] [PubMed] [Google Scholar]
- 37.Araki K, Shimura T, Suzuki H, Tsutsumi S, Wada W, Yajima T, Kobayahi T, Kubo N, Kuwano H. E/N-cadherin switch mediates cancer progression via TGF-beta-induced epithelial-to-mesenchymal transition in extrahepatic cholangiocarcinoma. Br J Cancer. 2011;105:1885–1893. doi: 10.1038/bjc.2011.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yook JI, Li XY, Ota I, Hu C, Kim HS, Kim NH, Cha SY, Ryu JK, Choi YJ, Kim J, Fearon ER, Weiss SJ. A Wnt-Axin2-GSK3beta cascade regulates Snail1 activity in breast cancer cells. Nat Cell Biol. 2006;8:1398–1406. doi: 10.1038/ncb1508. [DOI] [PubMed] [Google Scholar]
- 39.Gauger KJ, Chenausky KL, Murray ME, Schneider SS. SFRP1 reduction results in an increased sensitivity to TGF-beta signaling. BMC Cancer. 2011;11:59–11. doi: 10.1186/1471-2407-11-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kwon CY, Kim KR, Choi HN, Chung MJ, Noh SJ, Kim DG, Kang MJ, Lee DG, Moon WS. The role of serum response factor in hepatocellular carcinoma: implications for disease progression. Int J Oncol. 2010;37:837–844. doi: 10.3892/ijo_00000734. [DOI] [PubMed] [Google Scholar]
- 41.Doble BW, Woodgett JR. Role of glycogen synthase kinase-3 in cell fate and epithelial-mesenchymal transitions. Cells Tissues Organs. 2007;185:73–84. doi: 10.1159/000101306. [DOI] [PubMed] [Google Scholar]
- 42.Heuberger J, Birchmeier W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb Perspect Biol. 2010;2:a002915. doi: 10.1101/cshperspect.a002915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bolos V, Grego-Bessa J, de la Pompa JL. Notch signaling in development and cancer. Endocr Rev. 2007;28:339–363. doi: 10.1210/er.2006-0046. [DOI] [PubMed] [Google Scholar]
- 44.Sharma A, Paranjape AN, Rangarajan A, Dighe RR. A monoclonal antibody against human Notch1 ligand-binding domain depletes subpopulation of putative breast cancer stem-like cells. Mol Cancer Ther. 2012;11:77–86. doi: 10.1158/1535-7163.MCT-11-0508. [DOI] [PubMed] [Google Scholar]
- 45.Wang Z, Li Y, Ahmad A, Azmi AS, Banerjee S, Kong D, Sarkar FH. Targeting Notch signaling pathway to overcome drug resistance for cancer therapy. Biochim Biophys Acta. 2010;1806:258–267. doi: 10.1016/j.bbcan.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Timmerman LA, Grego-Bessa J, Raya A, Bertran E, Perez-Pomares JM, Diez J, Aranda S, Palomo S, McCormick F, Izpisua-Belmonte JC, de la Pompa JL. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004;18:99–115. doi: 10.1101/gad.276304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bao B, Wang Z, Ali S, Kong D, Li Y, Ahmad A, Banerjee S, Azmi AS, Miele L, Sarkar FH. Notch-1 induces epithelial-mesenchymal transition consistent with cancer stem cell phenotype in pancreatic cancer cells. Cancer Lett. 2011;307:26–36. doi: 10.1016/j.canlet.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48.Brabletz S, Bajdak K, Meidhof S, Burk U, Niedermann G, Firat E, Wellner U, Dimmler A, Faller G, Schubert J, Brabletz T. The ZEB1/miR-200 feedback loop controls Notch signalling in cancer cells. EMBO J. 2011;30:770–782. doi: 10.1038/emboj.2010.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang Y, Ahn YH, Gibbons DL, Zang Y, Lin W, Thilaganathan N, Alvarez CA, Moreira DC, Creighton CJ, Gregory PA, Goodall GJ, Kurie JM. The Notch ligand Jagged2 promotes lung adenocarcinoma metastasis through a miR-200-dependent pathway in mice. J Clin Invest. 2011;121:1373–1385. doi: 10.1172/JCI42579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Louro ID, Bailey EC, Li X, South LS, McKie-Bell PR, Yoder BK, Huang CC, Johnson MR, Hill AE, Johnson RL, Ruppert JM. Comparative gene expression profile analysis of GLI and c-MYC in an epithelial model of malignant transformation. Cancer Res. 2002;62:5867–5873. [PubMed] [Google Scholar]
- 51.Maitah MY, Ali S, Ahmad A, Gadgeel S, Sarkar FH. Up-regulation of sonic hedgehog contributes to TGF-beta1-induced epithelial to mesenchymal transition in NSCLC cells. PLoS One. 2011;6:e16068. doi: 10.1371/journal.pone.0016068. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Yoo YA, Kang MH, Lee HJ, Kim BH, Park JK, Kim HK, Kim JS, Oh SC. Sonic hedgehog pathway promotes metastasis and lymphangiogenesis via activation of Akt, EMT, and MMP-9 pathway in gastric cancer. Cancer Res. 2011;71:7061–7070. doi: 10.1158/0008-5472.CAN-11-1338. [DOI] [PubMed] [Google Scholar]
- 53.Joost S, Almada LL, Rohnalter V, Holz PS, Vrabel AM, Fernandez-Barrena MG, McWilliams RR, Krause M, Fernandez-Zapico ME, Lauth M. GLI1 inhibition promotes epithelial-to-mesenchymal transition in pancreatic cancer cells. Cancer Res. 2012;72:88–99. doi: 10.1158/0008-5472.CAN-10-4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bafna S, Singh AP, Moniaux N, Eudy JD, Meza JL, Batra SK. MUC4, a multifunctional transmembrane glycoprotein, induces oncogenic transformation of NIH3T3 mouse fibroblast cells. Cancer Res. 2008;68:9231–9238. doi: 10.1158/0008-5472.CAN-08-3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chaturvedi P, Singh AP, Chakraborty S, Chauhan SC, Bafna S, Meza JL, Singh PK, Hollingsworth MA, Mehta PP, Batra SK. MUC4 mucin interacts with and stabilizes the HER2 oncoprotein in human pancreatic cancer cells. Cancer Res. 2008;68:2065–2070. doi: 10.1158/0008-5472.CAN-07-6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mukhopadhyay P, Lakshmanan I, Ponnusamy MP, Chakraborty S, Jain M, Pai P, Smith LM, Lele SM, Batra SK. MUC4 overexpression augments cell migration and metastasis through EGFR family proteins in triple negative breast cancer cells. PLoS One. 2013;8:e54455. doi: 10.1371/journal.pone.0054455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ponnusamy MP, Singh AP, Jain M, Chakraborty S, Moniaux N, Batra SK. MUC4 activates HER2 signalling and enhances the motility of human ovarian cancer cells. Br J Cancer. 2008;99:520–526. doi: 10.1038/sj.bjc.6604517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ponnusamy MP, Lakshmanan I, Jain M, Das S, Chakraborty S, Dey P, Batra SK. MUC4 mucin-induced epithelial to mesenchymal transition: a novel mechanism for metastasis of human ovarian cancer cells. Oncogene. 2010;29:5741–5754. doi: 10.1038/onc.2010.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rachagani S, Macha MA, Ponnusamy MP, Haridas D, Kaur S, Jain M, Batra SK. MUC4 potentiates invasion and metastasis of pancreatic cancer cells through stabilization of fibroblast growth factor receptor 1. Carcinogenesis. 2012;33:1953–1964. doi: 10.1093/carcin/bgs225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Deng J, Wang L, Chen H, Li L, Ma Y, Ni J, Li Y. The role of tumour-associated MUC1 in epithelial ovarian cancer metastasis and progression. Cancer Metastasis Rev. 2013 doi: 10.1007/s10555-013-9423-y. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 61.Rajabi H, Jin C, Ahmad R, McClary C, Joshi MD, Kufe D. Mucin 1 Oncoprotein Expression Is Suppressed By The Mir-125b Oncomir. Genes Cancer. 2010;1:62–68. doi: 10.1177/1947601909357933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rajabi H, Alam M, Takahashi H, Kharbanda A, Guha M, Ahmad R, Kufe D. MUC1-C oncoprotein activates the ZEB1/miR-200c regulatory loop and epithelial-mesenchymal transition. Oncogene. 2013:10. doi: 10.1038/onc.2013.114. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roy LD, Sahraei M, Subramani DB, Besmer D, Nath S, Tinder TL, Bajaj E, Shanmugam K, Lee YY, Hwang SI, Gendler SJ, Mukherjee P. MUC1 enhances invasiveness of pancreatic cancer cells by inducing epithelial to mesenchymal transition. Oncogene. 2011;30:1449–1459. doi: 10.1038/onc.2010.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Horn G, Gaziel A, Wreschner DH, Smorodinsky NI, Ehrlich M. ERK and PI3K regulate different aspects of the epithelial to mesenchymal transition of mammary tumor cells induced by truncated MUC1. Exp Cell Res. 2009;315:1490–1504. doi: 10.1016/j.yexcr.2009.02.011. [DOI] [PubMed] [Google Scholar]
- 65.Rajabi H, Ahmad R, Jin C, Joshi MD, Guha M, Alam M, Kharbanda S, Kufe D. MUC1-C oncoprotein confers androgen-independent growth of human prostate cancer cells. Prostate. 2012;72:1659–1668. doi: 10.1002/pros.22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Majhi PD, Lakshmanan I, Ponnusamy MP, Jain M, Das S, Kaur S, Shimizu ST, West WW, Johansson SL, Smith LM, Yu F, Rolle CE, Sharma P, Carey GB, Batra SK, Ganti AK. Pathobiological implications of MUC4 in non-small-cell lung cancer. J Thorac Oncol. 2013;8:398–407. doi: 10.1097/JTO.0b013e3182829e06. [DOI] [PubMed] [Google Scholar]
- 67.Comamala M, Pinard M, Theriault C, Matte I, Albert A, Boivin M, Beaudin J, Piche A, Rancourt C. Downregulation of cell surface CA125/MUC16 induces epithelial-to-mesenchymal transition and restores EGFR signalling in NIH:OVCAR3 ovarian carcinoma cells. Br J Cancer. 2011;104:989–999. doi: 10.1038/bjc.2011.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Theriault C, Pinard M, Comamala M, Migneault M, Beaudin J, Matte I, Boivin M, Piche A, Rancourt C. MUC16 (CA125) regulates epithelial ovarian cancer cell growth, tumorigenesis and metastasis. Gynecol Oncol. 2011;121:434–443. doi: 10.1016/j.ygyno.2011.02.020. [DOI] [PubMed] [Google Scholar]
- 69.Bast RC, Jr, Spriggs DR. More than a biomarker: CA125 may contribute to ovarian cancer pathogenesis. Gynecol Oncol. 2011;121:429–430. doi: 10.1016/j.ygyno.2011.04.032. [DOI] [PubMed] [Google Scholar]
- 70.Gubbels JA, Belisle J, Onda M, Rancourt C, Migneault M, Ho M, Bera TK, Connor J, Sathyanarayana BK, Lee B, Pastan I, Patankar MS. Mesothelin-MUC16 binding is a high affinity, N-glycan dependent interaction that facilitates peritoneal metastasis of ovarian tumors. Mol Cancer. 2006;5:50. doi: 10.1186/1476-4598-5-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bara J, Chastre E, Mahiou J, Singh RL, Forgue-Lafitte ME, Hollande E, Godeau F. Gastric M1 mucin, an early oncofetal marker of colon carcinogenesis, is encoded by the MUC5AC gene. Int J Cancer. 1998;75:767–773. doi: 10.1002/(sici)1097-0215(19980302)75:5<767::aid-ijc17>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 72.Lesuffleur T, Kornowski A, Augeron C, Dussaulx E, Barbat A, Laboisse C, Zweibaum A. Increased growth adaptability to 5-fluorouracil and methotrexate of HT-29 sub-populations selected for their commitment to differentiation. Int J Cancer. 1991;49:731–737. doi: 10.1002/ijc.2910490517. [DOI] [PubMed] [Google Scholar]
- 73.Sylvester PA, Myerscough N, Warren BF, Carlstedt I, Corfield AP, Durdey P, Thomas MG. Differential expression of the chromosome 11 mucin genes in colorectal cancer. J Pathol. 2001;195:327–335. doi: 10.1002/path.951. [DOI] [PubMed] [Google Scholar]
- 74.Inaguma S, Kasai K, Ikeda H. GLI1 facilitates the migration and invasion of pancreatic cancer cells through MUC5AC-mediated attenuation of E-cadherin. Oncogene. 2011;30:714–723. doi: 10.1038/onc.2010.459. [DOI] [PubMed] [Google Scholar]
- 75.Yamazoe S, Tanaka H, Sawada T, Amano R, Yamada N, Ohira M, Hirakawa K. RNA interference suppression of mucin 5AC (MUC5AC) reduces the adhesive and invasive capacity of human pancreatic cancer cells. J Exp Clin Cancer Res. 2010;23:29, 53. doi: 10.1186/1756-9966-29-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Truant S, Bruyneel E, Gouyer V, De WO, Pruvot FR, Mareel M, Huet G. Requirement of both mucins and proteoglycans in cell-cell dissociation and invasiveness of colon carcinoma HT-29 cells. Int J Cancer. 2003;104:683–694. doi: 10.1002/ijc.11011. [DOI] [PubMed] [Google Scholar]
- 77.Valque H, Gouyer V, Gottrand F, Desseyn JL. MUC5B leads to aggressive behavior of breast cancer MCF7 cells. PLoS One. 2012;7:e46699. doi: 10.1371/journal.pone.0046699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Leir SH, Harris A. MUC6 mucin expression inhibits tumor cell invasion. Exp Cell Res. 2011;317:2408–2419. doi: 10.1016/j.yexcr.2011.07.021. [DOI] [PubMed] [Google Scholar]
- 79.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–4751. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–715. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D, zur HA, Brunton VG, Morton J, Sansom O, Schuler J, Stemmler MP, Herzberger C, Hopt U, Keck T, Brabletz S, Brabletz T. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487–1495. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
- 82.Tirino V, Camerlingo R, Bifulco K, Irollo E, Montella R, Paino F, Sessa G, Carriero MV, Normanno N, Rocco G, Pirozzi G. TGF-beta1 exposure induces epithelial to mesenchymal transition both in CSCs and non-CSCs of the A549 cell line, leading to an increase of migration ability in the CD133(+) A549 cell fraction. Cell Death Dis. 2013;4:e620. doi: 10.1038/cddis.2013.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gassenmaier M, Chen D, Buchner A, Henkel L, Schiemann M, Mack B, Schendel DJ, Zimmermann W, Pohla H. CXC chemokine receptor 4 is essential for maintenance of renal cell carcinoma-initiating cells and predicts metastasis. Stem Cells. 2013;31(8):1467–1476. doi: 10.1002/stem.1407. [DOI] [PubMed] [Google Scholar]
- 84.Hiraga T, Nakajima T, Ozawa H. Bone resorption induced by a metastatic human melanoma cell line. Bone. 1995;16:349–356. doi: 10.1016/8756-3282(94)00048-4. [DOI] [PubMed] [Google Scholar]
- 85.Hiraga T, Nakamura H. Imatinib mesylate suppresses bone metastases of breast cancer by inhibiting osteoclasts through the blockade of c-Fms signals. Int J Cancer. 2009;124:215–222. doi: 10.1002/ijc.23903. [DOI] [PubMed] [Google Scholar]
- 86.Iguchi H, Tanaka S, Ozawa Y, Kashiwakuma T, Kimura T, Hiraga T, Ozawa H, Kono A. An experimental model of bone metastasis by human lung cancer cells: the role of parathyroid hormone-related protein in bone metastasis. Cancer Res. 1996;56:4040–4043. [PubMed] [Google Scholar]
- 87.Giudice FS, Pinto DS, Jr, Nor JE, Squarize CH, Castilho RM. Inhibition of histone deacetylase impacts cancer stem cells and induces epithelial-mesenchyme transition of head and neck cancer. PLoS One. 2013;8:e58672. doi: 10.1371/journal.pone.0058672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Giannoni E, Bianchini F, Calorini L, Chiarugi P. Cancer associated fibroblasts exploit reactive oxygen species through a proinflammatory signature leading to epithelial mesenchymal transition and stemness. Antioxid Redox Signal. 2011;14:2361–2371. doi: 10.1089/ars.2010.3727. [DOI] [PubMed] [Google Scholar]
- 89.Sullivan NJ, Sasser AK, Axel AE, Vesuna F, Raman V, Ramirez N, Oberyszyn TM, Hall BM. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene. 2009;28:2940–2947. doi: 10.1038/onc.2009.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zavadil J, Bottinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764–5774. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- 91.Bates RC, Mercurio AM. Tumor necrosis factor-alpha stimulates the epithelial-to-mesenchymal transition of human colonic organoids. Mol Biol Cell. 2003;14:1790–1800. doi: 10.1091/mbc.E02-09-0583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci USA. 2008;105:6392–6397. doi: 10.1073/pnas.0802047105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Radisky DC, Levy DD, Littlepage LE, Liu H, Nelson CM, Fata JE, Leake D, Godden EL, Albertson DG, Nieto MA, Werb Z, Bissell MJ. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature. 2005;436:123–127. doi: 10.1038/nature03688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Park J, Schwarzbauer JE. Mammary epithelial cell interactions with fibronectin stimulate epithelial-mesenchymal transition. Oncogene. 2013 doi: 10.1038/onc.2013.118. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yamada N, Kitamoto S, Yokoyama S, Hamada T, Goto M, Tsutsumida H, Higashi M, Yonezawa S. Epigenetic regulation of mucin genes in human cancers. Clin Epigenetics. 2011;2:85–96. doi: 10.1007/s13148-011-0037-3. [DOI] [PMC free article] [PubMed] [Google Scholar]