

Abstract
Though long-term administration of proton pump inhibitor (PPI) imposed the risk of gastrointestinal track tumorigenesis by accompanied hypergastrinemia, no overt increases of colon cancer risk were witnessed after a long-term cohort study. Our recent investigation revealed that PPI prevented colitis-associated carcinogenesis through anti-inflammatory, anti-oxidative, and anti-mutagenic mechanisms in spite of hypergastrinemia. Therefore, we hypothesized that PPI might either antagonize the trophic action of gastrin on gastrointestinal tumorigenesis or synergize to exert augmented anti-tumorigenic actions. We challenged APCMin/+ mice with gastrin, PPI, PPI and gastrin together for 10 weeks and counted intestinal polyposis accompanied with molecular changes. Gastrin significantly increased intestinal polyposis, but combination of PPI and gastrin markedly attenuated intestinal polyposis compared to gastrin-promoted APCMin/+ mice (P < .001), in which significant β-catenin phosphorylation and inhibition of β-catenin nuclear translocation were observed with PPI alone or combination of PPI and gastrin, whereas gastrin treatment significantly increased β-catenin nuclear translocation. Significant footprints of apoptosis, G0/G1 accumulation, inactivation of p38 and extracellular signal-regulated kinase, decreased expressions of CD31, and inhibition of tumor necrosis factor-α and cyclooxygenase-2 were noted in the combination group. In vitro investigations were similar to in vivo findings as shown that PPI treatment inhibited the binding of gastrin to its receptor, inactivated β-catenin-associated signaling including Tcf/Lef and glycogen synthase kinase β, and paradoxically inhibited β-catenin-associated proliferative activities. Our investigations explain why colon cancer risk has not increased despite long-term use of PPIs and provide a rationale for using PPI to achieve anti-tumorigenesis beyond acid suppression.
Introduction
Gastrin, which is produced by G cells in the gastric antrum, is fundamentally responsible for the stimulation of acid secretion from parietal cells but also acts as a potent cell growth factor, leading to either the maintenance of the gastric mucosa and proliferation of enterochromaffin-like cells or notably neoplastic transformation [1]. Gastrin is known to bind to the cholecystokinin B receptor (CCKBR), which mediates proliferative action through signal transduction pathways. Because not only colorectal cancer cells but also nontransformed colon epithelium cells express the CCKBR [2], gastrin and their receptors are regarded as potential risk for gastrointestinal cancer.
The development of a proton pump inhibitor (PPI) and its widespread prescription for blocking acid secretion lead to hypergastrinemia, which can increase the risk of colorectal cancer, particularly in patients with Zollinger-Ellison syndrome, pernicious anemia, and Helicobacter pylori infection [3]. However, a recent clinical study of a large-scale cohort and prospective investigation reported that hypergastrinemia following long-term PPI therapy at a regular dose is not associated with an increased risk of colorectal cancer [4]. This is quite in contrast to results from animal models of colon carcinogenesis, in which hypergastrinemia clearly increased the incidence and growth rate of an epithelial neoplasm [5]. As a plausible explanation of the unexpected no risk of colon cancer in spite of hypergastrinemia, two speculations were possible. One is the double-edged sword role of gastrin in tumorigenesis; gastrin suppresses the growth of CCKBR expressing colon cancer cell by inducing apoptosis in vitro and in vivo [6], whereas it has trophic effects on colon carcinogenesis [5]. The other is preventing action of PPI on hypergastrinemia-associated tumorigenesis. Therefore, the exact role of gastrin in the control of cancer cell growth remains controversial and requires further investigation.
PPIs are potent blockers of gastric acid secretion and are widely prescribed for treatment of acid peptic disorders [7]. However, PPIs have been found to have additional anti-oxidant properties and direct effects on neutrophils, monocytes, endothelial cells, and epithelial cells that might prevent inflammation beyond authentic gastric acid-suppressive action through blocking efflux of hydronium in parietal cells. In detail, PPIs may mediate anti-inflammatory effects through the inhibition of angiogenic growth factors and the expression of the intercellular adhesion molecule-1 and the vascular cell adhesion molecule-1, as well as the endothelial-dependent neutrophil adhesion [8], and induce selective induction of apoptosis in cancer cells.
Our recent investigation shows that long-term PPI treatment inhibited colitis-associated carcinogenesis, supported with selective cancer cell apoptosis, anti-oxidative action, and anti-inflammatory actions as well as regulation of angiogenic factors including vascular endothelial growth factors (VEGFs) [9]. However, the question arose as to how hypergastrinemia consequent to long-term PPI contributed to colon anti-tumorigenesis. On the basis of this publication, we hypothesized that PPI might impose the antagonistic role of hypergastrinemia or exert anti-tumorigenesis through the paradoxically reciprocal augmented action of anti-tumorigenesis with gastrin. The combination of PPI and gastrin contributed to the attenuation of intestinal polyposis either through inhibition of β-catenin-associated proliferation or gastrin-associated inflammation in vitro and in vivo APCMin/+ mice model.
Materials and Methods
Materials and Cell Culture
Pantoprazole among PPIs including omeprazole, lansoprazole, and rabeprazole, was chosen after a preliminary study that pantoprazole was the most stable in inhibiting proton pump activities and was purchased from Amore-Pacific Pharma (Seoul, Korea). Gastrin was purchased from Sigma-Aldrich (St Louis, MO). The HCT116 human colorectal cancer cell line was obtained from American Type Culture Collection (ATCC, Manassas, VA) several years ago. The HCT116 cell line has been tested and authenticated with short tandem repeat profiling method in Korean Cell Line Bank in January 2013. The information on the HCT116 cell line was provided in the Supplemental data. The HCT116 human colorectal cancer cell line was cultured in McCoy's 5A medium containing 10% (vol/vol) FBS and 100 U/ml penicillin. Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2.
Antibodies
β-Actin, α-tubulin, β-catenin, phospho-extracellular signal-regulated kinase (ERK), ERK, survivin, CCKBR, and cyclooxygenase-2 (COX-2) antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Cyclin D1, cyclin E, cyclin A, cyclin-dependent kinase (CDK) 2, CDK4, phospho-p38, p38, phospho-c-Jun N-terminal kinase (JNK), JNK, phospho-β-catenin, Bcl-2, cleaved caspase-3, cleaved caspase-8, poly(ADP-ribose) polymerase (PARP), and phospho-glycogen synthase kinase 3β (GSK3β) antibodies were purchased from Cell Signaling Technology (Denver, MA).
Cell Proliferation Assay
In a cell proliferation assay using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT; Sigma-Aldrich), cells were seeded into 96-well plates at 1 x 104 to 2 x 104 cells per well overnight before drugs were added. The cells were incubated with different concentrations of gastrin for 12 hours. Cells were incubated with MTT for 2 hours and read at an OD of 570 nm.
Cell Cycle Analysis and Apoptosis Analysis
Cells were treated with gastrin, a PPI, the combination of gastrin and a PPI, or phosphate-buffered saline (PBS) as a control. After trypsinization, cells were fixed in 70% ethanol for 30 minutes at 4°C. The cells were washed twice with PBS and incubated for 30 minutes in the dark at 37°C in 1 ml of PBS containing 100 µg of propidium iodide and 100 µg of RNase A. Flow cytometric analysis was completed, and the proportion of cells was assessed by the histograms generated using the computer program Mod-Fit. Apoptosis was analyzed by FACS using the Annexin V-FITC Apoptosis Detection Kit (BD Biosciences, Bergen, NJ).
Reverse Transcription-Polymerase Chain Reaction and Real-Time Polymerase Chain Reaction
After treatment, the medium was removed by suction, and cells were washed with Dulbecco's PBS twice. RiboEX (500 µl; GeneAll, Seoul, Korea) was added to plates that were incubated for 10 minutes at 4°C. RiboEX was harvested and placed in a 1.5-ml tube, and 100 µl of chloroform was added and gently mixed. After incubation for 10 minutes in ice, samples were centrifuged at 10,000g for 30 minutes. Supernatants were extracted and mixed with 200 µl of isopropanol, and mixtures were incubated at 4°C for 1 hour. After centrifuging at 13,000g for 30 minutes, the pellet was washed with 70% (vol/vol) ethanol. After allowing the ethanol to evaporate completely, the pellet was dissolved in 100 µl of diethylene pyrocarbonate-treated water (Invitrogen Life Technologies, Carlsbad, CA). cDNA was prepared using a reverse transcriptase originating from Murine-Moloney leukemia virus (Promega, Madison, WI), according to the manufacturer's instructions. The polymerase chain reaction (PCR) primers used are shown in Table 1. PCR was performed using over 30 cycles of 94°C for 20 seconds, 58°C for 30 seconds, and 72°C for 45 seconds. Oligonucleotide primers were purchased from Bioneer (Seoul, Korea).
Table 1.
Primer Sequence for PCR.
| Gene Name | Sequence |
| Glyceraldehyde-3-phosphate dehydrogenase | Forward, 5′-AGG TCG GAG TCA ACG GAT TTG G-3′ |
| Reverse, 5′-AA GTC TTC TGG GTG GCA GTG ATG-3′ | |
| IL-8 | Forward, 5′-TGG GTG CAG AGG GTT GTG-3′ |
| Reverse, 5′-CAG ACT AGG GTT GCC AGA TTT A-3′ | |
| HIF-1α | Forward, 5′-ACA GCA GCC AGA CGA TCA TGT-3′ |
| Reverse, 5′-AAA TGA GCT GTC TGT GAT CCA GCA TT-3′ | |
| VEGF | Forward, 5′-TCG GGC CTC CGA AAC CAT G-3′ |
| Reverse, 5′-GGT TCC CGA AAC CCT GAG G-3′ | |
| PDGF | Forward, 5′-AGG AAG CCA TTC CCG CAG TT-3′ |
| Reverse, 5′-CTA ACC TCA CCT GGA CCT CT-3′ | |
| COX-2 | Forward, 5′-GGT CTG GTG CCT GGT CTG ATG ATG-3′ |
| Reverse, 5′-GTC CTT TCA AGG AGA ATG GTG C-3′ | |
| Mouse TNF-α | Forward, 5′-CTG AGG TCA ATC TGC CCA AGT AC-3′ |
| Reverse, 5′-CTT CAC AGA GCA ATG ACT CCA AAG-3′ | |
| Mouse β-actin | Forward, 5′-GGT GGG AAT GGG TCA GAA GG-3′ |
| Reverse, 5′-TCA CGC ACG ATT TCC CTC TC-3′ |
ELISA and Sandwich ELISA
Small intestine tissues were homogenized with 1 ml of cell lysis buffer (Cell Signaling Technology) containing 1 mM PMSF, incubated for 20 minutes, and centrifuged at 10,000g for 10 minutes. Supernatants were recentrifuged and collected. All samples were stored at -80°C until required. Mouse tumor necrosis factor-α (TNF-α) and mouse prostaglandin E2 (PGE2; R&D Systems, Minneapolis, MN) ELISAs were performed according to the manufacturer's instructions.
The sandwich ELISA measures the amount of antigen between two layers of antibodies (a capture and a detection antibody). A 96-well polyvinyl chloride plate containing the capture antibody, anti-cholecystokinin 2 receptor (Santa Cruz Biotechnology), at a concentration of 10 µg/ml in carbonate/bicarbonate buffer (pH 7.4) was covered with an adhesive plastic and incubated overnight at 4°C. The coating solution was removed, and the plate was washed. The remaining protein-binding sites were blocked in the coated wells by incubation with 5% BSA buffer overnight at 4°C. Cells were treated with gastrin, a PPI, or the combination of PPI and gastrin, or PBS as a control. Samples were added to each well and incubated for 90 minutes at 37°C. The samples were removed, the plates were washed, and the detection antibody, anti-gastrin (biotin; ABcam, Cambridge, MA), was added to each well and incubated for 2 hours at room temperature. The plate was washed, and the secondary antibody, HRP-conjugated streptavidin, was added. The plate was incubated for 2 hours at room temperature and washed. A 3,3′,5,5′-Tetramethylbenzidine solution was added to each well, the plate was incubated for 15 to 30 minutes, an equal volume of stopping solution (2 M H2SO4) was added to the plate, and the plate was read at an OD of 450 nm.
Luciferase Reporter Activity Assay
Cultured cells were seeded at a concentration achieving 80% confluence in 12-well plates for 24 hours before transfection. The cells were transiently transfected with 0.2 µg/well of a translucent Tcf/Lef-Luc reporter vector, which was designed to measure the transcriptional activity of Tcf/Lef-responsive genes. After transfection, the cells were treated with specific reagents such as gastrin, PPI, or the combination of PPI and gastrin in serum-free medium at the indicated time periods. The cell lysates were then obtained with 1x reporter lysis buffer (Promega). The luciferase activity was assayed using a Victor2 luminometer (PerkinElmer, Waltham, MA). The relative luciferase activity was calculated after normalization of cellular proteins. All values are expressed as the percentage of activity relative to basal activity.
Immunohistochemical Staining
After paraffin blocks were dewaxed and rehydrated with graded alcohol, tissue sections were heated in pressure jars filled with 10 mM/l citrate buffer in a microwave for 10 minutes. Slides were cooled in water for 15 minutes and washed in PBS. The slides were incubated overnight with the primary antibody. After incubation, a subsequent reaction was formed using a VECTOR kit (Vector Laboratories, Inc, Burlingame, CA). Finally, the slides were incubated with 3,3′-diaminobenzidine (Invitrogen Life Technologies) and counter-stained with hematoxylin (Sigma-Aldrich).
Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick End Labeling Staining
Apoptosis was visualized using a terminal deoxynucleotidyl transferase (TdT) fRAGel DNA Fragmentation Detection Kit (Oncogene Research Products, La Jolla, CA). After routine deparaffinization, rehydration, and washing in PBS (pH 7.4), tissues were digested with proteinase K (20 µg/ml in PBS) for 20 minutes at room temperature and washed. Tissues were incubated in equilibration buffer for 10 minutes and treated with TdT enzyme at 37°C for 1 hour. To determine the apoptotic index in each group, TdT-mediated dUTP nick end labeling (TUNEL)-immunostained sections were scanned under low-power magnification (x100) to locate the apoptotic hotspots.
Animals and Study Protocol
C57BL/6J-APCMin/+ mice on a pure C57BL/6 background were obtained from Jackson Laboratories (Bar Harbor, ME). APCMin/+ mice were verified as previously described by PCR using tail DNA extracted. The sequences of PCR primers were recommended by Jackson Laboratories. Five-week-old male C57BL/6J-APCMin/+ mice were fed sterilized commercial pellet diets (Biogenomics, Seongnam, South Korea) and sterile water ad libitum and housed in an air-conditioned biohazard room at a temperature of 24°C. One group of eight mice (group 1) was given intraperitoneal (i.p.) injections of PBS (iNtRON Biotechnology, Seongnam, South Korea) three times per week for 10 weeks. The second group (group 2) was given i.p. injections of gastrin (10 µg/kg; Sigma-Aldrich) three times per week for 10 weeks. The third group (group 3) was given i.p. injections of PPI (pantoprazole, 8.0 mg/kg; Amore-Pacific Pharma) three times per week for 10 weeks. The fourth group (group 4) was given i.p. injections of gastrin and PPI three times per week for 10 weeks (Figure 1A). Among PPIs, including omeprazole, lansoprazole, rabeprazole, and pantoprazole, we choose pantoprazole because in the preliminary study, the highest stability of proton pump inhibiting action in an aqueous condition was seen in pantoprazole. Animals were handled in an accredited animal facility in accordance with international policies of the Association for Assessment and Accreditation of Laboratory Animal Care. Following killing, the small intestine was resected, opened longitudinally, and washed with sterile PBS. The small intestine was divided by length into three equal sections (proximal, middle, and distal segments). Polyps on the intestinal segments were counted, and their sizes were measured with a caliper.
Figure 1.
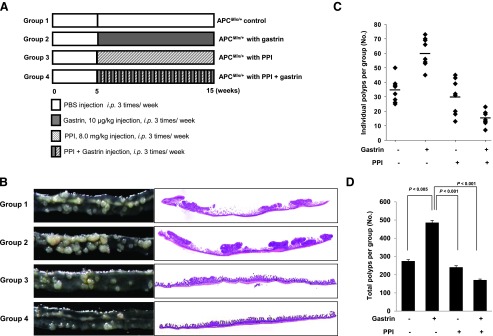
The gross morphology of intestinal polyposis according to group in an APCMin/+ mouse model. (A) The schematic overview of the experimental protocol of an animal model for small intestine polyposis using APCMin/+ mice. (B) Gross and microscopic pathologies according to group: control, gastrin, PPI alone, and the combination of PPI and gastrin. Representative gross and pathologic pictures (original magnification, x1). (C) The polyps in the small intestine of the individual mouse were counted according to group. Gastrin administration significantly increased the polyp number, but PPI treatment markedly decreased in groups 3 and 4. (D) Total polyps in the whole small intestine were counted according to group. Gastrin administration significantly increased polyp number (P < .005), but statistically significant decreases in polyp numbers were observed in groups 3 and 4 compared with group 2 (P < .001). Results are expressed as means ± SD (n = 8).
Statistical Analysis
All the experiments in this study except in vivo animal experiment were repeated more than thrice, and the results are expressed as the means ± SD. The data were analyzed by one-way analysis of variance, and the statistical significance between groups was determined by Duncan multiple range test. Statistical significance was accepted at P < .05.
Results
PPI Ameliorated Gastrin-Promoted Intestinal Polyposis in APCMin/+ Mice
APCMin/+ mice are known to develop multiple intestinal polyposis based on the accumulation of β-catenin owing to an apc gene mutation [10]. As shown in Figure 1, 15-week-old APCMin/+ mice developed multiple intestinal polyps, mostly on the small intestine. However, 10 µg/kg gastrin significantly accentuated intestinal polyposis (P < .005), confirming the trophic effect of gastrin on intestinal polyposis. A PPI alone (pantoprazole, 8 mg/kg) in the current study attenuated intestinal polyposis compared to the control and more significantly through the combination of PPI and gastrin (P < .001; Figure 1 and Table 2), signifying the augmented inhibitory action of intestinal polyposis with co-administration of PPI and gastrin. These results suggest that besides the direct anti-tumorigenic action of PPI, PPI might synergize with gastrin in inhibiting intestinal polyposis.
Table 2.
PPI Suppresses Intestinal Polyposis in the APCMin/+ Mouse.
| Group | Total | Proximal | Middle | Distal | |||
| >2 mm | <2 mm | >2 mm | <2 mm | >2 mm | <2 mm | ||
| Vehicle control (% control) | 275 ± 8.07 (100) | 2.1 ± 1.2 (100) | 1.1 ± 1.8 (100) | 5.8 ± 3.8 (100) | 8.1 ± 5.7 (100) | 6.4 ± 2.6 (100) | 10.9 ± 4.9 (100) |
| Gastrin (% control) | 486 ± 12.21* (176) | 5.4 ± 2.4 (257) | 1.1 ± 1.5* (100) | 14.9 ± 7.5 (256) | 11.7 ± 5.8* (144) | 14.2 ± 9.2 (221) | 11.2 ± 6.3† (102) |
| PPI (% control) | 241 ± 8.52‡ (87) | 2.1 ± 2.1 (100) | 0.6 ± 1.2§ (53) | 5.1 ± 3.8 (87) | 10.8 ± 6.7§ (79) | 6.4 ± 4.7¶ (100) | 5.1 ± 4.2¶ (46) |
| PPI + gastrin (% control) | 171 ± 5.08‡ (62) | 1.4 ± 1.4 (66) | 0.7 ± 1.1‡ (63) | 2.9 ± 2.8§,# (50) | 4.9 ± 2.4‡ (60) | 5.7 ± 5.6§ (89) | 3.4 ± 1.6¶ (31) |
P < .01 versus control.
P < .05 versus control.
P < .001 versus gastrin.
P < .01 versus gastrin.
P < .05 versus gastrin.
P < .05 versus PPI.
Combination of PPI and Gastrin Prevented β-Catenin Activation and Induced Apoptosis as Augmented Anti-Polyposis Mechanisms
To document how PPI blocked the gastrin-promoted trophic effect in intestinal polyposis, we hypothesized that the anti-polyposis effect of PPI alone or combination of PPI and gastrin might be due to increased phosphorylation of β-catenin and subsequently decreased nuclear translocation of β-catenin. As shown in Figure 2A, gastrin significantly decreased the phosphorylation of β-catenin in the whole lysates from intestinal mucosa of APCMin/+ mice (P < .05) in group 2, whereas phospho-β-catenin was increased in group 3 and preserved in group 4. Further confirmations of these β-catenin changes were done by immunohistochemistry staining (Figure 2B). As seen in groups 1 and 2, β-catenin was increasingly expressed in the colonocyte nucleus in APCMin/+ mice, whereas groups 3 and 4 showed staining of β-catenin in the cytoplasm and membrane of the colonocyte (P < .05; Figure 2B). Next, we have checked the expression of cyclins and CDK according to group. Similar to the reports, gastrin administration increased the expression of cyclin D1 and CDK4 (Figure 2C). Although PPI alone group did not change cyclin D1 and CDK4, the combination of PPI and gastrin decreased their expression. In addition, the treatment with combination of PPI and gastrin downregulated the expressions of cyclin E, CDK2, and CDK4 in APCMin/+ mice (Figure 2C). Our previous publications revealed that the anti-tumorigenic action of PPI was through selective induction of apoptosis in tumors. Therefore, we performed TUNEL assay as well as Western blot for apoptosis executors. TUNEL staining result showed significantly increased apoptotic index in either group 3 or group 4 even in polyp neighbor as well as polyp lesion (P < .05; Figure 2D). Western blot analysis was done to compare the expression of apoptotic molecules (including caspase-3, caspase-8, and PARP) and anti-apoptotic molecules (including Bcl-2 and survivin), all of which were known to be engaged in intestinal polyposis and carcinogenesis. PPI alone and the combination of PPI and gastrin induced the expression of cleaved caspase-3 and resulting PARP cleavage, whereas Bcl-2 expression was decreased (Figure 2E).
Figure 2.
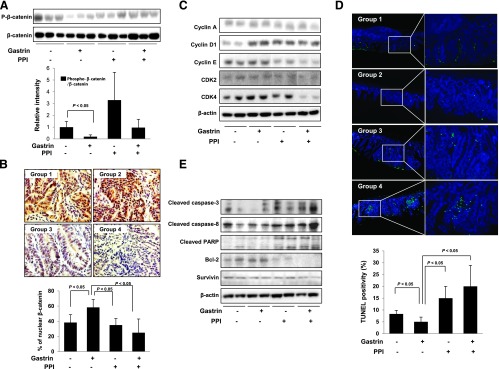
The changes in β-catenin, cell cycle, and apoptosis according to animal group. (A) Comparison of intestinal mucosal protein levels of phosphorylated β-catenin according to group. Phosphorylation of β-catenin in group 2 was significantly decreased (P < .05), but phosphorylation of β-catenin was increased in group 3. (B) β-Catenin expression using immunohistochemistry staining according to animal group. Compared to group 1, nuclear staining as well as cytoplasmic staining was greatly increased in group 2. However, the mean percentages of nuclear β-catenin staining were significantly increased in group 2 (P < .05) but significantly decreased in groups 3 and 4 (P < .05). (C) Equal amounts of total protein extracted from small intestine tissues were subjected to Western blot analysis using the indicated antibodies. (D) TUNEL staining to confirm the apoptosis. Compared to mean TUNEL-positive cells in groups 1 and 2, the numbers of TUNEL-positive cells were significantly increased in groups 3 and 4 (P < .05). All bars represent the mean and SD of triplicate values, respectively. (E) Equal amounts of total protein extracted from small intestine tissues were subjected to Western blot analysis using the indicated antibodies.
Co-Administration of PPI and Gastrin Showed Anti-Tumorigenic Action through Induction of Apoptosis and Inhibition of Angiogenic Activities, Mitogen-Activated Protein Kinase Activation, and Inflammatory Signaling
Because active angiogenesis is also one of the core mechanisms for the intestinal polyposis of APCMin/+ mice [11], we investigated the changes of angiogenic activity according to the group through CD31 expression. As seen in Figure 3A, induced expression of CD31 in APCMin/+ mice as well as increased polyposis in group 2 was all associated with significant increases than group 1 (P < .05). However, group 3 or group 4 significantly decreased the expression of CD31 in APCMin/+ mice (P < .005; Figure 3A). Because these angiogenic activities of APCMin/+ mice are associated with increased signal transduction activations, we have compared the active form of mitogen-activated protein kinases (MAPKs) according to group and found that gastrin activated the phosphorylation of p38, ERK, and JNK. However, the activation of MAPKs was all inhibited in either PPI alone or combination of PPI and gastrin (Figure 3B). Our preliminary cDNA microarray study demonstrated that gastrin increased the expression of inflammatory mediators, suggesting that gastrin might increase genes engaged in both pro-inflammatory and angiogenic actions (data not shown), among which we have checked the changes of TNF-α and COX-2, genes known to be principally engaged in intestinal polyposis as well as colon cancer [12,13]. Though statistically insignificant, the expression of TNF-α and COX-2 was increased in gastrin-treated group 2, but their expression was significantly decreased in groups 3 and 4 (Figure 3, C and D). Similar to COX-2 expression, the level of PGE2, a product of COX-2, was decreased in both PPI alone and combination of PPI and gastrin groups but not to a significant level (Figure 3E). The conclusion drawn from the in vivo mouse model showed that the combination of gastrin and PPI exerted significant anti-polyposis effect through β-catenin inactivation, increased apoptosis, anti-angiogenesis, and MAPK inactivation relevant to decreased levels of pro-inflammatory mediators. These novel findings were further documented in the following in vitro cellular models.
Figure 3.
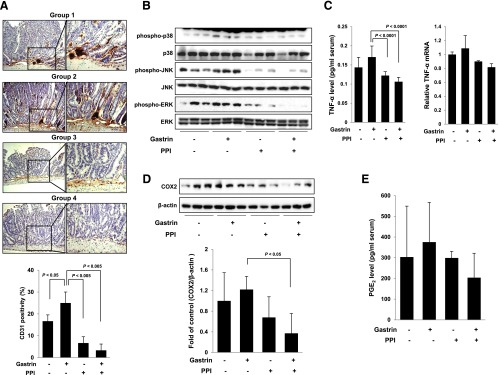
Inflammatory signaling pathway according to animal groups. (A) Gastrin administration in APCMin/+ mice (group 2) significantly increased the expression of CD31, whereas the expression of CD31 was significantly decreased in groups 3 and 4 (P < .005). (B) The phosphorylation levels of p38, JNK, and ERK were determined by Western blot analysis. The phosphorylation of MAPKs was significantly inhibited in groups 3 and 4. (C) Changes in TNF-α mRNA expression and serum protein levels. The treatment with PPI inhibited the levels of TNF-α induced by gastrin in groups 3 and 4. (D–E) The expression of COX-2 and the level of PGE2 were investigated by Western blot analysis and ELISA, respectively. Group 2 showed the induction of COX-2 and PGE2; however, PPI administration decreased the levels of indicated factors in groups 3 and 4.
Gastrin exerted trophic effects in a dose-dependent manner, significantly in higher dose than 100 nM gastrin in HCT116 colon cancer cells (P < .05; Figure 4A). PPI significantly decreased cellular viability in a dose-dependent manner, and PPI showed significantly higher cytotoxic effect than PPI alone in the presence of 100 nM gastrin (P < .05; Figure 4B). To investigate the anti-proliferative activity of PPI alone or the combination of PPI and gastrin, we examined the cell cycle distribution and apoptosis in HCT116 cells by flow cytometry. As shown in Figure 4C, quantitation of cells in the G0/G1, S, and G2/M phases of the cell cycle showed that HCT116 cells accumulated in the S phase after exposure to gastrin, whereas the cell population in G0/G1 was decreased. The distribution of cell cycle was restored after treatment with PPI alone or the combination of PPI and gastrin, suggesting that PPI addition either increased the G0/G1 phase or decreased the S phase, opening the possibility of cell apoptosis. Flow cytometry using propidium iodide (PI) and annexin V showed significantly decreased apoptotic cell death after treatment with gastrin, but PPI alone or the combination of PPI and gastrin significantly increased apoptosis (P < .05; Figure 4D). These findings from flow cytometry were further validated through Western blot analyses of cyclin D1, cyclin E, and CDK4 as well as caspase-3, caspase-8, PARP, Bcl-2, and survivin (Figure 4, E and F). Similar to the data from a previous animal model (Figure 2C), gastrin induced the expression of cyclin D1 and CDK4, but PPI alone and the combination of PPI and gastrin decreased the expression of cell cycle markers, consistent with the finding of flow cytometry shown in Figure 4C. Gastrin administration was associated with either inhibition of PARP cleavage or induction of Bcl-2, but PPI alone or the combination of PPI and gastrin increased cleavage of caspase-3, caspases-8, and PARP, whereas the expressions of Bcl-2 and survivin were decreased, indicating that PPI might inhibit the proliferation of HCT116 cells induced by gastrin through induction of apoptosis.
Figure 4.
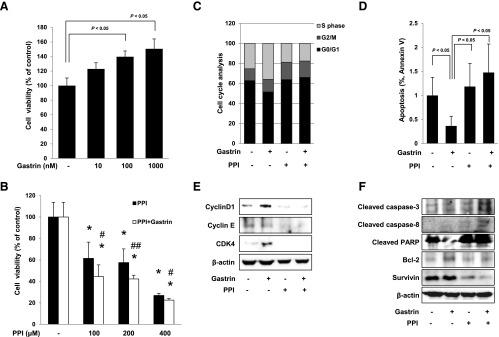
Changes of cell viability, cell cycle, and apoptosis in HCT116 cells. Cell viability was observed with MTT assay. (A) Gastrin significantly increased cell viability in a dose-dependent manner (P < .05). Conversely, PPI decreased cell viability in a dose-dependent manner (P < .05). (B) The combination of 100, 200, or 400 µM PPI and 100 nM gastrin paradoxically augmented significant decreases in cell viability (P < .05). All experiments were independently repeated at least three times. *P < .05 versus control; #P < .05 versus PPI; ##P < .01 versus PPI. (C) Flow cytometric analysis of HCT116 cells synchronized at the G1/S transition by serum starvation and released into the cell cycle with gastrin, PPI alone, or the combination of PPI and gastrin. The percentage of cells in different phases of the cell cycle was determined by using the Mod-Fit program. (D) HCT116 cells were treated with gastrin, PPI, or the combination for 12 hours and analyzed by flow cytometry for fluorescein isothiocyanate-annexin V and propidium iodide staining to verify the induction of apoptosis. The percentage of cells in apoptosis was determined by using the Mod-Fit program. (E) HCT116 cells were challenged with gastrin, PPI, or the combination of PPI and gastrin for 24 hours to investigate the expression of cell cycle markers. The expressions of cyclin D1, cyclin E, and CDK4 were assessed by Western blot analysis. (F) Western blot analysis was performed with antibodies of Bcl-2, cleaved caspase-3 and cleaved caspase-8, PARP, and survivin, respectively. PPI-treated cells led to apoptosis and cells with the combination of PPI and gastrin promoted more apoptosis than cells exposed to PPI alone.
Blockade of Gastrin Binding to Its Receptor and Inactivation of β-Catenin Signaling Might Explain the Anti-Tumorigenic Action of PPI
To determine whether the anti-proliferative effect of PPI was through antagonizing the trophic effect of gastrin, we examined the binding activity of CCKBR-gastrin. As shown in Figure 5A, PPI significantly antagonized gastrin-CCKBR binding (P < .05) and the combination of PPI and gastrin blocked gastrin-CCKBR binding higher than PPI alone. Because the ultimate trophic effect of gastrin is executed by Tcf/Lef binding relevant to β-catenin activation [14], we performed a luciferase reporter assay of Tcf/Lef after the treatment with gastrin alone, PPI alone, and the combination of PPI and gastrin. As shown in Figure 5B, gastrin significantly increased Tcf/Lef reporter activity, but the combination of PPI and gastrin markedly inhibited the Tcf/Lef promoter activity (P < .05). To prove these results by the combination of PPI and gastrin, we have determined the phosphorylation of GSK3β, another key regulator engaged in β-catenin activation. As shown in Figure 5C, gastrin administration abolished the phosphorylation of GSK3β to preserve the proliferative action of β-catenin. However, PPI alone and the combination of PPI and gastrin restored the phosphorylation of GSK3β. Because these trophic actions of gastrin were associated with the activation of MAPKs as seen in Figure 3B, we examined the levels of p-p38, p-JNK, and p-ERK. As shown in Figure 5D, treatment with gastrin induced the phosphorylation of p38 and ERK and these results are consistent with our in vivo findings. These activations of p38 and ERK were significantly inhibited in the presence of PPI (Figure 5D). Finally, since tissue analysis from APCMin/+ mice showed that gastrin significantly increased CD31 expression (Figure 3A), we assessed the angiogenic growth factors including interleukin-8 (IL-8), PDGF, hypoxia-inducible factor-1α (HIF-1α), COX-2, and VEGF. As shown in Figure 5E, gastrin increased the expression of all of these angiogenic factors except PDGF, whereas the expression of these angiogenic factors was all decreased with the combination of PPI and gastrin. Taken together, we suggest that the trophic effect of hypergastrinemia relevant to long-term PPI administration can be antagonized by paradoxically inhibitory action of PPI on blocking gastrin binding to CCKBR receptor, thereby facilitating the inactivation of β-catenin signaling pathway, G0/G1 arrest, and finally apoptosis in vitro and in vivo intestinal polyposis model.
Figure 5.
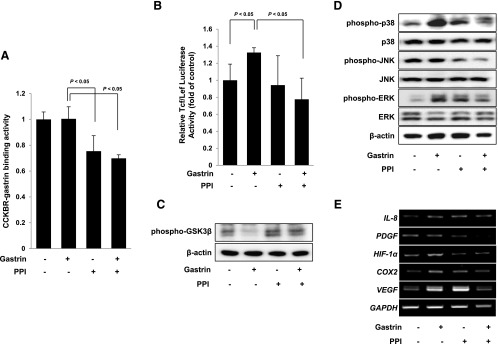
PPI suppresses gastrin-induced trophic effect through blockade of the binding of gastrin to its receptor. (A) To identify binding activity between CCKBR and gastrin, a sandwich ELISA was performed. PPI alone and the combination of PPI and gastrin significantly inhibited the binding between CCKBR and gastrin. (B) HCT116 cells were transiently transfected with the pTcf/Lef-Luc reporter vector. After transfection, the cells were cultured in serum-free medium with gastrin, PPI, or the combination of PPI and gastrin for 12 hours and the cell lysates were obtained to observe the luciferase activity. The combination of PPI and gastrin significantly decreased the luciferase reporter activity of Tcf/Lef. (C) Gastrin significantly inactivated GSK3β phosphorylation, but this inactivation was highly recovered with PPI treatment. (D) HCT116 cells were treated with gastrin, PPI, or the combination of PPI and gastrin for 12 hours and the phosphorylation of MAPKs was investigated. Gastrin induced the phosphorylation of p38 and ERK and the treatment with PPI downregulated their phosphorylations. (E) The changes in angiogenic factors including IL-8, PDGF, HIF-1α, COX-2, and VEGF were evaluated by reverse transcription-PCR. Gastrin induced the expression of angiogenic factors; however, PPI and the combination of PPI and gastrin inhibited the expression of angiogenic factors induced by gastrin.
Discussion
In this study, we found that the combination of PPI and gastrin paradoxically inhibited intestinal polyposis even under the mutation of the apc gene as well as amelioration of gastrin's trophic effect. PPI and gastrin synergistically inhibited β-catenin-associated proliferation signaling as well as gastrin-associated inflammatory mediators. Our study contains supporting evidence to explain both how long-term PPI administration plays the preventive role in colitis-associated carcinogenesis and the results of a cohort study aimed to reveal the risk of colon cancer with PPI-associated hypergastrinemia.
Although characterized as a stimulant of gastric acid secretion, the peptide hormone (gastrin) also exerts growth-promoting effect on normal and malignant gastrointestinal tissues [15]. Gastrin has also been shown to induce the growth of colonic gastric carcinoma both in vivo and in vitro, emphasizing the importance of gastrin as a growth factor for gastrointestinal neoplasms [1,16]. Several studies have concluded that gastrin is a trophic and mitogenic peptide for normal and neoplastic gastrointestinal mucosal cells and even cells outside the digestive system [17]. Studies in humans have also shown that higher levels of circulating gastrin are associated with hypertrophy of the gastric mucosa and hyperplasia of parietal and enterochromaffin-like cells [18,19] as well as direct growth-promoting effects of gastrin on transplanted human colon carcinoma and chemically induced colon tumors [20,21]. Several reports have demonstrated that activation of protein kinases in response to gastrin stimulation leads to the induction of cellular proliferation through a signal transduction pathway that involves the activation of ERKs [22]. In the present study, treatment of HCT116 colorectal cancer cells with gastrin (100 nM) led to significant proliferation and gastrin treatment induced the progression of the G1/S phase and the expression of CDK4 and cyclin D1. Progression from the G1 to the S phase of the cell cycle is regulated by the periodic expression of cyclin D1 and cyclin E. An increase in the expression of cyclin D1 is recognized as a critical event in the activation of the cell cycle machinery of the cell [23], which modulates the activity of the CDKs, including CDK4, CDK6, and CDK2 [24]. In addition to these changes, binding of gastrin to the CCKBR induces cell proliferation through promoting cell cycle passage in late G1 and the inhibition of the binding of gastrin to the CCKBR by YM022 or transient transfection of siRNA abrogates this effect. The β-catenin/Tcf-4 complex is transported into the nucleus where it acts as a transcription factor. Because Tcf-4-deficient mice have no proliferating cells in their intestinal crypts, the β-catenin/Tcf-4 complex is an important regulator of intestinal proliferation [14]. The identification of gastrin as a functionally relevant upstream target of the β-catenin/Tcf-4 pathway strengthens the importance of gastrin as a potential target for novel therapeutic modalities in the treatment of colorectal cancer. We additionally documented that PPI can be a good therapeutic for colorectal polyposis as well as cancer because it antagonized the trophic action of gastrin. We previously reported the overexpression of CCKBR in colorectal cancer HCT116 cells [25], which can explain the reason why the treatment with gastrin made little change of the binding of gastrin to the CCKBR compared with the vehicle control in HCT116 cells. To further investigate the role of PPI in the interaction of gastrin and CCKBR, we repeated sandwich ELISA in rat small intestinal epithelial IEC-6 cells. As expected, treatment of IEC-6 cells with gastrin induced the binding activity of gastrin to its receptor, whereas PPI treatment inhibited the interaction of gastrin and CCKBR (data not shown), suggesting the possibility that the structure of PPI might compete with gastrin to the active site(s) of CCKBR or interfere in the interaction between gastrin and CCKBR.
The trophic role of gastrin in intestinal polyps is further supported by polyposis in the APCMin/+ mice model, in which gastrin significantly increased intestinal polyposis as well as β-catenin nuclear translocation. The results from the gastrin-treated APCMin/+ mice showed a significant increase in polyposis compared with nontreated APCMin/+ mice, which further strengthened our finding that the combination of PPI and gastrin paradoxically decreased intestinal polyposis through inhibiting CCKBR-gastrin binding (Table 2). Because gastrin is not only important in colon cancer promotion but also important in the early stages of polyp development, long-term PPI administration allowed for efficient surveillance of colon tumorigenesis and a cohort study performed more than 10 years after PPI use clearly showed that there was no risk of colon cancer in spite of hypergastrinemia. Clinical studies have indicated that hypergastrinemia can be associated with an increased risk of colorectal cancer and concerns have been raised about colon cancer risk with hypergastrinemia caused by PPIs [26]. However, a recent study reported that hypergastrinemia following long-term PPI therapy at a regular dose is not associated with an increased risk of colorectal cancer [4]. In regard to the “colitis-associated carcinogenesis” model, we recently revealed that repeated colitis increased the expression of nitric oxide and TNF-α, which ultimately led to tumorigenesis, but omeprazole treatment efficiently attenuated the generation of nitric oxide and the expression of TNF-α [9]. However, since a past study reported that gastric mucosal hypertrophy is frequently associated with hypergastrinemia, and the degree of gastritis is related to the concentration of gastrin [27,28], surely hypersecretion of gastrin should be able to modify the epithelial structure by its well-known ability to promote cellular proliferation and also by regulating migration, invasion, and apoptosis [29]. Therefore, we hypothesized that PPI can provoke hypergastrinemia based on the nature of the drug and that real biology under the combination of PPI and gastrin differs from that of gastrin alone, signifying that PPIs exert considerable peculiar biologic actions.
Although PPIs are specifically targeted for blocking hydronium efflux by inhibiting proton pumps in the apical portion of parietal cells, PPIs have been shown to exert selective apoptosis-inducing actions against gastric or colon carcinogenesis, to attenuate Helicobacter pylori-associated angiogenesis, and to impose significant cytoprotective actions independent of gastric acid inhibition [30,31]. We have suggested that PPIs have anti-oxidative and cytoprotective actions through nuclear factor E2-related factor 2 activation and subsequent heme oxygenase-1 induction imposes protection from nonsteroidal anti-inflammatory drug-induced gastroduodenal damages [32].
Gastrin has been known to induce inflammation as evidenced with increased inflammatory mediators associated with hypergastrinemia. We also found that the levels of TNF-α and COX-2 were increased with gastrin administration in both in vitro colon cells and in vivo small intestine tissues, explaining the mutual influence on β-catenin activation and polyposis. As an additional explanation of the inhibition of polyposis through augmented apoptosis by PPIs, in spite of the trophic effect of gastrin, the apoptosis-inducing capacity even in the polyp or tumor prevails with PPIs. Because maintenance of intracellular or extracellular pH is very important for cell function and because cancer cells in vivo often exist in an ischemic microenvironment with a lower extracellular pH than that of the surrounding normal cells, the acidity in tumors is due to the increased production of acidic metabolites from rapid and large amounts of glycolysis and is provoked by the limited ability of the tumor vasculature to remove these acidic products [33,34]. To overcome this hypoxic microenvironment and prevent accumulation of the increased acidic metabolites, the ability to dispose intracellular protons is critical for cancer cell survival [34,35]. There are some mechanisms involved in the regulation of pH in tumor tissues. The main mechanism has reported that a proton (H+) is exported by the sodium-hydrogen anti-transport using the energy of the gradient of Na+ [36,37]. In this case, the blocking of H+ efflux by PPIs might contribute to attenuated polyposis. PPIs successfully suppress tumor cell viability by inducing apoptotic cell death. Therefore, these findings imply that blockage of another kind of proton pump predominantly expressed in tumor cells could be used as a promising anti-cancer drug.
Supplementary Material
Abbreviations
- COX
cyclooxygenase
- HIF
hypoxia-inducible factor
- IL
interleukin
- PPI
proton pump inhibitor
- TNF
tumor necrosis factor
- VEGF
vascular endothelial growth factor
Footnotes
This study was supported by a grant from the Ministry of Education, Science and Technology (2010-0002052), Korea. No potential conflicts of interest were disclosed.
This article refers to supplementary materials, which are designated by Figures W1 and W2 and are available online at www.neoplasia.com.
References
- 1.Rozengurt E, Walsh JH. Gastrin, CCK, signaling, and cancer. Annu Rev Physiol. 2001;63:49–76. doi: 10.1146/annurev.physiol.63.1.49. [DOI] [PubMed] [Google Scholar]
- 2.Kovac S, Xiao L, Shulkes A, Patel O, Baldwin GS. Gastrin increases its own synthesis in gastrointestinal cancer cells via the CCK2 receptor. FEBS Lett. 2010;584:4413–4418. doi: 10.1016/j.febslet.2010.09.046. [DOI] [PubMed] [Google Scholar]
- 3.Hartwich A, Konturek SJ, Pierzchalski P, Zuchowicz M, Labza H, Konturek PC, Karczewska E, Bielanski W, Marlicz K, Starzynska T, et al. Helicobacter pylori infection, gastrin, cyclooxygenase-2, and apoptosis in colorectal cancer. Int J Colorectal Dis. 2001;16:202–210. doi: 10.1007/s003840100288. [DOI] [PubMed] [Google Scholar]
- 4.Robertson DJ, Larsson H, Friis S, Pedersen L, Baron JA, Sorensen HT. Proton pump inhibitor use and risk of colorectal cancer: a population-based, case-control study. Gastroenterology. 2007;133:755–760. doi: 10.1053/j.gastro.2007.06.014. [DOI] [PubMed] [Google Scholar]
- 5.Colucci R, Blandizzi C, Tanini M, Vassalle C, Breschi MC, Del Tacca M. Gastrin promotes human colon cancer cell growth via CCK-2 receptor-mediated cyclooxygenase-2 induction and prostaglandin E2 production. Br J Pharmacol. 2005;144:338–348. doi: 10.1038/sj.bjp.0706053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muerkoster S, Isberner A, Arlt A, Witt M, Reimann B, Blaszczuk E, Werbing V, Folsch UR, Schmitz F, Schafer H. Gastrin suppresses growth of CCK2 receptor expressing colon cancer cells by inducing apoptosis in vitro and in vivo. Gastroenterology. 2005;129:952–968. doi: 10.1053/j.gastro.2005.06.059. [DOI] [PubMed] [Google Scholar]
- 7.Parikh N, Howden CW. The safety of drugs used in acid-related disorders and functional gastrointestinal disorders. Gastroenterol Clin North Am. 2010;39:529–542. doi: 10.1016/j.gtc.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kedika RR, Souza RF, Spechler SJ. Potential anti-inflammatory effects of proton pump inhibitors: a review and discussion of the clinical implications. Dig Dis Sci. 2009;54:2312–2317. doi: 10.1007/s10620-009-0951-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim YJ, Lee JS, Hong KS, Chung JW, Kim JH, Hahm KB. Novel application of proton pump inhibitor for the prevention of colitis-induced colorectal carcinogenesis beyond acid suppression. Cancer Prev Res (Phila) 2010;3:963–974. doi: 10.1158/1940-6207.CAPR-10-0033. [DOI] [PubMed] [Google Scholar]
- 10.Yamada T, Mori Y, Hayashi R, Takada M, Ino Y, Naishiro Y, Kondo T, Hirohashi S. Suppression of intestinal polyposis in Mdr1-deficient ApcMin/+ mice. Cancer Res. 2003;63:895–901. [PubMed] [Google Scholar]
- 11.Korsisaari N, Kasman IM, Forrest WF, Pal N, Bai W, Fuh G, Peale FV, Smits R, Ferrara N. Inhibition of VEGF-A prevents the angiogenic switch and results in increased survival of Apc+/min mice. Proc Natl Acad Sci USA. 2007;104:10625–10630. doi: 10.1073/pnas.0704213104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kwong E, Trzaskos JM, Evans JF, Taketo MM. Suppression of intestinal polyposis in ApcΔ716 knockout mice by inhibition of cyclooxygenase 2 (COX-2) Cell. 1996;87:803–809. doi: 10.1016/s0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]
- 13.Kim S, Domon-Dell C, Wang Q, Chung DH, Di Cristofano A, Pandolfi PP, Freund JN, Evers BM. PTEN and TNF-α regulation of the intestinal-specific Cdx-2 homeobox gene through a PI3K, PKB/Akt, and NF-κB-dependent pathway. Gastroenterology. 2002;123:1163–1178. doi: 10.1053/gast.2002.36043. [DOI] [PubMed] [Google Scholar]
- 14.Koh TJ, Bulitta CJ, Fleming JV, Dockray GJ, Varro A, Wang TC. Gastrin is a target of the β-catenin/TCF-4 growth-signaling pathway in a model of intestinal polyposis. J Clin Invest. 2000;106:533–539. doi: 10.1172/JCI9476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koh TJ, Chen D. Gastrin as a growth factor in the gastrointestinal tract. Regul Pept. 2000;93:37–44. doi: 10.1016/s0167-0115(00)00176-2. [DOI] [PubMed] [Google Scholar]
- 16.Walsh JH. Role of gastrin as a trophic hormone. Digestion. 1990;47(suppl 1):11–16. doi: 10.1159/000200509. discussion 49–52. [DOI] [PubMed] [Google Scholar]
- 17.Yassin RR. Signaling pathways mediating gastrin's growth-promoting effects. Peptides. 1999;20:885–898. doi: 10.1016/s0196-9781(99)00077-7. [DOI] [PubMed] [Google Scholar]
- 18.Singh P, Owlia A, Espeijo R, Dai B. Novel gastrin receptors mediate mitogenic effects of gastrin and processing intermediates of gastrin on Swiss 3T3 fibroblasts. Absence of detectable cholecystokinin (CCK)-A and CCK-B receptors. J Biol Chem. 1995;270:8429–8438. doi: 10.1074/jbc.270.15.8429. [DOI] [PubMed] [Google Scholar]
- 19.Bordi C, D'Adda T, Azzoni C, Pilato FP, Caruana P. Hypergastrinemia and gastric enterochromaffin-like cells. Am J Surg Pathol. 1995;19(suppl 1):S8–S19. [PubMed] [Google Scholar]
- 20.Crean GP, Marshall MW, Rumsey RD. Parietal cell hyperplasia induced by the administration of pentagastrin (ICI 50, 123) to rats. Gastroenterology. 1969;57:147–155. [PubMed] [Google Scholar]
- 21.McGregor DB, Jones RD, Karlin DA, Romsdahl MM. Trophic effects of gastrin on colorectal neoplasms in the rat. Ann Surg. 1982;195:219–223. doi: 10.1097/00000658-198202000-00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stepan VM, Tatewaki M, Matsushima M, Dickinson CJ, del Valle J, Todisco A. Gastrin induces c-fos gene transcription via multiple signaling pathways. Am J Physiol. 1999;276:G415–G424. doi: 10.1152/ajpgi.1999.276.2.G415. [DOI] [PubMed] [Google Scholar]
- 23.Yang K, Hitomi M, Stacey DW. Variations in cyclin D1 levels through the cell cycle determine the proliferative fate of a cell. Cell Div. 2006;1:32. doi: 10.1186/1747-1028-1-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palmero I, Peters G. Perturbation of cell cycle regulators in human cancer. Cancer Surv. 1996;27:351–367. [PubMed] [Google Scholar]
- 25.Han YM, Park JM, Park SH, Hahm KB, Hong SP, Kim EH. Gastrin promotes intestinal polyposis through cholecystokinin-B receptor-mediated proliferative signaling and fostering tumor microenvironment. J Physiol Pharmacol. 2013;64:429–437. [PubMed] [Google Scholar]
- 26.Yang YX, Hennessy S, Propert K, Hwang WT, Sedarat A, Lewis JD. Chronic proton pump inhibitor therapy and the risk of colorectal cancer. Gastroenterology. 2007;133:748–754. doi: 10.1053/j.gastro.2007.06.022. [DOI] [PubMed] [Google Scholar]
- 27.Burkitt MD, Varro A, Pritchard DM. Importance of gastrin in the pathogenesis and treatment of gastric tumors. World J Gastroenterol. 2009;15:1–16. doi: 10.3748/wjg.15.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reinhardt JD, McCloy RM, Blackwell CF. Autoimmune atrophic gastritis with hypergastrinemia. South Med J. 1976;69:1551–1553. doi: 10.1097/00007611-197612000-00010. [DOI] [PubMed] [Google Scholar]
- 29.Ibiza S, Alvarez A, Romero W, Barrachina MD, Esplugues JV, Calatayud S. Gastrin induces the interaction between human mononuclear leukocytes and endothelial cells through the endothelial expression of P-selectin and VCAM-1. Am J Physiol Cell Physiol. 2009;297:C1588–C1595. doi: 10.1152/ajpcell.00082.2009. [DOI] [PubMed] [Google Scholar]
- 30.Yeo M, Kim DK, Han SU, Lee JE, Kim YB, Cho YK, Kim JH, Cho SW, Hahm KB. Novel action of gastric proton pump inhibitor on suppression of Helicobacter pylori induced angiogenesis. Gut. 2006;55:26–33. doi: 10.1136/gut.2005.067454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yeo M, Kim DK, Kim YB, Oh TY, Lee JE, Cho SW, Kim HC, Hahm KB. Selective induction of apoptosis with proton pump inhibitor in gastric cancer cells. Clin Cancer Res. 2004;10:8687–8696. doi: 10.1158/1078-0432.CCR-04-1065. [DOI] [PubMed] [Google Scholar]
- 32.Lee HJ, Han YM, Kim EH, Kim YJ, Hahm KB. A possible involvement of Nrf2-mediated heme oxygenase-1 up-regulation in protective effect of the proton pump inhibitor pantoprazole against indomethacin-induced gastric damage in rats. BMC Gastroenterol. 2012;12:143. doi: 10.1186/1471-230X-12-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stubbs M, McSheehy PM, Griffiths JR. Causes and consequences of acidic pH in tumors: a magnetic resonance study. Adv Enzyme Regul. 1999;39:13–30. doi: 10.1016/s0065-2571(98)00018-1. [DOI] [PubMed] [Google Scholar]
- 34.Tannock IF, Rotin D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. 1989;49:4373–4384. [PubMed] [Google Scholar]
- 35.Lee AH, Tannock IF. Heterogeneity of intracellular pH and of mechanisms that regulate intracellular pH in populations of cultured cells. Cancer Res. 1998;58:1901–1908. [PubMed] [Google Scholar]
- 36.Karmazyn M, Gan XT, Humphreys RA, Yoshida H, Kusumoto K. The myocardial Na+-H+ exchange: structure, regulation, and its role in heart disease. Circ Res. 1999;85:777–786. doi: 10.1161/01.res.85.9.777. [DOI] [PubMed] [Google Scholar]
- 37.Wakabayashi S, Shigekawa M, Pouyssegur J. Molecular physiology of vertebrate Na+/H+ exchangers. Physiol Rev. 1997;77:51–74. doi: 10.1152/physrev.1997.77.1.51. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.