

Abstract
G protein-coupled receptors (GPCRs) signal through G protein α and βγ subunit families to regulate a wide range of physiological and pathophysiological processes. As such, GPCRs are major targets for therapeutic drugs. Downstream targets of GPCRs have also gained interest as a therapeutic approach to complex pathologies involving multiple GPCRs. One such approach involves targeting of the G proteins themselves. Several small molecule Gα and Gβγ modulators have been developed and been tested in various animal models of disease. Here we will discuss the requirements for targeting Gα and Gβγ subunits, the mechanisms of action of currently identified inhibitors, and focus on the potential utility of Gα and Gβγ inhibitors in the treatment of various cancers.
The G Protein-Coupled Receptor system as a therapeutic target
GPCRs comprise a large family of receptors for physiologically relevant ligands such as adrenalin or serotonin, which makes them attractive drug targets [1]. For each GPCR ligand there are often multiple subtypes involved in specific cellular functions. For example, there are 13 different GPCR family members that bind serotonin, but they are differentially expressed and couple to distinct signal transduction mechanisms [2]. Targeting of these specific GPCR subtypes holds the promise of being able to almost surgically manipulate the biology controlled by these receptors, and thus greatly limiting potential side effects of pharmacological therapy. A potential downside to targeting GPCR subtypes is that some pathologies, such as cancer and heart disease, are the result of dysregulation of a number of GPCR signaling pathways and circulating factors. Therefore, targeting one receptor may not be sufficient to be an effective treatment.
As an alternate approach to treatment of diseases involving multiple ligand-dependent signaling inputs, an interest has developed in more broad-based pharmacological targeting of key steps in common pathways downstream of multiple receptors that are directly involved in mediating a cellular disease pathway[3, 4]. This approach sacrifices specificity for the sake of increased efficacy, but for complex and deadly diseases like cancer, efficacy is of paramount concern. In the GPCR signaling system there are multiple second messenger cascades activated by G proteins downstream of GPCRs that have been considered as targets [3, 5]. In the classic G protein signaling system, GPCRs couple to heterotrimeric G proteins that, when activated by exchange of GDP for GTP on the G protein α subunit, are conformationally competent to interact with high affinity with second messenger generating enzymes or ion channels[1, 6]. Superimposed on this basic system are regulators of the G proteins (RGS proteins) that stimulate GTP hydrolysis on Gα subunits[7], and regulators of the GPCRs such as G protein coupled-receptor kinases (GRKs), arrestin, and associated pathways regulated by arrestin [8]. Many of these systems have been investigated as potential therapeutic avenues either with genetic or small molecule based approaches [9–13]. In this review we will discuss the potential for pharmacological targeting the G protein subunits directly with a particular focus on their utility in treating cancer.
Structural features of G proteins amenable to small molecule binding
Although peptide-based inhibitors of G protein α subunits and G protein βγ subunits have been developed [10, 14–16], we will focus on small molecule inhibitors because of their potential therapeutic utility, with the exception of one peptidic inhibitor that has potent actions in cellular systems. Gα and Gβγ represent distinct molecular problems with respect to small molecule binding. Gα subunits have a catalytic site and numerous clefts that have the potential to bind to small molecules [17, 18] and inhibit Gα nucleotide exchange and GTP hydrolysis activity. Gβγ does not have a catalytic pocket directly amenable to small molecule targeting but does have a concave surface at a protein-protein interaction “hot spot” that has proven amenable to small molecule binding [17, 19–21].
Structural features of the α subunit
The G protein α subunit consists of two distinct domains, a GTPase or Ras-like domain and an α helical domain connected by two flexible random coil linkers[22, 23] (Figure 1A). The guanine nucleotide binding site is comprised of amino acids contributed by both domains at the interface between these two domains. The mechanism for how alteration in GPCR conformation upon activation by ligand catalyzes the nucleotide exchange reaction on G proteins has been the subject of intense interest for many years [24, 25]. A recent breakthrough is the elucidation of the three dimensional structure of a complex of the agonist-bound β-adrenergic receptor and Gαsβ1γ2, by X-ray crystallography [26]. This structure reveals an agonist-dependent opening of the intracellular surface of the receptor allowing for extensive interaction between various intracellular receptor surfaces and various domains of the Gα subunit, leading to alterations in the Gα subunit GDP binding pocket. These GPCR-G protein interfaces could represent small molecule targets that have not yet been exploited, although short Gα-derived peptides can selectively disrupt this interface[27]. The detailed mechanism for how receptors transmit conformational information through the Gα subunit will not be discussed here, but an unexpected result, supported by complementary electron microscopy and dynamics studies, was that the α helical domain moves apart from the GTPase domain in the GPCR-activated transition state [26, 28–30]. This suggests that the release of GDP either requires these inter-domain movements, or that the movements are a consequence of GDP dissociation. Interestingly, peptides, such as GPR/goloco peptides[10], and some small molecules (discussed below) that inhibit GDP release seem to prevent the inter-domain movements of the G protein α subunit, suggesting that the inter-domain movements are required, rather than a consequence of GDP release.
Figure 1.
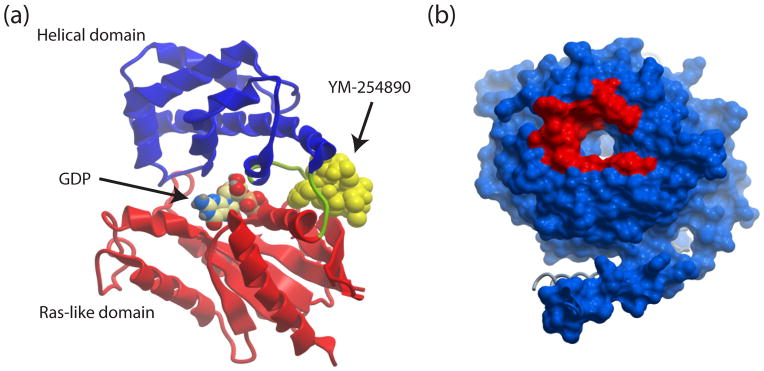
Structural representations of Gαq and Gβγ with potential sites for small molecule interactions. (a) Gαq bound to YM-254890. The α helical domain is in blue, the Ras-like domain is in red and the linkers connecting the two domains are in green. GDP is in CPK and YM-254890 is in space fill yellow. Rendered from PDB 3AH8. (b) Surface representation of Gβ. In red is the contact surface for the SIGK peptide used in a completion screen for small molecule discovery. Rendered from PDB 1XHM using MolSoft ICM.
Structural features of the Gβγ subunit
As discussed above, targeting Gβγ is more complicated because it involves inhibiting protein-protein interactions rather than a catalytic function. Protein-protein interactions are generally thought to be more difficult to block because of the often two-dimensional nature of the binding surfaces [31]. Unlike catalytic binding pockets that bind small molecule substrates with amino acid coordination in three dimensions, small molecule interaction with two dimensional protein-protein interaction surfaces may not derive sufficient binding energy from limited potential bonding interactions. The Gβ subunit is the prototype for the WD40 repeat, 7 bladed β-propeller protein family characterized by 7 blades, each consisting of 4 antiparallel β-sheets, arranged circularly with the last of the 7 blades packing against the first [5, 17, 19]. This arrangement results in formation of a toroid shape with a hole in the middle and a major protein binding surface on the top of the torus over the hole [5, 32] (Figure 1B). Thus, this protein interaction surface on Gβ is not flat but rather is formed by the ridge of the “donut” with a concave center projecting into the hole. This 3D surface may be more amenable to small molecule binding than a typical flat protein-protein interaction interface[5].
Mechanisms of small molecule actions on Gα subunits
Several small molecule G protein α subunit inhibitors have been developed that all have an apparently common mechanism of action. They bind to G protein α subunits to prevent intrinsic and receptor-stimulated GDP release.
Suramin
Developed by Bayer in 1916, suramin is a symmetric polysulphonated napthylamine-benzamide derivative that has been used to treat African Sleeping Disease and river blindness (Figure 2A). In 1996, Freissmuth and colleagues discovered that suramin is a relatively specific inhibitor of GDP release on the Gs family G protein α subunits (IC50 ~250 nM), and inhibited nucleotide exchange on Gαi and Gαo, at 20 and 10 fold higher concentrations, respectively[33]. Because these assays were conducted with purified G protein α subunits, suramin must be binding directly to G protein α subunits. Suramin and its analogues inhibit GPCR-stimulated nucleotide exchange on Gαs, and Gαs-dependent stabilization of high affinity agonist binding to adrenergic receptors, presumably by preventing GDP release thereby preventing formation of the high affinity Gα-receptor, nucleotide-free transition state. Derivatization of suramin led to identification of analogues that are highly selective antagonists for Gs relative to Gi or Gq [34].
Figure 2.

Structures of compounds that bind to Gα or Gβγ subunits.
In the absence of mutagenic mapping or structural data, the detailed molecular mechanism for suramin-dependent inhibition of GDP release is not understood. Suramin is large for a small molecule (1297.29 Da) and has the potential to occupy a large surface of Gαs. Interestingly, purified adenylyl cyclase reduced the ability of suramin to inhibit nucleotide exchange on purified Gαs, indicating that binding of adenylyl cyclase and suramin to Gαs are mutually exclusive[33]. This suggests that suramin exerts its effect by binding to the effector binding sites on Gα subunits, but other interpretations involving alterations in Gα conformation by adenylyl cyclase are possible. Suramin is highly sulfonated with a strong negative charge that limits its utility as a reagent or a drug because it cannot cross cell membranes. Nevertheless, it is the first example of an organic molecule-based approach to inhibition of G protein-dependent signaling, and it has an overall mechanism of action that is similar to other G protein α subunit small molecule inhibitors.
Imidazopirazines
The imidazo-pyrazine derivative BIM-46174 (Figure 2B) was identified in a cell-based differential screen in an MCF7 breast cancer cell line for compounds that would inhibit cholera-toxin-induced, but not forskolin-induced, cAMP accumulation[35]. This screening strategy was designed to find compounds that inhibit Gs but not receptors or adenylyl cyclase. BIM-46174 and a more stable derivative BIM-46187 inhibit activation of multiple G proteins by GPCRs and bind directly to α subunits to inhibit nucleotide exchange [35, 36]. BIM-46174 inhibition of nucleotide exchange on the free Gα subunits suggests that it inhibits the conformational changes associated with G protein α subunit GDP release. Again, the lack of structural or mutagenic data limits our understanding of the mechanistic details at structural level of how this compound acts [36].
BIM-46174 contains a free sulfhydryl group and BIM-46187 is a disulfide bridged dimer of BIM-46174 [36]. The developers argue that because both the free compound and the dimer inhibit G protein activity that the –SH group is not involved in the activity of the compound, but it is not clear how carefully the disulfide linkage was controlled in these experiments to ensure that no free BIM-46714 is present in the BIM-46I87 samples. Unlike suramin, BIM-46174 is effective in cells, allowing the cell biology of the compound to be assessed. The disulfide is likely to be in a reduced state in the cellular environment. In cells, these compounds block all signaling pathways initiated by GPCRs through heterotrimeric G protein families, and thus, although not directly tested on all the individual Gα subunit family members in vitro, this class of compounds appears to bind and inhibit all G protein α subunit families equally.
YM-254890, a selective Gαq inhibitor
A cyclic depsipeptide, YM-254890 (Figure 2C) was derived from Chromobacterium and has potent antithrombotic effects. Recently it has been shown to specifically inhibit Gq signaling[37, 38]. Although not technically a small organic molecule, but because it has actions in cells and animals, it has potential as a therapeutic or lead molecule. Similar to suramin and BIM-46174, YM-254890 inhibits Gαq by inhibiting the nucleotide exchange reaction by binding directly to the Gα subunit and preventing either spontaneous or receptor-stimulated GDP release[37]. The compound inhibits Gq signaling downstream of GPCR activation but does not inhibit activation of signaling stimulated by transfected Gq209L, a mutation that stabilizes the GTP bound form of the Gα subunit[37]. This suggests that compounds that inhibit Gα subunits by inhibiting GDP release will be not be effective at treating diseases resulting from constitutively activating G protein subunit mutations that block GTP hydrolysis but may be effective at treating cancers or inflammatory processes related to GPCR-dependent activation of G protein pathways.
YM-254890 binding to Gαq is the only example of a small molecule-G protein complex for which structural information is available, thus allowing for an atomic level understanding of its mechanism of action[39]. The compound interacts directly with a pocket of the α1 helix and β2 strands of the Ras-like GTPase domain and the αA helix of the α helical domain (Figure 1A). In addition, the compound interacts with the inter-domain linkers including switch 1. This binding mode might be expected to prevent inter-domain movements and provides a plausible mechanism for inhibiting GDP release. Many of the direct contacts for YM-254890 with Gαq are conserved amongst members of the Gαq family (Gαq, 11, 14, 15 and 16) but vary significantly in the other G protein α subunit family members. Thus, there is an opportunity for either rational design or high throughput screening (HTS) to identify α subunit subtype-selective inhibitors that capitalize on the amino acid diversity in this pocket.
Mechanisms of small molecule actions on Gβγ subunits
M119/gallein
Multiple classes of small molecule inhibitors have been identified that bind to G protein βγ subunits and modulate interactions with select groups of effectors downstream of Gβγ [40] (Box 1). We developed the concept that different classes of small molecules interact with Gβγ in unique binding modes that lead to differential modulation of pathways downstream of Gβγ. A major class of antagonist molecules identified in this screen is the M119 class which includes M119, gallein and M119K amongst others (Figure 2D) [40, 41]. In part because of the ready availability of gallein and M119, this class of compounds has been the most extensively validated, however no atomic level information detailing the interactions between G βγ and these molecules has been published. The nature of the screen used to identify these molecules (Box 1) indicates that the molecules bind to a protein interaction “hot spot” on the top surface of the β subunit WD40 repeat that is normally involved in interactions with of the switch II helix of Gα and various effectors[20, 40].
Box 1. Steps in screeningcreen for identification and characterization of Gβγ-binding small molecules.
Test libraries of compounds for their ability to compete for binding of SIGK peptide to Gβγ. Compounds that bind to Gβγ can be identified by screening for completion for the binding of SIGK-peptide to Gβγ. The structure of the SIGK-Gβγ complex has been solved and SIGK is known to bind to a site that overlaps with many Gβγ targets. Given the relative inflexibility of Gβγ it is likely that compounds that inhibit SIGK binding do so by directly competing for SIGK binding, thus some idea of the compound binding site is given a priori. For example, compounds of the M119 family (Figure 2D) were identified in a small scale screen of the NCI diversity set for competition for SIGK binding to Gβγ in an ELISA format.
Secondary assays for disruption of βγ-dependent effector regulation. Compounds identified in the primary screen with low μM IC50’s in the primary assay are chosen and tested in vitro for effects on Gβγ-dependent target regulation. For example, compounds can be tested for their ability to inhibit purified Gβγ-dependent activation of purified PLCβ enzymatic activity. If compounds inhibit the Gβγ-dependent component of these types of assays, without inhibiting intrinsic basal PLC (or other effector) activity it is good evidence that the compounds bind directly to Gβγ. Specificity for different Gβγ targets can be examined by comparison of compound potency and efficacy in a variety of in vitro assays or in cell based assays of Gβγ signaling as discussed below. For example, some key effectors that have been tested in vitro include PLCβ, adenylyl cyclase, phosphoinositide 3-kinase and GRK2.
Assays for effectiveness in cells. For small molecules that inhibit Gβγ-dependent PLCβ regulation a standard cell based assay for inhibition of this pathway is formyl-Met-Leu-Phe (fMLP)-dependent regulation of Ca2+ release in differentiated HL60-neutrophil like cells. fMLP signaling is representative of chemoattractant and chemokine receptor signaling that relies largely on Gβγ released from Gi heterotrimers to regulate multiple signaling pathways in neutrophils including PLC, PI3K and pREX activation. For high throughput analysis of compounds, cell based assays could be conducted prior to in vitro assays since a single cell based experiment can give a readout of multiple signaling pathways and identifies molecules that are cell permeable.
Gβγ-binding compounds have been identified that either inhibit or potentiate Gβγ signaling. The M119 class inhibits interactions with downstream targets. Conceptually, the nature of inhibitory interactions can be readily explained as direct competition for binding interactions. There is no evidence for allosteric interactions mediating the effects of the compounds. Gβγ is relatively inflexible, but NMR spectroscopy analysis of Gβγ in the presence and absence of protein or peptide binding partners reveals both small and large allosteric changes [42]. Analysis of small molecule binding using this same NMR-based approach revealed no allosteric alterations with binding of the M119 or gallein. A selective reduction in NMR peak intensity for a single position was found upon binding of one compound from another class (M201), indicating a change in local dynamics, but again no conformational alterations were detected. Thus it is likely that compounds that inhibit interactions with downstream targets do so by directly interfering with crucial contacts between Gβγ subunits and effectors.
How compounds potentiate Gβγ signaling is conceptually more complex. Several scenarios can be imagined. For example, compounds may interfere with Gα-βγ subunit interactions and release free βγ that can signal downstream without nucleotide exchange. Such a scenario would require that the compound interactions with Gβγ that drive subunit dissociation do not interfere with Gβγ effector interactions. This mechanism was identified for the peptide SIRK, which binds to Gβγ at the α/βγ interface and drives ERK activation in cells [43, 44]. Such compounds would not necessarily be expected to enhance Gβγ-dependent effector activation in vitro, and would not be expected to inhibit the particular target of Gβγ whose activation is enhanced in cells. Alternatively, compounds binding at the Gβγ-effector interface compounds could increase the affinity of Gβγ for the target molecule by simply acting as a “molecular glue”. Such a scenario is testable in vitro by monitoring either the apparent affinity or direct binding of targets to Gβγ. Finally, compounds could alter the dynamics or local conformation of the βγ-effector binding site in a way that would allow for more efficient effector activation.
Receptor specific biased Gβγ inhibition
An exciting recent study has identified a novel mechanism for selective inhibition of Gβγ signaling downstream of specific receptors [45]. In a screen for antagonists for the receptor for 5-oxo-6E,8Z,11Z,14Z-eicosatetraenoic acid (5-oxo-ETE) receptor (OXE-R), a compound was identified that inhibited Gβγ signaling without influencing Gα subunit signaling. Surprisingly this compound inhibits OXE-R dependent signaling pathways without affecting Gβγ signaling by other Gi coupled receptors, indicating that the compound binds to the OXE-R to selectively block Gβγ signaling. This is difficult to explain based on current models of Gβγ activation, where Gα activation and dissociation is thought to be the mechanism for Gβγ activation, and implies a more active role of specific receptor-Gβγ interactions in regulation of activation of Gβγ pathways. The existence of such a mechanism implies that other specific receptor ligands can be developed that bias Gα vs. Gβγ pathways.
Small molecule G protein inhibitors in the treatment of cancer
There is considerable evidence for the utility of small molecule G protein α and βγ subunit inhibitors as potential therapeutic agents in a number of scenarios including heart failure, inflammation and pain (Table I) that have been discussed in other reviews[5, 46]. An interesting potential application of G protein inhibitors is in the treatment of various cancers that are under the control of a complex array of GPCR ligands regulating multiple steps in the development of primary tumors and metastasis[47, 48]. Because there are several reviews on the role of GPCRs and G proteins in cancer, I will briefly discuss key conceptual points that are relevant to considering G proteins as therapeutic targets in cancer, and consider the empirical data supporting the utility of small molecule G protein inhibitors in cancer treatment.
GPCRs in cancer
G protein signaling has an underappreciated role in contributing to cancer processes. Many GPCRs are overexpressed in various cancer cell types where they respond to autocrine and paracrine signals and regulate tumor growth and metastasis[47]. For example, many inflammatory mediators released in the tumor microenvironment such as IL-8 and prostaglandin E2 (PGE2) bind to overexpressed GPCRs on tumor cells to regulate tumor cell growth and survival [49, 50]. Thrombin-dependent activation of protease-activated receptors (PARs) promotes growth and survival as well as release of matrix metalloproteases involved in tumor escape and cell migration[51]. PGE2 and lysophosphatidic acid (LPA) contribute to angiogenesis important for oxygen delivery to growing tumor[52]. Chemokines secreted by stromal cells and metastatic target tissues are crucial for migration and invasion processes associated with metastatic cancer[53].
In addition to roles in responding to the local growth environment, a recent high throughput analysis for somatic mutations in human cancers revealed a significant prevalence of mutations in GPCRs including adhesion GPCRs, chemokine receptors and metabotropic glutamate receptors [54]. As examples, the adhesion family receptor brain specific angiogenesis inhibitor 1 (BAI1), and type 1 and 8 metabotropic glutamate receptors were found frequently mutated in lung squamous and adenocarcinomas. Many of these mutations are in the coding regions of these genes suggesting they may have functional effects leading to tumor initiation.
In each cancer type a different complex array of GPCRs and GPCR ligands are required at multiple levels including: tumor initiation, tumor cell growth, survival, escape migration and invasion. A strategy that targets individual GPCRs in some cases may inhibit cancer progression, but a broad-based strategy that targets common factors downstream of GPCRs may be more effective at multiple stages of cancer development.
G proteins in cancer
Mutations in G protein α subunits drive particular cancers. Gαs mutations have been found to be associated with endocrine adenomas [55]. The first report described an activating mutation of Gαs at R201 to either C or H[56]. Both of these mutations inhibit the GTP hydrolytic activity of the G protein αs subunit resulting a constitutively GTP bound G protein subunit and constitutive cAMP production. More recently, the same high throughput analysis cited above for GPCRs, found Gαs R201 mutations to be prevalent in pancreatic cancers (12%), and Gαs upregulated in 12% of ovarian cancers and a significant percentage of breast cancers [54]. Additionally, a functional mutation in Gαo at R243H was found at high prevelance found in breast cancers (5–15%) [54]. The Gαo at R243H mutation was analyzed functionally and shown to be constitutively active due to a high basal ability to exchange GDP for GTP [57]. Compounds such as suramin or BIM-46174 would be expected to inhibit cancers driven by exchange activated Gα mutants such as Gαo R243H but not GTPase deficient mutants such as Gαs R201.
In another recent study, somatic mutations in Gαq or Gα11 were found to be prevalent in certain types of melanomas [58, 59]. Ocular uveal melanomas in particular have a high prevelance (83%) of either Gαq or Gα11 mutations. Both Q209L and R183C mutations were identified in either Gαq or Gα11 with the Q209L mutation being more prevalent. As discussed above, YM-254890 inhibits signaling by R183C but not Q209L, indicating that it would be useful for treatment for tumors containing the R183C mutation.
RGS proteins
One could easily envision that if GPCRs can turn on G proteins to regulate various aspects of cancer, then other proteins that regulate G protein activity could also be involved in cancer. Indeed, alterations in levels of RGS proteins have been shown to be associated with various cancer states[60]. RGS2, for example is downregulated in androgen-resistant prostate cancer[61] and in acute myelogenous leukemia[62]. Downregulation of RGS2 would be expected to enhance signaling through Gq dependent GPCR signaling pathways. RGS4 is downregulated in human breast cancer tissue, and overexpression of RGS4 inhibits chemokine receptor-dependent cell migration and invasion[63]. RGS4 downregulation would be expected to enhance signaling through Gαq, Gαi and Gβγ-dependent signaling pathways. RGS protein single nucleotide polymorphisms (SNPs) are associated with lung and bladder cancers [60, 64].
In many cancers, dysregulation of GPCR signaling occurs with upregulation of multiple GPCRs and GPCR ligands, which may be combined with alterations in RGS protein levels as well. Additionally, as discussed above, GPCRs signaling is involved at multiple levels of the cancer process including proliferation, cell survival, tumor escape, cell migration, invasion and tissue homing. In this complex scenario, molecules that target common G proteins, downstream of multiple GPCRs/RGS proteins, would likely be more efficacious than targeting single GPCRs. Cancers caused by mutations in the G protein α subunits are rarer, but again, targeting the mutant G protein itself could be efficacious.
Evidence for efficacy of small molecule G protein-targeting in cancer
The pan Gα GDP-release inhibitor BIM-46174, in the low to mid μM range, inhibits the growth of multiple types of human cancer cell lines and drug-resistant cancer cell lines in culture[35]. This effect seems to be through a compound-dependent enhancement of apoptosis rather than through altering cell cycle and mitosis, indicating that GPCR ligands in the culture medium are enhancing survival. BIM-46174 also inhibits invasion of cancer cells in response to Wnt3a and neurotensin. In vivo intraperitoneal (IP) injection of BIM-46174 partially inhibited growth of human lung and pancreatic tumor xenografts in nude mice, and was strongly synergistic with the common chemotherapy drug cisplatin.
Gαq, Gα12 and Gα13 have all been shown to promote tumorigenesis, however; BIM-45174 does not inhibit migration or invasion of cancer cells driven by overexpression of constitutively active Gα12, Gα13 or free Gβγ. This supports the notion that the compound acts by inhibiting GPCR-dependent nucleotide exchange on Gα subunits, not by directly inhibiting downstream signaling by Gα. Inhibition of nucleotide exchange on Gα subunits blocks Gβγ signaling by preventing Gβγ release from the Gαβγ heterotrimer. Thus, inhibition of Gβγ signaling could by a mechanism by which Gα inhibitors block some aspects of cancer cell function.
Considerable evidence indicates that Gβγ-signaling plays a role in cancer processes. The pan βγ inhibitor, GRK2ct (βARKct) inhibits growth of prostate cancer cells by virtue of its ability to inhibit Gβγ-dependent activation of ERK[65]. Small molecule Gβγ-binding could, in theory, act similarly but βγ binding compounds identified thus far do not influence GPCR- and Gβγ-dependent ERK activation in cells[21]. However, as new molecules are discovered and characterized, it seems likely that inhibitors of Gβγ-dependent ERK activation will be identified.
Gβγ release from heterotrimeric G proteins initiates key signaling events downstream of chemokine receptors, which are Gi-coupled receptors involved in multiple aspects of cancer. These receptors regulate migration and invasion processes underlying metastasis [53], but are also involved in tumor cell survival and proliferation[47]. One study examined effects of the M119 class of Gβγ inhibitors on migration and invasion of an MDA-MB-231 breast cancer cell line in response to a mixture of ligands in NIH3T3 cell culture medium[66]. M119K (Figure 2D) (at low μM concentrations) strongly inhibited cell migration and invasion. Close examination of the cells showed that M119K inhibited Rac-dependent lamellipodia formation, consistent with the ability of the M119 class of compounds to inhibit Gβγ-dependent activation of the Rac exchange factor pREX [41, 67]. The compound also strongly inhibited migration and lamellipodia formation in response to the CXCR4 chemokine receptor ligand CXCL12 (SDF1α), but only weakly inhibited epidermal growth factor (EGF)-stimulated migration, demonstrating specificity for GPCR- dependent signaling pathways. A separate study examining the role of Gβγ in breast cancer cell growth showed that M119 significantly inhibited the growth rate of MDA-MB-231 cells [68]. This study went on to show that inhibition of Gβγ signaling by expression of Gβγ sequestering proteins inhibited tumor growth and lung metastasis in mice, but the effects of Gβγ inhibitory compounds were not examined in this in vivo setting. Although multiple studies have examined the efficacy of Gβγ inhibitory compounds in other therapeutic models in vivo (Table 1), there are as yet no studies examining effects of the M119 class or other βγ inhibitors in in vivo cancer models.
Table 1. In addition to cancer Gβγ is a potential target for therapeutics in multiple conditions.
Experiments with knockouts of Gβγ targets, protein based inhibitors of Gβγ including the c-terminus of GPCR kinase 2 (GRK2), Gβγ-binding peptides, and small molecules implicate Gβγ as a therapeutic target in multiple conditions.
| Therapeutic Condition | Knockout, GRK2ct or peptide data | M119/Gallein |
|---|---|---|
| Inflammation | Gβγ plays a critical role in leukocyte migration in response to chemoattractants [70] | Gallein blocks acute inflammatory responses [41] |
| Pain | Gβγ-dependent regulation of PLC may inhibit opioid signaling. Blockade of this pathway could increase the potency of μOR agonists [71] | M119 and gallein potentiate the analgesic actions of morphine and limit tolerance and dependence [21, 80] |
| Heart Failure | GRK2ct prevents overload hypertrophy [72–74] | M119 and gallein protect mice from heart failure in a chronic adrenergic receptor stimulation model and a calsequestrin model [81] |
| Hypertension | Vascular GRK2 overexpression causes HTN, GRK2ct reverses [75] | Not tested |
| Drug Addiction | GRK2ct and peptide based Gβγ inhibitors inhibit synergistic cooperativity of addictive drugs [76, 77] | Not tested |
| Arterial restenosis | Adenoviral delivery of GRK2ct prevents restenosis in animal models [79]. | Not tested |
Specificity issues with G protein inhibitors and cancer
Current treatments for cancer are notoriously non-specific to the point of being toxic. Newer treatments such as Gleevec have relatively high specificity but only work on a subset of cancers. If single GPCRs could be found that are the major driving factors in certain cancers, they would be ideal high-specificity anticancer targets to exploit in an emerging era of personalized medicine. In this review we have discussed how various cancers may be controlled by a wide array of factors such that a more efficacious approach is one that has broad specificity. In the case of targeting Gα subunits there is of course the danger that inhibiting individual Gα subunit pathways will have a range of effects beyond the cancer cells that are targeted but this consideration would clearly be outweighed by the benefits of potential controlling or eliminating cancers.
Similar considerations exist for Gβγ subunit signaling where Gβγ is central to the functions of all GPCRs. Several lines of evidence indicate that the Gβγ inhibition strategy is far more specific than might be presumed. One key to the specificity of this approach is that the small molecule Gβγ inhibitors that have been characterized do not block all Gβγ functions. For example, Gβγ inhibitors do not block activation of Gα subunits by GPCRs despite a requirement for Gβγ in this process. Another key to specificity is that most GPCRs do not signal downstream via Gβγ, rather GαGTP is a primary mediator in most cases. Finally, Gβγ inhibitors only block a subset of downstream targets, so for the relatively limited subset of responses mediated by Gβγ in select tissues, only some of those responses are inhibited by the Gβγ inhibitors identified thus far.
Concluding remarks
In this review I discussed the progress in small molecule G protein inhibitor development and the potential for G protein subunit inhibitors in the treatment of complex diseases such as cancer. Pan-Gα inhibitors may be well suited for cancer treatment, but for other therapies, selective inhibition of Gα pathways may be required. The identification of a selective Gq class of inhibitor indicates that Gα subunit subtype inhibitor development is possible, and the structural data indicate a way forward for development of novel Gα subunit subtype selective inhibitors. Gβγ inhibitors have more selectivity than pan Gα subunit inhibitors because Gβγ signaling is most often downstream of Gi-coupled receptors, and the Gβγ inhibitors discovered thus far only inhibit some signals downstream of Gβγ [5, 69]. Nevertheless, Gβγ subtype-selective inhibitors would be highly desirable. The structural basis for development of Gβγ subtype-selective inhibitors is not clear but such molecules might be discovered as novel Gβγ inhibitors emerge.
Overall, these types of data provide support for the emerging idea that GPCR signaling can be a major driver of tumor growth and metastases. Targeting specific GPCRs may represent novel avenues for treatment of cancer but targeting pathways downstream of receptors may also be effective and in some cases more effective in treating certain types of cancer.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pierce KL, et al. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3:639–650. doi: 10.1038/nrm908. [DOI] [PubMed] [Google Scholar]
- 2.Brunton LL, Chabner BA, Knollmann BC. The Pharmacological Basis of Therapeutics. McGraw-Hill; 2011. [Google Scholar]
- 3.Ruckle T, et al. PI3Kγ inhibition: towards an ‘aspirin of the 21st century’? Nat Rev Drug Discov. 2006;5:903–918. doi: 10.1038/nrd2145. [DOI] [PubMed] [Google Scholar]
- 4.Fishman MC, Porter JA. Pharmaceuticals: A new grammar for drug discovery. Nature. 2005;437:491–493. doi: 10.1038/437491a. [DOI] [PubMed] [Google Scholar]
- 5.Lin Y, Smrcka AV. Understanding molecular recognition by G protein βγ subunits on the path to pharmacological targeting. Mol Pharmacol. 2011;80:551–557. doi: 10.1124/mol.111.073072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilman AG. G proteins: transducers of receptor-generated signals. Ann Rev Biochem. 1987;56:615–649. doi: 10.1146/annurev.bi.56.070187.003151. [DOI] [PubMed] [Google Scholar]
- 7.Ross EM, Wilkie TM. GTPase-activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS-like proteins. Annual Review of Biochemistry. 2000;69:795–827. doi: 10.1146/annurev.biochem.69.1.795. [DOI] [PubMed] [Google Scholar]
- 8.Shenoy SK, Lefkowitz RJ. β-arrestin-mediated receptor trafficking and signal transduction. Trends in Pharmacological Sciences. 2011;32:521–533. doi: 10.1016/j.tips.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sjogren B, Neubig RR. Thinking Outside of the “RGS Box”: New Approaches to Therapeutic Targeting of Regulators of G Protein Signaling. Molecular Pharmacology. 2010;78:550–557. doi: 10.1124/mol.110.065219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kimple AJ, et al. Regulators of G-Protein Signaling and Their Gα Substrates: Promises and Challenges in Their Use as Drug Discovery Targets. Pharmacol Rev. 2011;63:728–749. doi: 10.1124/pr.110.003038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noor N, et al. β-Arrestin: A signaling molecule and potential therapeutic target for heart failure. Journal of Molecular and Cellular Cardiology. 2011;51:534–541. doi: 10.1016/j.yjmcc.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rengo G, et al. GRK2 as a novel gene therapy target in heart failure. Journal of Molecular and Cellular Cardiology. 2011;50:785–792. doi: 10.1016/j.yjmcc.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kamal FA, et al. Taking the heart failure battle inside the cell: Small molecule targeting of Gβγ subunits. Journal of Molecular and Cellular Cardiology. 2011;51:462–467. doi: 10.1016/j.yjmcc.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blumer JB, et al. Mechanistic pathways and biological roles for receptor-independent activators of G-protein signaling. Pharmacology & therapeutics. 2007;113:488–506. doi: 10.1016/j.pharmthera.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scott JK, et al. Evidence that a protein-protein interaction ‘hot spot’ on heterotrimeric G protein βγ subunits is used for recognition of a subclass of effectors. EMBO J. 2001;20:767–776. doi: 10.1093/emboj/20.4.767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen J, et al. A region of adenylyl cyclase 2 critical for regulation by G protein βγ subunits. Science. 1995;268:1166–1169. doi: 10.1126/science.7761832. [DOI] [PubMed] [Google Scholar]
- 17.Wall MA, et al. The structure of the G protein heterotrimer Giα1β1γ2. Cell. 1995;83:1047–1058. doi: 10.1016/0092-8674(95)90220-1. [DOI] [PubMed] [Google Scholar]
- 18.Lambright DG, et al. The 2.0 Å crystal structure of a heterotrimeric G protein. Nature. 1996;379:311–319. doi: 10.1038/379311a0. [DOI] [PubMed] [Google Scholar]
- 19.Sondek J, et al. Crystal structure of a G-protein βγ dimer at 2.1Å resolution. Nature. 1996;379:369–374. doi: 10.1038/379369a0. [DOI] [PubMed] [Google Scholar]
- 20.Davis TL, et al. Structural Definition of a Preferred Protein Interaction Site in the G protein β1γ2 heterodimer. Biochemistry. 2005;44:10593–10604. doi: 10.1021/bi050655i. [DOI] [PubMed] [Google Scholar]
- 21.Bonacci TM, et al. Differential Targeting of Gβγ-Subunit Signaling with Small Molecules. Science. 2006;312:443–446. doi: 10.1126/science.1120378. [DOI] [PubMed] [Google Scholar]
- 22.Sprang SR. G protein mechanisms:Insights from structural analysis. Ann Rev Biochem. 1997;66:639–678. doi: 10.1146/annurev.biochem.66.1.639. [DOI] [PubMed] [Google Scholar]
- 23.Lambright DG, et al. Structural determinants for activation of the α-subunit of a heterotrimeric G protein. Nature. 1994;369:621–628. doi: 10.1038/369621a0. [DOI] [PubMed] [Google Scholar]
- 24.Rosenbaum DM, et al. The structure and function of G-protein-coupled receptors. Nature. 2009;459:356–363. doi: 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oldham WM, Hamm HE. How do Receptors Activate G Proteins? In: Stephen RS, editor. Advances in Protein Chemistry. Academic Press; 2007. pp. 67–93. [DOI] [PubMed] [Google Scholar]
- 26.Rasmussen SGF, et al. Crystal structure of the β2 adrenergic receptor-Gs protein complex. Nature. 2011;477:549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gilchrist A, et al. Gα COOH-Terminal Minigene Vectors Dissect Heterotrimeric G Protein Signaling. Sci STKE. 2002:pl1. doi: 10.1126/stke.2002.118.pl1. [DOI] [PubMed] [Google Scholar]
- 28.Chung KY, et al. Conformational changes in the G protein Gs induced by the β2 adrenergic receptor. Nature. 2011;477:611–615. doi: 10.1038/nature10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Westfield GH, et al. Structural flexibility of the Gαs α-helical domain in the β2-adrenoceptor Gs complex. Proc Natl Acad Sci U S A. 2011;108:16086–16091. doi: 10.1073/pnas.1113645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Eps N, et al. Interaction of a G protein with an activated receptor opens the interdomain interface in the alpha subunit. Proc Natl Acad Sci U S A. 2011;108:9420–9424. doi: 10.1073/pnas.1105810108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arkin MR, Whitty A. The road less traveled: modulating signal transduction enzymes by inhibiting their protein-protein interactions. Current opinion in chemical biology. 2009;13:284–290. doi: 10.1016/j.cbpa.2009.05.125. [DOI] [PubMed] [Google Scholar]
- 32.Lodowski DT, et al. Keeping G Proteins at Bay: A Complex Between G Protein-Coupled Receptor Kinase 2 and Gβγ. Science. 2003;300:1256–1262. doi: 10.1126/science.1082348. [DOI] [PubMed] [Google Scholar]
- 33.Freissmuth M, et al. Suramin analogues as subtype-selective G protein inhibitors. Mol Pharmacol. 1996;49:602–611. [PubMed] [Google Scholar]
- 34.Hohenegger M, et al. Gsα-selective G protein antagonists. Proc Natl Acad Sci U S A. 1998;95:346–351. doi: 10.1073/pnas.95.1.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prévost GP, et al. Anticancer Activity of BIM-46174, a New Inhibitor of the Heterotrimeric Gα/Gβγ Protein Complex. Cancer Research. 2006;66:9227–9234. doi: 10.1158/0008-5472.CAN-05-4205. [DOI] [PubMed] [Google Scholar]
- 36.Ayoub MA, et al. Inhibition of heterotrimeric G protein signaling by a small molecule acting on Gα subunit. J Biol Chem. 2009;284 doi: 10.1074/jbc.M109.042333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takasaki J, et al. A Novel Gαq/11-selective Inhibitor. J Biol Chem. 2004;279:47438–47445. doi: 10.1074/jbc.M408846200. [DOI] [PubMed] [Google Scholar]
- 38.Kawasaki T, et al. Antithrombotic and thrombolytic efficacy of YM-254890, a G q/11 inhibitor, in a rat model of arterial thrombosis. Thrombosis and haemostasis. 2003;90:406–413. doi: 10.1160/TH03-02-0115. [DOI] [PubMed] [Google Scholar]
- 39.Nishimura A, et al. Structural basis for the specific inhibition of heterotrimeric Gq protein by a small molecule. Proc Natl Acad Sci U S A. 2010;107:13666–13671. doi: 10.1073/pnas.1003553107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bonacci TM, et al. Differential targeting of Gβγ-subunit signaling with small molecules. Science. 2006;312:443–446. doi: 10.1126/science.1120378. [DOI] [PubMed] [Google Scholar]
- 41.Lehmann DM, et al. Small Molecule Disruption of G Protein βγ Subunit Signaling Inhibits Neutrophil Chemotaxis and Inflammation. Mol Pharmacol. 2008;73:410–418. doi: 10.1124/mol.107.041780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smrcka AV, et al. NMR analysis of G-protein βγ subunit complexes reveals a dynamic Gα-Gβγ subunit interface and multiple protein recognition modes. Proc Natl Acad Sci USA. 2010;107:639–644. doi: 10.1073/pnas.0909503107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ghosh M, et al. Receptor and nucleotide exchange independent mechanisms for promoting G protein subunit dissociation. J Biol Chem. 2003;273:34747–34750. doi: 10.1074/jbc.C300271200. [DOI] [PubMed] [Google Scholar]
- 44.Goubaeva F, et al. Stimulation of cellular signaling and G protein subunit dissociation by G protein βγ subunit binding peptides. J Biol Chem. 2003;278:19634–19641. doi: 10.1074/jbc.M300052200. [DOI] [PubMed] [Google Scholar]
- 45.Blattermann S, et al. A biased ligand for OXE-R uncouples Galpha and Gbetagamma signaling within a heterotrimer. Nature chemical biology. 2012;8:631–638. doi: 10.1038/nchembio.962. [DOI] [PubMed] [Google Scholar]
- 46.Smrcka AV, et al. G protein βγ subunits as targets for small molecule therapeutic development. Comb Chem High Throughput Screen. 2008;11:382–395. doi: 10.2174/138620708784534761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dorsam RT, Gutkind JS. G-protein-coupled receptors and cancer. Nat Rev Cancer. 2007:7. doi: 10.1038/nrc2069. [DOI] [PubMed] [Google Scholar]
- 48.Lappano R, Maggiolini M. G protein-coupled receptors: novel targets for drug discovery in cancer. Nat Rev Drug Discov. 2011;10:47–60. doi: 10.1038/nrd3320. [DOI] [PubMed] [Google Scholar]
- 49.Chang SH, et al. The prostaglandin E2 receptor EP2 is required for cyclooxygenase 2-mediated mammary hyperplasia. Cancer Res. 2005;65:4496–4499. doi: 10.1158/0008-5472.CAN-05-0129. [DOI] [PubMed] [Google Scholar]
- 50.Keane MP, et al. Depletion of CXCR2 inhibits tumor growth and angiogenesis in a murine model of lung cancer. J Immunol. 2004;172:2853–2860. doi: 10.4049/jimmunol.172.5.2853. [DOI] [PubMed] [Google Scholar]
- 51.Boire A, et al. PAR1 Is a Matrix Metalloprotease-1 Receptor that Promotes Invasion and Tumorigenesis of Breast Cancer Cells. Cell. 2005;120:303–313. doi: 10.1016/j.cell.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 52.Mills GB, Moolenaar WH. The emerging role of lysophosphatidic acid in cancer. Nat Rev Cancer. 2003;3:582–591. doi: 10.1038/nrc1143. [DOI] [PubMed] [Google Scholar]
- 53.Müller A, et al. Involvement of Chemokine Receptors in Breast Cancer Metastasis. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 54.Kan Z, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869–873. doi: 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- 55.Vallar L, et al. Altered Gs and adenylate cyclase activity in human GH-secreting pituitary adenomas. Nature. 1987;330:566–568. doi: 10.1038/330566a0. [DOI] [PubMed] [Google Scholar]
- 56.Weinstein LS, et al. Activating Mutations of the Stimulatory G Protein in the McCune–Albright Syndrome. New England Journal of Medicine. 1991;325:1688–1695. doi: 10.1056/NEJM199112123252403. [DOI] [PubMed] [Google Scholar]
- 57.Garcia-Marcos M, et al. Molecular basis of a novel oncogenic mutation in GNAO1. Oncogene. 2011;30:2691–2696. doi: 10.1038/onc.2010.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Van Raamsdonk CD, et al. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457:599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Van Raamsdonk CD, et al. Mutations in GNA11 in Uveal Melanoma. New England Journal of Medicine. 2010;363:2191–2199. doi: 10.1056/NEJMoa1000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hurst JH, Hooks SB. Regulator of G-protein signaling (RGS) proteins in cancer biology. Biochemical pharmacology. 2009;78:1289–1297. doi: 10.1016/j.bcp.2009.06.028. [DOI] [PubMed] [Google Scholar]
- 61.Cao X, et al. Regulator of G-protein signaling 2 (RGS2) inhibits androgen-independent activation of androgen receptor in prostate cancer cells. Oncogene. 2006;25:3719–3734. doi: 10.1038/sj.onc.1209408. [DOI] [PubMed] [Google Scholar]
- 62.Schwäble J, et al. RGS2 is an important target gene of Flt3-ITD mutations in AML and functions in myeloid differentiation and leukemic transformation. Blood. 2005;105:2107–2114. doi: 10.1182/blood-2004-03-0940. [DOI] [PubMed] [Google Scholar]
- 63.Xie Y, et al. Breast Cancer Migration and Invasion Depend on Proteasome Degradation of Regulator of G-Protein Signaling 4. Cancer Research. 2009;69:5743–5751. doi: 10.1158/0008-5472.CAN-08-3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Berman DM, et al. A Functional Polymorphism in RGS6 Modulates the Risk of Bladder Cancer. Cancer Research. 2004;64:6820–6826. doi: 10.1158/0008-5472.CAN-04-1916. [DOI] [PubMed] [Google Scholar]
- 65.Bookout AL, et al. Targeting Gβγ Signaling to Inhibit Prostate Tumor Formation and Growth. J Biol Chem. 2003;278:37569–37573. doi: 10.1074/jbc.M306276200. [DOI] [PubMed] [Google Scholar]
- 66.Kirui JK, et al. Gβγ signaling promotes breast cancer cell migration and invasion. J Pharmacol Exp Ther. 2010;333:393–403. doi: 10.1124/jpet.109.164814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao T, et al. Signaling requirements for translocation of P-Rex1, a key Rac2 exchange factor involved in chemoattractant-stimulated human neutrophil function. Journal of Leukocyte Biology. 2007;81:1127–1136. doi: 10.1189/jlb.0406251. [DOI] [PubMed] [Google Scholar]
- 68.Tang X, et al. A Critical Role of G®© in Tumorigenesis and Metastasis of Breast Cancer. J Biol Chem. 2011;286:13244–13254. doi: 10.1074/jbc.M110.206615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Smrcka AV. G protein βγ subunits: central mediators of G protein-coupled receptor signaling. Cell Mol Life Sci. 2008;65:2191–2214. doi: 10.1007/s00018-008-8006-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li Z, et al. Roles of PLCβ2 and β3 and PI3K in Chemoattractant-Mediated Signal Transduction. Science. 2000;287:1046–1049. doi: 10.1126/science.287.5455.1046. [DOI] [PubMed] [Google Scholar]
- 71.Xie W, et al. Genetic alteration of phospholipase Cβ3 expression modulates behavioral and cellular responses to μ opioids. Proc Natl Acad Sci U S A. 1999;96:10385–10390. doi: 10.1073/pnas.96.18.10385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Akhter SA, et al. Restoration of beta-adrenergic signaling in failing cardiac ventricular myocytes via adenoviral-mediated gene transfer. Proc Natl Acad Sci U S A. 1997:94. doi: 10.1073/pnas.94.22.12100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rockman HA, et al. Expression of a β-adrenergic receptor kinase 1 inhibitor prevents the development of myocardial failure in gene-targeted mice. Proc Natl Acad Sci U S A. 1998;95:7000–7005. doi: 10.1073/pnas.95.12.7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Koch WJ, et al. Cardiac function in mice overexpressing the β-adrenergic receptor kinase or a βARK inhibitor. Science. 1995;268:1350–1353. doi: 10.1126/science.7761854. [DOI] [PubMed] [Google Scholar]
- 75.Eckhart AD, et al. Vascular-targeted overexpression of G protein-coupled receptor kinase-2 in transgenic mice attenuates beta-adrenergic receptor signaling and increases resting blood pressure. Molecular Pharmacology. 2002:61. doi: 10.1124/mol.61.4.749. [DOI] [PubMed] [Google Scholar]
- 76.Yao L, et al. Addicting drugs utilize a synergistic molecular mechanism in common requiring adenosine and Gi-βγ dimers. Proc Natl Acad Sci US A. 2003;100:14379–14384. doi: 10.1073/pnas.2336093100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yao L, et al. betagamma Dimers mediate synergy of dopamine D2 and adenosine A2 receptor-stimulated PKA signaling and regulate ethanol consumption. Cell. 2002;109:733–743. doi: 10.1016/s0092-8674(02)00763-8. [DOI] [PubMed] [Google Scholar]
- 78.Daaka Y. G proteins in cancer: the prostate cancer paradigm. Sci STKE. 2004 doi: 10.1126/stke.2162004re2. [DOI] [PubMed] [Google Scholar]
- 79.Iaccarino G, Koch WJ. Transgenic mice targeting the heart unveil G protein-coupled receptor kinases as therapeutic targets. Assay Drug Dev Technol. 2003;1:347–355. doi: 10.1089/154065803321204484. [DOI] [PubMed] [Google Scholar]
- 80.Mathews JL, et al. A Novel Gβγ Subunit Inhibitor Selectively Modulates μ-Opioid-Dependent Antinociception and Attenuates Acute Morphine-Induced Antinociceptive Tolerance and Dependence. J Neurosci. 2008;28:12183–12189. doi: 10.1523/JNEUROSCI.2326-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Casey LM, et al. Small molecule disruption of G βγ signaling inhibits the progression of heart failure. Circ Res. 2010;107:532–539. doi: 10.1161/CIRCRESAHA.110.217075. [DOI] [PMC free article] [PubMed] [Google Scholar]