

Abstract
This chapter is focused on the iron metallome in eukaryotes at the cellular and subcellular level, including properties, utilization in metalloproteins, trafficking, storage, and regulation of these processes. Studies in the model eukaryote Saccharomyces cerevisiae and mammalian cells will be highlighted. The discussion of iron properties will center on the speciation and localization of intracellular iron as well as the cellular and molecular mechanisms for coping with both low iron bioavailability and iron toxicity. The section on iron metalloproteins will emphasize heme, iron-sulfur cluster, and non-heme iron centers, particularly their cellular roles and mechanisms of assembly. The section on iron uptake, trafficking, and storage will compare methods used by yeast and mammalian cells to import iron, how this iron is brought into various organelles, and types of iron storage proteins. Regulation of these processes will be compared between yeast and mammalian cells at the transcriptional, post-transcriptional, and post-translational levels.
Keywords: eukaryote, heme, iron metallome, iron-sulfur cluster, iron trafficking, metal homeostasis
1 Introduction
Organisms use a variety of transition metals as catalytic centers in proteins, including iron, copper, manganese, and zinc. Iron is well suited to redox reactions due to its capability to act as both an electron donor and acceptor. In eukaryotic cells, iron is a cofactor for a wide variety of metalloproteins involved in energy metabolism, oxygen binding, DNA biosynthesis and repair, synthesis of biopolymers, cofactors, and vitamins, drug metabolism, antioxidant function, and many others. Because iron is so important for survival, organisms utilize several techniques to optimize uptake and storage to ensure maintenance of sufficient levels for cellular requirements. However, the redox properties of iron also make it extremely toxic if cells have excessive amounts. Free iron can catalyze the formation of reactive oxygen species such as the hydroxyl radical, which in turn can damage proteins, lipids, membranes, and DNA. Cells must maintain a delicate balance between iron deficiency and iron overload that involves coordinated control at the transcriptional, post-transcriptional, and post-translational levels to help fine tune iron utilization and iron trafficking.
This review will focus on the iron metallome in eukaryotes from uptake to regulation. The physicochemical properties will be investigated, including bioavailability for uptake, chemical forms and subcellular locations of intracellular iron, and mechanisms of toxicity. In addition, the function and formation of iron metalloproteins such as heme and iron-sulfur clusters will be considered. Uptake systems, iron chaperones, and storage systems in yeast and mammalian cells will be compared. Regulation of these processes at the gene, mRNA, and proteins levels will be examined, again comparing yeast and mammalian cells.
2 Physicochemical Properties of the Iron Metallome
2.1 Intracellular Concentrations, Oxidation State, and Speciation
Iron is the most abundant metal on Earth, thus it is not surprising that almost all organisms have evolved to exploit the unique chemical properties of this ubiquitous transition metal. Iron primarily exists in either the ferrous (Fe2+) or ferric (Fe3+) oxidation state in biological systems. Due to its critical role in cell metabolism, iron constitutes a significant portion of the eukaryotic metallome [1]. Intracellular iron concentrations vary with cell type, environmental conditions, and disease state. The iron concentration of human erythroid cells was measured at 300–400 μM [2], while isolated rat hepatocytes maintain iron concentrations close to 1 mM [3]. Iron overload diseases caused by mutations in iron handling proteins can lead to 10- to 20-fold increases in these intracellular iron levels in specific tissues [4–6]. The local bioavailability of iron also strongly influences intracellular concentrations. For example, analysis of the single-celled model eukaryote S. cerevisiae demonstrated intracellular iron concentrations ranging from 250 μM to 600 μM depending on the iron content of the growth medium [1,7].
To better study the iron metallome, biophysical probes such as Mössbauer and electron paramagnetic resonance (EPR) have been recently employed to measure not only the absolute iron concentration, but also the types of iron and how this varies within specific organelles [8]. Lindahl and colleagues have used an integrated biophysical approach to characterize the iron speciation in S. cerevisiae whole cells and organelles under several growth conditions [7,9–11]. These studies clearly demonstrate that the mitochondria and vacuole are the two central hubs of iron metabolism in this organism. In general, yeast mitochondria contain 700–800 μM Fe. In respiring cells, most of this mitochondrial iron is present as the prosthetic groups of the respiratory complexes (~70% [4Fe-4S]2+ clusters and heme centers), with the remaining iron present as [2Fe-2S]1+ clusters in enzymes and as non-heme, high spin Fe2+ ions. Conversely, in fermenting cells the iron from respiratory complexes is reduced to ~30% of the total iron, there is an increase in non-heme iron, and the appearance of ferric phosphate nanoparticles. Mutations in Fe-S cluster assembly and trafficking proteins leads to increased concentration of these nanoparticles with a concomitant rise in reactive oxygen species [7,11]. The differences in iron speciation in each of these different situations are readily apparent from the Mössbauer spectra (Figure 1). The other major iron repository in yeast is the vacuole. Vacuoles isolated from fermenting yeast contain an average of 220 μM Fe in the ferric iron, which is expected given the acidic environment of this organelle (pH ~ 5). Vacuolar iron is present in both a soluble Fe(III) complex and insoluble, magnetically-interacting Fe(III) nanoparticles, which interconvert based on changes in pH [9].
Figure 1.

Mössbauer spectra comparing differing iron species found in (a) respiring mitochondria, (b) fermenting mitochondria, (c) Atm1-depleted mitochondria, and (d) whole fermenting yeast cells. (reprinted with permission from ref [8])
Characterization of iron speciation in other organelles and organisms is still in the initial stages as most published studies are based on a single technique, instead of verification by several methods. In addition, various techniques have been used to study different organisms, making comparison of results challenging. Iron can be found in a variety of different forms based on location (and thus pH and redox potential), available ligands, and cellular need. The integrated approach described above is one of the most promising for studying the iron metallome: by combining Mössbauer, EPR, X-ray absorption spectroscopy (XAS), electronic absorption spectroscopy, and electron microscopy, one can resolve different groups of iron species (such as Fe-S clusters, hemes, and nanoparticles) at a relatively low concentration. In particular, XAS techniques such as X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) give information about oxidation state, geometry, and ligation. All forms of iron can be detected and quantified, so changing levels of species can also be monitored [12].
Another promising method for future studies is X-ray fluorescence microscopy (XRFM), which provides information about metal distribution, oxidation state, and coordination. XRFM offers high spatial resolution of biological samples by detection of emitted X-rays from the sample after irradiation. Pairing this with XANES can provide more information about iron speciation and subcellular distribution. A combination of XRFM and XAS studies on brain tissue from Alzheimer’s disease patients showed an increased concentration of iron found mainly in the oxidized form [13]. Fluorescence intensity from XRFM studies is directly proportional to the element concentration, providing some quantitative analysis of samples. Quantification of transition metals in cells and organelles can also be accomplished using particle-induced X-ray emission (PIXE). Like XRFM, PIXE analysis detects X-rays that are emitted which are characteristic of elements in the sample. This technique is capable of detecting and quantifying trace elements (including Fe, Mn, Zn, and Cu) in the μg/g range [12].
2.2 Subcellular Distribution
As mentioned above, the majority of cellular iron is found in the mitochondria and the cytosol for utilization in iron-dependent proteins. While yeast store excess iron in the vacuole, mammals express iron storage proteins such as ferritin and mitochondrial ferritin for this purpose. In addition, iron is recycled in lysosomes after iron-containing proteins are degraded. For example, human liver and spleen cells from patients with hemochromatosis (an iron overload disease) were found to contain iron-loaded lysosomes (siderosomes), hemosiderin (a degradation product of ferritin), and ferritin [14]. There has not been a significant amount of research focused on the concentration and chemical nature of iron in the endoplasmic reticulum (ER). However, the existence of iron pools in this organelle is likely since a number of heme and non-heme iron proteins are located in the ER. For more information on iron-containing organelles and iron storage proteins, refer to Sections 4.2 and 4.3.
2.3 Bioavailability, Intracellular Labile Iron Pools
2.3.1 Iron Bioavailability
Although iron is one of the most abundant elements on Earth, the environment is usually oxygenated, non-acidic, and aqueous. Under these conditions, extracellular iron is predominantly found in the poorly soluble ferric (Fe3+) state. One way that organisms such as yeast improve iron bioavailability is by acidifying the local environment. The solubility of ferric iron is pH-dependent, changing from 10−18 M at pH 7 to 10−3 M at pH 2. By lowering the pH of the surrounding environment, organisms facilitate solubilization and uptake of iron. ATP-driven proton transporters move H+ ions from the cytosol across the plasma membrane to control the pH at the cell surface [15]. Humans also use an acidic environment to facilitate uptake of dietary iron. Uptake mainly occurs though enterocytes in the duodenum, which receives the acidic contents of the stomach. Iron can then be absorbed for storage in intestinal cells or delivery to other cells [15,16].
Many microorganisms, including some fungi, also secrete low molecular weight compounds known as siderophores into their surroundings, which form high-affinity (~10−33 M) complexes with ferric iron to make it bioavailable for uptake. Transporters on the cell surface then recapture the Fe3+-siderophores complexes. For infectious microorganisms, these molecules help the invading pathogen acquire iron from the host for survival. Interestingly, two recent reports suggest that mammalian cells may also synthesize their own siderophores [17,18]. In both cases, the siderophore-like compound was isolated by screening for molecules that bound to siderocalins, a class of lipocalins that specifically bind exogenous siderophores. Siderocalins are weapons in the immune system arsenal, designed to prevent the invading organisms from acquiring iron by sequestering Fe3+-bound siderophores [19]. However, these new studies suggest that siderocalins may also bind Fe3+ complexed with endogenous siderophores to facilitate iron trafficking. The candidate endogenous siderophore-like compounds isolated include catechol and catechol-like compounds [17], as well as a molecule with a 2,5-dihydroxybenzoic acid (2,5-DHBA) iron-binding moiety, which is isomeric to 2,3-DHBA found in the bacterial siderophore enterobactin (Figure 2) [18]. BDH2, a homologue of bacterial EntA (which catalyzes 2,3-DHBA production), was found to be responsible for 2,5-DHBA production. Knockdowns of BDH2 suggested that the 2,5-DHBA-containing mammalian siderophore is involved in regulating both cytosolic and mitochondrial iron levels [18].
Figure 2.
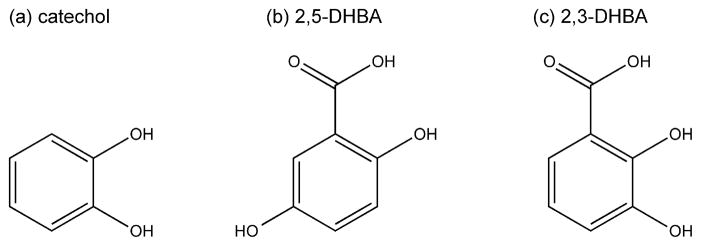
Structural comparison of Fe3+-binding siderophore moieties: (a) catechol and (b) 2,5-DHBA found in mammalian cells, and (c) 2,3-DHBA found in bacterial enterobactin.
2.3.2 Intracellular Labile Iron Pools
The vast majority of iron is bound to proteins and enzymes for use as a cofactor (for more information, see Section 3) and stored in ferritin, vacuoles, and lysosomes. The remaining iron in the cell is proposed to be part of a labile iron pool (LIP), also known as chelatable or free iron, which is most likely present as ferrous complexes given the neutral pH and reducing conditions inside the cell. The LIP is thought to constitute only 0.2–3% of total cellular iron [20]. Studies suggest that the LIP acts as a crossroads in iron trafficking, providing iron for incorporation into metalloenzymes, feeding pathways for heme and Fe-S biosynthesis, and directing excess iron towards storage or export proteins [21]. The LIP is also assumed to be dynamic in nature, shrinking and growing in response to the needs of the cell. Only recently has research focused on uncovering the chemical nature of iron in the cytosolic LIP [22]. At physiological conditions, small molecular weight ligands such as phosphates, citrate, cysteine, and glutathione (GSH) are available to bind Fe(II) [20]. While the predominant ligand for the LIP remains an open question, potentiometric and binding affinity studies suggest that only GSH is present at high enough concentrations with sufficient binding affinity to buffer the labile Fe(II) pool, forming the proposed pentaaqua-Fe(II)-GSH complex shown in Figure 3. This iron-GSH complex is suggested to be a way for cells to distinguish between Fe(II) and Mn(II), which have similar intracellular concentrations. In addition, the Fe(II)-GSH complex may play a role in iron trafficking based on the interaction of GSH with monothiol glutaredoxins, which are essential for iron regulation and trafficking (see Section 4.1) [23].
Figure 3.

Proposed structure of the glutathione-coordinating complex with Fe(II) found in the labile iron pool.
There is some evidence that a labile iron pool exists in individual organelles as well as the cytosol. Using fluorescent indicators, the LIP in mammalian mitochondria was measured between 1 and 16 μM depending on the cell type, which constitutes <0.4% of total mitochondrial iron [24–26]. Studies on mitochondrial iron speciation in yeast suggest that a somewhat larger pool of non-heme, high spin Fe2+ is used in assembly of hemes and Fe-S clusters. The exact nature of this iron is still unknown, although it is presumably loosely bound by low molecular weight ligands similar to the cytosol. In actively respiring mitochondria, this pool constitutes ~2% of total mitochondrial iron, but grows to 20% during fermentation when the rate of Fe-S cluster and heme biosynthesis decreases. Since the total mitochondrial iron in yeast is nearly identical in respiring vs. fermenting cells, these results demonstrate dynamic shifts in subcellular iron speciation rather than mitochondrial iron import in response to changes in energy metabolism [10].
2.4 Mechanisms of Iron Toxicity
2.4.1 Oxidative Stress and Formation of ROS
Although iron is required for many cellular processes, excess iron levels can be toxic to cells. Iron has a central role in the production of one of the most reactive oxygen species (ROS) found in the cell, namely the hydroxyl radical (HO•). Intracellular iron catalyzes formation of HO• non-enzymatically by reacting with the superoxide anion (O2•−) (Eq. 1) and hydrogen peroxide (H2O2) (Eq. 2) [27].
| (1) |
| (2) |
| (3) |
ROS such as H2O2 and O2•− are produced naturally in vivo through enzymatic reactions and auto-oxidation from endogenous compounds and have well-documented roles in signal transduction pathways and immune cell response [28]. However, when left unchecked, these molecules together with HO• have the ability to initiate oxidative damage to DNA, lipids, and proteins, all of which contribute to cell death, aging, and various diseases. Thus iron overload diseases are often characterized by elevated levels of biomarkers for oxidative stress, including protein carbonyls, DNA oxidation products, lipid peroxidation, advanced glycation end products, and malondialdehyde formation. Accumulation of iron in the brain coupled with oxidative stress is also a common feature of neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease [29].
2.4.2 Iron Interference in Other Metal Trafficking Pathways
Recent studies also suggest that iron toxicity may not be solely due to iron-catalyzed ROS formation. Kaplan and coworkers demonstrated that toxicity is not dependent on the presence of oxygen, since iron is toxic to yeast even under anaerobic growth conditions [30]. Alternatively iron toxicity may stem from the interference of excess iron in other metal trafficking pathways. Strong evidence for this hypothesis is the effect of excess iron on manganese trafficking to the antioxidant enzyme superoxide dismutase (SOD). Eukaryotic cells express an SOD in the mitochondria, SOD2, which preferentially binds manganese over iron under normal conditions. SOD2 is an essential antioxidant enzyme since deletion of the SOD2 gene leads to neonatal lethality in mouse models [31,32]. SOD2 activity requires the correct insertion of manganese into the enzyme, while misincorporation of iron renders it inactive [33,34]. Studies of SOD2 mismetallation in yeast revealed the presence of two distinct iron pools in the mitochondria, one being “SOD2-inert” and the other “SOD2-reactive”. Disruption of iron homeostasis increases the reactive pool (without significantly affecting total mitochondrial iron), allowing for iron incorporation into SOD2. In particular, disruptions in the late stages of mitochondrial Fe-S cluster biogenesis led to diversion of iron to SOD2 (see Section 3.2.2). A somewhat similar situation was observed in a mouse model of the iron overload disease hereditary hemochromatosis, albeit via a different mechanism. In this case, cytosolic iron overaccumulation was found to disrupt trafficking of copper, zinc, and manganese to mitochondria, leading to deficiencies of these essential metals in this organelle. Consequently, Mn-SOD2 activity was significantly reduced leading to lower respiratory activity and increased lipid peroxidation [35].
3 Iron Metalloproteins
3.1 Types of Iron Metalloproteins
3.1.1 Heme-Containing Proteins
Organisms utilize heme-containing proteins for a variety of processes, including sensing and transport of oxygen, energy metabolism, transcriptional regulation, and protein stability. Heme consists of iron bound to a porphyrin ring (shown in Figure 4), where the iron can act as an electron source or sink for redox and electron transfer processes. In mammals, heme is one of the most important iron cofactors. It is best known as an oxygen carrier when bound to hemoglobin in red blood cells. Cytochromes are hemoproteins involved in electron transfer reactions. For example, cytochrome c transfers electrons between Complexes III and IV in the electron transport chain of the mitochondria. The cytochrome P450 enzyme family catalyzes the oxidation of many organic compounds, including lipids, hormones, and xenobiotics. Catalases and peroxidases are both hemoprotein families that protect against peroxide damage. Catalase prevents H2O2 damage by catalyzing its decomposition to H2O and O2. Peroxidases use heme to convert peroxides into alcohols using electron donors and protons, again to prevent damage caused by reactive peroxides. Recently, it was shown that the nuclear receptor Rev-erbα binds heme and regulates circadian rhythmicity as well as other metabolic pathways [36].
Figure 4.

Heme cofactor. Structure of protoporphyrin IX with ferrous iron inserted.
3.1.2 Iron-Sulfur Cluster-Containing Proteins
Similar to the heme cofactor, organisms employ iron in the iron-sulfur cluster cofactor for its versatility in electron transfer reactions, with redox potentials ranging from −500 to +300 mV. Iron sulfur clusters form as complexes of iron (Fe2+ or Fe3+) and inorganic sulfide (S2−) in various arrangements, with two of the most common being [2Fe-2S] and [4Fe-4S] clusters (Figure 5). Certain prokaryotes and cyanobacteria also utilize larger, more complex clusters that incorporate additional metals such as molybdenum, nickel, and vanadium. For example, the nitrogenase enzyme contains a FeMoco cofactor cluster (MoFe7S9) and a P cluster (Fe8S7) in addition to a [4Fe-4S] cluster in the heterodimer form [37]. Evidence for these types of complex clusters in higher eukaryotes is lacking. Fe-S proteins can also convert between cluster forms. The enzyme aconitase is active in the [4Fe-4S] cluster form, while partial disassembly to a [3Fe-4S] cluster renders it reversibly inactive. Fe-S clusters are usually bound to proteins via coordination of the iron to sulfur from cysteine and nitrogen from histidine, although serine and arginine have also been shown to ligate Fe-S clusters. In addition to binding to protein residues, the iron can bind other small molecules such as GSH, homocitrate, CO, and CN−. Recent studies also demonstrate that a carbon atom coordinates all 6 Fe at the core of the FeMoco cluster in nitrogenase from nitrogen-fixing bacteria [38,39]. In addition to their well-known redox function in electron transfer reactions, Fe-S clusters are also involved in heme biosynthesis, DNA synthesis and repair, ribosome assembly, tRNA modification, nucleotide and amino acid metabolism, and biogenesis of Fe-S proteins [40].
Figure 5.

Common forms of iron-sulfur cluster cofactors: (a) [2Fe-2S] and (b) [4Fe-4S].
3.1.3 Mono- and Dinuclear Non-Heme Iron Proteins
While heme iron and Fe-S clusters are two of the most common ways that proteins use iron as a cofactor, it is found in other forms. Non-heme iron cofactors can be bound directly to proteins as mononuclear and dinuclear iron centers with a variety of amino acid ligands and bridging atoms adapted for specific roles. Many non-heme iron centers catalyze similar reactions to heme enzymes. For example, both heme and non-heme diiron enzymes can act as monooxygenases that insert oxygen atoms into substrate molecules. Non-heme iron proteins catalyze a wide array of reactions, such as converting nucleoside diphosphates (NDP) to deoxyNDPs (ribonucleotide reductase), catalyzing biomineralization of iron for intracellular storage (ferritin), sensing oxygen (prolyl hydroxylases), synthesizing eicosanoids (lipoxygenases), and modifying histones (lysine demethylases).
3.2 Assembly and Insertion of Iron Cofactors
3.2.1 Assembly and Insertion of Heme Cofactors
The steps involved in the synthesis of heme are well conserved from prokaryotes to eukaryotes (Figure 6). As mentioned previously, free iron is toxic to cells due to generation of ROS. Both porphyrin and heme are also toxic, generating oxygen radicals and peroxidase activity, respectively. To reduce the risk of these potentially toxic molecules, heme biogenesis is linked to intracellular iron concentrations and synthesis of hemoprotein precursors. The heme synthesis machinery is distributed in both the cytosol and the mitochondria, requiring intermediates in this pathway to be shuttled across membranes. Transport of these porphyrin intermediates must be tightly regulated, again to reduce the risk of toxic components accumulating in the cell [41].
Figure 6.

Heme biosynthesis pathway in eukaryotes. (a) Glycine is transported into the mitochondrial matrix via an unknown mechanism where it is combined with succinyl-CoA by ALA synthase (ALAS) to form ALA. (b) ALA is transported out to the cytosol where it is converted to CPgenIII through four conserved steps. (c) ABCB6 is the transporter proposed to import CPgenIII to the IMS where it is converted first to PPgenIX by CPOX, then to protoporphyrin IX (PPIX) by PPOX. (d) PPIX is transported to the matrix where iron is inserted by ferrochelatase (FECH). The proposed Fe(II) importers are Mrs3/4 (Mfrn1/2 in mammalian cells). (e) Assembled heme is inserted into target apo proteins to form hemoproteins in the mitochondria, possibly aided by FECH. (f) Heme is inserted into target apo proteins in the cytosol, possibly aided by GSTs.
The first phase in heme biosynthesis is formation of the pyrrole. Initially, ALA synthase (ALAS) catalyzes this condensation reaction between succinyl-CoA and glycine to form 5-aminolevulinic acid (ALA) in the mitochondrial matrix. ALA is then transported to the cytosol, possibly via exchange for glycine by the mitochondrial carrier protein SLC25A38, where aminolevulinate dehydratase (ALAD) catalyzes the condensation of two ALA molecules to form monopyrrole porphobilinogen. After formation of the monopyrrole is complete, porphobilinogen deaminase (PBGD) catalyzes assembly of the unstable tetrapyrrole hydroxymethylbilane (HMB) from four molecules of porphobilinogen. Formation of the tetrapyrrole macrocycle is completed by uroporphyrinogen III synthase (URO3S), which catalyzes the ring inversion and closure of HMB to make uroporphyrinogen III (UPgenIII) [42,43]. Once the tetrapyrrole is formed, the side chains need to be modified to form the correct porphyrin before insertion of iron. Uroporphyrinogen decarboxylase (UROD) catalyzes the removal of carboxyl groups from the acetic acid side chains of UPgenIII to form coproporphyrinogen III (CPgenIII). Coproporphyrinogen oxidase (CPOX) catalyzes the conversion of CPgenIII to protoporphyrinogen IX (PPgenIX) via oxidative decarboxylation of the pyrrole ring propionate groups to vinyl groups. CPOX is cytosolic in yeast, and located in the mitochondrial intermembrane space (IMS) in higher eukaryotes. Several studies suggest that the ATP-binding cassette transporter protein ABCB6 is either the CPgenIII transporter, or is somehow involved in the transport of CPgenIII to the IMS in mammals [44]. Protoporphyrinogen oxidase (PPOX) located on the outer surface of the mitochondrial inner membrane catalyzes oxidation of PPgenIX to protoporphyrin IX (PPIX) in the IMS [42].
The final step in forming heme is the insertion of ferrous iron into PPIX by ferrochelatase (FECH) in the mitochondrial matrix. There is some evidence that FECH physically interacts with PPOX across the mitochondrial inner membrane to allow substrate channeling of PPIX between these two enzymes [45]. Human and S. pombe forms of FECH contain a structural [2Fe-2S] cluster that is sensitive to nitric oxide, while S. cerevisiae and bacterial ferrochelatases lack the Fe-S cluster [46]. Studies indicate that ferrous iron may be imported into the matrix by Mrs3/4 importers (Mfrn1/2 or mitoferrin in mammalian cells). The yeast homologue of human frataxin, Yfh1, is proposed to act as an iron chaperone and donate Fe(II) to ferrochelatase for heme biosynthesis [47]. However, human frataxin does not seem to be involved in heme biosynthesis, although it may have a role in Fe-S protein assembly [40,48].
Once heme is fully assembled, it must be transported from FECH in the matrix across one or more membranes to target hemoproteins found in various organelles, such as the IMS, cytosol, nucleus, ER, and lysosomes [49]. FECH may act as a heme shuttle for proteins in the matrix that are in the same vicinity, such as cytochrome P450. For proteins outside the matrix, there is no known heme chaperone in mammals, although heme chaperones for cytochrome c have been identified in plants and bacteria [50]. Some cytosolic heme-binding proteins have also been suggested to have a role in heme transport, including glutathione S-transferases (GSTs) from liver and red blood cells [51]. Hemoproteins found in the secretory pathway may obtain heme in the ER, indicating a role for the ER in heme delivery. The ER and mitochondria have been shown to physically interact via a tethering complex that may provide a path for heme transport from the mitochondria [45].
3.2.2 Assembly and Insertion of Iron-Sulfur Clusters
There are two identified systems in eukaryotes for assembly of iron-sulfur clusters: the mitochondrial iron-sulfur cluster (ISC) assembly machinery and the cytosolic Fe-S protein assembly (CIA) machinery (Figure 7). In the mitochondria, Fe-S protein maturation occurs via two distinct stages. First, Fe(II) and sulfur are combined on a homodimeric scaffold protein (Isu) to form a labile Fe-S cluster. Once formed, the nascent Fe-S cluster is then transferred to its ultimate target protein via additional accessory proteins. In the initial stage, sulfur is obtained from free cysteine via the pyridoxal phosphate-dependent cysteine desulfurase Nfs1. Both yeast and human Nfs1 are mainly mitochondrial, although a portion of Nfs1 localizes to the nucleus and cytosol in both systems. In yeast, the mitochondrial form is essential for both mitochondrial and cytosolic cluster assembly, while the cytosolic form may act as a sulfur donor for other pathways requiring mobilized sulfur, such as cytosolic tRNA thio-modification [52]. Nfs1 function is dependent on formation of a stable complex with a partner protein named Isd11, although the specific role of Isd11 is not clear. The Nfs1-Isd11 complex produces sulfane sulfur (S0) via persulfide formation, thus a source of electrons is required to reduce this molecule to sulfide (S2−) for Fe-S cluster synthesis. This electron transfer is most likely performed by the ferredoxin-ferredoxin reductase pair Yah1 and Arh1 in yeast (FDX1/2 and FDXR in humans) using electrons from NADH [48,53].
Figure 7.

Mitochondrial and cytosolic Fe-S cluster assembly in yeast (top panel) and mammalian cells (bottom panel). (a) In the mitochondria, sulfur is obtained from the cysteine desulfurase Nfs1, interacting with Isd11. Nfs1 transfers sulfur as a persulfide to Isu1/2 (ISCU in humans). (b) Iron is imported to the mitochondria by the transporters Mrs3/4 (mitoferrins or Mfrn1/2 in humans) and possibly donated to Isu1/2 through frataxin (FXN, or Yfh1 in yeast). (c) Electrons are donated by NADH through the ferredoxin-ferredoxin reductase pair Yah1-Arh1 to reduce S0 to S2−. (d) The assembled Fe-S cluster is transferred to target proteins by a chaperone system consisting of Ssq1, Jac1, Mge1, and Grx5 (HSC20, HSC70, and GLRX5 in humans). (e) Isa1/2 specifically delivers clusters to aconitase-like proteins. (f) An unknown substrate produced by the ISC machinery is exported out to the cytosol by the transporter Atm1 (ABCB7 in humans). This process may also include the sulfhydryl oxidase Erv1 (ALR in humans), GSH, and Dre2 (CIAPIN1). (g) Fe-S clusters are assembled in the cytosol on the scaffold complex formed by Cfd1 and Nbp35 (NUBP2 and NUBP1 in humans, respectively). (h) The assembled cluster is transferred to target cytosolic and nuclear proteins by Nar1 and Cia1 (IOP1 and CIAO1 in humans). (i) Mammalian cells also express cytosolic versions of NFS1, ISD11, ISCU, HSC20, and possibly FXN that may also facilitate de novo assembly of Fe-S clusters outside the mitochondria.
It is clear that the Nfs1-Isd11 complex transfers sulfur to the Isu scaffold proteins (Isu1 and Isu2 in yeast, ISCU in humans). Similar to Nfs1, a small amount of human ISCU exhibits cytosolic localization where it may function as a scaffold for extramitochondrial Fe-S cluster biogenesis [48,53]. The mechanisms of cluster assembly on scaffold proteins, including the order of Fe and S binding and the sources of iron, remain open questions. As far as iron delivery, one proposal is that Yfh1, the yeast homologue of human frataxin, delivers iron to the Isu proteins. Yfh1 was shown to bind iron in vitro, and to bind Isu1/Nfs1 in an iron-dependent manner in the mitochondria, possibly enabling iron transfer [54]. More recently, frataxin was proposed to stabilize the active form of the Nfs1-Isd11-Isu complex thereby promoting sulfur transfer and enhancing Fe-S cluster formation [55].
Once the cluster is assembled, it must be transferred from the scaffold to the target apoprotein. Bacterial U-type scaffolds (IscU, NifU) are capable of making and transferring both [2Fe-2S] and [4Fe-4S] clusters, thus eukaryotic Isu proteins may also be able to assemble both types of clusters. The cluster transfer system is formed by the chaperone Ssq1 (an Hsp70 chaperone), a J-type accessory chaperone, Jac1, and the chaperone/nucleotide release factor, Mge1. While not required for cluster assembly on Isu proteins, depletion of these chaperones results in cluster accumulation on Isu1. This system most likely takes the transiently bound cluster from the Isu scaffold and inserts it into the target protein. The monothiol glutaredoxin Grx5 also may play a role in cluster transfer, as Grx5 depletion results in cluster accumulation on Isu1, although a specific role has not yet been determined. Studies on zebrafish and human forms of Grx5 show that it is important for cytosolic Fe-S assembly, and thus may regulate heme synthesis by facilitating Fe-S cluster assembly on IRP1 (see Section 5.2.2) [48,53]. Additional mitochondrial Fe-S biogenesis assembly factors (e.g. Isa1, Isa2, Iba57, BolA3, Nfu1, Ind1) are proposed to function as intermediate scaffold/delivery proteins between ISCU and specific subsets of target proteins [40].
In addition to the mitochondrial ISC system, yeast and mammalian cells have cytosolic iron-sulfur assembly (CIA) components that build clusters for cytosolic and nuclear proteins. Since the mitochondrial form of yeast Nfs1 is essential for both mitochondrial and cytosolic Fe-S cluster assembly, one theory is that the mitochondria exports a sulfur-containing substrate produced by the ISC machinery that is used by the CIA machinery to build and/or insert Fe-S clusters [40]. In yeast, the ABC transporter Atm1 (ABCB7 in humans) is proposed to export the unidentified sulfur-containing substrate from the mitochondria to the cytosol since depletion of Atm1 inhibits cytosolic Fe-S assembly. A recent study also implicates the mammalian ABC transporter ABCB8 in export of mitochondrial iron for cytosolic Fe-S cluster biogenesis [56]. The mitochondrial IMS-localized sulfhydryl oxidase Erv1 and the ubiquitous tripeptide GSH are also implicated in export of the sulfur-containing substrate from the mitochondria for cytosolic Fe-S cluster assembly, although their specific roles are nebulous [40]. However, a recent study reported that the cytosolic Fe-S assembly defect reported for Atm1-depleted strains was an artifact of the strain background used in the initial report, indicating that Atm1 activity is not required for cytosolic Fe-S cluster assembly in yeast [57]. In addition, studies in human cells suggest that the cytosolic Fe-S cluster assembly machinery is independent of the mitochondrial system [48].
Regardless of the specific details regarding initial Fe-S cluster assembly in the cytosol and the requirement of the mitochondrial ISC system, it is clear that a number of additional proteins are essential for assembling Fe-S clusters for cytosolic/nuclear proteins. Two potential scaffold proteins are the P-loop NTPases Cfd1 and Nbp35 that form a [4Fe-4S]-bridged heterotetramer. Yeast Nar1 (IOP1 and IOP2 in humans) is similar to bacterial iron hydrogenases, although it has no hydrogenase activity. Nar1 has two Fe-S clusters whose assembly requires the mitochondrial ISC systems, as well as Cfd1 and Nbp35. Depletion of mammalian IOP1 disrupts cytosolic Fe-S cluster assembly, and homology of Nar1/IOP1/2 to other hydrogenases has led to the suggestion that these proteins may act as electron donors in the CIA system, either in assembly or transfer of a cluster. A fourth member of the CIA machinery is Cia1, which might play a role late in biogenesis after Nbp35 and Nar1, possibly in cluster transfer to target proteins. In addition, Dre2, which is localized to both the cytosol and the mitochondrial intermembrane space, was recently found to be a possible link between the mitochondrial ISC and cytosolic CIA systems. Dre2 can be purified with both a [4Fe-4S] and a [2Fe-2S] cluster that seem to play catalytic or structural roles based on cluster stability. Since depletion of Dre2 impairs cytosolic Fe-S cluster assembly, it may work early in the CIA pathway and deliver the ISC system substrate necessary for cytosolic cluster assembly. The human homologue of Dre2, anamorsin (or CIAPIN1) is proposed to function in electron transfer in the CIA system, similar to Dre2. While the function appears conserved between yeast and humans, anamorsin only binds a [2Fe-2S] cluster [58]. Currently, the only proteins known to require the CIA system bind [4Fe-4S] clusters [40,59], thus there may exist a parallel pathway for assembly of cytosolic [2Fe-2S] clusters such as those found on the Grx3/4 proteins (more in Section 5.3) [60].
3.2.3 Assembly and Insertion of Non-Heme Iron Cofactors
Due to the multitude of different non-heme iron centers, there is not a singular system for assembly and insertion of these cofactors. In most cases, a specific set of proteins are required: a chaperone for iron delivery, redox proteins to maintain the oxidation state of iron, and enzymes involved in protein folding that allow for proper insertion of the metal. For metalloproteins that merely need their cofactor inserted (such as mononuclear iron), cells can minimize metal misincorporation by compartmentalizing proteins and using metal chaperones. The metal concentrations in different subcellular compartments can vary, and a metalloprotein’s metal affinity is usually tailored to these specific ranges. Here, we focus on one of the better-characterized assembly systems for incorporation and maturation of the dinuclear iron center of class Ia ribonucleotide reductases (RNRs).
Class I RNRs are divided into subgroups based on their metallocofactor. Class Ia RNRs contain a diferric-tyrosyl radical (Fe3+Fe3+-Y•) that is involved in the oxidation of a conserved active site cysteine to a thiyl radical (S•), which in turn reduces the substrate. Fe2+ first needs access to the metal binding site of the RNR that is buried in the protein. One hypothesis is that one subunit of the RNR acts as an iron chaperone to another subunit, then becomes part of the functional enzyme. Once the protein contains a diferrous center (Fe2+Fe2+), this can react with O2 to form a diferric peroxide. A conserved Trp residue reduces the diiron center to an Fe4+Fe3+ intermediate, producing a Trp cation radical (Trp+•). The intermediate can then oxidize the active site Tyr(Y), forming Fe3+Fe3+ and the Tyr radical (Y•) needed for catalysis [61]. Figure 8 shows diferric-tyrosyl radical cofactor formation (a) and proteins involved in the biosynthesis pathway (b).
Figure 8.
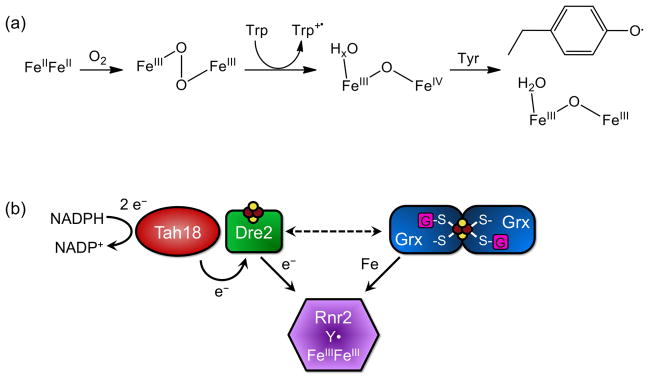
Formation of the class Ia RNR diferric-tyrosyl radical cofactor. (a) Formation reaction for the diferric-tyrosyl radical cofactor. (b) Model for the biosynthesis pathway of active Rnr2. Tah18 and Dre2 are proposed to donate electrons for diferric-tyrosyl radical formation on Rnr2. The [2Fe-2S] Grx3/4 homodimer most likely donates iron for the cofactor. Reducing equivalents from Dre2 may also be required for iron delivery (dashed line).
Recent studies on the cytosolic monothiol glutaredoxins, Grx3 and Grx4, and cytosolic Fe-S biogenesis factor Dre2 have revealed a possible role for these proteins in cofactor assembly on yeast Rnr2. Grx3/4 is involved in intracellular iron trafficking for the assembly of many iron cofactors, including Fe-S loading onto Dre2. Dre2 and the reductase Tah18 are postulated to act as an electron source for cytosolic Fe-S cluster assembly (analogous to the Yah1-Arh1 pair in the mitochondrial ISC system). Grx3/4 was shown to be the source of iron in RNR cofactor synthesis, either directly donating the iron, or indirectly by making it bioavailable for assembly. Dre2 also plays a role in RNR cofactor formation that seems to be separate from the role of Grx3/4. Although this specific function has not yet been determined, the Dre2-Tah18 pair may provide reducing equivalents to maintain the iron in its reduced state for delivery [62].
4 Iron Uptake, Trafficking, and Storage
4.1 Iron Uptake and Transport
4.1.1 Iron Uptake and Transport in S. cerevisiae
The yeast S. cerevisiae expresses an extensive system of membrane transporters for uptake of environmental iron (Figure 9). As previously mentioned, yeast have the ability to scavenge iron-loaded siderophores from their surroundings. Arn1, Taf1, Sit1, and Enb1 (also known as Arn1-4, respectively) are transporters specific for Fe3+-siderophore complexes. Environmental Fe3+ is reduced to the more soluble Fe2+ by the ferrireductases Fre1 and Fre2, which are also capable of reducing Cu2+. Once reduced, Fe2+ can be imported by the high-affinity uptake system encoded by FET3 and FTR1. Fet3 is a multicopper oxidase that oxidizes the Fre1/2-produced Fe2+ to Fe3+, which is then transferred across the plasma membrane by the transmembrane permease Ftr1. In addition, S. cerevisiae possesses low-affinity iron transporters (Fet4 and Smf1) that can also transport other transition metals. Fet4, in addition to being a low-affinity iron transporter, can import copper and zinc, and is responsible for most of the uptake under iron-replete conditions. Smf1, a member of the NRAMP transporter family found in both prokaryotes and eukaryotes, is a H+/M+ symporter that uses a pH gradient to import transition metals such as Fe2+, Mn2+, and Zn2+. Yeast express two other NRAMP proteins, Smf2 and Smf3, although they do not seem to play a significant role in environmental iron uptake. In addition to these uptake systems, the yeast genome encodes transmembrane ATP-driven H+ transporters to acidify the environment, which increases the solubility of Fe3+ [15,63].
Figure 9.
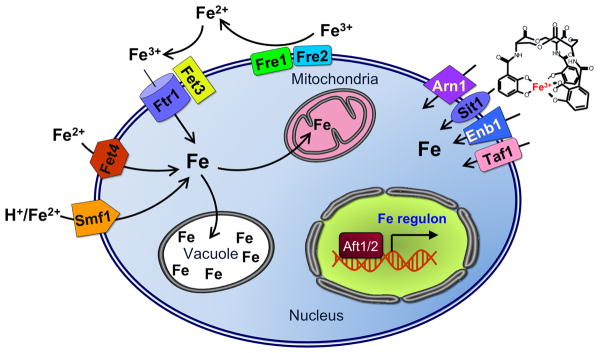
Iron uptake systems in S. cerevisiae. Arn1, Sit1, Enb1, and Taf1 are transporters for Fe3+-siderophore complexes. The ferrireductases Fre1/2 reduce environmental Fe3+ to Fe2+. Fet3 and Ftr1 form the high-affinity iron uptake system. Fet3 oxidizes Fe2+ to Fe3+ and Ftr1 transports this across the plasma membrane. Fet4 is a low-affinity transporter responsible for uptake under iron-replete conditions. Smf1, a member of the NRAMP family of transporters, is a H+/M+ symporter for some transition metals like Fe2+, Mn2+, and Zn2+. Aft1 and Aft2 are transcriptional regulators that activate expression of the iron regulon, including these iron uptake systems, under low iron conditions.
After cells have imported iron, it needs to be transported either for storage or utilization. Since free iron is redox-active and can damage proteins and membranes, it seems likely that cells would use a chaperone to bind and transport iron through the cell. In yeast, the cytosolic monothiol Grxs with a signature CGFS active site, Grx3 and Grx4, are suggested to have an essential role in cellular iron trafficking. Depletion of Grx3/4 leads to defects in iron-dependent proteins independent of induction of the Aft1 iron uptake system (see Regulation in Section 5.3 for more). Although these cells had excess cytosolic iron, iron delivery is impaired. Grx3/4 have previously been shown to bind a [2Fe-2S] cluster in a homodimeric complex. Not only is this Fe-S cluster essential for their role in iron trafficking, it is also required in iron sensing and regulation of the transcription factors Aft1 and Aft2 [23,64,65].
4.1.2 Iron Uptake and Transport in Mammalian Cells
In mammalian cells, absorption of dietary iron occurs in the intestine through the brush border of duodenal enterocytes. Inorganic iron mainly comes from vegetables, while heme iron comes from degradation of hemoglobin and myoglobin in red meat. The divalent metal transporter 1 (DMT1) also known as SLC11A1, is an NRAMP family protein that carries inorganic iron across the apical membrane into enterocytes. The ferric reductase Dcytb (duodenal cytochrome b) is required to reduce Fe3+ to Fe2+ prior to uptake by DMT1 since dietary inorganic iron is primarily in the oxidized form in the acidic environment of the duodenum. The mechanisms for uptake of dietary heme, which is absorbed more efficiently that inorganic iron, are somewhat nebulous. One possible candidate for dietary heme uptake is heme carrier protein HCP1 [66]. After internalization, heme iron is released into the enterocyte cytosol by heme oxygenase-1 (HO-1) via degradation of the heme molecule. Cytosolic iron is either stored in ferritin (see below) or exported across the basolateral membrane into the circulation by the ferrous iron exporter ferroportin (FPN). In enterocytes, FPN functions in concert with the multicopper oxidase hephaestin, which oxidizes exported Fe2+ to Fe3+ to facilitate iron loading onto the plasma iron carrier transferrin [15]. Many other mammalian cell types also have the ability to export iron via FPN, including macrophages and hepatocytes. The plasma multicopper oxidase ceruloplasmin functions with FPN in these cell types in an analogous manner to hephaestin [67].
Transferrin (Tf) binds two ferric ions with high affinity, providing iron for most human cell types (Figure 10). Iron-bound Tf is imported into the cell through the cell surface Tf receptor 1 (TfR1), forming a complex that is internalized by endocytosis. ATP-dependent proton pumps acidify the resulting endosome so iron is released from Tf, although this release likely also requires reduction of the iron to Fe2+. Members of the STEAP (six-transmembrane epithelial antigen of the prostate) protein family are ferric reductases that catalyze this reaction. Apo-Tf is reutilized after delivery to the plasma membrane and released back into the circulation. The freed ferrous iron is then transported from the endosome to the cytoplasm via DMT1 [67]. In addition to Tf-iron, some mammalian cells can utilize hemoglobin iron from degraded erythrocytes. Senescent red bloods cells (RBCs) are phagocytosed by macrophages in order to recycle body iron. Hemoglobin binds to the plasma protein haptoglobin, which then binds to CD163 on the surface of monocytes and macrophages. Similar to uptake of Tf-iron, this complex undergoes endocytosis before being broken down for iron release. The released heme iron can then bind to another plasma protein, hemopexin, which is endocytosed following binding to the CD91 receptor of certain cells [45]. The transmembrane protein HRG-1 has also been implicated in heme import [68].
Figure 10.

Iron import and export in a generic mammalian cell. The plasma protein transferrin (Tf) binds 2 ferric ions for uptake by the transferrin receptors (TfR1 and TfR2). TfR1 is found in all cell types, while TfR2 is limited to liver, intestinal, and red blood cells. Tf(Fe3+)2-bound TfR is internalized by endocytosis and ferric iron is released in the acidic environment of the endosome. Tf and TfR are recycled to the plasma membrane. DMT1 is involved in release of Tf iron from the endosome following reduction of Fe3+ to Fe2+ by STEAP ferrireductases. Dcytb (cytochrome b-like ferrireductase) reduces dietary Fe3+ to Fe2+, which is imported by DMT1 at the plasma membrane. The plasma proteins haptoglobin and hemopexin bind hemoglobin and free heme, respectively, produced by erythrocyte destruction. Haptoglobin-hemoglobin and heme-hemopexin complexes are recognized by CD163 and CD91 receptors, respectively, for subsequent endocytosis. Heme is also imported via HCP1 and HRG1. HO-1 (heme oxygenase-1) catalyzes degradation of the heme to remove iron. Imported iron can be stored in ferritin or trafficked to the mitochondria for synthesis of heme and Fe-S clusters. FPN is the iron exporter, transporting Fe2+ out of the cell. Ceruloplasmin (Cp) oxidizes this Fe2+ to Fe3+ for binding to Tf.
Once iron is delivered to the cytoplasm, it must be transferred to various sites for utilization and storage. When iron concentrations exceed cellular need, iron is deposited in the iron storage protein ferritin (see below). Iron is most likely trafficked via a chaperone so that it cannot prematurely react with cellular components. While no general iron chaperone has been confirmed yet, several possible candidates have been identified. Poly (rC) binding protein 1 (PCBP1) was shown to facilitate iron loading to ferritin; however, the exact mechanism of delivery is not yet characterized [54]. In addition, PCBP1 and its homologue PCBP2 were shown to act as iron chaperones to the iron-dependent enzymes HIF prolyl hydroxylases and factor inhibiting HIF (FIH1). As with the ferritin-PCBP1 interaction, the mechanism of iron delivery is not known; however, it seems likely that the PCBPs interact with their targets post-translationally for iron incorporation. Whether this takes place in the nucleus or the cytosol remains to be seen [69]. Cellular iron trafficking in yeast by [2Fe-2S]-bridged Grx3/4 homodimers has already been described (see above) [23]. The human ortholog of yeast Grx3/4, namely GLRX3, shares high sequence similarity with its yeast counterparts and forms analogous [2Fe-2S] bridged homodimers [70,71], thus human GLRX3 may play a similar role as an iron chaperone.
4.2 Iron Storage Proteins
Intracellular iron that is not immediately utilized is directed to the cytosolic iron storage protein ferritin. Ferritin self-assembles into a protein nanocage composed of 24 heavy (H) and light (L) subunits housing ferric oxide biominerals, with an average of 1000–1500 iron atoms stored per ferritin cage. During intracellular iron overload, ferritin iron levels can reach 3000–4000 iron atoms/nanocage, leading to formation of insoluble material from damaged ferritin known as hemosiderin. Several recent studies have unveiled the unique structural properties of ferritin that facilitate mineral nucleation during iron entry [72]. Each H subunit binds two ferrous ions in the active site that combine with O2 to form the di-Fe(III)O mineral precursor. Crystal growth is achieved as the mineral precursors from each active site coalesce during movement into the central cavity. Release of ferritin iron back into bioavailable cytosolic pools occurs via proteasomal or lysosomal degradation of the ferritin nanocage [73]. In addition, iron may also be released from ferritin via a non-destructive pathway involving reduction and dissolution of the ferric oxide biomineral [72].
A mitochondrial version of ferritin is also expressed in some human cell types. Mitochondrial ferritin is similar in sequence and structure to ferritin, but possesses relatively weak ferroxidase activity [74]. Nevertheless, mitochondrial ferritin overexpression was shown to reduce production of reactive oxygen species while promoting mitochondrial iron loading and cytosolic iron depletion [75]. However, the increased mitochondrial iron in cells overexpressing mitochondrial ferritin is less bioavailable for heme and Fe-S cluster biogenesis [76]. Interestingly, mitochondrial ferritin transcription does not seem to be regulated by iron, but rather by the oxidative metabolic activity demand. Taken together, these studies thus suggest that mitochondrial ferritin functions to sequester redox-active iron in this organelle and highlight the importance of the mitochondrion in regulation of whole cell iron metabolism [76,77].
4.3 Iron Entry and Exit into Extramitochondrial Organelles
4.3.1 Yeast Vacuoles
As mentioned earlier, in addition to mitochondria, the vacuole is the other major hub of iron metabolism in S. cerevisiae. As yeast do not possess ferritin for iron storage, the vacuole is designed to serve this purpose. The acidic pH in vacuoles, which ranges from 4.5–5.5, permits increased iron solubility, and is regulated by ATP-driven proton pumps. Iron is imported from the cytosol to the vacuole through the transporter Ccc1, found in the vacuolar membrane. In addition to being an iron (and manganese) storage organelle, vacuoles help protect cells from the toxic effects of excess iron by sequestering it in an unreactive form. The protein complexes involved in iron export from the vacuole are comparable to the cell surface iron import machinery. A member of the FRE family, Fre6, is localized to the vacuole where it acts as a ferrireductase in exporting iron from the vacuole. Fre6 works in combination with the Fet5-Fth1 ferrous iron transport complex, which is analogous to the cell surface Fet3-Ftr1 import complex. Smf3, another member of the NRAMP family and homologous to Smf1, is also found in the vacuolar membrane. Smf3 and Fet5-Fth1 all work downstream from Fre6 in iron export from the vacuole [63,78–80].
4.3.2 Lysosomes
In mammalian cells, lysosomes play a significant role in iron recycling as the primary site for degradation of cytosolic ferritin. These organelles containing acidic hydrolases that store and degrade biological waste produced by the cell, including damaged proteins and organelles. Much of the material taken up by lysosomes contains iron, mainly derived from ferritin and respiratory complexes. Iron concentrations in lysosomes can vary greatly depending on how much material has been taken up, and if iron-rich compounds have recently been degraded. Lysosomes with higher iron content tend to be more susceptible to oxidative stress and destabilization. Fenton chemistry becomes more likely under the favorable lysosomal conditions of an acidic pH and the availability of reducing equivalents. Oxidative damage to membranes can cause leaky lysosomes, which exposes other cellular components to the harmful materials contained in this organelle [81].
4.3.3 Endoplasmic Reticulum
As mentioned earlier, studies suggest that the ER may acquire heme via direct contact with the mitochondrion [45]. The existence of non-heme iron enzymes in the mammalian ER, such as prolyl and lysyl hydroxylases, suggests that ionic iron is also transported into the ER and/or Golgi. Although the yeast genome does not encode homologues for these enzymes, prolyl hydroxylases heterologously expressed in S. cerevisiae are correctly targeted and enzymatically active, demonstrating iron delivery to the secretory pathway [82]. In addition, iron-free ferritin monomers are translocated into the ER in yeast and mammalian cells [83], and iron-loaded ferritin is detected in the secretory pathway of Drosophila cells [84]. Taken together, these studies provide evidence that ionic iron is transported into the ER/Golgi compartment in both lower and higher eukaryotes [83]. However, specific ER iron transporters for either yeast or mammalian cells have not yet been identified.
4.3.4 Nucleus
Numerous nuclear proteins contain heme, non-heme iron, or Fe-S cluster cofactors that are necessary for their activity. Examples of heme-binding nuclear proteins include the nuclear receptor Rev-ervα and the transcriptional repressor Bach1 [49], while diiron-binding nuclear proteins include RNRs and HIF prolyl hydroxylases. A growing list of Fe-S proteins involved in DNA repair, DNA replication, and transcriptional elongation are also localized to the nucleus [40]. In each of these cases, it is not known whether iron cofactor acquisition and assembly occurs before or after entry in the nucleus. Nevertheless, a chelatable, labile iron pool similar in concentration to the cytosolic labile iron pool has been detected in mammalian nuclei [20], thus nuclear iron is bioavailable for insertion into iron metalloproteins if necessary. Nuclear ferritin has also been detected in some mammalian cells types, demonstrating the capacity of this organelle to store and sequester DNA-damaging, redox-active iron [85]. The specific mechanisms by which iron enters the nucleus are unclear. Labile iron bound to low molecular weight ligands may freely diffuse through nuclear pores. Alternatively, there is evidence that iron is actively transported across the nuclear membrane via an ATP-dependent transport system [86].
5 Regulation of the Iron Metallome
5.1 Regulation at the Transcriptional Level
5.1.1 Yeast Iron-Responsive Transcription Factors
5.1.1.1 S. cerevisiae Hap1
S. cerevisiae regulate iron homeostasis via transcriptional, post-transcriptional, and post-translational mechanisms. At the transcriptional level, yeast utilize several regulatory factors that respond not only to cellular iron status, but also oxygen levels. Yeast use heme as an oxygen sensor to differentiate between aerobic and anaerobic growth conditions. Heme is well-suited to this role since it not only binds to oxygen, but the synthesis of porphyrin for heme depends on oxygen availability. Lower oxygen levels lead to decreased heme synthesis, which in turn decreases synthesis of respiratory proteins. The transcriptional activator Hap1 controls this process in S. cerevisiae. Hap1 is a heme-binding protein that activates transcription of respiratory proteins under normal oxygen levels, as well as the repressor Rox1. Rox1 then represses transcription of genes involved in anaerobic growth. Under low or no oxygen conditions when heme synthesis is attenuated, Hap1 is inactive, thus aerobic metabolism genes are turned off and anaerobic genes are turned on. Neither Hap1 nor Rox1 has any effect on transcription of the iron regulon (more below) [87].
5.1.1.2 S. cerevisiae Aft1/Aft2
In Saccharomyces cerevisiae, expression of high affinity ionic iron and siderophore transporters is primarily controlled by the transcriptional activator Aft1 and to a lesser extent, its paralog Aft2. In addition to these transporters, Aft1 and Aft2 control several other genes that together comprise the iron regulon. The iron regulon includes genes encoding the high affinity iron transport system (FET3, FTR1, CCC2, ATX1, FRE1-FRE6), the siderophore transport system (ARN1-4, FIT1-3), and vacuolar iron export systems (SMF3, FET5, FTH1) [15]. Aft1/2 nuclear localization is controlled by the cellular iron status: in iron-starved cells, Aft1/2 accumulates in the nucleus, while under iron-replete conditions, Aft1/2 is shuttled to the cytosol by the exportin Msn5. Aft1/2 localization is regulated by a complex formed of Grx3/4, Fra1, and Fra2 proteins, which transmits an inhibitory signal that is dependent on the synthesis of mitochondrial Fe-S clusters (for more, see Section 5.3.1) [87]. Aft mutants are unable to grow on iron-poor media, although this is not primarily due to a malfunctioning iron uptake system, but rather the lack of control of intracellular iron use. This misuse of iron also renders cells more sensitive to oxidative stress, most likely due to metal toxicity and formation of ROS [88–90].
Aft1 and Aft2 have both overlapping and independent functions. Many iron regulon genes are induced by both Aft1 and Aft2; although in most cases, Aft1 activation elicits a stronger response. Aft1 and Aft2 regulate gene expression by binding to iron-responsive elements (FeREs) with the conserved sequence 5′-CACCC-3′. However, Aft1 binds more selectively than Aft2 since it preferentially binds 5′-TGCACC-3′, while Aft2 prefers 5′-G/ACACC-3′. Transcriptional analysis of Aft1 and Aft2 target genes suggest that Aft1 is primarily involved in cellular iron uptake, while Aft2 specifically regulates intracellular trafficking to vacuoles and mitochondria [90,91]. However, biophysical analysis of iron speciation in yeast mutants that express constitutively active forms of Aft1 or Aft2 suggests that the Aft proteins do not regulate trafficking of cytosolic iron into mitochondria and vacuoles [7].
5.1.1.3 S. cerevisiae Yap5
The regulatory functions of the low iron-sensing transcriptional activators Aft1/Aft2 are also complemented by a high iron-sensing transcriptional activator named Yap5. Under high iron conditions, Yap5 activates expression of the vacuolar iron transporter Ccc1 resulting in increased iron transport into the vacuole, which effectively lowers cytosolic iron levels [92]. Yap5 binds to the CCC1 promoter independent of iron levels, but only induces expression under high-iron conditions via formation of an intramolecular disulfide bond [92,93]. A recent microarray study identified a handful of Yap5-regulated genes, including TYW1, GRX4, and CUP1. Tyw1 is an Fe-S enzyme involved in modification of tRNA bases; however, the catalytic function of Tyw1 is not implicated in iron response. Instead, the data suggests that Yap5 protects the cell from excess iron by increasing Tyw1 levels, which sequesters iron into Fe-S clusters. It is possible that upregulation of Grx4 expression has a similar effect since this [2Fe-2S]-binding protein is implicated in iron trafficking [23]. CUP1 encodes a Cu-binding metallothionein that is important for resistance to copper toxicity [94]. Interestingly, biophysical studies indicate that Cup1 also binds four Fe2+ atoms/monomer in vitro [95], and thus may also play a role in iron sequestration under toxic iron conditions. Taken together, these studies suggest that Yap5 responds to high cellular iron levels by decreasing free cytosolic iron through sequestration in vacuoles or incorporation into Fe- or Fe-S cluster-binding proteins [92].
5.1.2 Mammalian HIF-2α
Unlike the transcriptional regulation of iron uptake and storage genes in yeast, studies suggest that mammalian cellular iron homeostasis is primarily regulated by post-transcriptional control of mRNA translation and stability (see Section 5.2.2). However, iron-dependent regulation at the transcriptional level was recently implicated in control of intestinal iron absorption. As described in Section 4.2.1, mammalian enterocytes control iron import and export by regulating levels of the ferric reductase DcytB and iron importer DMT1 located at the apical membrane, as well as the basolateral iron exporter FPN. While the peptide hormone hepcidin is the main regulator of FPN (see Section 5.3.2.1), the hypoxia-inducible factor HIF-2α (also known as EPAS1) controls expression of DctyB and DMT1. Under iron deficiency or hypoxic conditions, HIF-2α forms a heterodimeric complex with HIF-1β, also known as aryl hydrocarbon nuclear receptor (ARNT), that induces transcription of both DcytB and DMT1. When iron levels are sufficient, HIF-2α is degraded (see Section 5.3.2.2) and does not activate expression of the iron absorption genes [96,97].
5.2 Regulation at the Post-Transcriptional Level
5.2.1 Iron-Responsive mRNA-Binding Proteins in Yeast
In addition to activation of iron uptake and transport genes by Aft1 and Aft2, iron deficiency in S. cerevisiae also leads to reprogramming of iron-dependent metabolic pathways in order to preserve limited iron pools for essential pathways [15,98]. This metabolic reprogramming is primarily accomplished by two mRNA binding proteins, Cth1 and Cth2, whose expression is upregulated by Aft1/2 under low iron conditions. Both proteins bind to the 3′ untranslated regions of mRNAs encoding iron-utilizing proteins in non-essential pathways, thereby facilitating their degradation [99,100]. Cth1 and its paralog Cth2 are tandem zinc finger (TZF) proteins related to the mammalian protein tristetraprolin (TTP). Similar to TTP, Cth1/2 binds to AU-rich elements (ARE) in target mRNAs. Cth2 binds to mRNAs that encode enzymes in heme biosynthesis, Fe-S cluster biosynthesis, the TCA cycle, the electron transport chain, and components of fatty acid metabolism pathways. Gene targets of Cth1 are mainly mitochondrial proteins involved in respiration and amino acid synthesis. Cth1 shares a subset of target genes with Cth2, including some involved in oxidative phosphorylation [99–101].
Cth1 and Cth2 regulation helps to redistribute iron to vital processes under iron starvation. One of the enzymes that receives the limited iron is ribonucleotide reductase (RNR). RNR is composed of a large R1 subunit that houses the catalytic site and a small R2 subunit that contains the diferric tyrosyl radical cofactor. One facet of RNR regulation in yeast involves subcellular localization, since the R1 subunit is primarily localized to the cytosol while the R2 subunit residues in the nucleus under normal conditions. During genotoxic stress, the iron-containing R2 subunit is exported to the cytosol allowing assembly of the active, holo enzyme [102]. Nuclear localization of the R2 subunit depends on an interaction with the nuclear protein Wtm1. Interestingly, the R2 subunit is also shuttled to the cytoplasm under iron deficient conditions, suggesting that the limited available iron is partly funneled to this essential enzyme. Iron-responsive transport of R2 to the cytoplasm is dependent on Cth1 and Cth2, which are responsible for binding to AREs in the WTM1 mRNA and degrading it in response to low iron. Cth1 and Cth2 enhance RNR activity not only by facilitating relocalization, but also by degrading mRNA of nonessential iron-dependent pathways to increase available iron. In addition, Cth1 and Cth2 degrade new RNR transcripts to optimize the use of the limited iron available in the redistributed RNR enzymes [103].
5.2.2 Iron-Responsive mRNA-Binding Proteins in Mammalian Cells
In contrast to yeast, iron uptake and storage in mammalian cells is mainly regulated at the post-transcriptional level through two iron regulatory proteins, IRP1 and IRP2 [73,104,105]. Under iron-depleted conditions, IRP1 and IRP2 bind to iron response elements (IREs) that form hairpin structures in the 5′ or 3′ untranslated regions (UTRs) of their target mRNAs. IREs are found in mRNAs encoding a variety of proteins involved in iron metabolism, including ferritin, TfR, heme synthesis, DMT1, and FPN. IRP binding to mRNA either stabilizes it or sterically blocks its translation depending on the location of the IRE. When IREs are found in the 3′ UTR, such as in TfR, IRP binding protects the mRNA from degradation, allowing transcription and subsequent iron uptake. When IREs are found in the 5′ UTR, such as with ferritin mRNA, IRP binding blocks translation and thus decreases iron storage. Under iron replete conditions when IRP1 and IRP2 lose their IRE-binding activity, mRNAs with IREs in the 3′ UTR are degraded while mRNAs with IREs in the 5′ UTR are stable and freely translated [105]. Figure 11 illustrates how various target mRNAs are regulated by IRPs.
Figure 11.

mRNA regulation of iron trafficking and utilization factors by the IRE-IRP system. In iron-deplete cells, IRP1 lacks the [4Fe-4S] and IRP2 is stabilized, thus both IRPs are able to bind the target IREs. Translation of mRNAs with 5′ IREs (e.g. ferritin) is blocked, while mRNAs with 3′ IREs (e.g. TfR1) are stabilized and translated. In iron-replete cells, IRP1 binds a [4Fe-4S] cluster that precludes IRE binding, while IRP2 is degraded by the proteasome. IREs of the target RNAs are unoccupied. mRNAs with IREs in the 5′ UTR are translated, while RNase degrades mRNAs with IREs in the 3′ UTR.
The IRPs are cytoplasmic proteins that belong to the aconitase family of isomerases. IRP1 is capable of switching from mRNA-binding activity to aconitase activity by binding a [4Fe-4S] cluster that prevents IRE binding. When cells are iron-depleted, the cluster is degraded and IRP1 gains mRNA-binding activity. In addition to being regulated by iron availability, IRP1 activity is influenced by Fe-S cluster biogenesis: IRE-binding activity is increased when the Fe-S assembly machinery is impaired. This cluster can also be degraded by exposure to hydrogen peroxide or nitric oxide. Although IRP1 and IRP2 are 57% identical, IRP2 does not bind an Fe-S cluster. Instead, it is synthesized and stable under low iron conditions, and degraded under high iron conditions. In addition to regulation by iron, IRP2 is also inhibited by ROS, RNS, and phosphorylation. IRP2 appears to be the main regulator of iron homeostasis as it can compensate for a loss of IRP1, while IRP1 cannot necessarily compensate for loss of IRP2 [105,106].
5.3 Regulation at the Post-Translational Level
5.3.1 Post-Translational Iron Regulation in S. cerevisiae
Recent studies have started to unveil the post-translational mechanisms that govern the iron-dependent activity of the S. cerevisiae transcription factors Aft1 and Aft2 (see Section 5.1.1.2 for more on Aft1/2). Genetic studies suggest that Aft1/2 does not directly sense iron, but rather responds indirectly to mitochondrial Fe-S cluster assembly [107]. Interpretation of the mitochondrial Fe-S signal is dependent on a signaling pathway involving four cytosolic proteins: Grx3, Grx4, Fra1, and Fra2, which all have homologues in mammalian cells. Grx3 and Grx4 are multidomain CGFS monothiol glutaredoxins that form [2Fe-2S] cluster-bound homodimers with a role in intracellular iron trafficking (see Section 4.1.1) [23,65]. Grx3 and Grx4 perform redundant functions since deletion of GRX3 or GRX4 singly has little or no phenotypic consequence, while grx3Δgrx4Δ double mutants are severely growth impaired or inviable [23,108]. Fra1 is an aminopeptidase P-like protein that is also implicated in regulation of vacuolar iron uptake [109]. Fra2 is a member of the BolA protein family of unknown function, although recent work has linked both prokaryotic and eukaryotic BolA homologues to Fe-S cluster biogenesis [110]. Iron-dependent regulation of Aft1/2 occurs at the protein level since Aft1/2 consistently localizes to the nucleus and binds its target promoters in the absence of the Fra1/Fra2/Grx3/Grx4 signaling pathway [64].
A variety of protein-protein interactions control signaling in this pathway. Grx3/4 interacts with Aft1 and Aft2 via a conserved CDC motif in Aft1/2. In addition, Grx3/4 and Fra1 both bind to Fra2. Biochemical and spectroscopic studies revealed that Grx3/4 and Fra2 form [2Fe-2S]-bridged heterodimers, while mutagenesis studies confirmed that the amino acids necessary for cluster binding in vitro were also required for inhibition of Aft1/2 activity in vivo [60,65]. Thus, Aft1/2 is proposed to sense the cellular iron status based on the ability of Grx3/4 and Fra2 to bind a [2Fe-2S] cluster [110]. Interestingly, Fe-S binding by Grx3/4 in vivo does not require the CIA machinery (see Section 3.2.2), indicating that a parallel pathway exists for insertion of the [2Fe-2S] cluster on Grx3/4. Under Fe replete conditions when Fra2-Grx3/4 binds an Fe-S cluster, the Fra1/Fra2/Grx3/4 signaling pathway is proposed to induce multimerization of Aft1/2. This conformational change in turn facilitates interaction with the exportin Msn5, leading to cytosolic localization of Aft1/2 and deactivation of the iron regulon. If the Fe-S signal is not received (iron-deplete conditions), the complex is unable to inhibit Aft1/2 activity, and these transcription factors move to the nucleus and induce the iron regulon to increase cellular iron levels [60,64]. However, the iron regulon is not fully activated in iron-sufficient medium in fra1Δ or fra2Δ mutants or upon disruption of mitochondrial Fe-S assembly pathways, suggesting that a separate iron signal may partially inhibit Aft1/2 activity in these mutants. The specific molecular mechanisms for inhibiting Aft1/2 activity via the Grx3/Grx4/Fra1/Fra2 signaling pathway or an alternate iron signal are still elusive. Figure 12 shows a model for regulation of Aft1 and Aft2 based on the available information for this system.
Figure 12.

Proposed model for regulation of Aft1/2 in S. cerevisiae. Under iron replete conditions (left panel), the [2Fe-2S]-bridged Fra2-Grx3/4 complex (and possibly Fra1) relays the cellular iron status to Aft1/2, leading to Aft1/2 oligomerization. Aft1/2 is consequently shuttled to the cytosol by the exportin Msn5, deactivating the iron regulon. Under low iron conditions (right panel), Grx3/4 and Fra2 cannot bind an Fe-S cluster, and Aft1/2 does not receive the Fe-S signal. Msn5 does not recognize Aft1/2 and it accumulates in the nucleus where it activates the iron regulon.
5.3.2 Post-Translational Iron Regulation in Mammalian Cells
5.3.2.1 Hepcidin
Systemic iron homeostasis in mammals requires communication between cells that acquire (enterocytes), recycle (macrophages), store (hepatocytes), and utilize iron (developing erythrocytes). The coordination of systemic iron acquisition and usage is mainly regulated post-translationally by the peptide hormone hepcidin. Hepcidin is a cysteine-rich, 25-amino acid peptide secreted by the liver and its only known target is the protein FPN. Hepcidin circulates in the plasma and binds to FPN found on the surface of intestinal duodenal cells and macrophages, thereby promoting FPN phosphorylation and subsequent degradation. Since FPN is the only known mammalian iron exporter, its degradation leads to decreased iron absorption from the intestine and disruption of iron recycling from macrophages [111]. Hepcidin expression is regulated at the transcriptional level by several stimuli, including iron availability, inflammation, and hypoxia. Basal transcriptional expression of the hepcidin gene requires C/EBPα (CCAAT enhancer-binding protein α) binding to a CCAAT sequence in the hepcidin promoter. Iron-dependent regulation of hepcidin expression occurs in response to two factors: iron stores and circulating iron. Hepatic iron stores activate hepcidin expression through bone morphogenic protein (BMP) signaling via an unknown mechanism. BMP binding to the BMP receptor activates a cascade that leads to hepcidin transcription. While several BMPs can induce hepcidin expression, BMP6 seems to be the most relevant as its expression is regulated by hepatic iron stores. Plasma iron levels can also regulate hepcidin expression through the major histocompatibility complex class 1-like protein HFE and TfR. HFE is a membrane protein that interacts with TfR1 and TfR2. When plasma iron levels are low, TfR1 is proposed to bind HFE. When iron-bound Tf increases, it displaces HFE from TfR1, allowing HFE to interact with TfR2, which activates a signaling pathway leading to hepcidin expression. Inflammatory response and ER stress can also induce hepcidin expression through the BMP and C/EBPα pathways [73,112].
5.3.2.2 PHD Regulation of HIF-2α
As previously mentioned, HIF-2α controls iron absorption in enterocytes via transcriptional activation of iron uptake genes. The mechanism of activation is indirectly dependent on intracellular iron levels: HIF-2α protein levels are controlled by a prolyl hydroxylase (PHD) that requires O2 and Fe2+ for activity. Under normoxia conditions with sufficient iron, PHD is active and hydroxylates HIF-2α, resulting in ubiquitination by the VHL (von Hippel-Lindau) ubiquitin ligase. Ubiquitinated HIF-2α is subsequently targeted for degradation by the proteasome. In hypoxic conditions and/or under iron deficiency, PHD is inactive, thus HIF-2α is not hydroxylated and degraded, and instead forms a heterodimeric complex with HIF-1β that induces transcription of DcytB and DMT1 [96]. Thus, HIF-2α directly links iron absorption with both oxygen and iron metabolism. The mRNA for HIF-2α also contains an IRE in the 5′ UTR that is a target for IRP1. HIF-2α is therefore post-transcriptionally regulated in an iron-dependent fashion. The relationship between PHDs, HIF-2α, and IRP1 demonstrates feedback regulation between iron and oxygen metabolism that involves transcriptional, post-transcriptional, and post-translational control mechanisms [113].
5.3.2.3 FBXL5 Regulation of IRP2
IRP2 plays a role in iron homeostasis by controlling the stability of mRNAs that encode proteins involved in iron trafficking and iron utilization. While it is clear that IRP2 is degraded under high iron conditions by a ubiquitin ligase, the identity of this enzyme was only recently uncovered. The SCF (SKP1-CUL1-F-box) ubiquitin ligases were found to be specific for IRP2 when customized by the F-box protein FBXL5. FBXL5 forms an SCF complex that physically interacts with IRP2 in an iron-dependent fashion. This regulation does not occur at the translational level, as FBXL5 mRNA levels were not affected by changes in iron. Rather, the FBXL5 protein is targeted for degradation based on iron availability. FBXL5 stability also depends on intracellular oxygen concentrations since exposure to oxygen leads to degradation. These observations are explained by the fact that FBXL5 contains a hemerythrin domain that binds iron and oxygen. Many iron-bound hemerythrin domains function as oxygen sensors and metal storage sites. The ability of the FBXL5 hemerythrin domain to bind iron seems to be indicator of stability. If an iron center cannot be bound either due to insufficient iron or hypoxic conditions, the protein is targeted for degradation. The stability of FBXL5 in turn regulates IRP2. Degradation of FBXL5 prevents formation of the SCF ubiquitin ligase, thus IRP2 is not targeted for degradation, and can bind IREs. Under iron-replete conditions, the F-box domain of FBXL5 can bind its iron cofactor and form the SCF ligase complex, which then promotes degradation of IRP2 by ubiquitination [114,115].
6 Concluding Remarks and Future Directions
While a great deal of progress has been made in characterizing the iron metallome of eukaryotes, there is still more left to uncover. Recent progress in spectroscopic techniques has aided in revealing the various forms of cellular iron. Future studies will focus on the nature of labile iron pools, including location, iron ligands, and cellular concentrations. How does iron vary among eukaryotes, and under different growth conditions? The overall assembly of heme and Fe-S cluster cofactors is well-established, but there are some holes in our knowledge here as well. Even though some common iron chaperones have been identified, these need to be studied in more detail. Future work in this area will determine how iron is delivered for cofactor assembly, and how the assembled cofactors are trafficked to target proteins. One intriguing area of study is the identity of the product exported from the mitochondrial ISC machinery for the cytosolic CIA machinery. In addition, assembly of non-heme iron cofactors demands more investigation, as it seems this process is unique to the target protein. Several iron transporters have been identified and categorized to different organelles, particularly the mitochondria and vacuoles. However, just as chaperones to transfer iron for cofactor assembly are not well-characterized, neither are chaperones for intracellular trafficking. In addition, while transport proteins have been identified for iron import, the mechanisms of import must be further characterized. Iron storage proteins are another focus for future studies, especially the mechanisms for releasing iron once cellular need increases. Are there specific enzymes that target storage proteins for degradation or facilitate dissolution of ferritin biominerals? How is this regulated?
One of the most important topics for future study is regulation of the iron metallome. From yeast to humans, eukaryotes have several complex mechanisms to maintain iron levels, from transcriptional to post-transcriptional to post-translational control. There appears to be connections not only between these different regulatory levels, but also to oxygen metabolism. For example, hypoxia plays a regulatory role in controlling the expression and stability of several proteins: the hypoxia-inducible factors control iron uptake, and hypoxic conditions may induce expression of the hormone hepcidin. Several forms of regulation depend on indirect sensing of cellular iron levels, such as yeast Aft1 and Aft2. What is the mechanism for interpreting these cellular iron signals and how are Fe-S clusters involved? Furthermore, failures in regulation have been linked to many iron-related diseases, including hemochromatosis and anemia. A deeper understanding of the regulatory intricacies of iron metabolism will help in the treatment of these diseases.
Acknowledgments
This work was funded by the National Institutes of Health Grants ES013780 and GM086619 and the South Carolina Research Foundation. The authors would like to thank Dr. L. Celeste for helpful discussions.
Abbreviations
- ABC
ATP-binding cassette
- ALA
δ-aminolevulinic acid
- ALAD
aminolevulinate dehydratase
- ALAS
ALA synthase
- ARE
AU-rich elements
- BMP
bone morphogenic protein
- C/EBPα
CCAAT enhancer-binding protein α
- CIA
cytosolic Fe-S protein assembly
- Cp
ceruloplasmin
- CPgenIII
coproporphyrinogen III
- CPOX
coproporphyrinogen oxidase
- DHBA
dihydroxybenzoic acid
- dNDP
deoxynucleoside diphosphate
- dNTP
deoxynucleoside triphosphate
- EPR
electron paramagnetic resonance
- ER
endoplasmic reticulum
- EXAFS
extended X-ray absorption fine structure
- FECH
ferrochelatase
- FeRE
iron-responsive element
- Fe-S
iron-sulfur
- FIH1
factor inhibiting HIF
- FPN
ferroportin
- Grx
glutaredoxin
- GSH
glutathione
- GST
glutathione S-transferase
- HCP1
Heme carrier protein 1
- HIF
hypoxia-inducible factor
- HMB
hydroxymethylbilane
- HO-1
heme oxygenase-1
- HRG
heme responsive gene
- IMS
intermembrane space
- IRE
iron regulatory element
- IRP
iron regulatory protein
- ISC
iron-sulfur cluster
- LIP
labile iron pool
- Mfrn
mitoferrin
- NDP
nucleoside diphosphate
- NRAMP
natural resistance-associated macrophage protein
- NTP
nucleoside triphosphate
- PBGD
porphobilinogen deaminase
- PCBP
poly (rC) binding protein
- PHD
prolyl hydroxylase
- PIXE
particle-induced X-ray emission
- PPgenIX
protoporphyrinogen IX
- PPIX
protoporphyrin IX
- PPOX
protoporphyrinogen oxidase
- RNR
ribonucleotide reductase
- ROS
reactive oxygen species
- RNS
reactive nitrogen species
- SCF
SKP1-CUL1-F-box
- SOD
superoxide dismutase
- STEAP
six-transmembrane epithelial antigen of the prostate
- Tf
transferrin
- TfR
Tf receptor
- TTP
tristetraprolin
- TZF
tandem zinc finger
- UPgenIII
uroporphyrinogen III
- URO3S
uroporphyrinogen III synthase
- UROD
uroporphyrinogen decarboxylase
- UTR
untranslated region
- VHL
von Hippel-Lindau
- XANES
X-ray absorption near edge structure
- XAS
X-ray absorption spectroscopy
- XRFM
X-ray fluorescence microscopy
References
- 1.Eide DJ, Clark S, Nair TM, Gehl M, Gribskov M, Guerinot ML, Harper JF. Genome Biol. 2005;6:R77. doi: 10.1186/gb-2005-6-9-r77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Epsztejn S, Glickstein H, Picard V, Slotki IN, Breuer W, Beaumont C, Cabantchik ZI. Blood. 1999;94:3593–3603. [PubMed] [Google Scholar]
- 3.Petrat F, Rauen U, de Groot H. Hepatology. 1999;29:1171–1179. doi: 10.1002/hep.510290435. [DOI] [PubMed] [Google Scholar]
- 4.Ceccarelli D, Gallesi D, Giovannini F, Ferrali M, Masini A. Biochem Biophys Res Commun. 1995;209:53–59. doi: 10.1006/bbrc.1995.1469. [DOI] [PubMed] [Google Scholar]
- 5.Gao X, Qian M, Campian JL, Marshall J, Zhou Z, Roberts AM, Kang YJ, Prabhu SD, Sun XF, Eaton JW. Free Radic Biol Med. 2010;49:401–407. doi: 10.1016/j.freeradbiomed.2010.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petrak J, Myslivcova D, Man P, Cmejla R, Cmejlova J, Vyoral D. Am J Physiol Gastrointest Liver Physiol. 2006;290:G1059–1066. doi: 10.1152/ajpgi.00469.2005. [DOI] [PubMed] [Google Scholar]
- 7.Miao R, Holmes-Hampton GP, Lindahl PA. Biochemistry. 2011;50:2660–2671. doi: 10.1021/bi102015s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lindahl PA, Holmes-Hampton GP. Curr Opin Chem Biol. 2011;15:342–346. doi: 10.1016/j.cbpa.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cockrell AL, Holmes-Hampton GP, McCormick SP, Chakrabarti M, Lindahl PA. Biochemistry. 2011;50:10275–10283. doi: 10.1021/bi2014954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holmes-Hampton GP, Miao R, Garber Morales J, Guo Y, Munck E, Lindahl PA. Biochemistry. 2010;49:4227–4234. doi: 10.1021/bi1001823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miao R, Kim H, Koppolu UM, Ellis EA, Scott RA, Lindahl PA. Biochemistry. 2009;48:9556–9568. doi: 10.1021/bi901110n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ortega R, Deves G, Carmona A. J R Soc Interface. 2009;6(Suppl 5):S649–658. doi: 10.1098/rsif.2009.0166.focus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fahrni CJ. Curr Opin Chem Biol. 2007;11:121–127. doi: 10.1016/j.cbpa.2007.02.039. [DOI] [PubMed] [Google Scholar]
- 14.Iancu TC, Deugnier Y, Halliday JW, Powell LW, Brissot P. J Hepatol. 1997;27:628–638. doi: 10.1016/s0168-8278(97)80079-7. [DOI] [PubMed] [Google Scholar]
- 15.Kaplan CD, Kaplan J. Chem Rev. 2009;109:4536–4552. doi: 10.1021/cr9001676. [DOI] [PubMed] [Google Scholar]
- 16.Hentze MW, Muckenthaler MU, Andrews NC. Cell. 2004;117:285–297. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- 17.Bao G, Clifton M, Hoette TM, Mori K, Deng SX, Qiu A, Viltard M, Williams D, Paragas N, Leete T, Kulkarni R, Li X, Lee B, Kalandadze A, Ratner AJ, Pizarro JC, Schmidt-Ott KM, Landry DW, Raymond KN, Strong RK, Barasch J. Nat Chem Biol. 2010;6:602–609. doi: 10.1038/nchembio.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Devireddy LR, Hart DO, Goetz DH, Green MR. Cell. 2010;141:1006–1017. doi: 10.1016/j.cell.2010.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Correnti C, Strong RK. J Biol Chem. 2012;287:13524–13531. doi: 10.1074/jbc.R111.311829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petrat F, de Groot H, Rauen U. Biochem J. 2001;356:61–69. doi: 10.1042/0264-6021:3560061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hentze MW, Muckenthaler MU, Galy B, Camaschella C. Cell. 2010;142:24–38. doi: 10.1016/j.cell.2010.06.028. [DOI] [PubMed] [Google Scholar]
- 22.Hider RC, Kong XL. Biometals. 2011;24:1179–1187. doi: 10.1007/s10534-011-9476-8. [DOI] [PubMed] [Google Scholar]
- 23.Muhlenhoff U, Molik S, Godoy JR, Uzarska MA, Richter N, Seubert A, Zhang Y, Stubbe J, Pierrel F, Herrero E, Lillig CH, Lill R. Cell Metab. 2010;12:373–385. doi: 10.1016/j.cmet.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Petrat F, Weisheit D, Lensen M, de Groot H, Sustmann R, Rauen U. Biochem J. 2002;362:137–147. doi: 10.1042/0264-6021:3620137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rauen U, Springer A, Weisheit D, Petrat F, Korth HG, de Groot H, Sustmann R. Chembiochem. 2007;8:341–352. doi: 10.1002/cbic.200600311. [DOI] [PubMed] [Google Scholar]
- 26.Sturm B, Bistrich U, Schranzhofer M, Sarsero JP, Rauen U, Scheiber-Mojdehkar B, de Groot H, Ioannou P, Petrat F. J Biol Chem. 2005;280:6701–6708. doi: 10.1074/jbc.M408717200. [DOI] [PubMed] [Google Scholar]
- 27.Kehrer JP. Toxicology. 2000;149:43–50. doi: 10.1016/s0300-483x(00)00231-6. [DOI] [PubMed] [Google Scholar]
- 28.Rovira II, Finkel T, Masters BS, Dickman MB, Lee J, Ragsdale SW, Lee CC. In: Redox Biochemistry. Banerjee R, Becker DF, Dickman MB, Gladyshev VN, Ragsdale SW, editors. John Wiley & Sons, Inc; Hoboken, NJ, USA: 2007. [Google Scholar]
- 29.Jomova K, Valko M. Toxicology. 2011;283:65–87. doi: 10.1016/j.tox.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 30.Lin H, Li L, Jia X, Ward DM, Kaplan J. J Biol Chem. 2011;286:3851–3862. doi: 10.1074/jbc.M110.190959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lebovitz RM, Zhang H, Vogel H, Cartwright J, Jr, Dionne L, Lu N, Huang S, Matzuk MM. Proc Natl Acad Sci USA. 1996;93:9782–9787. doi: 10.1073/pnas.93.18.9782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Nat Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 33.Naranuntarat A, Jensen LT, Pazicni S, Penner-Hahn JE, Culotta VC. J Biol Chem. 2009;284:22633–22640. doi: 10.1074/jbc.M109.026773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang M, Cobine PA, Molik S, Naranuntarat A, Lill R, Winge DR, Culotta VC. EMBO J. 2006;25:1775–1783. doi: 10.1038/sj.emboj.7601064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jouihan HA, Cobine PA, Cooksey RC, Hoagland EA, Boudina S, Abel ED, Winge DR, McClain DA. Mol Med. 2008;14:98–108. doi: 10.2119/2007-00114.Jouihan. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yin L, Wu N, Curtin JC, Qatanani M, Szwergold NR, Reid RA, Waitt GM, Parks DJ, Pearce KH, Wisely GB, Lazar MA. Science. 2007;318:1786–1789. doi: 10.1126/science.1150179. [DOI] [PubMed] [Google Scholar]
- 37.Johnson MK. Curr Opin Chem Biol. 1998;2:173–181. doi: 10.1016/s1367-5931(98)80058-6. [DOI] [PubMed] [Google Scholar]
- 38.Lancaster KM, Roemelt M, Ettenhuber P, Hu Y, Ribbe MW, Neese F, Bergmann U, DeBeer S. Science. 2011;334:974–977. doi: 10.1126/science.1206445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spatzal T, Aksoyoglu M, Zhang L, Andrade SL, Schleicher E, Weber S, Rees DC, Einsle O. Science. 2011;334:940. doi: 10.1126/science.1214025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sheftel A, Stehling O, Lill R. Trends Endocrinol Metab. 2010;21:302–314. doi: 10.1016/j.tem.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 41.Hamza I. ACS Chem Biol. 2006;1:627–629. doi: 10.1021/cb600442b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ajioka RS, Phillips JD, Kushner JP. Biochim Biophys Acta. 2006;1763:723–736. doi: 10.1016/j.bbamcr.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 43.Shoolingin-Jordan PM, Al-Dbass A, McNeill LA, Sarwar M, Butler D. Biochem Soc Trans. 2003;31:731–735. doi: 10.1042/bst0310731. [DOI] [PubMed] [Google Scholar]
- 44.Krishnamurthy PC, Du G, Fukuda Y, Sun D, Sampath J, Mercer KE, Wang J, Sosa-Pineda B, Murti KG, Schuetz JD. Nature. 2006;443:586–589. doi: 10.1038/nature05125. [DOI] [PubMed] [Google Scholar]
- 45.Schultz IJ, Chen C, Paw BH, Hamza I. J Biol Chem. 2010;285:26753–26759. doi: 10.1074/jbc.R110.119503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dailey HA. Biochem Soc Trans. 2002;30:590–595. doi: 10.1042/bst0300590. [DOI] [PubMed] [Google Scholar]
- 47.Park S, Gakh O, O’Neill HA, Mangravita A, Nichol H, Ferreira GC, Isaya G. J Biol Chem. 2003;278:31340–31351. doi: 10.1074/jbc.M303158200. [DOI] [PubMed] [Google Scholar]
- 48.Rouault TA. Dis Model Mech. 2012;5:155–164. doi: 10.1242/dmm.009019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Severance S, Hamza I. Chem Rev. 2009;109:4596–4616. doi: 10.1021/cr9001116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spielewoy N, Schulz H, Grienenberger JM, Thony-Meyer L, Bonnard G. J Biol Chem. 2001;276:5491–5497. doi: 10.1074/jbc.M008853200. [DOI] [PubMed] [Google Scholar]
- 51.Harvey JW, Beutler E. Blood. 1982;60:1227–1230. [PubMed] [Google Scholar]
- 52.Nakai Y, Umeda N, Suzuki T, Nakai M, Hayashi H, Watanabe K, Kagamiyama H. J Biol Chem. 2004;279:12363–12368. doi: 10.1074/jbc.M312448200. [DOI] [PubMed] [Google Scholar]
- 53.Lill R, Muhlenhoff U. Annu Rev Biochem. 2008;77:669–700. doi: 10.1146/annurev.biochem.76.052705.162653. [DOI] [PubMed] [Google Scholar]
- 54.Subramanian P, Rodrigues AV, Ghimire-Rijal S, Stemmler TL. Curr Opin Chem Biol. 2011;15:312–318. doi: 10.1016/j.cbpa.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bridwell-Rabb J, Iannuzzi C, Pastore A, Barondeau DP. Biochemistry. 2012;51:2506–2514. doi: 10.1021/bi201628j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ichikawa Y, Bayeva M, Ghanefar M, Potini V, Sun L, Mutharasan RK, Wu R, Khechaduri A, Jairaj Naik T, Ardehali H. Proc Natl Acad Sci USA. 2012;109:4152–4157. doi: 10.1073/pnas.1119338109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bedekovics T, Li H, Gajdos GB, Isaya G. J Biol Chem. 2011;286:40878–40888. doi: 10.1074/jbc.M111.270082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Banci L, Bertini I, Ciofi-Baffoni S, Boscaro F, Chatzi A, Mikolajczyk M, Tokatlidis K, Winkelmann J. Chem Biol. 2011;18:794–804. doi: 10.1016/j.chembiol.2011.03.015. [DOI] [PubMed] [Google Scholar]
- 59.Sharma AK, Pallesen LJ, Spang RJ, Walden WE. J Biol Chem. 2010;285:26745–26751. doi: 10.1074/jbc.R110.122218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li H, Mapolelo DT, Dingra NN, Keller G, Riggs-Gelasco PJ, Winge DR, Johnson MK, Outten CE. J Biol Chem. 2011;286:867–876. doi: 10.1074/jbc.M110.184176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cotruvo JA, Stubbe J. Annu Rev Biochem. 2011;80:733–767. doi: 10.1146/annurev-biochem-061408-095817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y, Liu L, Wu X, An X, Stubbe J, Huang M. J Biol Chem. 2011;286:41499–41509. doi: 10.1074/jbc.M111.294074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bleackley MR, Macgillivray RT. Biometals. 2011;24:785–809. doi: 10.1007/s10534-011-9451-4. [DOI] [PubMed] [Google Scholar]
- 64.Kumanovics A, Chen OS, Li L, Bagley D, Adkins EM, Lin H, Dingra NN, Outten CE, Keller G, Winge D, Ward DM, Kaplan J. J Biol Chem. 2008;283:10276–10286. doi: 10.1074/jbc.M801160200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li H, Mapolelo DT, Dingra NN, Naik SG, Lees NS, Hoffman BM, Riggs-Gelasco PJ, Huynh BH, Johnson MK, Outten CE. Biochemistry. 2009;48:9569–9581. doi: 10.1021/bi901182w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shayeghi M, Latunde-Dada GO, Oakhill JS, Laftah AH, Takeuchi K, Halliday N, Khan Y, Warley A, McCann FE, Hider RC, Frazer DM, Anderson GJ, Vulpe CD, Simpson RJ, McKie AT. Cell. 2005;122:789–801. doi: 10.1016/j.cell.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 67.Anderson GJ, Vulpe CD. Cell Mol Life Sci. 2009;66:3241–3261. doi: 10.1007/s00018-009-0051-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rajagopal A, Rao AU, Amigo J, Tian M, Upadhyay SK, Hall C, Uhm S, Mathew MK, Fleming MD, Paw BH, Krause M, Hamza I. Nature. 2008;453:1127–1131. doi: 10.1038/nature06934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nandal A, Ruiz JC, Subramanian P, Ghimire-Rijal S, Sinnamon RA, Stemmler TL, Bruick RK, Philpott CC. Cell Metab. 2011;14:647–657. doi: 10.1016/j.cmet.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Haunhorst P, Berndt C, Eitner S, Godoy JR, Lillig CH. Biochem Biophys Res Commun. 2010;394:372–376. doi: 10.1016/j.bbrc.2010.03.016. [DOI] [PubMed] [Google Scholar]
- 71.Li H, Mapolelo DT, Randeniya S, Johnson MK, Outten CE. Biochemistry. 2012;51:1687–1696. doi: 10.1021/bi2019089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Theil EC. Curr Opin Chem Biol. 2011;15:304–311. doi: 10.1016/j.cbpa.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang J, Pantopoulos K. Biochem J. 2011;434:365–381. doi: 10.1042/BJ20101825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bou-Abdallah F, Santambrogio P, Levi S, Arosio P, Chasteen ND. J Mol Biol. 2005;347:543–554. doi: 10.1016/j.jmb.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 75.Santambrogio P, Erba BG, Campanella A, Cozzi A, Causarano V, Cremonesi L, Galli A, Della Porta MG, Invernizzi R, Levi S. Haematologica. 2011;96:1424–1432. doi: 10.3324/haematol.2011.042952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Richardson DR, Lane DJ, Becker EM, Huang ML, Whitnall M, Suryo Rahmanto Y, Sheftel AD, Ponka P. Proc Natl Acad Sci USA. 2010;107:10775–10782. doi: 10.1073/pnas.0912925107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang ML, Lane DJ, Richardson DR. Antioxid Redox Signal. 2011;15:3003–3019. doi: 10.1089/ars.2011.3921. [DOI] [PubMed] [Google Scholar]
- 78.Li L, Chen OS, McVey Ward D, Kaplan J. J Biol Chem. 2001;276:29515–29519. doi: 10.1074/jbc.M103944200. [DOI] [PubMed] [Google Scholar]
- 79.Philpott CC, Protchenko O. Eukaryot Cell. 2008;7:20–27. doi: 10.1128/EC.00354-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Singh A, Kaur N, Kosman DJ. J Biol Chem. 2007;282:28619–28626. doi: 10.1074/jbc.M703398200. [DOI] [PubMed] [Google Scholar]
- 81.Kurz T, Eaton JW, Brunk UT. Int J Biochem Cell Biol. 2011;43:1686–1697. doi: 10.1016/j.biocel.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 82.Toman PD, Chisholm G, McMullin H, Giere LM, Olsen DR, Kovach RJ, Leigh SD, Fong BE, Chang R, Daniels GA, Berg RA, Hitzeman RA. J Biol Chem. 2000;275:23303–23309. doi: 10.1074/jbc.M002284200. [DOI] [PubMed] [Google Scholar]
- 83.De Domenico I, Vaughn MB, Paradkar PN, Lo E, Ward DM, Kaplan J. Cell Metab. 2011;13:57–67. doi: 10.1016/j.cmet.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 84.Missirlis F, Kosmidis S, Brody T, Mavrakis M, Holmberg S, Odenwald WF, Skoulakis EM, Rouault TA. Genetics. 2007;177:89–100. doi: 10.1534/genetics.107.075150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Alkhateeb AA, Connor JR. Biochim Biophys Acta. 2010;1800:793–797. doi: 10.1016/j.bbagen.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 86.Gurgueira SA, Meneghini R. J Biol Chem. 1996;271:13616–13620. doi: 10.1074/jbc.271.23.13616. [DOI] [PubMed] [Google Scholar]
- 87.Kaplan J, McVey Ward D, Crisp RJ, Philpott CC. Biochim Biophys Acta. 2006;1763:646–651. doi: 10.1016/j.bbamcr.2006.03.008. [DOI] [PubMed] [Google Scholar]
- 88.Blaiseau PL, Lesuisse E, Camadro JM. J Biol Chem. 2001;276:34221–34226. doi: 10.1074/jbc.M104987200. [DOI] [PubMed] [Google Scholar]
- 89.Rutherford JC, Bird AJ. Eukaryot Cell. 2004;3:1–13. doi: 10.1128/EC.3.1.1-13.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Rutherford JC, Jaron S, Winge DR. J Biol Chem. 2003;278:27636–27643. doi: 10.1074/jbc.M300076200. [DOI] [PubMed] [Google Scholar]
- 91.Courel M, Lallet S, Camadro JM, Blaiseau PL. Mol Cell Biol. 2005;25:6760–6771. doi: 10.1128/MCB.25.15.6760-6771.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li L, Jia X, Ward DM, Kaplan J. J Biol Chem. 2011;286:38488–38497. doi: 10.1074/jbc.M111.286666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Li L, Bagley D, Ward DM, Kaplan J. Mol Cell Biol. 2008;28:1326–1337. doi: 10.1128/MCB.01219-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Winge DR, Nielson KB, Gray WR, Hamer DH. J Biol Chem. 1985;260:14464–14470. [PubMed] [Google Scholar]
- 95.Ding XQ, Bill E, Trautwein AX, Hartmann HJ, Weser U. Eur J Biochem. 1994;223:841–845. doi: 10.1111/j.1432-1033.1994.tb19060.x. [DOI] [PubMed] [Google Scholar]
- 96.Mastrogiannaki M, Matak P, Keith B, Simon MC, Vaulont S, Peyssonnaux C. J Clin Invest. 2009;119:1159–1166. doi: 10.1172/JCI38499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shah YM, Matsubara T, Ito S, Yim SH, Gonzalez FJ. Cell Metab. 2009;9:152–164. doi: 10.1016/j.cmet.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Philpott CC, Leidgens S, Frey AG. Biochim Biophys Acta. 2012 doi: 10.1016/j.bbamcr.2012.01.012. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Puig S, Vergara SV, Thiele DJ. Cell Metab. 2008;7:555–564. doi: 10.1016/j.cmet.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Puig S, Askeland E, Thiele DJ. Cell. 2005;120:99–110. doi: 10.1016/j.cell.2004.11.032. [DOI] [PubMed] [Google Scholar]
- 101.Kaplan CD, Kaplan J. Cell Metab. 2005;2:4–6. doi: 10.1016/j.cmet.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 102.Yao R, Zhang Z, An X, Bucci B, Perlstein DL, Stubbe J, Huang M. Proc Natl Acad Sci USA. 2003;100:6628–6633. doi: 10.1073/pnas.1131932100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sanvisens N, Bano MC, Huang M, Puig S. Mol Cell. 2011;44:759–769. doi: 10.1016/j.molcel.2011.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Recalcati S, Minotti G, Cairo G. Antioxid Redox Signal. 2010;13:1593–1616. doi: 10.1089/ars.2009.2983. [DOI] [PubMed] [Google Scholar]
- 105.Rouault TA. Nat Chem Biol. 2006;2:406–414. doi: 10.1038/nchembio807. [DOI] [PubMed] [Google Scholar]
- 106.Pantopoulos K. Ann N Y Acad Sci. 2004;1012:1–13. doi: 10.1196/annals.1306.001. [DOI] [PubMed] [Google Scholar]
- 107.Rutherford JC, Ojeda L, Balk J, Muhlenhoff U, Lill R, Winge DR. J Biol Chem. 2005;280:10135–10140. doi: 10.1074/jbc.M413731200. [DOI] [PubMed] [Google Scholar]
- 108.Pujol-Carrion N, Bellí G, Herrero E, Nogues A, de la Torre-Ruiz MA. J Cell Sci. 2006;119:4554–4564. doi: 10.1242/jcs.03229. [DOI] [PubMed] [Google Scholar]
- 109.Li L, Murdock G, Bagley D, Jia X, Ward DM, Kaplan J. J Biol Chem. 2010;285:10232–10242. doi: 10.1074/jbc.M109.096859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li H, Outten CE. Biochemistry. 2012 doi: 10.1021/bi300393z. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kaplan J, Ward DM, De Domenico I. Int J Hematol. 2011;93:14–20. doi: 10.1007/s12185-010-0760-0. [DOI] [PubMed] [Google Scholar]
- 112.Gkouvatsos K, Papanikolaou G, Pantopoulos K. Biochim Biophys Acta. 2012;1820:188–202. doi: 10.1016/j.bbagen.2011.10.013. [DOI] [PubMed] [Google Scholar]
- 113.Sanchez M, Galy B, Muckenthaler MU, Hentze MW. Nat Struct Mol Biol. 2007;14:420–426. doi: 10.1038/nsmb1222. [DOI] [PubMed] [Google Scholar]
- 114.Salahudeen AA, Thompson JW, Ruiz JC, Ma HW, Kinch LN, Li Q, Grishin NV, Bruick RK. Science. 2009;326:722–726. doi: 10.1126/science.1176326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vashisht AA, Zumbrennen KB, Huang X, Powers DN, Durazo A, Sun D, Bhaskaran N, Persson A, Uhlen M, Sangfelt O, Spruck C, Leibold EA, Wohlschlegel JA. Science. 2009;326:718–721. doi: 10.1126/science.1176333. [DOI] [PMC free article] [PubMed] [Google Scholar]