

Abstract
Poly(ADP-ribose) polymerase-1 (PARP-1) binds intermediates of base excision repair (BER) and becomes activated for poly(ADP-ribose) (PAR) synthesis. PAR mediates recruitment and functions of the key BER factors XRCC1 and DNA polymerase β (pol β) that in turn regulate PAR. Yet, the molecular mechanism and implications of coordination between XRCC1 and pol β in regulating the level of PAR are poorly understood. A complex of PARP-1, XRCC1 and pol β is found in vivo, and it is known that pol β and XRCC1 interact through a redox-sensitive binding interface in the N-terminal domain of XRCC1. We confirmed here that both oxidized and reduced forms of XRCC1 are present in mouse fibroblasts. To further understand the importance of the C120-C20 oxidized form of XRCC1 and the interaction with pol β, we characterized cell lines representing stable transfectants in Xrcc1−/− mouse fibroblasts of wild-type XRCC1 and two mutants of XRCC1, a novel reduced form with the C12-C20 disulfide bond blocked (C12A) and a reference mutant that is unable to bind pol β (V88R). XRCC1-deficient mouse fibroblasts are extremely hypersensitive to methyl methanesulfonate (MMS), and transfected wild-type and C12A mutant XRCC1 proteins similarly reversed MMS hypersensitivity. However, after MMS exposure the cellular PAR level was found to increase to a much greater extent in cells expressing the C12A mutant than in cells expressing wild-type XRCC1. PARP inhibition resulted in very strong MMS sensitization in cells expressing wild-type XRCC1, but this sensitization was much less in cells expressing the C12A mutant. The results suggest a role for the oxidized form of XRCC1 in the interaction with pol β in 1) controlling the PAR level after MMS exposure and 2) enabling the extreme cytotoxicity of PARP inhibition during the MMS DNA damage response.
1. Introduction
The base excision repair (BER) pathway is the primary mechanism of repair of single base lesions in DNA, and the primary lesions resulting from treatment of cells with the methylating agent methyl methanesulfonate (MMS) are methylated bases. In experiments conducted in vitro, single-nucleotide BER can be accomplished by four enzymes, i.e., a DNA glycosylase, AP endonuclease 1, DNA polymerase β (pol β) and DNA ligase. In cellular repair, BER becomes far more complex by virtue of important, but poorly understood, roles of various accessory factors, including XRCC1 and PARP-1.
Poly(ADP-ribose) polymerase-1 (PARP-1) has roles in cellular DNA damage recognition and repair. As a result of binding to abasic sites, nicks and strand-breaks in DNA, PARP-1 becomes catalytically activated synthesizing poly(ADP-ribose) (PAR) polymers onto itself and other repair and chromatin remodeling factors. As a result of self-poly(ADP-ribosyl)ation, other BER proteins, e.g., XRCC1, pol β and DNA ligase IIIα (lig-IIIα), are recruited to sites of DNA damage. Following cellular damage, XRCC1 recruitment is mediated by binding of its BRCT I domain (Fig. 1) to the PAR polymers of PARP-1, and in PARP-1-deficient cells, there is little appearance of XRCC1 at DNA damage-induced foci [1].
Fig. 1.
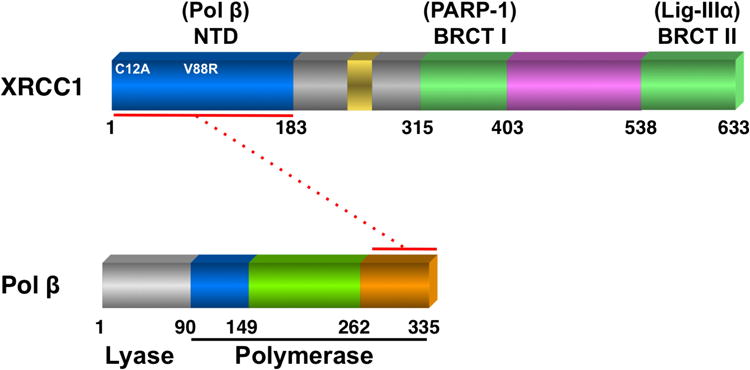
Domain organization of mouse XRCC1 and pol β proteins. Indicated is the location of the two N-terminal domain (NTD) mutant XRCC1 proteins (C12A) and (V88R) utilized in this study. The NTD of XRCC1 interacts with the C-terminal end of the polymerase domain of pol β as shown by the dotted red line. XRCC1 also interacts with PARP-1 via its BRCT I domain and DNA ligase IIIα via its BRCT II domain.
In the presence of a PARP inhibitor, PARP-1 binds at sites of DNA damage, but self-poly(ADP-ribosyl)ation is prevented. Under these conditions, it is proposed that BER enzyme recruitment is hindered, but binding of PARP-1 to DNA is stabilized, impeding DNA repair [2] and resulting in stalling of replication fork progression [3]. Wild-type mouse fibroblasts are dramatically sensitized to the cytotoxic effects of MMS by combined treatment with the PARP inhibitor 4-amino-1,8-naphthalimide (4-AN) [4, 5]. PARP inhibition combined with MMS results in apoptotic cell death linked to formation of replication-dependent double-strand breaks [6].
XRCC1 is a multi-domain protein (Fig. 1) with no known catalytic activity. It interacts with a number of key repair proteins and is proposed to act as a scaffold able to modulate and coordinate the various steps of BER. XRCC1-deficient mouse fibroblasts are extremely hypersensitive to MMS, and this is linked to repair deficiency as measured by the comet assay [5]. The protein-protein interaction between XRCC1 and the 31 kDa polymerase domain of pol β (Fig. 1) is critical for BER [7]. Point mutations in the human XRCC1 NTD, including V86R prevent binding to pol β [8, 9], and the human V86R XRCC1 protein fails to completely complement the MMS hypersensitivity phenotype of XRCC1-deficient CHO cells [10].
Oxidized and reduced conformations of the XRCC1 N-terminal domain (NTD) have been identified in crystallographic and NMR studies of the NTD peptide [11]. Both the reduced and oxidized forms of the XRCC1 NTD interact with pol β, however, the pol β:NTD interaction is stronger with the oxidized form of the NTD. Here, the presence of the C12-C20 oxidized form of full-length XRCC1 is confirmed in mouse fibroblast cells. We show that the level of the reduced and oxidized forms is similar in untreated and MMS-treated cells. However, in the presence of PARP inhibitor, the level of the reduced form is higher consistent with less avid binding to pol β. Alanine substitution at C12 (C12A) removes the potential for this disulfide bond formation and is expected to stabilize the protein in the reduced form with lower pol β binding affinity. We confirm this expectation in the present work and further evaluate the importance of XRCC1 and pol β binding in protection against MMS-induced toxicity. We first characterize the structural integrity of the full-length and NTDs of wild-type and C12A XRCC1 proteins, and then compare the pol β binding affinities of reduced wild-type and C12A NTDs as described previously [11]. We then characterize cell lines representing stable transfectants in Xrcc1−/− mouse fibroblast cells with full-length wild-type and C12A forms of XRCC1. The results point to a critical role of the oxidized form of XRCC1 in 1) controlling the PAR response after MMS treatment and 2) enabling the extreme cell killing response to the combination of MMS-induced DNA damage plus PARP inhibitor.
2. Materials and Methods
2.1. Preparation of stable XRCC1 mutant cell variants
Xrcc1+/+ and Xrcc1−/− p53-deficient mouse embryonic fibroblasts were obtained from Dr. Robert Tebbs [12]. These cells were maintained in low glucose Dulbecco's Modified Eagle's medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) (HyClone, Logan, UT) in a 10% CO2 incubator at 37°C. Mycoplasma testing was performed routinely using a MycoAlert® Mycoplasma detection kit (Lonza, Rockland, ME).
A clone containing the full-length open reading frame of mouse XRCC1 was obtained from Invitrogen. The cDNA was subcloned into the pDONR221 vector then the pEF-DEST51 vector utilizing Gateway technology (Invitrogen). XRCC1 mutants, C12A and V88R, were introduced by site-directed mutagenesis of the pDONR221 vector and subcloned into pEF-DEST51. The resulting mammalian cell expression vectors, pXR1 (wild-type), pXRE (C12A) and pXV (V88R) were sequence verified.
One day before transfection, 2 × 105 Xrcc1−/− cells were seeded in six-well dishes in 2 ml of growth medium so that cells would be 95% confluent at time of transfection. Cells were transfected with DNA complex in serum-free medium using Lipofectamine™2000 (Invitrogen). After transfection, cells were split into fresh growth medium containing 10% FBS. Selection with blasticidin (10 μg/ml; Invitrogen) was started the following day and single cell clones were isolated and screened for XRCC1 expression by western blotting.
2.2. Purification and characterization of XRCC1 wild-type and C12A mutant NTD
The gene for human XRCC1 NTD (amino acids 1-155) optimized for E. coli expression was obtained from Genscript and then cloned into a pUC 18 vector. The gene was amplified using primers from IDT that were designed to add a C-terminal hexa-histidine affinity tag along with restriction sites for subcloning into a pET21a plasmid. The C12A mutation was made using Agilent's QuickChange Site-Directed Mutagenesis Kit. The wild-type and C12A mutant vectors were transformed into BL21-DE3-RIL cells. Bacteria were grown on 15N-labeled M9 minimal media supplemented with 15NH4Cl and 2.5 ml/l 15N-Bioexpress (Cambridge Isotopes). Cells were grown to an OD ∼0.6, induced with 1 mM IPTG, then expressed overnight at 20°C.
Cells were pelleted (5,000 g/20 min at 4°C), resuspended in a buffer containing 20 mM imidazole, 500 mM NaCl, 20 mM Tris-HCl, pH 7.6, and protease inhibitor cocktail (Roche), sonicated, then centrifuged at 30,000 g for 20 min at 4°C to produce a clear lysate. The His-tagged proteins were purified using immobilized metal affinity chromatography. Lysate was loaded onto a 3 ml bed of Ni+2 charged NTA-resin (Amersham). Protein was eluted with a stepped gradient of 20, 75, 400 mM imidazole buffer containing 100 mM NaCl, 20 mM Tris, pH 7.6. Proteins eluted with the 400 mM imidazole buffer. Protein was further purified using a Superdex 26/60 S75 preparative grade gel filtration column (GE Amersham).
For NMR studies, 120 μM protein was exchanged into buffer (140 mM NaCl, 20 mM Tris-d11, 1 mM EDTA, 5 mM TCEP, 0.25 mM azide, 10% D2O, pH 7.5) using a Zeba column (Pierce). The 1H-15N HSQC experiments were performed at 25°C on a Varian UNITY INOVA 600 MHz NMR spectrometer, equipped with a 5 mM 1H triple resonance probe with actively shielded z-axis gradients. The NMR data were processed using NMRPipe [13] and the spectra were analyzed using NMRView [14]. Melting temperatures were analyzed by circular dichroism (CD) at 225 nm on 5 μM of U-[15N]-labeled wild-type and C12A NTD in buffer containing 140 mM NaCl, 20 mM, Tris, 5 mM TCEP, pH 7.35.
2.3. Measurement of pol β binding affinities with XRCC1 NTDs
The P300C pol β catalytic domain mutant was purified as previously described [11]. For fluorescence experiments, the purified P300C pol β catalytic domain mutant was reduced with 1 mM TCEP before being applied to a Superdex S75 10/300 GL column equilibrated with 20 mM Tris, pH 8.0 and 150 mM NaCl. Fractions containing the P300C mutant protein were collected. Aminosulfonylbenzofuran (ABD)-F was immediately added in a 3-fold molar excess and allowed to incubate overnight before being reapplied to a Superdex S75 10/300 GL column equilibrated with 20 mM Tris, pH 8.0 and 150 mM NaCl. Steady-state fluorescence measurements were carried out on a Horiba FluoroLog Fluorimeter at 20°C. ABD-labeled P300C protein was at a concentration of 20 nM in 20 mM Tris, pH 8.0 and 150 mM NaCl buffer for fluorescence measurements. For reduced conditions, all buffers and samples additionally contained 1 mM TCEP. An excitation wavelength of 389 nm was utilized for all measurements and the emission at 500 nm was recorded as a function of either wild-type XRCC1 NTD or the C12A mutant.
2.4. Cytotoxicity studies
Cells were seeded at a density of 5-10,000 cells per well in six-well dishes in medium without selection antibiotics. The next day, cells were treated for 1 h with a range of concentrations of MMS (Sigma-Aldrich) or hydrogen peroxide (H2O2; Fisher Scientific). In some studies, cells were also treated with 4-AN (5 μM; Acros) for a total of 24 h as indicated. After washing with Hanks' Balanced Salt Solution (HBSS), control growth medium was added and the cells were allowed to grow until untreated control cells reached 80% confluence [15]. At this time, triplicate wells for each concentration were counted by a cell lysis procedure, and results were expressed as % control growth [16].
2.5. Western blot analysis
For preparation of whole cell extracts [17], cells were washed with phosphate-buffered saline (PBS), scraped, collected into PBS and centrifuged. Cell pellets were flash frozen in dry ice and thawed by resuspension in Buffer I (10 mM Tris-HCl, pH 7.8, 0.2 M KCl, 25 mM NaF and complete protease inhibitor cocktail (Roche)). An equal volume of Buffer II (10 mM Tris-HCl, pH 7.8, 0.2 M KCl, 25 mM NaF, 2 mM EDTA, 40% glycerol, 0.2% NP-40 and 2 mM dithiothreitol (DTT)) was then added as described previously. The suspension was rotated for 1 h at 4°C and extracts were clarified by centrifugation in a microcentrifuge at full speed for 15 min at 4°C. Total protein concentration of extracts was determined by the Bio-Rad assay using bovine serum albumin (BSA) as protein standard.
Extract protein samples (60 μg) were loaded onto 4-12% Bis-Tris NuPAGE gels (Invitrogen) and electrophoresed in NuPAGE MES running buffer at 4°C. Proteins were transferred to nitrocellulose filters at 25 V overnight, also in the cold. Following transfer, filters were blocked overnight at 4°C in 5% nonfat dry milk in TBS with 0.1% Tween 20 (TBST). Filters were first incubated for 2 h at room temperature or overnight at 4°C with mouse monoclonal anti XRCC1 primary antibody (Thermo Fisher Scientific, 33-2-5). After washing with TBST, filters were further incubated with anti-mouse IgG-horseradish peroxidase (HRP) conjugated secondary antibody (1:2,000-1:20,000 dilution, Bio-Rad) and visualized using Super Signal chemiluminescent detection (Thermo Scientific) according to the manufacturer's instructions. Where blots were probed with multiple primary antibodies, they were stripped for up to 30 min at 37°C or room temperature in Restore Western Blot Stripping Buffer (Thermo Scientific), washed three times in TBST, then blocked in 5% nonfat dry milk/TBST overnight. Other primary antibodies utilized were pol β 18S monoclonal, Ligase III-1F3 (GeneTex GTX70143), Ligase 1-0H5 (GeneTex MS GTX7041), PARP-1 (BD Pharmingen 51-669GR) and GAPDH (Alpha Diagnostic G3PDH11-M) or tubulin (Sigma T 9026) used as a loading control.
2.6. Co-immunoprecipitation
Cells were washed in PBS and collected by scraping, suspended in two volumes of lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 25 mM NaF, 0.1 mM sodium orthovanadate, 0.2% Triton X-100, and 0.3% NP-40) containing protease inhibitors, 0.1 mM PMSF, 1 μg/ml aprotinin, and 5 μg/ml leupeptin and incubated on ice for 30 min. After agitating the tubes briefly, the lysates were centrifuged at 20,800 × g for 30 min at 4°C and the supernatant fraction was removed [18]. Protein concentrations were determined using the Bio-Rad protein assay.
For co-immunoprecipitations with XRCC1, an equal amount (1 mg protein) of cell extract was mixed with 1-2 μg of anti-XRCC1 antibody (Santa Cruz H-300). The mixture was incubated with rotation for 4 h at 4°C. The immunocomplexes were adsorbed onto protein A-sepharose and protein G-agarose beads (1:1 mixture) by incubating the mixture for 16 h at 4°C. The beads were then washed four times with lysis buffer containing protease inhibitors. Finally, the beads were resuspended in SDS sample buffer, heated at 95°C for 5 min, and briefly centrifuged. The soluble proteins were separated by electrophoresis on 4-12% SDS-PAGE gels in MOPS buffer and then transferred to a nitrocellulose membrane for 2 h. The membranes were blocked then probed with anti pol β antibody (Abcam ab3181) or pol β 18S monoclonal antibody, and anti-XRCC1 antibody, also anti-ligase III and anti-PARP-1 monoclonal antibodies as described above. In control experiments, the immunoprecipitating antibody was substituted with agarose-conjugated IgG.
2.7. Treatment of cells for analysis of cellular redox status of XRCC1
Cells were plated and treated with MMS (0.25 mM, 1 h), 4-AN (5 μM, constant exposure), or a combination of MMS and 4-AN when near confluent. All cells were harvested 4 h after dosing. An excess of iodoacetamide (∼1 M) was added immediately to the PBS for washing and in the lysis buffer after scraping. The XRCC1 co-immunoprecipitation procedure was similar to that described previously except iodoacetamide was added fresh at each step to maintain its concentration in the lysates and the work was carried out in the dark since iodoacetamide is photosensitive. The co-immunoprecipitated protein samples were loaded onto two 4-12% Bis-Tris NuPage polyacrylamide gels and electrophoresed in MES buffer. One gel (inputs and co-ip samples) was immunoblotted with XRCC1 primary antibody (Thermo Fisher Scientific 33-2-5) to confirm the presence of immunoprecipitated XRCC1 protein of the correct size. The second gel was stained with Simply Blue safe stain (Invitrogen), then destained with water and used for mass spectrometry.
2.8. In-gel digestion and mass spectrometry
Regions from polyacrylamide gels containing XRCC1 (as determined by western blotting) were minced and digested with trypsin (Promega) for 8 h in an automated fashion with a Progest robotic digester from Genomic Solutions. Briefly minced gel bands were incubated twice for 15 min in 100 μl of 25 mM ammonium bicarbonate, 50% (v/v) acetonitrile. The gel was then dehydrated by a 20 min incubation in 100 μl of acetonitrile followed by drying under a nitrogen stream. 250 ng of trypsin (Promega) were added, followed by an 8 h incubation at 37°C. The resulting peptides were extracted by first collecting the digest supernatant, then incubating the gel with 50 μl of water for 20 min and collecting the resulting supernatant and, finally, collecting the supernatant from 2 independent 20 min incubations of the gel in 50 μl of 5% (v/v) formic acid, 50% (v/v) acetonitrile. All supernatants were pooled during the collection process and were lyophilized to dryness. The lyophilized samples were resuspended in 40 μl of 0.1% formic acid and spiked with 20 fmoles and 100 fmoles of the carbamidomethylated and unmodified synthetic peptides respectively.
Synthetic peptides utilized were 13C-, 15N-lysine labeled peptides corresponding to the unmodified and carbamidomethylated (C12 and C20) forms of tryptic peptide 3 of mouse XRCC1 (residues 8-26; HVVSCSSQDSTHCAENLLK) purchased from Sigma-Aldrich and used without further purification. These synthetic peptides were analyzed by nanoLC-ESI-MS and MS/MS at various concentrations (0.1 fmole to 1.0 nmole) to ensure they behaved similarly to the native peptides from XRCC1 that had been observed in previous qualitative analyses and to determine linearity of mass spectrometer response.
NanoLC-ESI-MS/MS analyses were then performed using an Agilent 1100 nanoLC system on-line with an Agilent XCT Ultra ion trap mass spectrometer with the Chip Cube Interface. 20 μl of the peptide mixture from the in-gel digest were loaded onto an Agilent C18 chip (75 μm × 43 mm) followed by a 15 min wash of 5% acetonitrile, 0.1% formic acid. Peptides were eluted by applying a linear gradient from 5% acetonitrile, 0.1% formic acid to 50% acetonitrile, 0.1% formic acid to the column over 45 min. This was followed by a 5 min gradient from 50% acetonitrile, 0.1% formic acid to 95% acetonitrile, 0.1 % formic acid and then a 10 min hold at 95% acetonitrile, 0.1% formic acid. The mass spectrometer was used in the positive ion, standard enhanced mode and included settings of a mass range from 200 to 2200 m/z, an ionization potential of 2.1 kV, 100 ms accumulation time, and a 1.0 V fragmentation amplitude. Data were acquired using the MRM feature of the Agilent XCT Ultra mass spectrometer with isolations of m/z 686.0, 689.0, 725.0, and 728.0 (corresponding to the (M+3H)+3 ions of the natural abundance disulfide-bonded, “heavy” disulfide-bonded, natural abundance carbamidomethylated, and “heavy” carbamidomethylated, respectively) with 3 m/z windows. Using the DataAnalysis software package from Agilent, extracted ion chromatograms (EICs) were generated for ions m/z 910, 914, 969, and 973 (doubly charged b17 ions of natural abundance disulfide-bonded (see Fig. S1 for MS/MS), “heavy” disulfide-bonded, natural abundance carbamidomethylated, and “heavy” carbamidomethylated, respectively) with 2 m/z windows (see Fig. S2). Areas under the peak of the EICs were calculated using the “Find Compounds” tool in DataAnalysis. Approximate relative amounts of the “oxidized” and “reduced” forms of the native XRCC1 peptides were calculated using the ratios of the natural abundance areas to the areas from the appropriate synthetic peptide. Experiments were performed in triplicate. Further details can be found in the Supplementary figures.
2.9. Determination of total cellular PAR
Cellular PAR levels were quantified using the PARP in vivo Pharmacodynamic Assay 2nd Generation (PDA II) kit (4520-096-K, Trevigen) as previously [19]. Briefly, cells were seeded in 60 mm dishes at 106 cells per dish and treated the next day with 10 mM MMS at 4°C in growth medium supplemented with 25 mM HEPES. After 20 min, they were washed in ice-cold HBSS and fresh, warm growth medium was added. Dishes were incubated for the indicated time at 37°C in a 10% CO2 incubator, then immediately placed on ice and lysed according to the kit protocol. Control cells were treated similarly and collected after 10 min. Cell counts were verified to ensure no growth differential between cell lines. After cell lysis and DNA digestion, total protein amounts were determined for each sample, and 4 μg of Xrcc1−/− cell extract and 10 μg of Xrcc1+/+, XRE8 and XV2 cell extracts were added to pre-coated capture antibody plates and incubated overnight at 4°C. The following morning, wells were washed four times in PBS with Tween-20 (PBST), and then 1:250 dilution of PAR polyclonal detecting antibody was added and incubated at room temperature for 2 h. Wells were again washed four times in PBST, then 1:250 dilution of goat anti-rabbit IgG-HRP was added and incubated for 1 h. Cells were washed again four times in PBST, then a 1:1 mixture of PARP PeroxyGlow™ A and B was added and luminescence was measured with a Tropix TR717 Microplate Luminometer.
2.10. Micro-irradiation and immunofluorescence
Cells were seeded in 35 mm glass bottomed petri dishes containing an etched grid (MatTek, Ashland, MA) at 2 × 105 cells per dish and incubated in growth medium supplemented with 10 μM BrdU for 24 h. After 24 h, samples were imaged using a 40× C-Apochromat (numerical aperture 1.2) water immersion objective coupled to a Zeiss LSM510 META confocal microscope (Carl Zeiss MicroImaging). DNA single strand breaks (SSBs) and DNA base lesions were introduced by UV laser micro-irradiation at 364 nm [19].
Previous studies have established that peak XRCC1 and pol β recruitment occurs 1 min after micro-irradiation [19]. Here, cells were fixed in 4% paraformaldehyde 1 min after UV damage. After fixation, cells were permeabilized with 0.25% Triton X-100 in PBS for 10 min, washed three times in PBS, then further permeabilized and blocked with PBS + 1% BSA for 30 min. Cells were then incubated with anti-XRCC1 antibody (1:50; Abcam ab1838) and anti-PADPR antibody (1:100; Abcam ab14460) for 1 h. Cells were washed three times with PBS, then incubated in Alexa 488 conjugated anti-mouse and Alexa 647 conjugated anti-chicken (1:2,000; Life Technologies) for 1 h. For pol β imaging, cells were permeablized in 1% SDS as previously described [20], and then incubated with anti-pol β antibody (1:200, Abcam ab26343) for 1 h. Cells were washed three times with PBS, then incubated in Alexa 546 conjugated anti-rabbit (1:2,000; Life Technologies). Fluorescence images were acquired with the same 40× water immersion objective on the LSM510. Recruitment of XRCC1 and pol β, and synthesis of PAR at the site of DNA damage was measured using IMAGEJ as described previously [19].
3. Results
3.1. Complementation of the MMS hypersensitivity phenotype of Xrcc1−/− mouse fibroblasts
XRCC1-deficient (Xrcc1−/−) mouse fibroblasts are hypersensitive (>8-fold) to the monofunctional methylating agent MMS compared with wild-type (Xrcc1+/+) cells (Fig. S3A) [5]. MMS, and other alkylating agent hypersensitivity phenotypes, have been reported for XRCC1-deficient Chinese hamster ovary (CHO) cells [21, 22]. Complementation of MMS hypersensitivity in mouse Xrcc1−/− cells expressing the wild-type XRCC1 protein has been demonstrated previously [5, 23]. Here, we isolated and evaluated several clones of Xrcc1−/− mouse fibroblast cells expressing different levels of the wild-type (WT) XRCC1 protein. One of these clones (WT17) expressed a very similar level of XRCC1 protein as the Xrcc1+/+ cell line (Fig. S3B, compare lanes 1 and 4 top panel) and as previously described this resulted in significant complementation (Fig. S3A). It was also confirmed that Xrcc1−/− cells as well as a vector control (pef3) expressed no XRCC1 protein (Fig. S3B, lanes 2 and 3).
It is known that the BRCT II domain of XRCC1 (Fig. 1) physically interacts with lig-IIIα forming a heterodimer [24-26], and that this well characterized interaction is required to stabilize the cellular level of lig-IIIα protein [12, 27]. As expected, there were extremely low levels of lig-IIIα in Xrcc1−/− and empty vector-transfected cells (Fig. S3B, lanes 2 and 3). Interestingly, lig-IIIα was increased only minimally in WT17 cells (Fig. S3B, lane 4), yet MMS hypersensitivity was substantially reversed.
A series of clones expressing varying levels of wild-type XRCC1 protein were characterized for MMS sensitivity (Fig. S4B). The XRCC1 protein expression level was ascertained by western blotting (Fig. S4A, top panel). A low level of XRCC1 expression (WT2) resulted in only minor reversal of the MMS hypersensitivity phenotype, whereas near wild-type XRCC1 levels correlated with significant reversal (clones WT16 and 5). Wild-type expression level (WT17) resulted in almost complete reversal. Interestingly, clone WT15 over-expressing XRCC1 protein was more hypersensitive then WT17 (Fig. S4B and C) and expression of lig-IIIα comparable to the level found in Xrcc1+/+ cells was seen only in this over-expressing clone (Fig. S4A, compare lanes 4 and 7). Taken together, the results demonstrate a strong correlation between XRCC1 expression level and MMS resistance, although high over-expression (clone WT15) showed lesser resistance than WT17 (Fig. S4C). The clear correlation between XRCC1 expression level and MMS resistance is in contrast to previous data obtained in transfected mouse fibroblasts where expression corresponding to 10% of the Xrcc1+/+ wild-type protein level showed only a ∼2-fold MMS hypersensitivity compared with Xrcc1+/+ cells [23]. These differences may result from the entirely different analysis methods used. In a comparison of MMS sensitivity of bone marrow cells isolated from Xrcc1+/+ and Xrcc1+/− mice, the heterozygote cells demonstrated a small, but significant, reduction in viability relative to wild-type cells [28].
3.2. Characterization of full-length mouse XRCC1 wild-type and C12A mutant proteins
Full-length mouse wild-type and C12A mutant proteins were expressed and purified, and the structural integrity of the C12A mutant was evaluated. Controlled proteolysis was first conducted on both proteins, as described in “Supplementary Materials and Methods.” There were no detectable differences between the wild-type and C12A proteins (Fig. S5A). In addition, CD spectroscopy was performed with these full-length proteins. No differences in protein folding were observed by comparison of the CD spectra (Fig. S5B). These results suggest that any cellular effects attributed to the C12A mutation were the result of blocking C12-C20 disulfide bond formation, rather than a gross structural folding defect in the C12A mutant protein.
3.3. Characterization of XRCC1 wild-type and C12A NTDs by NMR
Crystal structures of reduced wild-type and mutant oxidized forms of the XRCC1 NTD complexed with pol β have been solved, and these results were an underpinning for the current study [11]. The oxidized form of XRCC1 is characterized by a disulfide bond between residues C12 and C20 and variations in secondary structure and folding. We expected that alanine mutation at C12 of XRCC1 (C12A) (Fig. 1) would abolish formation of the disulfide bond, thus holding the protein in the reduced state. To confirm this expectation NMR spectroscopy was used to examine the structures of the reduced wild-type NTD and C12A NTD proteins (Fig. 2).
Fig. 2.
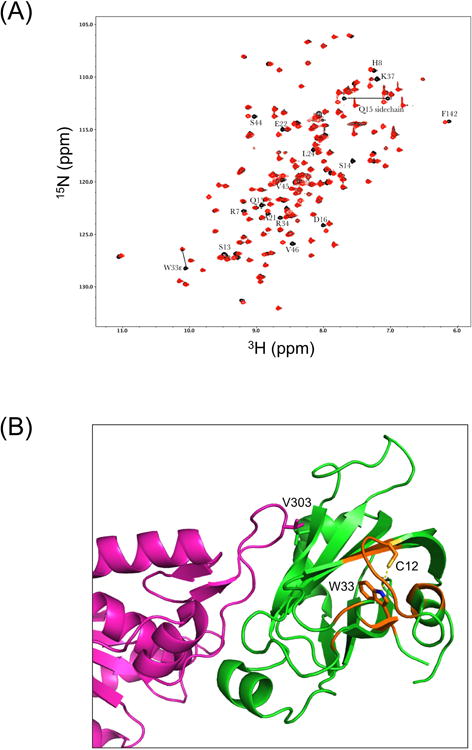
NMR characterization of XRCC1 NTDs. (A) Effect of C12A mutation on the 1H-15N HSQC spectrum of the XRCC1 NTD. The overlayed 1H-15N HSQC spectra for the reduced wild-type (black) and C12A (red) analogs are shown. The W33ε amide shows a large perturbation, consistent with the loss of an H-bonding interaction with the C12 sulfur. Most of these perturbed resonances arise from residues located near C12 and W33, so that changes in the position of W33 will impact the shifts of the amide resonances. In addition to the backbone shifts, a side chain NH2, assigned to Q15 based on its proximity to C12, also exhibits a significant shift perturbation. (B) Perturbed resonances in C12A NTD mapped onto the structure of the reduced wild-type XRCC1 NTD (green):pol β (catalytic domain-magenta) complex, pdb file 3K75. Locations of the perturbed amide resonances are indicated in orange, and the C12 and W33 side chains are also shown.
When compared with reduced wild-type protein, the 1H-15N HSQC NMR spectrum of the C12A mutant NTD showed some localized perturbation in the region of the mutation (Fig. 2A). However, this was not sufficient to indicate a significantly altered secondary structure compared with the structure of the reduced form of the wild-type NTD. The W33ε amide showed a perturbation, consistent with loss of an H-bonding interaction with the C12 sulfur. The loss of this H-bond not only perturbs the resonance, but also leads to a more dynamic W33ε side chain, and this was probably the main reason for the more generalized chemical shift perturbation observed. Other significantly perturbed amide resonances correspond to: R7, H8, C12, S13, S13, Q15, D16, A21, E22, L24, R34, K37, S44, V45, V46 and F132. In addition to the backbone shifts, a side chain NH2 assigned to Q15, based on its proximity to C12, also exhibited a significant shift. Since the altered region is on the opposite side of the protein to the pol β V303 loop interface (Fig. 2B), it seems unlikely that C12A would have a significantly altered pol β interaction than that observed with the reduced form of the wild-type NTD. Overall, these results confirmed the presence of the C12A mutation and the structural integrity of the C12A protein. The structural integrity of the C12A NTD was further evaluated by CD spectroscopy. The spectra of the reduced wild-type and C12A NTD proteins were indistinguishable (data not shown). Protein melting curves obtained by CD spectroscopy revealed that the proteins were slightly different, in relation to unfolding (Fig. 3A). The melting temperature (Tm) of C12A was ∼ 4°C below that of the wild-type NTD protein (50.6°C and 54.3°C, respectively). These observations were consistent with the NMR spectroscopy data.
Fig. 3.
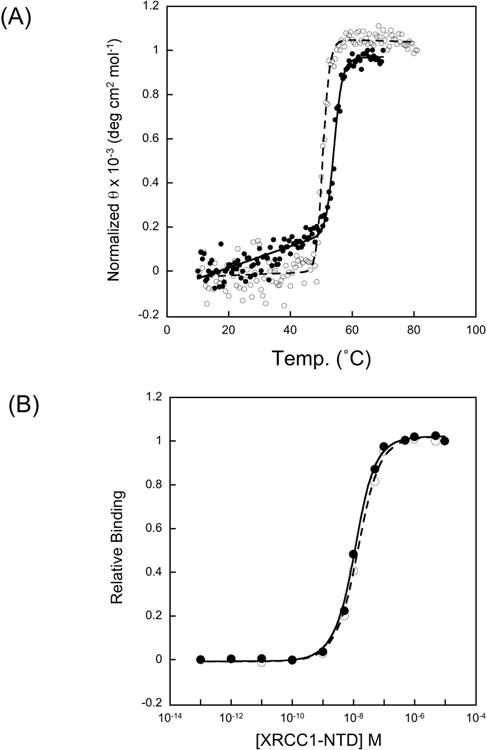
Characterization of XRCC1 NTDs. (A) CD melting curves (at 225 nm) for reduced wild-type XRCC1 NTD and C12A NTD. The Tm was 54.3 °C for wild-type (closed symbols) and 50.6 °C for C12A (open symbols). Studies were performed on 5 μM protein in 20 mM Tris, pH 7.35, 140 mM NaCl, 5 mM TCEP. (B) Determination of pol β catalytic domain/XRCC1 NTD binding affinities. Titration of aminosulfonylbenzofuran (ABD)-labeled pol β P300C mutant with reduced wild-type XRCC1 NTD (closed symbols) and mutant C12A XRCC1 NTD (open symbols). Curves are fit to a two-state binding isotherm.
3.4. Comparison of pol β binding affinity for the XRCC1 wild-type and C12A NTD proteins
A comparison of the binding of fluorophore-tagged P300C pol β catalytic domain with the reduced wild-type NTD and with the C12A mutant is shown in Figure 3B. As is apparent from this figure, the perturbation resulting from the C12A mutation is minimal. This similarity of the pol β binding affinities of the two NTDs is thus consistent with the NMR data showing that the C12A mutation introduces only local structural perturbations that are distal to the surface that is involved in pol β binding (Fig. 2B). Taken together, our results indicate that the C12A mutant XRCC1 is a good model for studies of the reduced form of the wild-type protein. As noted previously, the reduced form of the XRCC1 NTD has approximately 10-fold less avid binding affinity for pol β than the oxidized form (11).
3.5. Effect of blocking C12-C20 disulfide bond formation in cellular XRCC1
The C12A protein mammalian expression vector pXRE was prepared and the protein was stably expressed in Xrcc1−/− cells. One of the selected clones (XRE8) had a protein expression level similar to those in Xrcc1+/+ and WT17 cells (Fig. 4A, top panel). The expression of C12A protein significantly reversed the MMS hypersensitivity phenotype of Xrcc1−/− cells (Fig. 4B). Co-immunoprecipitation experiments revealed that the C12A disulfide blocked reduced protein was fully able to interact with lig-IIIα and PARP-1, but had a lesser interaction with pol β (Fig. 4C, compare lanes 1 and 3). This result with full-length reduced XRCC1 protein is consistent with in vitro NTD experiments where the C12-C20 disulfide blocked C12A protein demonstrated XRCC1-pol β affinity equivalent to reduced wild-type protein (Fig. 3B).
Fig. 4.
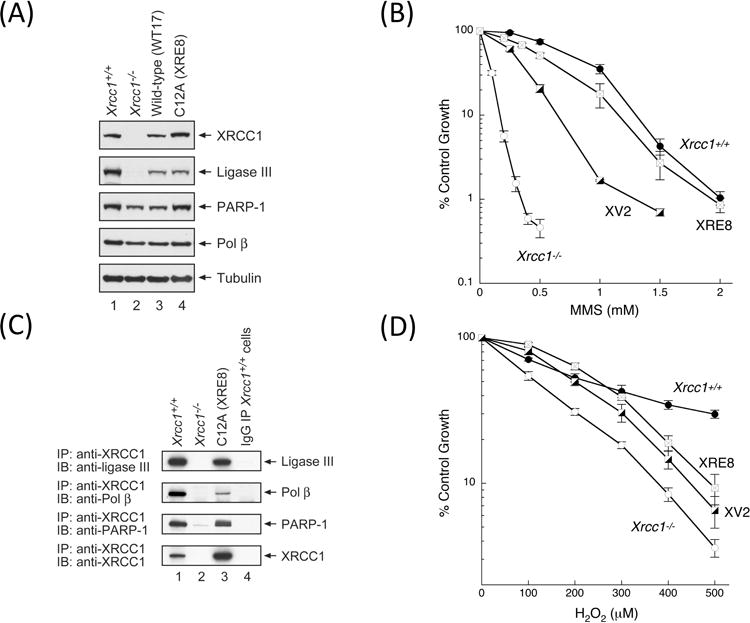
Characterization of XRE8 cells expressing disulfide blocked reduced XRCC1 protein. (A) Western blotting analysis of XRCC1 and other repair proteins in XRE8 cells compared with Xrcc1+/+, Xrcc1−/− and WT17. (B) Comparison of complementation of MMS hypersensitivity of Xrcc1−/− cells by stable expression of C12A (XRE8 cells) or V88R protein (XV2 cells). Full methods for growth inhibition assays are described in Section 2. (C) Equal amounts of cell extracts as indicated were immunoprecipitated with anti-XRCC1 antibody then immunoblotted with ligase III, pol β, PARP-1 and XRCC1 antibodies. In lane 4, immunoprecipitation was of Xrcc1+/+ cells with pre-immune IgG as a negative control. Full methods are described in Section 2. (D) Comparison of complementation of H2O2 hypersensitivity of Xrcc1−/− cells in XRE8 cells or XV2 cells measured by growth inhibition assay.
Pol β interacts with the NTD of XRCC1 [29, 30] (see Fig. 1), and the V86R point mutant in the human XRCC1 NTD, is known to abolish binding to human pol β [8, 9]. The corresponding mouse XRCC1 mutant protein (V88R, Fig. 1) was expressed in Xrcc1−/− mouse fibroblasts. The interaction between wild-type XRCC1 and pol β and the absence of such interaction between V88R and pol β (in XV2 cells) was confirmed in co-immunoprecipitation experiments (Fig. S6A). Clone XV2 expressing similar levels of XRCC1 (V88R) protein as Xrcc1+/+ cells and WT17 (Fig. S6B) exhibited decreased complementation compared with wild-type protein (Fig. 4B), confirming the importance of the XRCC1/pol β interaction in protection against MMS cytotoxicity [10].
3.6. Hydrogen peroxide hypersensitivity phenotype of Xrcc1−/− mouse fibroblasts
Only minor H2O2 hypersensitivity was observed in Xrcc1−/− cells compared with results obtained with MMS (compare Fig. 4B and D). Similar low-level H2O2 hypersensitivity has also been reported in XRCC1-deficient CHO cells [1, 7]. Additionally human breast cancer cells transfected with XRCC1 siRNA resulted in only low-level H2O2 hypersensitivity [31]. Cell lines XRE8 and XV2, with stable expression of C12A and V88R XRCC1 mutantations, respectively resulted in only partial complementation of the H2O2 hypersensitivity phenotype of Xrcc1−/− cells (Fig. 4D). These V88R results are consistent with those obtained with human V86R expressed in XRCC1-deficient CHO cells [7], suggesting that the loss of the pol β interaction is important to the protective effect of XRCC1. The similar results with the C12A mutant again were consistent with the importance of the stronger pol β and XRCC1 interaction in the protective effect against H2O2 exposure.
3.7. Cell cycle analysis after exposure to MMS
Flow cytometry was used to assess S phase DNA synthesis (incorporation of BrdU or EdU) in cells 2 h after treatment with MMS as described previously [15]. In Xrcc1+/+ cells, there was a rapid (observed at 2 h) MMS concentration-dependent inhibition of DNA synthesis (Fig. S7A). This inhibition observed after the highest doses of MMS (2 and 3 mM) was persistent to 4 h (data not shown). Similar results were obtained in WT17, XRE8 and XV2 cells (Fig. S7, panels C-E). In each case, the effect on DNA synthesis was persistent after 2 mM MMS exposure. In the same experiments in Xrcc1−/− cells, no inhibition of DNA synthesis was observed even at highly cytotoxic MMS concentrations (Fig. S7B). These results suggested a role for XRCC1 in the DNA synthesis inhibition, but this role was not dependent on C12-C20 disulfide bond formation or interaction with pol β. The results also confirm the integrity of the C12A and V88R mutant proteins.
3.8. Recruitment of XRCC1 and pol β following low energy laser-induced DNA damage
Micro-irradiation has been utilized to examine recruitment of repair proteins to nuclear sites of UV-induced damage as a function of XRCC1 expression [32]. With low laser power (∼0.17μJ), we have demonstrated formation of SSBs and base lesions rather than double strand breaks in irradiated cells [19]. Wild-type, C12A and V88R XRCC1 proteins (i.e., XRE8 and XV2 cells, respectively), were recruited to sites of DNA damage at 1 min after irradiation, and as expected, no XRCC1 recruitment was observed in Xrcc1−/− cells (Fig. 5A). These data were quantified in each case, and the results are plotted graphically in Figure 5C. PAR synthesis was demonstrated in all five treated cell lines (Fig. 5A and D). The least PAR synthesis was observed in Xrcc1+/+ and WT 17 cells, with a small increase apparent in Xrcc1−/−, XRE8 and XV2 cells (Fig. 5D). In Xrcc1+/+ cells, recruitment of pol β was observed at 1 min after irradiation (Fig. 5B and E). In contrast in Xrcc1−/− cells, there was no recruitment of pol β to irradiated sites, whereas PAR synthesis was observed. These results confirm previous evidence that XRCC1 is required for pol β recruitment to sites of DNA damage [32, 33]. Interestingly in XRE8 cells, pol β recruitment was observed at 1 min and at a slightly higher level than that in Xrcc1+/+ cells (Fig. 5D), despite the lower pol β binding affinity of the reduced C12A protein (Fig. 5C). The results suggest that XRCC1 disulfide bond formation is not required for recruitment of pol β. In XV2 cells, there was no observable recruitment of pol β (Fig. 5E) indicating that an interaction between pol β and XRCC1 is necessary for pol β recruitment (Fig. 5B and E).
Fig. 5.
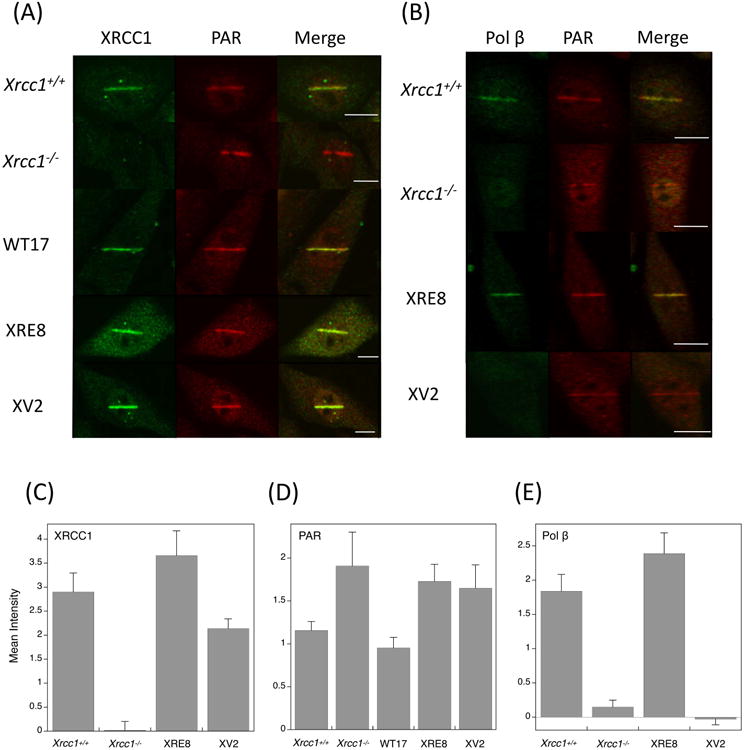
Immunofluorescence imaging of (A) XRCC1 and PAR and (B) pol β and PAR in XRCC1 cell variants. Experiments are as described in Section 2 and typical results are shown. Cell lines were irradiated in stripes, then allowed to repair for 1 min before assessment of XRCC1 and pol β recruitment, and PAR synthesis as indicated. Bars represent 10 μm. (D-F) Quantification of recruitment and synthesis data at 1 min after laser damage. Between 10 and 20 cells were analyzed in each case, error bars represent SEM.
3.8. PAR accumulation following exposure to MMS
Total cellular PAR was measured in cells treated with 10 mM MMS for 20 min at 4°C. These conditions allow accumulation of methylated base damage with minimal repair until cells are switched to warm MMS-free growth medium [5]. MMS survival curves after this treatment protocol (20 min, 4°C) are shown in Figure S8. The MMS sensitivities of the mutant XRCC1 cell lines relative to Xrcc1−/− are similar to those obtained after 1 h exposure at 37°C (compare Fig. 4B and S8), but Xrcc1+/+ cells were significantly more MMS resistant under the cold treatment protocol (15- vs 8-fold). The amount of PAR measured at any time will be the net result of synthesis by PARP enzymes in response to DNA damage, and degradation and removal of PAR chains by poly(ADP-ribose) glycohydrolase (PARG) recruited to damage sites by PARP activity [34, 35]. The lack of specific cell-permeable PARG inhibitors hinders study of the role of cellular PARG.
In Xrcc1+/+ cells, there was minimal PAR accumulation over the 30 min period after MMS exposure (Fig. 6); in fact, the levels were very similar to those measured in control non-treated (N/T) cells. A higher PAR level was observed in non-treated Xrcc1−/− cells, and after MMS exposure of these cells, the PAR level increased notably at the 10 min time point (Table 1). In the cell line complemented with wild-type XRCC1 protein (WT17), there was an intermediate level of PAR in N/T cells and, similar to results observed in Xrcc1+/+, this did not change significantly following MMS treatment (Fig. 6). In the non-treated XRE8 and XV2 cells, PAR levels were higher than those in Xrcc1−/− cells, but after MMS exposure, PAR levels in these cells were dramatically increased at 10 min (Table 1 and Fig. 6) and even immediately after MMS exposure (0 min). Since cells with C12A reduced XRCC1 protein show lower affinity for pol β (Fig. 4C), and the V88R protein in XV2 cells have no XRCC1 and pol β interaction (Fig. S6B), the increase in the level of PAR appears to correlate with a disruption in the interactions between PARP, XRCC1 and pol β after MMS-induced damage.
Fig. 6.
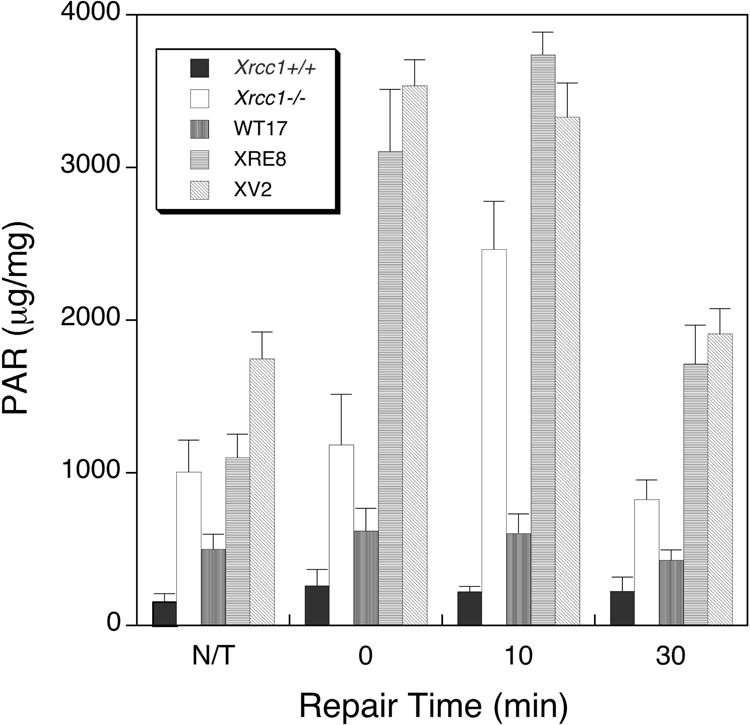
Total cellular PAR in XRCC1 variant cells following treatment with cold MMS. Cell lines were exposed to 10 mM MMS for 20 min at 4°C, then incubated in drug-free medium for up to 30 min in a 37°C incubator before PAR analysis as described in Section 2. Experiments were repeated at least 3 times and error bars reflect SEM.
Table 1. Relative cellular PAR 10 min after MMS*.
| Cell line | Relative PAR level |
|---|---|
| Xrcc1+/+ | 1 |
| WT17 | 3 |
| XV2 | 15 |
| XRE8 | 17 |
| Xrcc1−/− | 11 |
MMS treatment 10 mM for 20 min at 4°C
3.9. Effect of PARP inhibition on MMS sensitivity
We had demonstrated previously that inhibition of PARP activity by 4-AN strongly sensitizes Xrcc1+/+ and Xrcc1−/− cell lines to the cytotoxicity of agents resulting in DNA damage where repair is initiated by a monofunctional glycosylase [5]. It has been proposed that intermediates of single-nucleotide BER are critical for PARP-1 binding and for the 4-AN-mediated sensitization [36]. In experiments using cells expressing wild-type XRCC1 protein (WT17), very similar MMS sensitization was obtained as in Xrcc1+/+ cells (data not shown). Interestingly, Xrcc1−/− cells were sensitized by 4-AN approximately one-half that of the Xrcc1+/+ cells (i.e., 13- and 22-fold, respectively; Table 2), suggesting involvement of XRCC1 in the DNA damage response to PARP inhibition.
Table 2. Fold 4-AN-mediated sensitization to MMS*.
| MMS IC50 (mM) | Fold | ||
|---|---|---|---|
| Cell line | − 4-AN | + 4-AN | sensitization |
| Xrcc1+/+ | 1.5 | 0.06 | 22 |
| XV2 | 0.6 | 0.05 | 12 |
| XRE8 | 1.1 | 0.2 | 5 |
| Xrcc1−/− | 0.2 | 0.02 | 13 |
1 h exposure to MMS, 24 h exposure to 4-AN (5 μM)
Expression of the C12A disulfide-blocked XRCC1 mutant protein (XRE8 cells) resulted in almost complete complementation of MMS hypersensitivity under these treatment conditions (Fig. 4B), but 4-AN produced a dramatically lower (only 5-fold) sensitization effect (Fig. 7 and Table 2). These results suggest that the sensitization effect of PARP inhibition requires the XRCC1 redox function, with a requirement for at least transient formation of oxidized XRCC1 for the strongest sensitization effect. Expression of the V88R mutant XRCC1 protein (XV2 cells) resulted in only partial complementation of the MMS hypersensitivity phenotype (Fig. 3B), and XV2 cells were sensitized only 12-fold by the PARP inhibitor 4-AN to the level seen in Xrcc1+/+ cells with 4-AN (Table 2). In both XRE8 and XV2 cell lines, the lower 4-AN sensitization correlates with a decreased or absent XRCC1 and pol β protein interaction. However, the extreme decrease in sensitization of XRE8 cells (Table 2) suggests a strong effect of the block in XRCC1 oxidation in these cells.
Fig. 7.
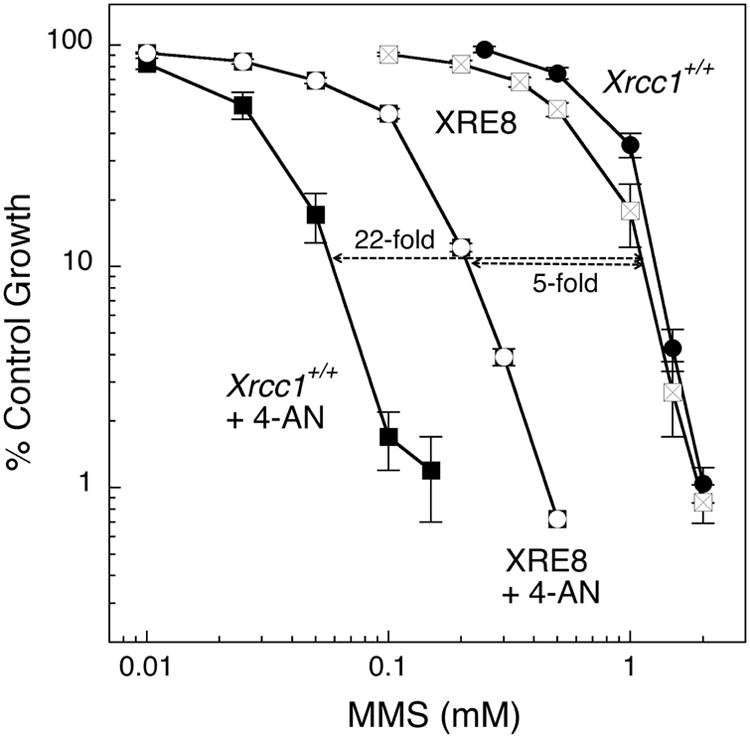
Sensitization to MMS by the PARP inhibitor 4-AN. Cells were treated with MMS for 1 h in the absence of inhibitor or in the presence of 4-AN (5 μM for 24 h). Full methods for growth inhibition assays are described in Section 2. Shown are Xrcc1+/+ (closed circles) sensitized 22-fold by 4-AN (closed squares) and XRE8 cells sensitized 5-fold by 4-AN (open symbols).
3.10. Redox status of XRCC1 protein in mouse fibroblasts
In light of the results described above for the XRE8 cell line, it was important to examine the redox status of the XRCC1 protein in control non-treated XRCC1+/+ mouse fibroblast cells. Initial qualitative mass spectrometry experiments for characterizing XRCC1 from mouse fibroblasts suggested that tryptic peptide 3, residues 8-26 (HVVSCSSQDSTHCAENLLK), could be readily detected as a mixture of reduced and disulfide-bonded forms. It was unclear if the C12-C20 disulfide-bonded form of this peptide was physiologically relevant as peptides that contain two cysteines readily form intra-peptide disulfides on the benchtop. Therefore, in attempts to capture the in vivo redox state(s) of XRCC1 in cells, samples were generated in the presence of an excess of iodoacetamide from the point of cell lysis until SDS-PAGE. This was done in an effort to carbamidomethylate all free sulfhydryls while any cysteines involved in a disulfide bond would be protected from carbamidomethylation. Hence, in these experiments the carbamidomethylated cysteines on XRCC1 are believed to represent cysteines that are reduced in the cell, whereas cysteines that are not carbamidomethylated should reflect those cysteines that are involved in disulfides or are otherwise protected from iodoacetamide. We acknowledge that the lack of carbamidomethylation is not a definitive marker for the presence of a disulfide bond between C12 and C20 of XRCC1, but rather a reflection of inaccessibility of these residues. Based upon the findings of Cuneo and London [11], however, we do believe a significant portion of the unmodified tryptic peptide 3 of XRCC1 is due to disulfide bonding. In efforts to gain information on the relative abundances of the unmodified and carbamidomethylated forms of tryptic peptide 3 of XRCC1, isotopically labeled forms of the carbamidomethylated and disulfide-bonded forms of the peptide were employed similarly as described by Kirkpatrick et al. [37].
The relative proportion of C12-C20 unmodified (54%) and carbamidomethylated XRCC1 (46%) was similar in untreated (N/T) Xrcc1+/+ cells (Fig. 8). Treatment with MMS had no effect on these levels consistent with the ability of both wild-type and mutant C12A XRCC1 proteins to complement the MMS hypersensitivity phenotype of Xrcc1−/− cells (Fig. S3A and 4B). In contrast, treatment of cells with 4-AN resulted in an increase in the carbamidomethylated (reduced) form of XRCC1 (70%), with a corresponding decrease in the unmodified (oxidized) protein (30%; Fig. 8). Treatment with the combination of MMS plus 4-AN gave similar results to 4-AN alone. These results suggest that treatment with 4-AN decreases the proportion of C12 and C20 that are protected from modification, presumably by a decrease in the amount of disulfide-bonded (oxidized) cellular XRCC1 protein. Taken together our observations suggest that redox of XRCC1 plays a role in the mechanism of 4-AN-mediated sensitization to MMS. These results are consistent with MMS sensitization by 4-AN being considerably less effective in XRE8 cells expressing XRCC1 protein locked in the reduced form (Fig. 7).
Fig. 8.
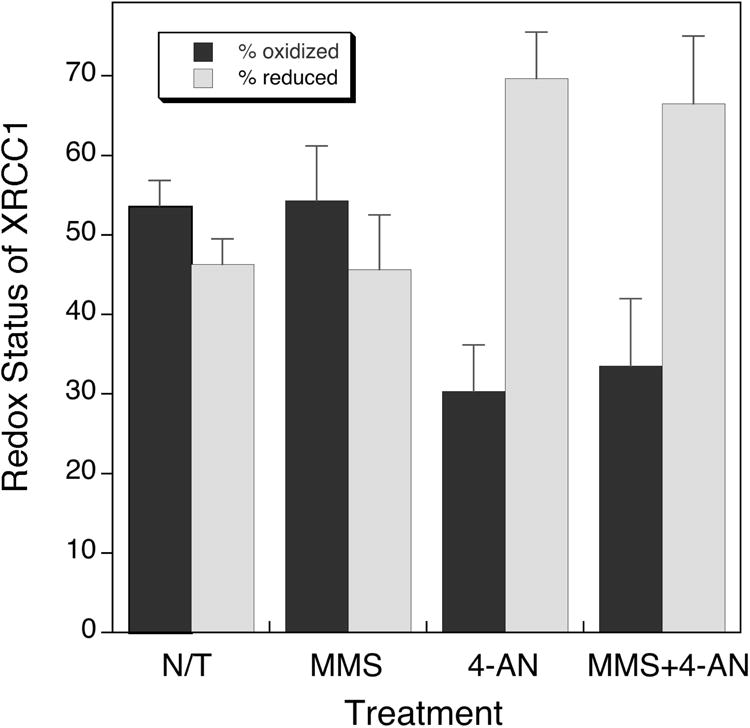
The redox status of XRCC1 protein in Xrcc1+/+ cells after various cell treatments as indicated. Measurements were by mass spectrometry as described in detail in Section 2. Relative amounts of the C12-C20 unmodified (oxidized) and carbamidomethylated (reduced) forms of the native XRCC1 peptides were calculated using the ratios of the natural abundance areas to the areas from the appropriate non-modified and carbamidomethylated synthetic peptides.
Discussion
Cellular methylated base lesions formed after exposure to MMS are repaired primarily by the single-nucleotide BER pathway in mouse fibroblasts. In this study we explored XRCC1 roles by expression in Xrcc1−/− cells of wild-type XRCC1 protein and two XRCC1 mutants with a focus on preventing oxidation of XRCC1 and decreasing the XRCC1 interaction with pol β. Consistent with results in another cell type [10], the interaction of XRCC1 with pol β is required for full complementation of MMS hypersensitivity in Xrcc1−/− mouse fibroblast cells (Fig. 4B). Cells with the disulfide bond-blocked form of XRCC1 protein (XRE8), and with the similar lower affinity for pol β as reduced wild-type protein (Fig. 3B), have substantially complemented MMS resistance (Fig. 4B). The similar MMS sensitivity of Xrcc1+/+ and XRE8 cells suggests equivalent formation and removal of the cytotoxic 5′-dRP intermediate of BER by pol β dRP lyase activity [36].
Evaluating sites of laser-induced damage in cells, XRCC1 wild-type and mutant proteins (C12A and V88R) were rapidly recruited to lesion sites (Fig. 5A and C), most likely by interaction with PAR polymer adducted PARP-1. There was significant recruitment of pol β in Xrcc1+/+, but not in Xrcc1−/− cells, 1 min after irradiation (Fig. 5B and E). Recruitment of pol β in XRE8 cells was equivalent to that in Xrcc1+/+ cells (Fig. 5E), in agreement with the high degree of complementation of MMS hypersensitivity by the C12A reduced protein (Fig 4B), but seemingly inconsistent with the observation of reduced pol β binding affinity of this protein (Fig. 4C). We propose that despite the reduced binding affinity of the C12A XRCC1 protein in damaged XRE8 cells, continual pol β recruitment is sufficient for repair of damaged sites. Yet, recruitment of pol β was not visible at this time in the XV2 cells (Fig. 5B and E).
Elevated PAR was observed in untreated repair-deficient Xrcc1−/− cells, as well as directly after MMS treatment, and the PAR level was increased strongly at 10 min, before returning to untreated levels at 30 min (Fig. 6 and Table 1). These results are similar to those reported previously in pol β-deficient cells [19]. Since PAR levels remained constant and low in the wild-type cells and in WT17 complemented cells (Fig. 6), it appeared that elevated PAR levels might be a reporter of decreased BER capacity in the Xrcc1−/− cells as in pol β null cells [19]. We find that PAR is also elevated in untreated XRE8 and XV2 cells and dramatically increases directly after MMS treatment and at 10 min after treatment to levels considerably higher than that observed in Xrcc1−/− cells (Fig. 6 and Table 1). These mutant XRCC1 cells demonstrate higher resistance to MMS than Xrcc1−/− cells using two different MMS treatment protocols (Fig. 4B and Fig S8) but are markedly less resistant to MMS than Xrcc1+/+ cells after the cold treatment protocol used for the PAR assay (Fig S8). These results are analogous to the increased PAR levels observed in Xrcc1−/−, XRE8 and XV2 cells after laser-induced damage (Fig. 5D).
In cells with DNA base damage, PAR serves to recruit XRCC1 that in turn binds and recruits pol β, the primary DNA polymerase of the BER pathway. A feature in common for the C12A and V88R mutant proteins is decreased or abolished binding affinity, respectively, for pol β as shown by co-immunoprecipitation (Fig. 4C and S6A). Wild-type level recruitment of pol β to laser-damaged DNA was observed in the disulfide-blocked XRE8 cells (Fig. 5B and E). But, as expected, in cells lacking XRCC1 (Xrcc1−/−) or the XRCC1 and pol β interaction (XV2), there was no recruitment of pol β to sites of laser-delivered DNA damage (Fig. 5B and E). The observed elevation in PAR with the C12A protein may be a reporter for sub-optimal interaction with pol β at sites of damage.
Considering the difference observed in PAR metabolism, we next determined the effects of PARP inhibition on MMS sensitivity. WT17 cells, with similar MMS sensitivity as Xrcc1+/+ cells, were similarly sensitized by 4-AN (data not shown), but Xrcc1−/− cells were sensitized to a lesser degree than wild-type XRCC1-expressing cells (Table 2). Thus, it is clear that XRCC1 protein has a role in the degree of 4-AN sensitization and recruitment of XRCC1 may be integral to the 4-AN sensitization mechanism. In studies of the XRE8 and XV2 cells, neither cell line was sensitized by 4-AN to the degree observed in Xrcc1+/+ cells (Fig. 7 and Table 2). Interestingly, the cell lines exhibiting the highest PAR levels after MMS exposure (Xrcc1−/−, XRE8 and XV2) (Table 1), had the lowest 4-AN-mediated sensitization (Fig. 7 and Table 2). Taken together, the data imply a role for the XRCC1 and pol β interaction in 4-AN-mediated sensitization to MMS. The relatively low level 4-AN sensitization observed in the XRE8 cells suggests that the redox status of XRCC1 is important to consider. If the PARP inhibitor sensitization is linked to replication fork collision and collapse, as proposed [3] increased stability of the PARP complex at a lesion site may lead to enhanced cytotoxicity. The lower affinity of the reduced XRCC1 and pol β interaction may somehow reduce PARP stability at the lesion site.
Finally, in Xrcc1+/+ cells, mass spectrometry revealed there are equivalent levels of unmodified (oxidized) and carbamidomethylated (reduced) XRCC1 protein in untreated (N/T) cells or those treated with MMS alone (Fig. 8). Treatment with 4-AN alone or MMS plus 4-AN significantly increased the proportion of reduced XRCC1 protein in cells. The results imply that oxidation of XRCC1 protein (not possible in XRE8 cells) is associated with 4-AN-induced damage signaling. In XRE8 cells, XRCC1 protein is locked in the reduced state and unable to undergo redox-dependent changes in protein structure as a result of 4-AN exposure [11]. As noted above, the ability for transient disulfide bond formation and attendant oxidation in wild-type XRCC1 appears to be required for the stronger 4-AN-mediated sensitization to MMS. In that this sensitization is proposed to reflect double-strand break formation secondary to replication block, it should be interesting to determine if XRE8 cells exhibit differences in replication stalling as a function of 4-AN and MMS treatment.
Supplementary Material
Highlights.
C12-C20 oxidized and reduced forms of XRCC1 were verified in mouse fibroblasts.
Pol β binding affinity to the XRCC1 NTD decreased with the reduced C12A mutant.
Folding properties of full-length C12A and wild-type XRCC1 were indistinguishable.
MMS-induced cellular PAR level was highly elevated in C12A-expressing cells.
PARP inhibitor sensitization to MMS was compromised in C12A-expressing cells.
Acknowledgments
The authors thank Dr. William Beard and Lois Wyrick for help with figure preparation and Zachary Weiner and Rebecca Childress for technical assistance. We thank Drs Vladimir Poltoratsky and Michelle Heacock, and members of the Protein Expression Core Facility for assistance with expression vector construction. We thank Dr. Robert Petrovich of the Protein Expression Core Facility for assistance with CD analysis. We thank Katina Johnson and Andrea Adams of the NIEHS Protein Microcharacterization Core for assistance with mass spectrometry analysis, C. Jeff Tucker at the NIEHS Fluorescence Microscopy and Imaging Center for assistance with micro-irradiation studies, and Dr. Carl Bortner and Maria Sifre for assistance with flow cytometry. The Office of Biological and Environmental Research supported research at the Oak Ridge National Laboratory, using facilities supported by the Scientific User Facilities Division, Office of Basic Energy Sciences, United States Department of Energy.
Grant Support: This work was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (project numbers Z01 ES050158 and ES050159 to S.H.W. and Z01 ES050147 to R.E.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.El-Khamisy SF, Masutani M, Suzuki H, Caldecott KW. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003;31:5526–5533. doi: 10.1093/nar/gkg761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murai J, Huang SyN, Das BB, Renaud A, Zhang Y, Doroshow JH, Ji J, Takeda S, Pommier Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012;72:5588–5599. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haince JF, Rouleau M, Hendzel MJ, Masson JY, Poirier GG. Targeting poly(ADP-ribosyl)ation: a promising approach in cancer therapy. Trends Mol Med. 2005;11:456–463. doi: 10.1016/j.molmed.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 4.Horton JK, Stefanick DF, Naron JM, Kedar PS, Wilson SH. Poly(ADP-ribose) polymerase activity prevents signaling pathways for cell cycle arrest following DNA methylating agent exposure. J Biol Chem. 2005;280:15773–15785. doi: 10.1074/jbc.M413841200. [DOI] [PubMed] [Google Scholar]
- 5.Horton JK, Watson M, Stefanick DF, Shaughnessy DT, Taylor JA, Wilson SH. XRCC1 and DNA polymerase β in cellular protection against cytotoxic DNA single-strand breaks. Cell Res. 2008;18:48–63. doi: 10.1038/cr.2008.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heacock ML, Stefanick DF, Horton JK, Wilson SH. Alkylation DNA damage in combination with PARP inhibition results in formation of S-phase-dependent double-strand breaks. DNA Repair. 2010;9:929–936. doi: 10.1016/j.dnarep.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dianova II, Sleeth KM, Allinson SL, Parsons JL, Breslin C, Caldecott KW, Dianov GL. XRCC1-DNA polymerase β interaction is required for efficient base excision repair. Nucleic Acids Res. 2004;32:2550–2555. doi: 10.1093/nar/gkh567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marintchev A, Gryk MR, Mullen GP. Site-directed mutagenesis analysis of the structural interaction of the single-strand-break repair protein, X-ray cross-complementing group 1, with DNA polymerase β. Nucleic Acids Res. 2003;31:580–588. doi: 10.1093/nar/gkg159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berquist BR, Singh DK, Fan J, Kim D, Gillenwater E, Kulkarni A, Bohr VA, Ackerman EJ, Tomkinson AE, Wilson DM. Functional capacity of XRCC1 protein variants identified in DNA repair-deficient Chinese hamster ovary cell lines and the human population. Nucleic Acids Res. 2010;38:5023–5035. doi: 10.1093/nar/gkq193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wong HK, Wilson DM., 3rd XRCC1 and DNA polymerase β interaction contributes to cellular alkylating-agent resistance and single-strand break repair. J Cell Biochem. 2005;95:794–804. doi: 10.1002/jcb.20448. [DOI] [PubMed] [Google Scholar]
- 11.Cuneo MJ, London RE. Oxidation state of the XRCC1 N-terminal domain regulates DNA polymerase β binding affinity. Proc Natl Acad Sci U S A. 2010;107:6805–6810. doi: 10.1073/pnas.0914077107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tebbs RS, Flannery ML, Meneses JJ, Hartmann A, Tucker JD, Thompson LH, Cleaver JE, Pedersen RA. Requirement for the Xrcc1 DNA base excision repair gene during early mouse development. Dev Biol. 1999;208:513–529. doi: 10.1006/dbio.1999.9232. [DOI] [PubMed] [Google Scholar]
- 13.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 14.Johnson BA, Blevins RA. NMR View: A computer program for the visualization and analysis of NMR data. J Biomol NMR. 1994;4:603–614. doi: 10.1007/BF00404272. [DOI] [PubMed] [Google Scholar]
- 15.Horton JK, Joyce-Gray DF, Pachkowski BF, Swenberg JA, Wilson SH. Hypersensitivity of DNA polymerase β null mouse fibroblasts reflects accumulation of cytotoxic repair intermediates from site-specific alkyl DNA lesions. DNA Repair. 2003;2:27–48. doi: 10.1016/s1568-7864(02)00184-2. [DOI] [PubMed] [Google Scholar]
- 16.Butler WB. Preparing nuclei from cells in monolayer cultures suitable for counting and for following synchronized cells through the cell cycle. Anal Biochem. 1984;141:70–73. doi: 10.1016/0003-2697(84)90426-3. [DOI] [PubMed] [Google Scholar]
- 17.Biade S, Sobol RW, Wilson SH, Matsumoto Y. Impairment of proliferating cell nuclear antigen-dependent apurinic/apyrimidinic site repair on linear DNA. J Biol Chem. 1998;273:898–902. doi: 10.1074/jbc.273.2.898. [DOI] [PubMed] [Google Scholar]
- 18.Kedar PS, Kim SJ, Robertson A, Hou E, Prasad R, Horton JK, Wilson SH. Direct interaction between mammalian DNA polymerase β and proliferating cell nuclear antigen. J Biol Chem. 2002;277:31115–31123. doi: 10.1074/jbc.M201497200. [DOI] [PubMed] [Google Scholar]
- 19.Gassman NR, Stefanick DF, Kedar PS, Horton JK, Wilson SH. Hyperactivation of PARP triggers nonhomologous end-joining in repair-deficient mouse fibroblasts. PLoS ONE. 2012;7:e49301. doi: 10.1371/journal.pone.0049301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilson DM, III, Bianchi C. Improved immunodetection of nuclear antigens after sodium dodecyl sulfate treatment of formaldehyde-fixed cells. J Histochem Cytochem. 1999;47:1095–1100. doi: 10.1177/002215549904700814. [DOI] [PubMed] [Google Scholar]
- 21.Thompson LH, Brookman KW, Dillehay LE, Carrano AV, Mazrimas JA, Mooney CL, Minkler JL. A CHO-cell strain having hypersensitivity to mutagens, a defect in DNA strand-break repair, and an extraordinary baseline frequency of sister-chromatid exchange. Mutat Res. 1982;95:427–440. doi: 10.1016/0027-5107(82)90276-7. [DOI] [PubMed] [Google Scholar]
- 22.Zdzienicka MZ, van der Schans GP, Natarajan AT, Thompson LH, Neuteboom I, Simons JW. A Chinese hamster ovary cell mutant (EM-C11) with sensitivity to simple alkylating agents and a very high level of sister chromatid exchanges. Mutagenesis. 1992;7:265–269. doi: 10.1093/mutage/7.4.265. [DOI] [PubMed] [Google Scholar]
- 23.Tebbs RS, Thompson LH, Cleaver JE. Rescue of Xrcc1 knockout mouse embryo lethality by transgene-complementation. DNA Repair. 2003;2:1405–1417. doi: 10.1016/j.dnarep.2003.08.007. [DOI] [PubMed] [Google Scholar]
- 24.Zhang X, Morera S, Bates PA, Whitehead PC, Coffer AI, Hainbucher K, Nash RA, Sternberg MJ, Lindahl T, Freemont PS. Structure of an XRCC1 BRCT domain: a new protein-protein interaction module. EMBO J. 1998;17:6404–6411. doi: 10.1093/emboj/17.21.6404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dulic A, Bates PA, Zhang X, Martin SR, Freemont PS, Lindahl T, Barnes DE. BRCT domain interactions in the heterodimeric DNA repair protein XRCC1-DNA ligase III. Biochemistry. 2001;40:5906–5913. doi: 10.1021/bi002701e. [DOI] [PubMed] [Google Scholar]
- 26.Cuneo MJ, Gabel SA, Krahn JM, Ricker MA, London RE. The structural basis for partitioning of the XRCC1/DNA ligase III-alpha BRCT-mediated dimer complexes. Nucleic Acids Res. 2011;39:7816–7827. doi: 10.1093/nar/gkr419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taylor RM, Wickstead B, Cronin S, Caldecott KW. Role of a BRCT domain in the interaction of DNA ligase III-α with the DNA repair protein XRCC1. Curr Biol. 1998;8:877–880. doi: 10.1016/s0960-9822(07)00350-8. [DOI] [PubMed] [Google Scholar]
- 28.McNeill DR, Lin PC, Miller MG, Pistell PJ, de Souza-Pinto NC, Fishbein KW, Spencer RG, Liu Y, Pettan-Brewer C, Ladiges WC, Wilson DM. XRCC1 haploinsufficiency in mice has little effect on aging, but adversely modifies exposure-dependent susceptibility. Nucleic Acids Res. 2011;39:7992–8004. doi: 10.1093/nar/gkr280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kubota Y, Nash RA, Klungland A, Schar P, Barnes DE, Lindahl T. Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase β and the XRCC1 protein. EMBO J. 1996;15:6662–6670. [PMC free article] [PubMed] [Google Scholar]
- 30.Marintchev A, Robertson A, Dimitriadis EK, Prasad R, Wilson SH, Mullen GP. Domain specific interaction in the XRCC1-DNA polymerase β complex. Nucleic Acids Res. 2000;28:2049–2059. doi: 10.1093/nar/28.10.2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brem R, Hall J. XRCC1 is required for DNA single-strand break repair in human cells. Nucleic Acids Res. 2005;33:2512–2520. doi: 10.1093/nar/gki543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lan L, Nakajima S, Oohata Y, Takao M, Okano S, Masutani M, Wilson SH, Yasui A. In situ analysis of repair processes for oxidative DNA damage in mammalian cells. Proc Natl Acad Sci U S A. 2004;101:13738–13743. doi: 10.1073/pnas.0406048101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hanssen-Bauer A, Solvang-Garten K, Sundheim O, Peña-Diaz J, Andersen S, Slupphaug G, Krokan HE, Wilson DM, Akbari M, Otterlei M. XRCC1 coordinates disparate responses and multiprotein repair complexes depending on the nature and context of the DNA damage. Environ Mol Mutagen. 2011;52:623–635. doi: 10.1002/em.20663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bonicalzi ME, Haince JF, Droit A, Poirier GG. Regulation of poly(ADP-ribose) metabolism by poly (ADP-ribose) glycohydolase: where and when?, CMLS. Cell Mol Life Sci. 2005;62:739–750. doi: 10.1007/s00018-004-4505-1. [DOI] [PubMed] [Google Scholar]
- 35.Mortusewicz O, Fouquerel E, Jean-Christophe A, Leonhardt H, Schreiber V. PARG is recruited to DNA damage sites through poly(ADP-ribose)- and PCNA-dependent mechanisms. Nuc Acids Res. 2011;39:5045–5056. doi: 10.1093/nar/gkr099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Horton J, Wilson S. Predicting enhanced cell killing through PARP inhibition. Mol Cancer Res. 2013 doi: 10.1158/1541-7786.MCR-12-0512. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kirkpatrick DS, Gerber SA, Gygi SP. The absolute quantification strategy: a general procedure for the quantification of proteins and post-translational modifications. Methods. 2005;35:265–273. doi: 10.1016/j.ymeth.2004.08.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.