

Abstract
Objective
To discuss the diagnosis and treatment of peripheral primitive neuroectodermal tumors of the pancreas based on our case and all the cases in the world.
Methods
The first case of peripheral primitive neuroectodermal tumors of the pancreas in Asia was preliminarily reported by our group in 2006. The patient underwent three operations for the primary tumor and recurrences over 41 months prior to the patient’s death in November 2007. All 14 reported cases of pancreatic PNETs in the world were analyzed. The corresponding literatures on its diagnosis and treatment of were reviewed.
Results
A 13 year-old female patient was diagnosed with pancreatic PNETs by the clinical, microscopic, immunohistochemical features, and cytogenetic analysis after the resection of the tumor located in the uncinate process of the pancreas at PUMC Hospital. During the follow-up course, radiotherapy and chemotherapy were given after the first operation. Two additional operations were performed 10 months and 25 months after the first one, respectively, because of tumor recurrence. The patient died 41 months after the initial diagnosis with the recurrence and metastasis that were not suitable for a further surgery. Primitive neuroectodermal tumors of the pancreas are extremely rare. A review of the world’s literature on this tumor identified fourteen cases with a mean survival time of 12 months (ranging from 6 to 50 months). These patients often have no specific clinical symptoms, but most do present with abdominal pain and/or jaundice. The diagnosis is established by small round tumor cells seen on light microscopy, immunohistochemical features of positive P30/32MIC2 with at least two positive neuronal markers., and cytogenetic analysis showing characteristic translocation of t[11;22][q24;q12]. Since pancreatic PNETs are highly aggressive, early diagnosis, immediate surgical resection and re-resection if possible, early radiotherapy and chemotherapy and close follow-up are required.
Conclusions
Peripheral primitive neuroectodermal tumors can arise in pancreas. The diagnosis and treatment should be made as early as possible, aggressive surgeries for the primary and recurrences may help to improve the prognosis.
Key Words: Peripheral primitive neuroectodermal tumors, pancreatic tumor, small round cell tumors, Ewing’s sarcoma
Introduction
Peripheral primitive neuroectodermal tumors (pPNETs) are a group of embryonic malignant tumors characterized as small round cell tumors. They are often considered as part of the Ewing’s sarcoma family of tumors, which exhibit neuroepithelial differentiation. Peripheral primitive neuroectodermal tumors seldom arise in organs, and it’s occurrence in the pancreas is extremely rare. The onset of the tumor is insidious and patients often have no specific clinical symptoms. The diagnosis of peripheral primitive neuroectodermal tumors of pancreas requires the combination of clinical symptoms, pathological characteristics, immunohistochemical features and cytogenetic analysis. Surgical resection followed by radiation and chemotherapy is the currently accepted treatment, although the prognosis remains poor due to the highly aggressive nature of this cancer. We report here the first case of pancreatic peripheral primitive neuroectodermal tumors in Asia (1) and analyze the world literature reported a total of thirteen cases.
Case report
Clinical symptoms
A thirteen year-old female patient was admitted to Peking Union Medical College Hospital (PUMCH) in June 2004. The patient suffered from mild abdominal pain, anorexia, polyuria, polydipsia, and weight loss of 10 kg (22 pounds) over the preceding two months. The patient denied fever, nausea, vomiting, constipation, diarrhea, jaundice, and irregularities of the autonomic nervous system. On exam, the patient was without jaundice or scleral icterus. No superficial lymph nodes were found. An immobile firm mass that was tender to deep palpation and without clear margins was identified in the right upper quadrant by palpation.
Laboratory and radiological results
The patient was found to have severe hyperglycemia ketoacidosis on laboratory test. Her random blood glucose was 17 mmol/L (306 mg/dL) and urine glucose was 55 mmol/L (990 mg/dL), while urine acetone bodies were more than 7.8 mmol/L with trace urine-protein. On CT scan of the abdomen, a large mass of 9 cm × 11 cm × 17 cm in size was seen between the liver, the pancreas and the right kidney with focal irregular intensification in the arterial period (Figure 1). Somatostatin receptor scan was negative for neuroendocrine tumors, and methoxyisobutyl isonitrile (131I-MIBI) scan was negative for pheochromocytoma.
Figure 1.
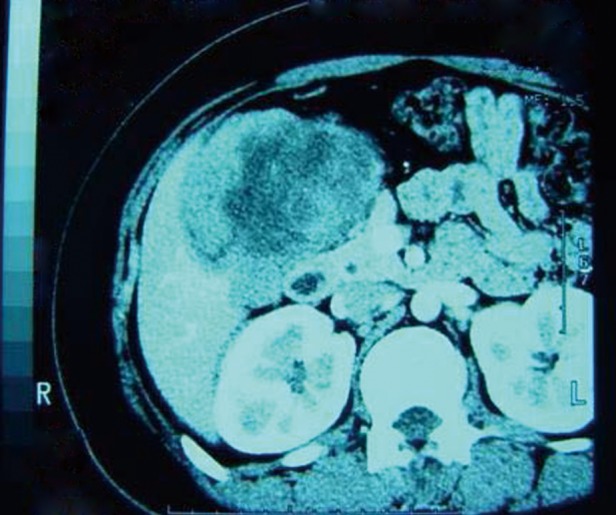
Abdominal CT appearance of the primary lesion. A mass of 9 cm × 11 cm × 17 cm located between the liver, the pancreas, and the right kidney with focal intensification in the arterial period
Diagnosis and treatment
The patient underwent surgical resection of the mass with an initial diagnosis of unspecified pancreatic tumor on June 19th, 2004. At laparotomy, a 15 cm × 15 cm × 10 cm mass was found on the uncinate process of the pancreas. The tumor had grown superiorly to the infrahepatic space, postero-lateral aspect of the duodenum, and inferiorly to the hepatic flexure of colon. The tumor was a firm lobular mass with clear demarcation. Since invasion of the colon at the hepatic flexure was found, resection of the uncinate process of the pancreas, while preserving the duodenum, and partial resection of transverse colon were performed. The gross pathology presented a thin and flat neoplasm which was 3.5 cm × 2.5 cm × l.0 cm in size and arose in the uncinate process of pancreas. The cross-section of the neoplasm is grey-red, grey-pink in color and exquisite in texture. The surgical margins were free of tumors as reported by frozen section. No metastases were found in pancreaticduodenal lymph nodes. The greater omentum was invaded by tumor nodules which were grey-white, grey-pink in color and soft in texture with small hemorrhages. The tumor was found to extend into the submucosa of the transverse colon.
The tumor cells were characterized by light microscopy as small round and oval cells with scant cytoplasm. The tumor was separated by fibrous connective tissue into the folial parts. Granular nuclear chromatin and karyokinesis phenomenon with unclear nucleoli were found in the tumor cells. There were no Homer-Wright rosettes in the tumor cells (Figure 2A). The immunohistochemical staining was performed on paraffin-embedded tissues. The tumor cells were found positive for CD99, NSE (neuron specific enolase), while negative for NF (neurofilament protein), TDT (terminal deoxynucleotidyl transferase), LCA (leukocyte common antigen), CgA (chromogranin A), AEl/AE3 (cytokeratin) and PAS (Figure 2B,C,D). Cytogenetic analysis confirmed t[11;22][q24;q12] translocation. Therefore, the diagnosis was made as pancreatic peripheral primitive neuroectodermal tumor.
Figure 2.

Pathology and Immunohistochemical staining of the tumor. A. Hematoxylin and eosin stain (original magnification ×150) of a PNET (primitive neuroectodermal tumor) of the pancreas. These neoplasms were composed of sheets of small round cells with hyperchromatic round to oval nuclei. The cells had no Homer-Wright rosettes; B. Immunohistochemical stain (original magnification ×150) for CD99 (p30/32MIC2). The neoplastic cells showed strong cytoplasmic membrane positive to the MIC-2 glycoprotein; C. Immunohistochemical stain (original magnification ×150) for neuron-specific enolase showing strong focal positive and demonstrating the neuronal differentiation of these small round cell tumors; D. Immunohistochemical stain (original magnification ×150) for cytokeratin (AE1/AE3) showing negative
Follow-up cause and additional surgeries
Her post-operative course was uneventful. Her blood glucose initially normalized without any medications for two months postoperatively, but it began to increase after the third month. The random blood glucose returned to 17 mmol/L (306 mg/dL) and hemoglobin A1C (HbA1C) reached 7.3%. The islet cell antibody (ICA) was weakly positive with ratio 1:10, and the glutamic acid decarboxylase (GAD) was also positive at 26 RU/mL. The PBG was controlled with preprandial short-acting insulin.
The patient was referred for radiation and chemotherapy one month post-operation. Radiotherapy by 3-D position fixing of the operative field was given one month postoperatively with the dose of 2 Gy per day 5 days per week until the total dose accumulated to 46 Gy. Four courses of VAC chemotherapy were given two months postoperatively with cyclophosphamide 1.0 g Day 1, vincristine 2 mg Day 1, epirubicin 60 mg Day 1-2 by continuous intravenous infusion, and interferon (Recombinant Interferon α-2a) 300 mg Day 8,10,12,15,17,19 by intramuscular injection were given monthly. Follow-up abdominal ultrasound exam, thoracic-abdominal CT scan, and bone scan showed no evidence of metastasis eight months post-operatively with increasing body weight and normalizing random blood glucose.
Nine months post operatively, the patient re-presented with severe hyperglycemia with fasting blood glucose (FBG) reaching 10 mmol/L (180 mg/dL) and random blood glucose reaching 14 mmol/L (252 mg/dL). Abdominal ultrasound revealed a mass 4 cm × 3 cm in size located near the left lobe of the liver. Ten months post operatively a repeat abdominal ultrasound showed a mass 3.2 cm × 3 cm in size located near the left lobe of liver and diaphragm (Figure 3). Ten days later, an, exploratory laparotomy was performed. A 6 cm × 5 cm × 5 cm mass was located near the diaphragmatic dome and invaded the diaphragm and left lobe of the liver. The mass was removed with sufficient hepatic margin. The gross pathology presented a thin and flat neoplasm which was 5 cm × 4 cm × 3 cm in size. There were observed metastasis in striated muscles and connective tissue of the diaphragm. The tumor cells were again found positive for CD99 and NSE, negative for AE1/AE3 on immunohistochemical stain.
Figure 3.
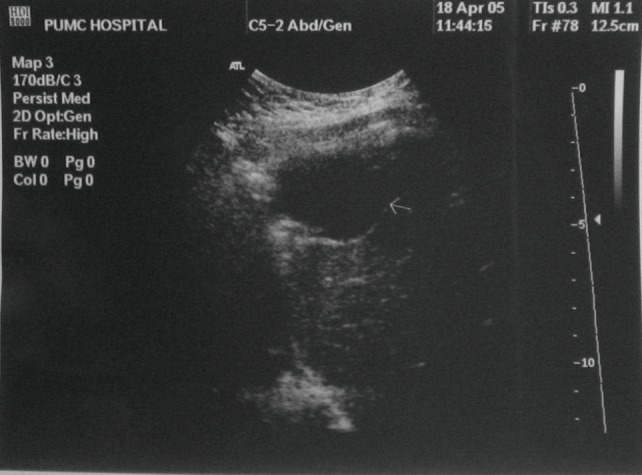
Abdominal ultrasound showing a mass 4 cm × 3 cm located near the left lobe of liver
Two months after the second operation, ascites was identified by abdominal ultrasound. After drainage, ascites analysis showed that the total number of cells was 5,180/mm3, with WBC of 180/mm3 (62% monocyte, and 38% PMN). The total protein quantity of the ascites was 44 G/L. Tumor cells were found in the ascites fluid by cytology.
The patient was given seven courses chemotherapy of MAID from July to December 2005. The MAID included: Ifosfamide (IFO) 3 g Day 1-4, 2 g Day 5; Mesna, 0.6 g tid Day 1-4, 0.4 g Tid Day 5; Epirubicin Hydrochloride (EIP) 40 mg Day 1-3; 5-(3,3-dimethyl-1-triazeno) Imidazole-4-carbox-amide (DTIC) 500 mg Day 1-3 by continuous intravenous infusion. The patient experienced nausea, vomiting and neutropenia as side effect and was provided symptomatic treatment. There was no ascites after the fourth course of MAID chemotherapy. The patient’s fasting blood glucose went back to 8 mmol/L (144 mg/dL). The HbA1C dropped to 7.3%. The patient recovered well and went back to school.
The patient developed nausea and light abdominal pain 26 months after the second operation (36 months after the first operation) and again presented to the hospital. Laboratory tests revealed that the patient again had severe hyperglycemia. Random blood glucose was more than 16 mmol/L (288 mg/dL), while HbA1C was 9%. Abdominal CT scan showed a large mass about 10 cm × 10 cm × 8 cm in size located in the retro-peritoneum. Somatostatin receptor scan did not indicate neuroendocrine tumors. Somatostatin receptor scan showing a large mass located in the right abdomen which was negative for neuroendocrine tumors (Figure 4).
Figure 4.

Somatostatin receptor scan showing a large mass located in the right abdomen, which was negative for neuroendocrine tumors
An exploratory laparotomy was again performed. The mass was located at the right upper abdomen, 10 cm × 10 cm × 8 cm in size. The mass abutted the liver without invasion however it invaded the peritoneum of the right kidney including the renal capsule. The tumor also invaded the hepatic flexure of colon with extension to the transverse colon. The mass was completely resected with the additional right hemicolectomy. The right kidney was preserved by removing only part of the renal capsule and leaving the renal parenchyma intact.
The gross pathology showed a thin and flat neoplasm invading the colon to the level of the muscularis. The lymph nodes were negative for metastasis. The tumor cells were found positive for CD99 and negative for LCA on immunohistochemical stain. The patient had uneventful recovering requiring insulin for glucose control.
Four months after the most recent operation, the patient experienced urinary retention. Abdominal CT scan demonstrated recurrence or metastasis of the cancer in the right kidney with hydronephrosis (Figure 5). A thoracic MRI demonstrated an invasive mass on the third thoracic vertebra, compressing to the spinal cord, which was thought to be metastasis (Figure 6). The patient accepted five courses of radiotherapy without relief of symptoms, and she subsequently died 41 months after the initial operation.
Figure 5.
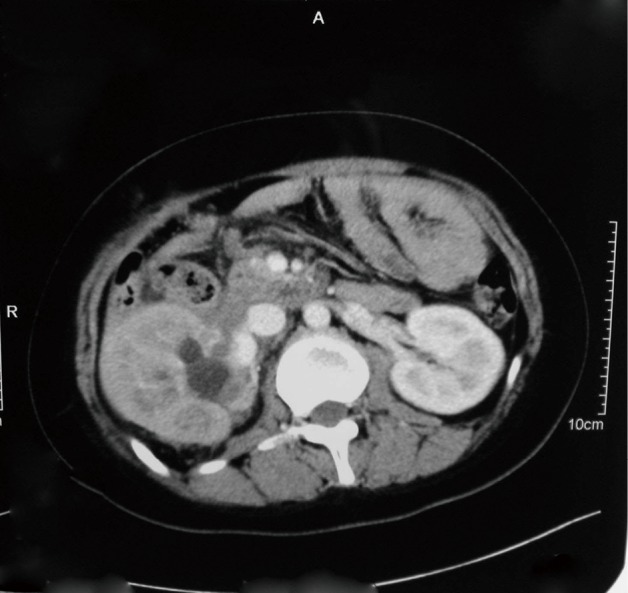
A recurrence or metastasis of the tumor in right kidney with hydronephrosis
Figure 6.
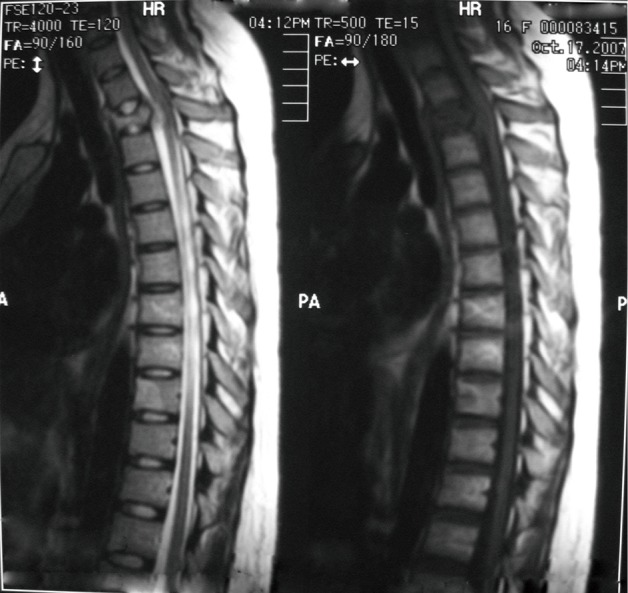
An invasive mass on the third thoracic vertebra, compressing the spinal cord, which was thought to be a tumor metastasis
Discussion
Peripheral primitive neuroectodermal tumors are a group of embryonic malignant tumors characterized as small round cell tumors (2). The cellular origin of the tumor is still controversial, but recent evidence suggests that primitive neuroepithelial stem cells are the most likely possibility (3). Although pPNETs seldom arise in solid organs, they have been reported to arise in a variety of organs such as kidney, urinary bladder, uterus, gall bladder, lung, and vagina (2-6). PNETs arising in the pancreas are extremely rare. In a report by Lüttges (6), only two cases among 600 primary pancreatic neoplasma (0.3%) were diagnosed as pancreatic PNETs. Previous to this report, there are only 13 cases of the pancreatic PNETs reported in the world’s literature (2-7). By analyzing all 14 cases (including ours), we found the patients ranged in age from 6 to 31 year-old without significant sex difference. The site of the tumor was in the head of the pancreas in eleven of the fourteen cases. This proclivity to the head of the pancreas can be explained, as the primitive neuroepithelial stem cells that develop into the head of the pancreas are different from those that give rise to the body and tail of the pancreas. All other pancreatic pPNETs are reported in the US or Europe, this is the first report of a pancreatic PNET in Asia (Table 1).
Table 1. Clinical features of world’s 14 cases of primitive neuroectodermal tumors of the pancreas (1-5).
| Case No. | Age (years) | Sex | Presenting symptoms | Size (cm) | Cytogenetic analysis | Diagnosis procedure | Treatment | Metastasis | Clinical follow-up |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 17 | M | Jaundice, Abdominal pain | 9.0 | + | Whipple resection | CHE | N/A | 33 mo; NED |
| 2 | 20 | M | Jaundice, Abdominal pain | 3.5 | + | Whipple resection | N/P | N/A | 27 mo; AWD |
| 3 | 25 | F | Abdominal pain | N/A | N/A | Biopsy | N/A | N/A | N/A |
| 4 | 21 | F | Abdominal pain | N/A | +* | Whipple resection | N/A | N/A | DOC |
| 5 | 25 | F | Jaundice, Abdominal pain | 8.0 | -* | Biopsy | N/A | N/A | N/A |
| 6 | 13 | M | Abdominal pain | 6.0 | N/A | Biopsy | N/A | N/P | 43 mo; NED |
| 7 | 6 | M | Jaundice, Abdominal pain | 3.5 | +* | Whipple resection | CHE | 48 mo; Recurrence | 48 mo; DOD |
| 8 | 31 | M | Abdominal pain fever | 10.0 | - | Whipple resection | RAD CHE | 4 mo, Recurrence; 24 mo/36 mo, lung | 50 mo; DOD |
| 9 | 6 | F | Abdominal pain Anemia | 5.5 | -** | Whipple resection | N/P | 6 mo;Recurrence | 6 mo; DOD |
| 10 | 13 | F | Dyspepsia Vomiting | 22 | N/A | Whipple resection | CHE | N/A | N/A |
| 11 | 31 | M | Abdominal pain Loss of appetite | N/A | N/A | Biopsy | CHE | N/A | N/A |
| 12 | 17 | M | Abdominal pain | 9 | + | Whipple resection # | RAD CHE | N/P | 8 mo; AWD |
| 13 | 33 | M | Abdominal pain | 15 | + | Laparotomy | RAD CHE | Simultaneously, liver, spleen | 12 mo;AWD |
| 14# | 13 | F | Abdominal pain Diabetes mellitus 2 | 3.5 | + | RUPT | RAD CHE | 9/36 mo, Recurrence; 12 mo, ascites | 41 mo; AWD |
Sex, M, male; F, female; N/A, not available; N/P, not performed; Cytogenetic analysis (+), t[11;22][q24;12]; *RT-PCR, reverse transcriptase Polymerase Chain Reaction data; RUPT, resection of the uncinate process tumor, preserving the duodenum; #Whipple resection, pylorus preserving pancreatoduodenectomy; -**, loss of cosmids F7 and E4 distal of the EWSR1 breakpoint in nearly all cells; NED, no evidence of disease; AWD, alive with disease; DOC: died of postoperative complications; DOD, died of disease; mo, month; Metastasis, post operation time and position of the metastasis; RAD, radiotherapy; CHE, chemotherapy; #14 our case
There are no specific clinical symptoms of pancreatic pPNETs, although all patients suffer from abdominal pain with the palpation of the abdominal mass. Half of the patients have jaundice, with obstructive jaundice being more common. Severe anemia or hemorrhage of the upper gastrointestinal tract can also be the presenting symptom (Table 1). Our case is the only one in which hyperglycemia was reported. As to the positive relationship between the elevated blood glucose level and the recurrence of the tumor, this case suggests that pancreatic PNETs may cause endocrine dysfunction. This could be explained by the relationship between the neuroepithelial stem cells that give rise to neurons that have a role in regulating pancreatic islet function. Most pancreatic PNET patients do not have exocrine dysfunction on laboratory tests, and most tumors are located in the head of the pancreas and ranged in size ranged from 3.5 cm to 10 cm. Abdominal CT and MRI are the most useful modalities to reveal the tumors. A mass located in the head of the pancreas with clear margin is the most common radiographic presentation. Abdominal CT scan demonstrates a tumor of variable density due to the tumor necrosis. Focal intensification in the arterial period on CT may be present; however, most tumors do not have a close relationship with arteries and will be without enhancement in the arterial phase. The metastasis and invasion to other organs and tissues may be found in the advance stages of disease (8) (Table 1).
There are no established pathological criteria for the diagnosis of the pancreatic PNETs. The combination of clinical symptoms, pathological characteristics, immunohistochemical features and cytogenetic analysis are well accepted. In principle, direct histological evidence of the PNETs and distinction from other small round cell tumors are the most essential points. Although Homer-Wright (H-W) rosette or atypical rosette array of the cells are specific characters of the PNETs in light microscopy, these characteristics are rarely present in pancreatic PNETs. Cell cytoplasm containing neuronal secretory granules, neurofilaments, and pyknic nucleus granules are the important criteria for the diagnosis of pPNETs (2-6,9).
The criteria of immunohistochemical features of PNET are based on two groups of markers: the product of MIC-2 gene on the X chromosome, which is known as P30/32MlC2, and at least two kinds of neuronal markers. A group of monoclonal antibodies, such as CD99, O13, HBA71, 12E7, RFB1 are also available for testing. None of these monoclonal antibodies are specific for PNETs, nor are their positive predictive value (PPV) the same in PNETs. But the PPV is 100% for MIC-2 gene testing with these monoclonal antibodies (2-6). The PPV of neural markers in reports also range widely with NSE (11/13 cases), CgA (1/13 cases), Syn (4/13 cases). Neural markers such as desmin, actin, S-100, insulin, glucagons, somatostatin are rarely positive in these reports (2-6) (Table 2).
Table 2. Immunohistochemical features of world’s 14 cases of primitive neuroectodermal tumors of the pancreas (1-5).
| No. ICH features | Case |
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | ||
| P30/32MIC-2 | CD99 | P | SP | P | P | ||||||||||
| O13 | P | P | P | P | P | P | P | P | P | ||||||
| HBA71 | P | ||||||||||||||
| 12E7 | P | ||||||||||||||
| RFB1 | |||||||||||||||
| AE1/AE3 | P | P | P | P | N | P | SP | N | P | P | N | ||||
| NSE | P | P | N | P | P | P | P | P | P | P | P | P | P | ||
| CHR | N | N | P | N | N | N | N | N | N | N | N | N | |||
| SYN | N | N | N | P | N | N | P | SP | P | N | N | N | P | N | |
| EMA | P | N | P | P | |||||||||||
| DES | N | N | N | N | N | SP | N | N | N | ||||||
| INS | N | N | N | N | |||||||||||
| GLU | N | N | N | N | |||||||||||
| SOM | N | N | N | N | |||||||||||
| S-100 | N | SP | N | P | |||||||||||
| VIM | P | N | P | P | P | ||||||||||
| Leu7 | P | ||||||||||||||
| LCA | N | N | N | ||||||||||||
| HMB-45 | N | N | N | ||||||||||||
| PAS | N | N | |||||||||||||
| ACT | N | ||||||||||||||
| TDT | N | ||||||||||||||
| CD79a | N | ||||||||||||||
| CEA | N | N | |||||||||||||
| CD43 | N | ||||||||||||||
| CD3 | N | ||||||||||||||
| NF | N | ||||||||||||||
P, positive; N, negative; SP, sporadic positive. AE1/AE3, cytokeratin AE1/AE3; NSE, neuron specific enolase; CHR/CgA, chromogranin A; SYN, synaptophysin; EMA, epithelial membrane antigen; VIM, vimentin; DES, desmin; ACT, actin; INS, insulin; GLU, glucagon; SOM, somatostatin; NF, neurofilament protein; TDT, terminal deoxynucleotidyl transferase; LCA, leukocyte common antigen; PAS, Periodic Acid Schiff stain; HMB-45; CD43; CD3; LEU7; 14 our case
The development of PNETs has close relationship with particular chromosomal translocations. The fusion gene product from chromosome translocation is not only a stable structure, but it also has specificity for the type of tumors. In Ewing sarcoma/PNETs, 85% of the karyotype are t[11;22][q24;q12], which come from the EWS-FLII gene infusion while 10% of them are t[21;22][q22;q12], which come from EWS-ERG gene infusion (10-12). In reports of pancreas PNETs, there are six cases which chromosome translocation are t[11;22][q24; q12] while no cases with t[21;22][q22;q12] and just one case of loss of cosmids F7 and E4 distal of the EWS-R1 breakpoint in nearly all cells (2-6).
Primary pancreas PNETs should be distinguished from five groups of diseases. These groups of diseases mainly include: (I) pancreatic metastasis of PNETs from other primary locations; (II) small round cell tumors identified by histological characteristics; (III) tumors with similar immunohistochemical features; (IV) tumors related to the EWS gene; and (V) other pancreas malignant tumors such as pancreatic endocrine tumors and non-epithelial pancreatic malignant tumors (12-14). The final diagnosis is based on the combination of immunohistochemical features and cytogenetic analysis.
pPNETs are highly aggressive malignant tumors with almost inevitable recurrence and metastasis. Metastasis to bone, bone marrow, lymph nodes, lung, liver and other organs have all been reported. In reported cases of pancreatic pPNETs, there are four cases with local recurrence and one case with right lung metastasis (2-6). Currently, the accepted treatments of pPNETs are surgical resection with associated chemotherapy and high dose radiation therapy. None of the other reported cases underwent three major surgical resections in an attempt to control the primary and its recurrences. Chemotherapy protocols such as CAV (cyclophosphamide, adriamycin, vincristine) and neoadjuvant chemotherapy protocol (vincristine, actinomycin, adriamycin, cyclophosphamide, isophosphamide, etoposide) are adopted to promote therapeutic efficacy. Local radiation therapy is used with some therapeutic efficacy. Unfortunately, all of these treatments are unsatisfactory (15).
PNETs are highly aggressive. The longest follow-up of pancreatic pPNETs is only 50 months (2-6). The case herein reported with 41 months follow up is the third longest survival in the world and this may be attributed to the aggressive surgical intervention. The factors influencing prognosis are still unclear. However, research has shown that high degree of neural differentiation and increased immune response, represented by Leu27 (also known as CD57, HNK-1 glycoprotein), may be a positive predictor of prognosis (2,16,17).
In summary, pPNETs can arise in the pancreas primarily but are extremely rare. Most pancreatic pPNETs are identified in children to adolescence. Small round cells with malignant presentation are its histologic character. Presence of P30/32 MlC2, the product of MIC-2 gene on X chromosome, and the expression of at least two kinds of neuronal markers are the immunohistochemical features of PNETs. Chromosome translocation at t[11;22][q24;q12] is the most common translocation seen on cytogenetics. The combination of clinical symptoms, pathological characteristics, immunohistochemical features, as well as cytogenetic analysis is required to identify the diagnosis of pancreatic PNET. Surgical resection with associated chemoradiotherapy is the current standard of treatment. Aggressive surgical resection in combination with chemotherapy and radiation therapy may extend survival. Pancreas PNETs are highly aggressive tumors with metastasis and/or recurrence being extremely common, thus the prognosis of this malignant disease is very poor.
Acknowledgements
Disclosure: The authors declare no potential conflicts of interest.
References
- 1.Sang XT, Liang NX, Mao YL, et al. Diagnosis and treatment of primitive neuroectodermal tumors of pancreas. Zhongguo Yi Xue Ke Xue Yuan Xue Bao 2006;28:191-5 [PubMed] [Google Scholar]
- 2.Movahedi-Lankarani S, Hruban RH, Westra WH, et al. Primitive neuroectodermal tumors of the pancreas: a report of seven cases of a rare neoplasm. Am J Surg Pathol 2002;26:1040-7 [DOI] [PubMed] [Google Scholar]
- 3.Bülchmann G, Schuster T, Haas RJ, et al. Primitive neuroectodermal tumor of the pancreas. An extremely rare tumor. Case report and review of the literature. Klin Padiatr 2000;212:185-8 [DOI] [PubMed] [Google Scholar]
- 4.Perek S, Perek A, Sarman K, et al. Primitive neuroectodermal tumor of the pancreas. A case report of an extremely rare tumor. Pancreatology 2003;3:352-6 [DOI] [PubMed] [Google Scholar]
- 5.Danner DB, Hruban RH, Pitt HA, et al. Primitive neuroectodermal tumor arising in the pancreas. Mod Pathol 1994;7:200-4 [PubMed] [Google Scholar]
- 6.Lüttges J, Pierré E, Zamboni G, et al. Malignant non-epithelial tumors of the pancreas. Pathologe 1997;18:233-7 [DOI] [PubMed] [Google Scholar]
- 7.O’Sullivan MJ, Perlman EJ, Furman J, et al. Visceral primitive peripheral neuroectodermal tumors: a clinicopathologic and molecular study. Hum Pathol 2001;32:1109-15 [DOI] [PubMed] [Google Scholar]
- 8.Obuz F, Kovanlikaya A, Olgun N, et al. MR imaging of pancreatic metastasis from extraosseous Ewing’s sarcoma. Pancreas 2000;20:102-4 [DOI] [PubMed] [Google Scholar]
- 9.Papierz W, Alwasiak J, Kolasa P, et al. Primitive neuroectodermal tumors: ultrastructural and immunohistochemical studies. Ultrastruct Pathol 1995;19:147-66 [DOI] [PubMed] [Google Scholar]
- 10.Pagani A, Macrí L, Rosolen A, et al. Neuroendocrine differentiation in Ewing’s sarcomas and primitive neuroectodermal tumors revealed by reverse transcriptase-polymerase chain reaction of chromogranin mRNA. Diagn Mol Pathol 1998;7:36-43 [DOI] [PubMed] [Google Scholar]
- 11.Sandberg AA, Bridge JA. Updates on cytogenetics and molecular genetics of bone and soft tissue tumors: Ewing sarcoma and peripheral primitive neuroectodermal tumors. Cancer Genet Cytogenet 2000;123:1-26 [DOI] [PubMed] [Google Scholar]
- 12.Grier HE. The Ewing family of tumors. Ewing’s sarcoma and primitive neuroectodermal tumors. Pediatr Clin North Am 1997;44:991-1004 [DOI] [PubMed] [Google Scholar]
- 13.Mulligan ME, Fellows DW, Mullen SE. Pancreatic metastasis from Ewing’s sarcoma. Clin Imaging 1997;21:23-6 [DOI] [PubMed] [Google Scholar]
- 14.Parham DM. Neuroectodermal and neuroendocrine tumors principally seen in children. Am J Clin Pathol 2001;115:S113-28 [DOI] [PubMed] [Google Scholar]
- 15.Zogopoulos G, Teskey L, Sung L, et al. Ewing sarcoma: favourable results with combined modality therapy and conservative use of radiotherapy. Pediatr Blood Cancer 2004;43:35-9 [DOI] [PubMed] [Google Scholar]
- 16.Eralp Y, Bavbek S, Başaran M, et al. Prognostic factors and survival in late adolescent and adult patients with small round cell tumors. Am J Clin Oncol 2002;25:418-24 [DOI] [PubMed] [Google Scholar]
- 17.Welsch T, Mechtersheimer G, Aulmann S, et al. Huge primitive neuroectodermal tumor of the pancreas: report of a case and review of the literature. World J Gastroenterol 2006;12:6070-3 [DOI] [PMC free article] [PubMed] [Google Scholar]