

Abstract
Chronic alcohol intake decreases adiponectin and sirtuin 1 (SIRT1) expressions, both of which have been implicated in various biological processes including inflammation, apoptosis and metabolism. We have previously shown that moderate consumption of alcohol aggravates liver inflammation and apoptosis in rats with pre-existing nonalcoholic steatohepatitis (NASH). This study investigated whether moderate alcohol intake alters SIRT1 activity, adiponectin/Adiponectin receptor (AdipoR)-related signaling and lipid metabolism in a pre-existing NASH status. Sprague-Dawley rats were fed with a high-fat diet (71% energy from fat) for 6 weeks to induce NASH then subsequently divided into 2 sub-groups: fed either a modified high-fat diet (HFD, 55% energy from fat) or a modified high-fat alcoholic diet (HFA, 55% energy from fat and 16% energy from ethanol) for an additional 4 weeks. We observed in comparison to HFD group, HFA increased hepatic nuclear SIRT1 protein but decreased its deacetylase activity. SREBP-1c protein expression and FAS mRNA levels were significantly upregulated, while DGAT1/2 and CPT-I mRNA levels were downregulated in the livers of HFA compared to HFD. Although hepatic AdipoR1 decreased, HFA did not alter AdipoR2 and their downstream signaling. There were no significant changes in plasma adiponectin and free fatty acids (FFA), as well as adiponectin expression in adipose tissue between the two groups. The present study indicates that suppression in SIRT1 deacetylase activity contributes to alcohol-exacerbated hepatic inflammation and apoptosis in rats with pre-existing NASH. In addition, moderate alcohol intake did not modulate adiponectin/AdipoR signaling axis in this model.
Keywords: Adiponectin, alcohol consumption, nonalcoholic steatohepatitis (NASH), rats, sirtuin-1 (SIRT1)
Introduction
Non-alcoholic fatty liver disease (NAFLD), ranging from steatosis to non-alcoholic steatohepatitis (NASH), fibrosis, and cirrhosis, is one of the most common forms of chronic liver disease in the United States (1,2). It affects as much as 30% of the adult population and is strongly associated with obesity and diabetes (3). The pathogenesis of the NAFLD has been suggested as a two-hit hypothesis, with steatosis as first hit and prolonged oxidative stress, lipotoxicity and other insults as second hit for the development and the progression of this disease (2,4-6). Recently, in the rats with pre-existing NASH induced by high-fat diet, we showed that even moderate alcohol intake (equivalent to 1-2 drinks⁄day) trigged in more hepatic inflammation and cellular apoptosis without significant steatosis progression (7). Thus, the molecular mechanism(s) that leads to NASH progression under moderate alcohol consumption needs further investigation.
Sirtuin 1 (SIRT1), the mammalian ortholog of yeast sirtuin silent information regulator 2 (Sir2), is a NAD+-dependent protein deacetylase that has been implicated in various biological processes, including inflammation, apoptosis and metabolism (8-11). SIRT1 displaces acetyl groups from histones and other proteins, thereby controlling their activity (8). SIRT1 has been proposed as an important target of ethanol in the liver (12-17). Chronic ethanol exposure has been shown to decrease hepatic SIRT1 gene and protein expressions and inhibits SIRT1 deacetylase activity (13). Recent study has showed that hepatic SIRT1 signaling can be upregulated by enhancing adiponectin/AdipoR in alcoholic fatty liver model (14), suggesting a relationship between adiponectin and SIRT1 as an important hepatic axis involved in fatty liver disease progression. It remains unknown whether SIRT1 expression and its activity, as well as adiponectin/AdipoR signaling, could be modulated by moderate alcohol consumption during NASH progression.
In the present study, we evaluated whether moderate alcohol consumption aggravates steatohepatitis by interfering with SIRT1 and adiponectin/AdipoRs signaling axis, while altering hepatic lipid metabolism in rats with pre-existing NASH.
Methods
Animal and experimental model
The diet composition, animal and experimental model have been previously described (7). To explore the mechanism which moderate alcohol was involved with more liver inflammation in pre-existing NASH (7), we have used samples from two groups: high-fat diet-induced NASH (as a control group) and NASH plus alcohol. Low-fat group was not included because high-fat diet-induced NASH has been confirmed before. Briefly, eight-week old male Sprague-Dawley rats (Charles River Co., Wilmington, MA) were fed ad libitum with a high-fat diet (71% energy from fat; Dyets Inc., Bethlehem, PA) for 6 weeks to develop pre-existing NASH (18). The rats were then randomized into 2 sub-groups and fed either a modified high-fat diet (HFD, 55% energy from fat; n=10) or a modified high-fat alcoholic diet (HFA, 55% energy from fat and 16% energy from ethanol; n=10) for 4 weeks. To avoid differences in the energy provided, fat removed from high-fat diet was replaced by dextrin maltose to HFD. The protein contents between the two diets were identical.
Rats were housed in individual cages, in a temperature (24±2 °C) and humidity (55%±5%) controlled environment on a 12-12 h light dark cycle. At the end of experiment, all rats were killed by cardiac exsanguinations and vital organ harvest under deep isoflurane anesthesia. Plasma was collected by cardiac puncture, and rat livers were excised, snap frozen in liquid nitrogen and stored at –80 °C. The Institutional Animal Care and Use Committee at the USDA Human Nutrition Research Center on Aging approved the animal protocol.
Plasma and liver analysis
The plasma adiponectin was analyzed using ELISA kit (Adiponectin Cat. #EZRADP-62K-Millipore Corporation, CA). Plasma and hepatic triacylglycerol (TG) and free fatty acids (FFA) levels were determined by a colorimetric method (L-Type triglyceride M and HR Series NEFA-HR kits, respectively, Wako Diagnostics, Richmond, VA, USA).
mRNA expression by real-time PCR
Total RNA were extracted from liver tissues using the TriPure reagent (Roche Applied Science), following the manufacture’s protocol. 400 ng of total RNA were used for the synthesis of 20 µL of complementary DNA (cDNA) by random primer Moloney Murine Leukemia Virus reverse transcriptase (Invitrogen). Plasma membrane protein fatty acid translocase CD36 (FAT/CD36), fatty acid transport proteins (FATP), diacylglycerol acyltransferase 1 and 2 (DGAT1 and DGAT2), fatty acid synthase (FAS) and carnitine palmitoyltransferase I (CPT-I) mRNA concentration were determined by qRT-PCR. Primers were designed using the Primer Express version 2.0 (Applied Biosystems) software. The sequence for FAT/CD36 (NCBI reference sequence: AF072411.1) were: forward 5'-AATGAGCCCAACAGTTCCGA-3' and reverse 5'-GAATTAAGTTGAAACCAGGCCACA-3'; For FATP (NCBI reference sequence: U89529.1) were: forward 5'-CCCAAGTGGATACAACAGGCA-3' and reverse 5'-GGTCTAGAAAGAAGAGCCGGTC-3'; For DGAT1 (NCBI reference sequence: NM_053437.1) were: forward 5'-CCACCAGGATGCCATACTTGAT-3' and reverse 5'-GACAGCGGTTTCAGCAATTACC-3'; For DGAT2 (NCBI reference sequence: NM_001012345.1) were: forward 5'-CCTGCAGTGTCATCCTCATGTA-3' and reverse 5'-TGATCTCCTGCCACCTTTCTT-3'; For FAS (NCBI reference sequence: NM_017332.1) were: forward 5'-TGGATCCATGGCAGCTGTTG-3' and reverse 5'-TCATTCACTGCAGCCTGAGGTC-3'; For CPT-I (NCBI reference sequence: L07736.1) were: forward 5'-TCTTGCGATCATGCCCAG-3' and reverse 5'-TCATCCGGTTCAAGAATGGC-3'. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (NCBI reference sequence: NM_017008.3) was used as internal control, and the sequences were: forward 5'-ATCACCATCTTCCAGGAGCGA-3' and reverse 5'-AGCCTTCTCCATGGTGGTGAA-3'. Quantitative measurements were performed using the SYBR Green RT-qPCR kit (Invitrogen) according to the manufacturer’s instructions on an Applied Biosystems 7000 sequence detection system. The real-time cycler conditions were as follows: enzyme activation at 50 °C for 2 min and after denaturation at 95 °C for 10 min, the cDNA products were amplified with 40 cycles, each cycle consisting of denaturation at 95 °C for 15 seconds and annealing/extension at 60 °C for 1 min. Product purities were confirmed by dissociation curve analysis. mRNA expressions were quantified relative to the values of the control group after adjusting for GAPDH by the 2-∆∆CT method as described previously.
Protein expression by Western Blotting
Whole liver homogenates and nuclear fraction were prepared from frozen liver samples. Liver protein extracts (50 µg each) were resolved on sodium dodecyl sulfate-polyacrylamide (SDS-PAGE) gel electrophoresis. After membrane-blocking, immunoblottings were performed based on the manufacturer’s instruction for each of the primary antibodies against AdipoR1 (Santa Cruz Biotechnology, CA, USA; Catalog number sc-99183), AdipoR2 (Santa Cruz Biotechnology, CA, USA; Catalog number sc-46755), phosphorylated AMPKα (p-AMPKα-Thr172) (Cell Signaling, Beverly, MA, USA; Catalog number 2531), phosphorylated ACC (p-ACC-Ser79) (Cell Signaling, Beverly, MA, USA; Catalog number 3661), SREBP-1c (Santa Cruz Biotechnology, CA, USA; Catalog number sc-367), SIRT1 (Santa Cruz Biotechnology, CA, USA; Catalog number sc-15404) and PPAR-α (Santa Cruz Biotechnology, CA, USA; Catalog number sc-9000). Membranes were then incubated with the secondary antibodies against rabbit, mouse or goat (Bio-Rad Laboratory). Anti-GAPDH (Millipore Corporation, CA, USA; Catalog number MAB374) was used as internal control. Total protein expression of AMPK (Cell Signaling, Beverly, MA, USA; Catalog number 2532) and ACC (Cell Signaling, Beverly, MA, USA; Catalog number 3662) were used as internal controls for p-AMPK and p-ACC, respectively.
SIRT1 deacetylase activity
SIRT1 deacetylase activity was analyzed in nuclear extract from liver using the Cyclex SIRT1/Sir2 Deacetylase Fluorometric Assay Kit (Cyclex, Nagano, Japan; Catalog number CY-1151). The final reaction mixture (50 µL) contained 50 mM Tris-HCL (pH 8.8), 4 mM MgCL2, 0.5 mM DTT, 0.25 mA/mL Lysyl endopeptidase, 1 µM Trichostatin A, 200 µM NAD+ and 5 µL of nuclear extraction. The mixtures were mixed well and incubated for 10 min at room temperature and the reaction was stopped for adding 50 µL of 2X Stop Solution. The fluorescence intensity (ex. 340 nm, em. 460 nm) was measured (end-point) using a 1420-multilabel counter (Wallac Victor 2; Perkin-Elmer Life Sciences, Boston, MA) and normalized by the protein content.
Statistical analysis
The results are expressed as means ± SD. Comparison between groups were realized by student’s unpaired t-test. Statistical significance was assumed for P<0.05.
Results
General appearance and histological examination
General appearance and histological examination have been published in our previous study (7). Briefly, there were no significant differences on body (HFD =535.6±34.8 g vs. HFA =515.5±29.6 g, P>0.05) or liver (HFD =11.3±1.2 g vs. HFA =11.6±1.5 g, P>0.05) weight between HFD and HFA. The semi-quantitatively histological grading for steatosis showed there was no difference in steatosis (HFD =2.5±0.2 g vs. 3.0±0.4 g, P>0.05) between HFA and HFD groups. The semi-quantitatively examination of inflammatory foci showed that feeding rats with HFA led to a significantly higher degree of inflammation (HFD =1.7±0.4 g vs. 2.5±0.3 g, P>0.05) when compared to HFD.
HFA increased hepatic nuclear SIRT1 protein level while reduced SIRT1 activity
While there was no difference on hepatic total SIRT1 levels (whole cell extract) between two groups (data not shown), moderate alcohol consumption significantly increased the nuclear localization of SIRT1 protein compared to HFD (Figure 1A). However, nuclear SIRT1 deacetylase activity was significantly reduced in the HFA group as compared with that of the HFD (Figure 1B).
Figure 1.
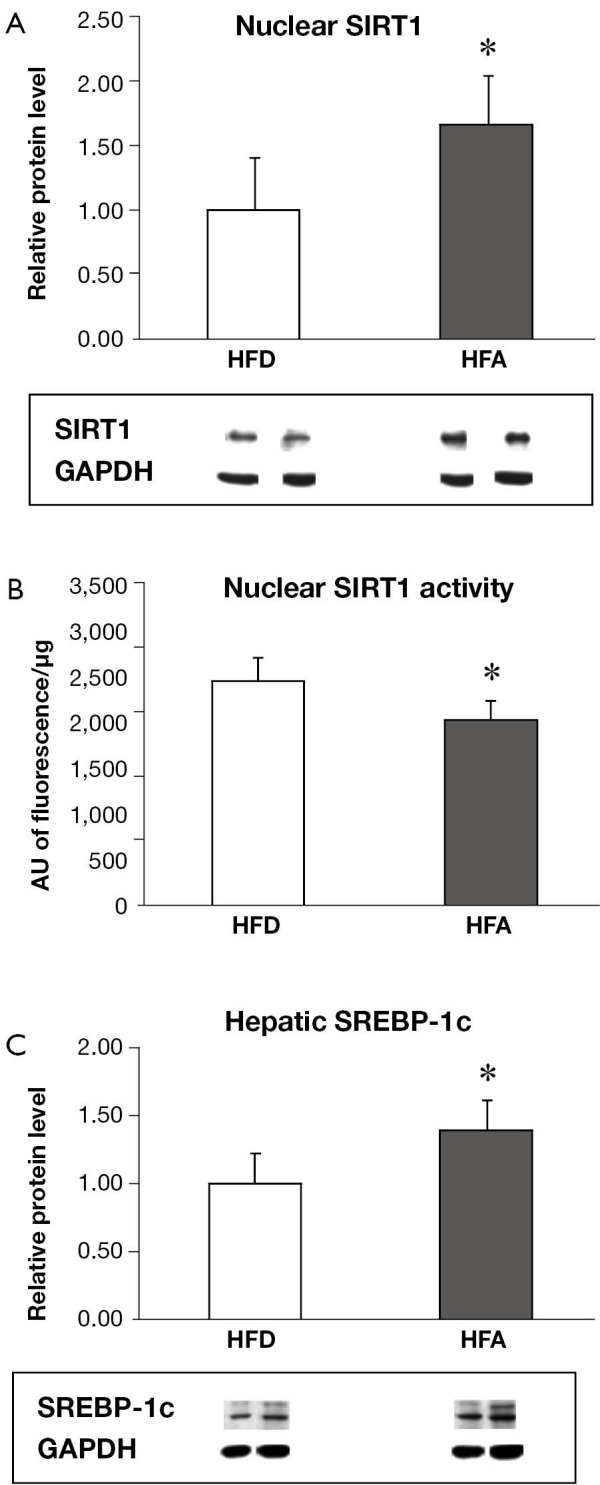
Hepatic nuclear SIRT1 relative protein expression. (A) And deacetylase activity; (B) And hepatic relative protein expression of SREBP-1c; (C) Means ± SD are shown. Statistical analyzes was performed by student’s unpaired t-test (*P<0.05 between groups). SIRT1, sirtuin 1; SREBP-1c, sterol regulatory element-binding protein-1c; HFD, high fat diet; HFA, high fat diet plus alcohol.
HFA upregulated hepatic genes responsible for fatty acid synthesis and influx while downregulated hepatic enzymes involved in triglyceride synthesis and fatty acids mitochondrial transport
HFA significantly upregulated the protein levels of SREBP-1c (Figure 1C) and the mRNA expressions of FAS and FATP (Figure 2), which are involved in fatty acid synthesis and influx, respectively. There was no difference between groups for mRNA expression of FAT/CD36, a plasma membrane fatty acid transport proteins (Figure 2). The mRNA expression of DGAT1 and 2, enzymes that are responsible for triglyceride synthesis, were significantly downregulated in HFA when compared with HFD (Figure 2). The mRNA level of CPT-I, an enzyme that mediates fatty acid transport into mitochondria for oxidation, was also downregulated in HFA (Figure 2).
Figure 2.

Hepatic mRNA relative expression of hepatic metabolism-related enzymes. Means ± SD are shown. Statistical analyzes was performed by student’s unpaired t-test (*P<0.05 between groups). FAT/CD36, plasma membrane protein fatty acid translocase CD36; FATP, fatty acid transport proteins; DGAT1 and DGAT2, diacylglycerol acyltransferase 1 and 2; FAS, fatty acid synthase; CPT-I, carnitine palmitoyltransferase I; HFD, high fat diet; HFA, high fat diet plus alcohol.
Plasma adiponectin and hepatic and plasma concentration of free fatty acid and TG
HFA group displayed a lower level (20% reduction) of plasma adiponectin than HFD (Figure 3), with the results almost reaching statistical significance (P=0.06). There were no significant differences in the plasma and hepatic FFA and TG concentrations between two groups (data no shown).
Figure 3.

Plasma adiponectin and hepatic relative protein expression of AdipoR1 and AdipoR2. Means ± SD are shown. Statistical analyzes was performed by student’s unpaired t-test (*P<0.05 between groups). AdipoR1, adiponectin receptor 1; AdipoR2, adiponectin receptor 2; HFD, high fat diet; HFA, high fat diet plus alcohol.
HFA downregulated the protein levels of hepatic AdipoR1 but not AdipoR2 and their downstream signaling
The protein expression of AdipoR1 was significantly reduced in HFA in relation to HFD, while AdipoR2 expression was similar between the two groups (Figure 3). There were no significant differences in the phosphorylation of AMPK and ACC, as well as PPAR-α protein expression between HFD and HFA groups (data not shown).
Discussion
In this study, moderate alcohol treatment significantly increased nuclear SIRT1 protein accumulation while decreased SIRT1 deacetylase activity (Figure 1). These results provide new insights in regards to the role of SIRT1 protein levels, its localization and its deacetylase activity with respect to the relationship between NASH and moderate alcohol consumption. SIRT1 is mainly localized in the nucleus (8). Although it has been recently shown that dietary alcohol can decrease the abundance of hepatic nuclear SIRT1 protein in ob/ob mice (12), our results indicated that SIRT1 activity, rather than nuclear SIRT1 protein levels, is more relevant in the progression of liver pathology induced by moderate alcohol feeding in rats with pre-existing NASH. SIRT1 has been demonstrated to negatively regulate inflammation and apoptosis (8-11). Thus, a reduction in SIRT1 activity induced by moderate alcohol intake may contribute to hepatic inflammation and apoptosis progression in rats with pre-exiting NASH, as reported previously (7).
The precise mechanism by which moderate alcohol consumption increase SIRT1 protein but decrease SIRT1 deacetylase activity is unclear. Ethanol is metabolized in the liver by cytosolic alcohol dehydrogenase and mitochondrial aldehyde dehydrogenase, both of which consume NAD+ as a cofactor (19). Since SIRT1 is a NAD+—dependent enzyme and hepatic ethanol metabolism can deplete NAD+ in hepatocytes, the NAD:NADH ratio and can contribute to the decreased SIRT1 deacetylase activity in the HFA group. It is reasonable to believe that this alcohol-induced reduction in SIRT1 activity may provide a positive feedback signal to increase SIRT1 protein expression, which explains the uncoupled relationship between the protein expression and the activity of SIRT1 observed in this study.
Interestingly, the alcohol-induced decrease in SIRT1 deacetylase activity was associated with the increased SREBP-1c protein expression. SIRT-1 has been shown to negatively modulate SREBP-1c expression, an important transcription factor that regulates de novo fatty acid synthesis in liver, thus leading to the decrease in transcriptional activities of SREBP-1c regulated genes (17). This was supported by our observation that the lower SIRT1 activity-linked SREBP-1c overexpression was associated with the mRNA upregulation of its targeted gene FAS (Figure 2). In parallel, moderate alcohol consumption also downregulated CPT-1 gene expression, an enzyme that mediates fatty acid transport into mitochondria for fatty acid oxidation, as well as DGAT1 and DGAT2, of which are enzymes that are responsible for triglyceride synthesis. This result helps to understand our previous observation that the moderate alcohol intake was not involved with steatosis progression (7). Together, these results suggest that moderate alcohol consumption may ultimately lead to further hepatic lipid metabolic alteration in pre-existing NASH.
Unexpectedly, moderate alcohol consumption was not associated with significant alteration in adiponectin/AdipoR axis in this model, except by downregulating AdipoR1. Adiponectin and its hepatic receptor 1 and 2 have been demonstrated to implicate the pathogenesis of NAFLD (20-26). Suppression of AdipoR1 and AdipoR2 expression in liver has been involved in hepatic lipid metabolism dysregulation by decreasing PPAR-α levels and phosphorylation of AMPK (22-24). Our results suggest that adiponectin/AdipoR axis may not be essential for the progression of moderate alcohol intake-associated steatohepatitis. This notion was supported by our observations that there were no significant changes on hepatic levels of AdipoR2, pAMPK, pACC and PPARα, as well as mRNA expression of adiponectin in adipose tissue (data not shown).
In summary, this present study provided further insights into the cellular mechanisms responsible for the adverse effects of moderate alcohol consumption on the progression of pre-existing NASH. The possible mechanisms could be associated with the suppression in nuclear SIRT1 deacetylase activity linked to alcohol consumption. Although moderate alcohol intake did not significantly alter hepatic lipid metabolism and Adiponectin/AdipoR signaling, further studies in evaluating the role of prolonged moderate alcohol consumption in NASH progression is needed.
Acknowledgements
The authors thank Kang-Quan Hu for her technical assistance. The work was supported by US Department of Agriculture grant 1950-51000-064S and grants of the Dietmar Hopp Foundation, the Manfred Lautenschläger Foundation, Heidelberg, Germany. Any opinions, findings, conclusion, or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect the view of the U.S. Department of Agriculture. A.F. Nascimento (Process number: FAPESP 2011/21664-9) and R.A.M Luvizotto (Process number: FAPESP 2011/22786-0) received post-doc fellowship from Sao Paulo Research Foudantion.
Disclosure: The authors declare no conflict of interest.
References
- 1.Angulo P.Nonalcoholic fatty liver disease. N Engl J Med 2002;346:1221-31 [DOI] [PubMed] [Google Scholar]
- 2.Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science 2011;332:1519-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegel AB, Zhu AX. Metabolic syndrome and hepatocellular carcinoma: two growing epidemics with a potential link. Cancer 2009;115:5651-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stefan N, Kantartzis K, Häring HU. Causes and metabolic consequences of fatty liver. Endocr Rev 2008;29:939-60 [DOI] [PubMed] [Google Scholar]
- 5.Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: the central role of nontriglyceride fatty acid metabolites. Hepatology 2010;52:774-88 [DOI] [PubMed] [Google Scholar]
- 6.Feldstein AE, Bailey SM. Emerging role of redox dysregulation in alcoholic and nonalcoholic fatty liver disease. Antioxid Redox Signal 2011;15:421-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y, Seitz HK, Wang XD. Moderate alcohol consumption aggravates high-fat diet induced steatohepatitis in rats. Alcohol Clin Exp Res 2010;34:567-73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol 2012;13:225-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joo HY, Woo SR, Shen YN, et al. SIRT1 interacts with and protects glyceraldehyde-3-phosphate dehydrogenase (GAPDH) from nuclear translocation: implications for cell survival after irradiation. Biochem Biophys Res Commun 2012;424:681-6 [DOI] [PubMed] [Google Scholar]
- 10.Gillum MP, Kotas ME, Erion DM, et al. SirT1 regulates adipose tissue inflammation. Diabetes 2011;60:3235-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schug TT, Li X. Sirtuin 1 in lipid metabolism and obesity. Ann Med 2011;43:198-211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Everitt H, Hu M, Ajmo JM, et al. Ethanol administration exacerbates the abnormalities in hepatic lipid oxidation in genetically obese mice. Am J Physiol Gastrointest Liver Physiol 2013;304:G38-47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liang X, Hu M, Rogers CQ, et al. Role of SIRT1-FoxO1 signaling in dietary saturated fat-dependent upregulation of liver adiponectin receptor 2 in ethanol-administered mice. Antioxid Redox Signal 2011;15:425-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen Z, Liang X, Rogers CQ, et al. Involvement of adiponectin-SIRT1-AMPK signaling in the protective action of rosiglitazone against alcoholic fatty liver in mice. Am J Physiol Gastrointest Liver Physiol 2010;298:G364-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.You M, Liang X, Ajmo JM, et al. Involvement of mammalian sirtuin 1 in the action of ethanol in the liver. Am J Physiol Gastrointest Liver Physiol 2008;294:G892-8 [DOI] [PubMed] [Google Scholar]
- 16.Lieber CS, Leo MA, Wang X, et al. Effect of chronic alcohol consumption on Hepatic SIRT1 and PGC-1alpha in rats. Biochem Biophys Res Commun 2008;370:44-8 [DOI] [PubMed] [Google Scholar]
- 17.You M, Cao Q, Liang X, et al. Mammalian sirtuin 1 is involved in the protective action of dietary saturated fat against alcoholic fatty liver in mice. J Nutr 2008;138:497-501 [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Ausman LM, Russell RM, et al. Increased apoptosis in high-fat diet-induced nonalcoholic steatohepatitis in rats is associated with c-Jun NH2-terminal kinase activation and elevated proapoptotic Bax. J Nutr 2008;138:1866-71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zakhari S, Li TK. Determinants of alcohol use and abuse: Impact of quantity and frequency patterns on liver disease. Hepatology 2007;46:2032-9 [DOI] [PubMed] [Google Scholar]
- 20.Polyzos SA, Kountouras J, Zavos C, et al. The role of adiponectin in the pathogenesis and treatment of non-alcoholic fatty liver disease. Diabetes Obes Metab 2010;12:365-83 [DOI] [PubMed] [Google Scholar]
- 21.Peng Y, Rideout D, Rakita S, et al. Downregulation of adiponectin/AdipoR2 is associated with steatohepatitis in obese mice. J Gastrointest Surg 2009;13:2043-9 [DOI] [PubMed] [Google Scholar]
- 22.Yamauchi T, Kamon J, Minokoshi Y, et al. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med 2002;8:1288-95 [DOI] [PubMed] [Google Scholar]
- 23.Yamauchi T, Kamon J, Ito Y, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 2003;423:762-9 [DOI] [PubMed] [Google Scholar]
- 24.Yamauchi T, Nio Y, Maki T, et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat Med 2007;13:332-9 [DOI] [PubMed] [Google Scholar]
- 25.Iwabu M, Yamauchi T, Okada-Iwabu M, et al. Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. Nature 2010;464:1313-9 [DOI] [PubMed] [Google Scholar]
- 26.Xu A, Wang Y, Keshaw H, et al. The fat-derived hormone adiponectin alleviates alcoholic and nonalcoholic fatty liver diseases in mice. J Clin Invest 2003;112:91-100 [DOI] [PMC free article] [PubMed] [Google Scholar]