

Abstract
Significance: Mechanical activation of reactive oxygen species (ROS) and reactive nitrogen species (RNS) occurs in striated muscle and affects Ca2+ signaling and contractile function. ROS/RNS signaling is tightly controlled, spatially compartmentalized, and source specific. Recent Advances: Here, we review the evidence that within the contracting myocyte, the trans-membrane protein NADPH oxidase 2 (Nox2) is the primary source of ROS generated during contraction. We also review a newly characterized signaling cascade in cardiac and skeletal muscle in which the microtubule network acts as a mechanotransduction element that activates Nox2-dependent ROS generation during mechanical stretch, a pathway termed X-ROS signaling. Critical Issues: In the heart, X-ROS acts locally and affects the sarcoplasmic reticulum (SR) Ca2+ release channels (ryanodine receptors) and tunes Ca2+ signaling during physiological behavior, but excessive X-ROS can promote Ca2+-dependent arrhythmias in pathology. In skeletal muscle, X-ROS sensitizes Ca2+-permeable sarcolemmal “transient receptor potential” channels, a pathway that is critical for sustaining SR load during repetitive contractions, but when in excess, it is maladaptive in diseases such as Duchenne Musclar dystrophy. Future Directions: New advances in ROS/RNS detection as well as molecular manipulation of signaling pathways will provide critical new mechanistic insights into the details of X-ROS signaling. These efforts will undoubtedly reveal new avenues for therapeutic intervention in the numerous diseases of striated muscle in which altered mechanoactivation of ROS/RNS production has been identified. Antioxid. Redox Signal. 20, 929–936.
Introduction
Reactive oxygen species (ROS) or reactive nitrogen species (RNS) modulate numerous biochemical processes through the targeted modification of specific protein residues. In striated muscle, contractile activity and/or stretch increases ROS/RNS signaling and modulates a host of biochemical processes, including glucose uptake, gene expression, calcium signaling, and contractility. In pathological conditions, ROS/RNS signaling excess or dysfunction contributes to contractile dysfunction and arrythmogensesis in the failing heart as well as contractile dysfunction and myopathy in skeletal muscle.
Much progress has been made in unraveling the sources and molecular targets of ROS/RNS in the cardiovascular and skeletal muscle systems (i.e., myocyte, endothelium, and vascular smooth muscle). It is obvious that extra-myocyte-generated ROS and RNS demonstrate paracrine signaling to myocytes, and several excellent reviews have been published that consider these pathways (12, 15, 40, 41, 55). Much less is known, however, about the mechanisms by which ROS/RNS is generated within the myocyte during contraction or stretch (34, 54). Here, we provide a brief review of the mechanosignaling during contractile activity or via stretch, the subsequent ROS/RNS production, and oxidative/nitrosative signaling within the striated muscle myocyte. These pathways are now being revealed with single-cell studies that are enabled by new methods and reagents, enhancing our understanding of how mechanosensing via ROS/RNS contributes to physiological and pathophysiological processes within striated muscle.
ROS/RNS Signaling in Contracting Muscle Cells
Striated muscle generates superoxide as the primary ROS species and NO as a parental species from which RNS are generated. Each arises from several sources, and the content of each is increased with contractile activity. Once generated, each leads to the formation of secondary ROS or RNS, each with specific signaling roles. A basic schematic is presented in Figure 1 with greater detail provided in the later text.
FIG. 1.

Schematic representation of RNS and ROS production on the striated myocyte. See main text for details. ROS, reactive oxygen species; RNS, reactive nitrogen species; PLA2, phospholipase A2; TRP, transient receptor potential; RyR, ryanodine receptor; DHPR, dihydropyridine receptor; SOD, superoxide dismutase; XO, xanthine oxidase; jSR, junctional sarcoplasmic reticulum; t-tubule, transverse tubule; nNOS, neuronal nitric oxide synthase; NO, nitric oxide; ONOO−, peroxynitrite; Nox2, NADPH oxidase 2. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Superoxide
Superoxide is free-radical oxygen that is generated by the addition of a single electron to ground-state oxygen (O2•−) (73). Superoxide is a highly reactive, unstable species that is rapidly dismuted by superoxide dismutase (Cu/ZnSOD) to hydrogen peroxide (H2O2), a weaker but more stable reactive oxidant which is not a “free radical,” as it does not contain an unpaired electron. H2O2 is highly diffusible within and between cells, activates multiple signaling pathways, and is decomposed by either catalase or glutathione peroxidase to water and oxygen (52, 71). H2O2 is also the substrate for the extremely reactive hydroxyl radical (OH) that is generated via the Fenton Reaction with divalent metal cations, mainly ferrous iron (Fe2+) (35).
Within proteins, sulfhydryl groups on cysteine molecules are preferential targets for oxidation or the formation of disulfide bonds. Here, the sulfhydryl (–SH) group may initially be reversibly oxidized to a sulfenic acid (protein–SOH). This group can then be further oxidized to a sulfinic acid (protein-SOH+(O•)→protein SO2H) or irreversibly oxidized to sulfonic acid (protein-SO2H+(O•)→protein SO3H). The OH• can then form irreversibly oxidized protein sulfhydryl groups that are indicative of protein damage.
Mitochondria
The production of superoxide occurs in many locations within the cell. The most studied is the mitochondria where superoxide is produced within the electron transport chain (ETC) (3). Earlier studies suggested that 2%–5% of the total oxygen consumed underwent one electron reduction in the ETC (43); therefore, as mitochondrial activity increases >50-fold with aerobicaly sustained contractile activity, a >50-fold increase in activity-dependent ROS generation could theoretically be accounted for by mitochondrial sources (75). Recent data, however, have led to a downward revision in both the magnitude of ROS produced during steady-state contraction (approximately two to fourfold increase) and the percentage of the ROS generated by electron flow through the ETC (<1%) (66). It is now thought that little superoxide is contributed by the mitochiondria during steady-state contractions [see Powers and Jackson (52) for review]. This estimate is in line with our recent finding that the redox state of the mitochondria (a surrogate measure of ROS generation) is unchanged during steady-state contractions (47) as well as with recent direct measures of ROS within the mitochondria of contractile muscle fibers (61).
Xanthine oxidase
The enzyme xanthine oxidase (XO) has also been shown to produce superoxide in response to contractile activity in rodent muscle (69, 73). Available evidence, however, supports either a vascular source of XO-generated superoxide due to contractile shear stress or an increase in XO activity secondary to anaerobic metabolism that increases the availability of XO substrates (20, 21, 69). In either case, neither can account for steady-state ROS production during aerobic metabolism, and the latter supports only a mechanism that increases superoxide after exhaustive or fatiguing contractions.
Phospholipase A2
Superoxide anion is produced by phospholipase A2 (PLA2)-dependent processes (53). A Ca2+-independent PLA2 mechanism may act under resting conditions (22), whereas a Ca2+-dependent PLA2 may contribute to ROS production during contraction when cytosolic [Ca2+] is elevated (19, 33, 60). In both cases, PLA2 is believed to act by arachidonic acid-dependent activation of NADPH oxidase (24, 70).
NADPH oxidase
Superoxide is generated by NADPH oxidase (nicotinamide adenine dinucleotide phosphate-oxidase; Nox), a multimeric enzyme complex that generates superoxide by transferring electrons from NADPH to oxygen. Several Nox isoforms are expressed in striated muscle and located within the sarcoplasmic reticulum (SR), the sarcolemma, and the transverse tubules (t-tubules) (63, 65).
The Nox family of oxidases comprises seven members, each of which is based on a distinct core catalytic subunit. Striated muscle cells express NADPH oxidase 2 (Nox2) and 4, each of which bind p22phox, a small subunit that is essential for enzyme activity (65). Nox4 is constitutively active and does not require association with regulatory subunits, with regulation considered as occurring mainly by changes in expression level. Therefore, Nox4 likely contributes to the basal rate of ROS production in the myocyte. In contrast, Nox2 (also known as gp91phox) is activated by specific agonists (e.g., G-protein-coupled receptor agonists such as angiotensin II, growth factors, and cytokines) and mechanical/contractile stress, which induce the association of regulatory subunits (p47phox, p67phox, p40phox, and Rac1) and activation of the enzyme (1, 63).
Recent work in striated muscle by our group and others has implicated the Nox pathway as the major source of superoxide ROS during repetitive contraction in both skeletal and cardiac muscle cells (47, 61). ROS production with contraction has been shown to arise from the sarcolemma and t-tubules by activation of transmembrane Nox2 (14, 38, 57, 61). It was further suggested that Nox2-generated superoxide then promoted Nox4 activation within the mitochondria and generated ROS that was released into the cytosol (80, 81); however, experiments which directly address this hypothesis have not supported that idea (61).
Nitric oxide
In striated mucle cells, nitric oxide (NO) is generated by the nitric oxide synthases (NOSs). These enzymes are the products of three different genes: neuronal nitric oxide synthase (nNOS, or NOS1), endothelial nitric oxide synthase (eNOS, or NOS3), and inducible nitric oxide synthase (iNOS, or NOS2) (45, 68). All three enzymes catalyze the production of NO from the precursor L-arginine (15, 68). nNOS and eNOS contain a calmodulin (CaM)-binding domain, making them Ca2+ sensitive and therefore responsive to contractile activity (68, 71). iNOS contains constutively bound CaM at resting levels of Ca2+, and its activity is inhibited by association with kalirin or NAP110 (68). All NOSs generate NO and S-nitrosothiols (SNO) (68). NO primarily functions to stimulate soluble guanylate cyclase to produce cyclic guanosine monophosphate (cGMP), which has numerous downstream targets through cGMP-dependent kinases and phosphodiesterases (9). NO also readily reacts with superoxide to form perioxynitrite (NO•+O2•−→ONOO−), which, in turn, reacts potently with thiol groups to S-nitrosylate-specific protein targets. The formation of peroxynitrite also decreases the bioavailability of NO and superoxide, thus modifying the redox balance in the myocyte (36, 37).
X-ROS Signaling: Mechanotransduction-Activated Nox2-Dependent ROS Production
The concept of cellular “tensegrity” proposes that the stabilized microtubule (MT) network resists mechanical perturbations in cells and in doing so acts as a mechanotransducer (26, 67, 77, 78). Experiments and methods used to mechanically manipulate enzymatically isolated heart cells have been pioneered by several groups (7, 17, 42) and have provided a means to assess the “tensegrity” concepts in muscle cells. Our initial hypothesis that the MT network was a mechanosignaling element in the heart arose from elegant work in the literature (7, 8) and from experiments between our group and that of G. Iribe and P. Kohl (29). As a demonstration of our initial findings, Figure 2 shows a single isolated cardiomyocyte loaded with a calcium indicator dye to monitor Ca2+ sparks (local Ca2+ release from the ryanodine receptor; RyR2) and attached with carbon filaments that allowed cell stretch to be applied. Having demonstrated that an acute stretch of the myocyte caused a rapid increase of spark occurrence only in the stretched portion of the cell, we reasoned that the signal for stretch-dependent activation of the RyR should be rapid, reversible, and highly localized.
FIG. 2.

Time course of Fluo-4 signal intensity in a ventricular cardiomyocyte subjected to a half-cell stretch. The back panel shows an image of a cell, averaged from 10 confocal XY scans before the application of stretch. The low intensity areas indicate points of CF attachment at resting length (scale bar=20 μm). The front panel represents a pseudo color, pseudo 3D rendering of XT images temporally from back to front. Note that the shading across the image and the CF's fluorescence troughs indicate the stretch of one-half of the cell (right half) and that Ca2+ spark signals arise only in the stretched portion of the cell. This figure was used with permission from Iribe et al. (30). CF, carbon filament. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Since we had evidence from electron microscopy that MT filaments interdigitate with t-tubule/SR junctions (29), our initial hypothesis speculated on an interaction of MTs with clusters of calcium channels that somehow altered channel sensitivity in a stretch-dependent fashion. To support this hypothesis, we demonstrated that MT destabilization with colchicine was solely sufficient to abrogate the stretch-dependent activation of local calcium signals (i.e., Ca2+ sparks) in cardiomyocytes. Further experimentation, however, led us to refine our hypothesis.
Working with the knowledge that Nox2 is activated by contraction (i.e, contraction/compression followed by relaxation/stretch), osmotic stress (stretch or compression (31, 46, 64, 74)), or stretch (49), we focused our attention on a hypothesis by which an MT-dependent mechanotransduction pathway could translate a mechanical signal to Nox2 to activate ROS. To address this question within our own laboratories, we developed new tools and methods (38, 57), enabling us to establish a model of stretch-activated mechanotransduction in striated muscle that avoids potential confounders of contraction, membrane-damaging force (eccentric injury), or non-physiologic stress such as osmotic shock.
Using these new methods coupled with high-speed confocal fluorescence imaging approaches, we revealed that in cardiomyocytes loaded with the ROS indicator dichlorofluorescein (DCF), a brief, acute physiologic stretch elicited a burst of ROS production (Fig. 3). Since DCF is a non-specific ROS indicator, we used selective inhibitors and genetic approaches to reveal that the MT network acted as a mechanotransducer to activate Nox2-dependent ROS generation, a pathway termed X-ROS signaling (38, 57). In heart cells, X-ROS leads to post-translational modification of RyR2s either directly (56, 57) or, possibly, indirectly through a second messenger such as Ca2+-CaM kinase II (76). Regardless of the mechanism, X-ROS increases the sensitivity of RyR2s to [Ca2+]i and, thus, promotes the fidelity of excitation-contraction coupling (Fig. 4).
FIG. 3.

Kinetic response of the ROS signal generated during a brief cardiomyocyte stretch. Data are stylized based on experiments of a single cardiomyocyte loaded with the ROS indicator DCF and subjected to an 8% sarcomere length excursion (black trace) (57). DCF fluorescence intensity (blue trace) rises rapidly on the initiation of stretch and terminates rapidly with the relaxation of the stretch. The new level of DCF fluorescence after stretch reflects DCF as a non-reversible ROS indicator. The rate of ROS production estimated by the first derivative of the DCF fluorescence profile (green trace) indicates a burst of ROS production at the initiation of stretch that decays monotonically until relaxation. DCF, dichlorofluorescein. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
FIG. 4.
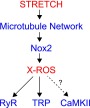
Graphical representation of the X-ROS activation in striated muscle. The stress of mechanical stretch is transmitted through the filamentous microtubule network to Nox2, which generates superoxide and secondary ROS species (see review). We have demonstrated that the downstream targets of Nox2-derived ROS (i.e., X-ROS) result in sensitization of RyR's in heart and TRP channels in skeletal muscle. Other potential targets include CaMKII (76). CaMKII, calmodulin kinase II. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Many of the features of X-ROS signaling in the heart are also found in skeletal muscle (38, 51), but the signaling involves additional molecular components (38). One prominent new component in skeletal muscle is a calcium-conducting sarcolemmal “transient receptor potential” (TRP) channels (2, 6, 18, 48, 82) whose opening is enhanced by Nox2-derived ROS (Fig. 4). This signaling system is an important pathological component in Duchenne muscular dystrophy, where an increase in MT network density and Nox2 expression leads to a detrimental enhancement of X-ROS signaling (38). In addition, whether X-ROS may affect cardiac TRP channels or skeletal RyR1 under dystrophic conditions or with dynamic stretching paradigms (58) remains to be determined.
One striking finding was the speed at which X-ROS production was enhanced with mechanical stretch in muscle (Fig. 3), a finding that likely was due to specialization within the signaling pathway. It is well established that Nox2 and its regulatory subunits are localized to the t-tubule membranes in the heart (57, 62) and skeletal muscle (25). MTs bisect this junctional space (29) and may interact with the Nox2 subunit Rac (4). Since ROS are short-lived species that are confined to interaction within their immediate vicinity, this tight spatial organization is likely critical for the rapid and controlled signaling reported here (38, 57); signaling fidelity is necessary for mechanoactivation of X-ROS during the contraction cycle.
Nox2 activation also typically requires phosphorylation and recruitment of multiple cytosolic regulatory subunits to its transmembrane catalytic subunit (gp91phox). The rapid nature of X-ROS signaling likely necessitates pre-assembly of the Nox2 complex to minimize steps that activate ROS production. In support of this, Nox2 subunits may tether to sarcolemmal membranes in hetero-multimeric complexes primed for rapid activation (50). With pre-assembly, mechanoactivation of this complex would then require an MT-dependent “switch” for activation. Rac is an MT-associated protein that associates with gp91phox separately from other regulatory subunits (50). Rac1 is a required subunit for Nox2 activation, and we have demonstrated that inhibition of Rac1 blocks stretch-activated X-ROS signaling (57).
Based on our results, we propose a minimal model in which MT-associated Rac1 is a critical activator of a pre-assembled Nox2 complex that generates superoxide during mechanical stress of contraction. While being exciting, this model requires validation and expansion in many key areas. Experimental demonstration of stretch-dependent Rac1 recruitment to gp91phox in an MT-dependent fashion is needed. In fact, no mechanistic detail is available that explains how the MT network acts as a dynamic mechanotransduction element in the muscle. Advances in other areas of MT biology will need to be exploited and adapted to address these key issues in striated muscle.
Future Directions
Most of these insights on contraction-generated ROS/RNS have been made possible by the real-time detection of these species in single, intact living cells that were either electrically activated to elicit contraction or mechanically stretched to assay passive mechanical responses. The combination of mechanical control of length and the measure of contractility in single isolated myocytes was pioneered by several groups (8, 10, 16, 27, 28, 29, 42, 79) and recently advanced in our laboratories (38, 57). While the approaches are still being refined, the availability of new commercial systems (38, 57) will undoubtedly increase the pace of research in this area.
Our laboratory and many other laboratories have used DAF-FM to monitor RNS/NO, and DCF (or CMDCF) to monitor ROS (i.e., superoxide) in single-cell preparations (33, 55, 59). While DAF is fairly species specific, DCF is known to be sensitive to a number of RNS and ROS species, including H2O2, superoxide, and peroxynitrite, and, thus, requires careful experimentation and controls (56). Each of these probes, however, also suffers from time-dependent mitochondrial compartmentalization (72), light-induced oxidation of the dye (5), and limited signal:noise, all of which complicates the interpretation of results. This represents a limitation in the field, and there is a great need for more selective and rapidly responsive indicators.
New advances in ROS/RNS detection are now available with probes to permit the monitoring of ROS in specific cell compartments. For example, redox-sensitive green fluorescent protein (ro-GFP) (13) and glutaredoxin-tagged ro-GFP (23) allow the monitoring of local subcellular redox potentials. In addition, genetically encoded probes which are specific for H2O2 (Hy-Per) that can be targeted to cytosol, mitochondria, or nuclei are now available (44). However, it remains to be determined whether these genetically encoded sensors are suitable for experiments that demand the highest temporal resolution.
An important goal for this research is the identification of specific targets for therapeutic intervention within the ROS/RNS pathway. In diseases of both the heart (i.e., cardiomyopathy, arrythmia) and skeletal muscle (i.e., muscular dystrophy), pre-clinical studies targeting either the MT cytoskeketon (i.e., MT network destabilization) (11, 32, 38, 39) or Nox-dependent ROS production (i.e., non-specific Nox inhibition) (38, 83, 84) have shown benefit and, thus, provide proof of concept for these approaches. The use of new techniques, reagents, and models will undoubtedly reveal new mechanistic details on the mechanoactivation of ROS/RNS production and oxidative/nitrosative signaling that will translate to new more specific targets for therapeutic intervention. For example, can ROS production be reduced by specifically targeting Nox2? Can the mechanisms that are responsible for the increase in MT density be targeted, thus reducing the mechanoactivation of Nox2? One thing is clear, the efforts to understand the mechanoactivation of ROS/RNS production have yielded, and will continue to yield, exciting and important rewards.
Abbreviations Used
- Ca2+
calcium
- CaM
calmodulin
- CF
carbon filament
- cGMP
cyclic guanosine monophosphate
- DCF
dichlorofluorescein
- DHPR
dihydropyridine receptor
- eNOS
endothelial nitric oxide synthase
- ETC
electron transport chain
- H2O2
hydrogen peroxide
- iNOS
inducible nitric oxide synthase
- jSR
junctional sarcoplasmic reticulum
- MT
microtubule
- nNOS
neuronal nitric oxide synthase
- NO
nitric oxide
- Nox2
NADPH oxidase 2
- ONOO−
peroxynitrite
- PLA2
phospholipase A2
- RNS
reactive nitrogen species
- ro-GFP
redox-sensitive green fluorescent protein
- ROS
reactive oxygen species
- RyR
ryanodine receptor
- SOD
superoxide dismutase
- SR
sarcoplasmic reticulum
- TRP
transient receptor potential
- t-tubule
transverse tubule
- XO
xanthine oxidase
- X-ROS signaling
Nox2 dependent ROS generation during mechanical stretch; a pathway termed X-ROS signaling
Acknowledgments
This work was supported by NIH grants R01 HL106059, P01 HL67849, R01 HL36974, RC2 NR011968, 1R01HL105239, and U54HD053177. B.L.P. was partly supported by the Training Program in Cardiovascular Cell Biology (T32 HL072751) and K99 HL114879. The research leading to these results has received funding from the European Community's Seventh Framework Program FP7/2007–2013 under grant agreement No. HEALTH-F2-2009-241526, EUTrigTreat. The authors would like to acknowledge support from Leducq North American-European Atrial Fibrillation Research Alliance.
References
- 1.Akki A, Zhang M, Murdoch C, Brewer A, and Shah AM. NADPH oxidase signaling and cardiac myocyte function. J Mol Cell Cardiol 47: 15–22, 2009 [DOI] [PubMed] [Google Scholar]
- 2.Allen DG, Gervasio OL, Yeung EW, and Whitehead NP. Calcium and the damage pathways in muscular dystrophy. Can J Physiol Pharmacol 88: 83–91, 2010 [DOI] [PubMed] [Google Scholar]
- 3.Bayeva M. and Ardehali H. Mitochondrial dysfunction and oxidative damage to sarcomeric proteins. Curr Hypertens Rep 12: 426–432, 2010 [DOI] [PubMed] [Google Scholar]
- 4.Best A, Ahmed S, Kozma R, and Lim L. The Ras-related GTPase Rac1 binds tubulin. J Biol Chem 271: 3756–3762, 1996 [DOI] [PubMed] [Google Scholar]
- 5.Bilski P, Belanger AG, and Chignell CF. Photosensitized oxidation of 2′,7′-dichlorofluorescin: singlet oxygen does not contribute to the formation of fluorescent oxidation product 2′,7′-dichlorofluorescein. Free Radic Biol Med 33: 938–946, 2002 [DOI] [PubMed] [Google Scholar]
- 6.Brinkmeier H. TRP channels in skeletal muscle: gene expression, function and implications for disease. Adv Exp Med Biol 704: 749–758, 2011 [DOI] [PubMed] [Google Scholar]
- 7.Calaghan SC, Belus A, and White E. Do stretch-induced changes in intracellular calcium modify the electrical activity of cardiac muscle? Prog Biophys Mol Biol 82: 81–95, 2003 [DOI] [PubMed] [Google Scholar]
- 8.Calaghan SC, Le Guennec JY, and White E. Cytoskeletal modulation of electrical and mechanical activity in cardiac myocytes. Prog Biophys Mol Biol 84: 29–59, 2004 [DOI] [PubMed] [Google Scholar]
- 9.Carnicer R, Crabtree MJ, Sivakumaran V, Casadei B, and Kass DA. Nitric oxide synthases in heart failure. Antioxid Redox Signal 18: 1078–1099, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cazorla O, Le Guennec JY, and White E. Length-tension relationships of sub-epicardial and sub-endocardial single ventricular myocytes from rat and ferret hearts. J Mol Cell Cardiol 32: 735–744, 2000 [DOI] [PubMed] [Google Scholar]
- 11.Cheng G, Kasiganesan H, Baicu CF, Wallenborn JG, Kuppuswamy D, and Cooper G. Cytoskeletal role in protection of the failing heart by beta-adrenergic blockade. Am J Physiol Heart Circ Physiol 302: H675–H687, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donoso P, Sanchez G, Bull R, and Hidalgo C. Modulation of cardiac ryanodine receptor activity by ROS and RNS. Front Biosci 16: 553–567, 2011 [DOI] [PubMed] [Google Scholar]
- 13.Dooley CT, Dore TM, Hanson GT, Jackson WC, Remington SJ, and Tsien RY. Imaging dynamic redox changes in mammalian cells with green fluorescent protein indicators. J Biol Chem 279: 22284–22293, 2004 [DOI] [PubMed] [Google Scholar]
- 14.Espinosa A, Leiva A, Pena M, Muller M, Debandi A, Hidalgo C, Carrasco MA, and Jaimovich E. Myotube depolarization generates reactive oxygen species through NAD(P)H oxidase; ROS-elicited Ca2+ stimulates ERK, CREB, early genes. J Cell Physiol 209: 379–388, 2006 [DOI] [PubMed] [Google Scholar]
- 15.Ferreira LF. and Reid MB. Muscle-derived ROS and thiol regulation in muscle fatigue. J Appl Physiol 104: 853–860, 2008 [DOI] [PubMed] [Google Scholar]
- 16.Gannier F, White E, Garnier, and Le Guennec JY. A possible mechanism for large stretch-induced increase in [Ca2+]i in isolated guinea-pig ventricular myocytes. Cardiovasc Res 32: 158–167, 1996 [PubMed] [Google Scholar]
- 17.Gannier F, White E, Lacampagne A, Garnier D, and Le Guennec JY. Streptomycin reverses a large stretch induced increases in [Ca2+]i in isolated guinea pig ventricular myocytes. Cardiovasc Res 28: 1193–1198, 1994 [DOI] [PubMed] [Google Scholar]
- 18.Gervasio OL, Whitehead NP, Yeung EW, Phillips WD, and Allen DG. TRPC1 binds to caveolin-3 and is regulated by Src kinase - role in Duchenne muscular dystrophy. J Cell Sci 121: 2246–2255, 2008 [DOI] [PubMed] [Google Scholar]
- 19.Gissel H. The role of Ca2+ in muscle cell damage. Ann N Y Acad Sci 1066: 166–180, 2005 [DOI] [PubMed] [Google Scholar]
- 20.Gomez-Cabrera MC, Borras C, Pallardo FV, Sastre J, Ji LL, and Vina J. Decreasing xanthine oxidase-mediated oxidative stress prevents useful cellular adaptations to exercise in rats. J Physiol 567: 113–120, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gomez-Cabrera MC, Close GL, Kayani A, McArdle A, Vina J, and Jackson MJ. Effect of xanthine oxidase-generated extracellular superoxide on skeletal muscle force generation. Am J Physiol Regul Integr Comp Physiol 298: R2–R8, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gong MC, Arbogast S, Guo Z, Mathenia J, Su W, and Reid MB. Calcium-independent phospholipase A2 modulates cytosolic oxidant activity and contractile function in murine skeletal muscle cells. J Appl Physiol 100: 399–405, 2006 [DOI] [PubMed] [Google Scholar]
- 23.Gutscher M, Pauleau AL, Marty L, Brach T, Wabnitz GH, Samstag Y, Meyer AJ, and Dick TP. Real-time imaging of the intracellular glutathione redox potential. Nat Methods 5: 553–559, 2008 [DOI] [PubMed] [Google Scholar]
- 24.Henderson LM, Chappell JB, and Jones OT. Superoxide generation is inhibited by phospholipase A2 inhibitors. Role for phospholipase A2 in the activation of the NADPH oxidase. Biochem J 264: 249–255, 1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hidalgo C, Sanchez G, Barrientos G, and Aracena-Parks P. A transverse tubule NADPH oxidase activity stimulates calcium release from isolated triads via ryanodine receptor type 1 S -glutathionylation. J Biol Chem 281: 26473–26482, 2006 [DOI] [PubMed] [Google Scholar]
- 26.Ingber DE. Tensegrity and mechanotransduction. J Bodyw Mov Ther 12: 198–200, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iribe G, Helmes M, and Kohl P. Force-length relations in isolated intact cardiomyocytes subjected to dynamic changes in mechanical load. Am J Physiol Heart Circ Physiol 292: H1487–H1497, 2007 [DOI] [PubMed] [Google Scholar]
- 28.Iribe G. and Kohl P. Axial stretch enhances sarcoplasmic reticulum Ca2+ leak and cellular Ca2+ reuptake in guinea pig ventricular myocytes: experiments and models. Prog Biophys Mol Biol 97: 298–311, 2008 [DOI] [PubMed] [Google Scholar]
- 29.Iribe G, Ward CW, Camelliti P, Bollensdorff C, Mason F, Burton RA, Garny A, Morphew MK, Hoenger A, Lederer WJ, and Kohl P. Axial stretch of rat single ventricular cardiomyocytes causes an acute and transient increase in Ca2+ spark rate. Circ Res 104: 787–795, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.This reference has been deleted.
- 31.Isaeva EV, Shkryl VM, and Shirokova N. Mitochondrial redox state and Ca2+ sparks in permeabilized mammalian skeletal muscle. J Physiol 565: 855–872, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ishibashi Y, Takahashi M, Isomatsu Y, Qiao F, Iijima Y, Shiraishi H, Simsic JM, Baicu CF, Robbins J, Zile MR, and Cooper Gt. Role of microtubules versus myosin heavy chain isoforms in contractile dysfunction of hypertrophied murine cardiocytes. Am J Physiol Heart Circ Physiol 285: H1270–H1285, 2003 [DOI] [PubMed] [Google Scholar]
- 33.Jackson MJ. Control of reactive oxygen species production in contracting skeletal muscle. Antioxid Redox Signal 15: 2477–2486, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jackson MJ. and McArdle A. Age-related changes in skeletal muscle reactive oxygen species generation and adaptive responses to reactive oxygen species. J Physiol 589: 2139–2145, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jackson MJ, Pye D, and Palomero J. The production of reactive oxygen and nitrogen species by skeletal muscle. J Appl Physiol 102: 1664–1670, 2007 [DOI] [PubMed] [Google Scholar]
- 36.Judge DP, Kass DA, Thompson WR, and Wagner KR. Pathophysiology and therapy of cardiac dysfunction in duchenne muscular dystrophy. Am J Cardiovasc Drugs 11: 287–294, 2011 [DOI] [PubMed] [Google Scholar]
- 37.Kass DA, Takimoto E, Nagayama T, and Champion HC. Phosphodiesterase regulation of nitric oxide signaling. Cardiovasc Res 75: 303–314, 2007 [DOI] [PubMed] [Google Scholar]
- 38.Khairallah RJ, Shi G, Sbrana F, Prosser BL, Borroto C, Mazaitis MJ, Hoffman EP, Mahurkar A, Sachs F, Sun Y, Chen YW, Raiteri R, Lederer WJ, Dorsey SG, and Ward CW. Microtubules underlie dysfunction in duchenne muscular dystrophy. Sci Signal 5: ra56, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koide M, Hamawaki M, Narishige T, Sato H, Nemoto S, DeFreyte G, Zile MR, Cooper GI, and Carabello BA. Microtubule depolymerization normalizes in vivo myocardial contractile function in dogs with pressure-overload left ventricular hypertrophy. Circulation 102: 1045–1052, 2000 [DOI] [PubMed] [Google Scholar]
- 40.Kuster GM, Hauselmann SP, Rosc-Schluter BI, Lorenz V, and Pfister O. Reactive oxygen/nitrogen species and the myocardial cell homeostasis: an ambiguous relationship. Antioxid Redox Signal 13: 1899–1910, 2010 [DOI] [PubMed] [Google Scholar]
- 41.Lamb GD. and Westerblad H. Acute effects of reactive oxygen and nitrogen species on the contractile function of skeletal muscle. J Physiol 589: 2119–2127, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Le Guennec JY, White E, Gannier F, Argibay JA, and Garnier D. Stretch-induced increase of resting intracellular calcium concentration in single guinea-pig ventricular myocytes. Exp Physiol 76: 975–978, 1991 [DOI] [PubMed] [Google Scholar]
- 43.Loschen G, Azzi A, Richter C, and Flohe L. Superoxide radicals as precursors of mitochondrial hydrogen peroxide. FEBS Lett 42: 68–72, 1974 [DOI] [PubMed] [Google Scholar]
- 44.Malinouski M, Zhou Y, Belousov VV, Hatfield DL, and Gladyshev VN. Hydrogen peroxide probes directed to different cellular compartments. PLoS One 6: e14564, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malinski T. Understanding nitric oxide physiology in the heart: a nanomedical approach. Am J Cardiol 96: 13i–24i, 2005 [DOI] [PubMed] [Google Scholar]
- 46.Martins AS, Shkryl VM, Nowycky MC, and Shirokova N. Reactive oxygen species contribute to Ca2+ signals produced by osmotic stress in mouse skeletal muscle fibres. J Physiol 586: 197–210, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Michaelson LP, Shi G, Ward CW, and Rodney GG. Mitochondrial redox potential during contraction in single intact muscle fibers. Muscle Nerve 42: 522–529, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Millay DP, Goonasekera SA, Sargent MA, Maillet M, Aronow BJ, and Molkentin JD. Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc Natl Acad Sci U S A 106: 19023–19028, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murdoch CE, Zhang M, Cave AC, and Shah AM. NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc Res 71: 208–215, 2006 [DOI] [PubMed] [Google Scholar]
- 50.Nauseef WM. Assembly of the phagocyte NADPH oxidase. Histochem Cell Biol 122: 277–291, 2004 [DOI] [PubMed] [Google Scholar]
- 51.Palomero J, Pye D, Kabayo T, and Jackson MJ. Effect of passive stretch on intracellular nitric oxide and superoxide activities in single skeletal muscle fibres: influence of ageing. Free Radic Res 46: 30–40, 2012 [DOI] [PubMed] [Google Scholar]
- 52.Powers SK. and Jackson MJ. Exercise-induced oxidative stress: cellular mechanisms and impact on muscle force production. Physiol Rev 88: 1243–1276, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Powers SK, Nelson WB, and Hudson MB. Exercise-induced oxidative stress in humans: cause and consequences. Free Radic Biol Med 51: 942–950, 2011 [DOI] [PubMed] [Google Scholar]
- 54.Powers SK, Smuder AJ, and Judge AR. Oxidative stress and disuse muscle atrophy: cause or consequence? Curr Opin Clin Nutr Metab Care 15: 240–245, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Powers SK, Talbert EE, and Adhihetty PJ. Reactive oxygen and nitrogen species as intracellular signals in skeletal muscle. J Physiol 589: 2129–2138, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Prosser BL, Khairallah RJ, Ziman AP, Ward CW, and Lederer WJ. X-ROS signaling in heart and skeletal muscle: stretch-dependent local ROS regulates [Ca(2+)](i). J Mol Cell Cardiol 58: 172–181, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Prosser BL, Ward CW, and Lederer WJ. X-ROS signaling: rapid mechano-chemo transduction in heart. Science 333: 1440–1445, 2011 [DOI] [PubMed] [Google Scholar]
- 58.Prosser BL, Ward CW, and Lederer WJ. X-ROS signaling is enhanced and graded by cyclic cardiomyocyte stretch. Cardiovasc Res 98: 307–314, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Reid MB. Free radicals and muscle fatigue: of ROS, canaries, and the IOC. Free Radic Biol Med 44: 169–179, 2008 [DOI] [PubMed] [Google Scholar]
- 60.Russell ST, Eley H, and Tisdale MJ. Role of reactive oxygen species in protein degradation in murine myotubes induced by proteolysis-inducing factor and angiotensin II. Cell Signal 19: 1797–1806, 2007 [DOI] [PubMed] [Google Scholar]
- 61.Sakellariou G, Vasilaki A, Palomero J, Kayani A, Zibrik L, McArdle A, and Jackson MJ. Studies of mitochondrial and non-mitochondrial sources implicate NADPH oxidase(s) in the increased skeletal muscle superoxide generation that occurs during contractile activity. Antioxid Redox Signal 18: 603–621, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sanchez G, Escobar M, Pedrozo Z, Macho P, Domenech R, Hartel S, Hidalgo C, and Donoso P. Exercise and tachycardia increase NADPH oxidase and ryanodine receptor-2 activity: possible role in cardioprotection. Cardiovasc Res 77: 380–386, 2008 [DOI] [PubMed] [Google Scholar]
- 63.Santos CX, Anilkumar N, Zhang M, Brewer AC, and Shah AM. Redox signaling in cardiac myocytes. Free Radic Biol Med 50: 777–793, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shkryl VM, Martins AS, Ullrich ND, Nowycky MC, Niggli E, and Shirokova N. Reciprocal amplification of ROS and Ca(2+) signals in stressed mdx dystrophic skeletal muscle fibers. Pflugers Arch 458: 915–928, 2009 [DOI] [PubMed] [Google Scholar]
- 65.Sirker A, Zhang M, and Shah AM. NADPH oxidases in cardiovascular disease: insights from in vivo models and clinical studies. Basic Res Cardiol 106: 735–747, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.St-Pierre J, Buckingham JA, Roebuck SJ, and Brand MD. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem 277: 44784–44790, 2002 [DOI] [PubMed] [Google Scholar]
- 67.Stamenovic D, Mijailovich SM, Tolic-Norrelykke IM, Chen J, and Wang N. Cell prestress. II. Contribution of microtubules. Am J Physiol Cell Physiol 282: C617–C624, 2002 [DOI] [PubMed] [Google Scholar]
- 68.Stamler JS. and Meissner G. Physiology of nitric oxide in skeletal muscle. Physiol Rev 81: 209–237, 2001 [DOI] [PubMed] [Google Scholar]
- 69.Stofan DA, Callahan LA, Di MA, Nethery DE, and Supinski GS. Modulation of release of reactive oxygen species by the contracting diaphragm. Am J Respir Crit Care Med 161: 891–898, 2000 [PubMed] [Google Scholar]
- 70.Supinski G, Nethery D, Stofan D, and DiMarco A. Extracellular calcium modulates generation of reactive oxygen species by the contracting diaphragm. J Appl Physiol 87: 2177–2185, 1999 [DOI] [PubMed] [Google Scholar]
- 71.Supinski GS. and Callahan LA. Free radical-mediated skeletal muscle dysfunction in inflammatory conditions. J Appl Physiol 102: 2056–2063, 2007 [DOI] [PubMed] [Google Scholar]
- 72.Swift LM. and Sarvazyan N. Localization of dichlorofluorescin in cardiac myocytes: implications for assessment of oxidative stress. Am J Physiol Heart Circ Physiol 278: H982–H990, 2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tsutsui H, Kinugawa S, and Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol 301: H2181–H2190, 2011 [DOI] [PubMed] [Google Scholar]
- 74.Ullrich ND, Fanchaouy M, Gusev K, Shirokova N, and Niggli E. Hypersensitivity of excitation-contraction coupling in dystrophic cardiomyocytes. Am J Physiol Heart Circ Physiol 297: H1992–H2003, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Urso ML. and Clarkson PM. Oxidative stress, exercise, and antioxidant supplementation. Toxicology 189: 41–54, 2003 [DOI] [PubMed] [Google Scholar]
- 76.Wagner S, Rokita AG, Anderson ME, and Maier LS. Redox regulation of sodium and calcium handling. Antioxid Redox Signal 18: 1063–1077, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wang N, Butler JP, and Ingber DE. Mechanotransduction across the cell surface and through the cytoskeleton. Science 260: 1124–1127, 1993 [DOI] [PubMed] [Google Scholar]
- 78.Wang N, Naruse K, Stamenovic D, Fredberg JJ, Mijailovich SM, Tolic-Norrelykke IM, Polte T, Mannix R, and Ingber DE. Mechanical behavior in living cells consistent with the tensegrity model. Proc Natl Acad Sci U S A 98: 7765–7770, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.White E, Le Guennec JY, Nigretto JM, Gannier F, Argibay JA, and Garnier D. The effects of increasing cell length on auxotonic contractions; membrane potential and intracellular calcium transients in single guinea-pig ventricular myocytes. Exp Physiol 78: 65–78, 1993 [DOI] [PubMed] [Google Scholar]
- 80.Whitehead NP, Streamer M, Lusambili LI, Sachs F, and Allen DG. Streptomycin reduces stretch-induced membrane permeability in muscles from mdx mice. Neuromuscul Disord 16: 845–854, 2006 [DOI] [PubMed] [Google Scholar]
- 81.Whitehead NP, Yeung EW, and Allen DG. Muscle damage in mdx (dystrophic) mice: role of calcium and reactive oxygen species. Clin Exp Pharmacol Physiol 33: 657–662, 2006 [DOI] [PubMed] [Google Scholar]
- 82.Williams IA. and Allen DG. Intracellular calcium handling in ventricular myocytes from mdx mice. Am J Physiol Heart Circ Physiol 292: H846–H855, 2007 [DOI] [PubMed] [Google Scholar]
- 83.Xu Q, Dalic A, Fang L, Kiriazis H, Ritchie RH, Sim K, Gao XM, Drummond G, Sarwar M, Zhang YY, Dart AM, and Du XJ. Myocardial oxidative stress contributes to transgenic beta(2)-adrenoceptor activation-induced cardiomyopathy and heart failure. Br J Pharmacol 162: 1012–1028, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhao Y, McLaughlin D, Robinson E, Harvey AP, Hookham MB, Shah AM, McDermott BJ, and Grieve DJ. Nox2 NADPH oxidase promotes pathologic cardiac remodeling associated with Doxorubicin chemotherapy. Cancer Res 70: 9287–9297, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]