

Abstract
Starting from a singleton chromanone high throughput screening (HTS) hit, we describe a focused medicinal chemistry optimization effort leading to the identification of a novel series of phenoxymethyl-dihydrothiazolopyridone derivatives as selective positive allosteric modulators (PAMs) of the metabotropic glutamate 5 (mGlu5) receptor. These dihydrothiazolopyridones potentiate receptor responses in recombinant systems. In vitro and in vivo drug metabolism and pharmacokinetic (DMPK) evaluation allowed us to select compound 16a for its assessment in a preclinical animal screen of possible antipsychotic activity. 16a was able to reverse amphetamine-induced hyperlocomotion in rats in a dose-dependent manner without showing any significant motor impairment or overt neurological side effects at comparable doses. Evolution of our medicinal chemistry program, structure activity, and properties relationships (SAR and SPR) analysis as well as a detailed profile for optimized mGlu5 receptor PAM 16a are described.

INTRODUCTION
Schizophrenia is a mental disorder that affects approximately 1% of the world population.1 This severely limiting illness is characterized by a combination of so-called positive (e.g., hallucinations and delusions), negative (e.g., anhedonia and poverty of speech), and cognitive symptoms.1 Currently available therapies, “typical” and “atypical” antipsychotics, rely on dopamine 2 (D2) receptor antagonism to exert their action, and, despite being highly efficacious in addressing positive symptoms, they are of limited value for the treatment of the other core symptoms of the disease. Furthermore, their clinical use is associated with a plethora of severe side-effects (e.g., Parkinson-like extrapyramidal symptoms (EPS), prolactin release, weight gain, or cardiac risk), and as such, patient compliance is extremely poor. As a result, during the past 10–15 years, several alternative mechanisms of action that do not involve direct interaction with dopaminergic receptors have been proposed and investigated.2,3 The ultimate hope is that different biological pathways may lead to effective antipsychotic medications with both increased efficacy versus all three core symptom domains and improved tolerability due to the lack of direct D2 receptor blockade.
One of the proposed pathophysiological causes that may lead to schizophrenia is a compromised function of N-methyl-d-aspartate (NMDA) receptors. This NMDA receptor hypo-function triggers a cascade of events, i.e., reduced activation of inhibitory γ-aminobutyric acidergic (GABAergic) interneurons in thalamus and cortex with a consequent disinhibition of excitatory glutamatergic pathways.4 On the basis of these observations, it has been proposed that compounds that enhance NMDA receptor function may have beneficial effects in schizophrenia. Unfortunately, direct agonists of the NMDA receptor tend to be neurotoxic; hence, alternative strategies to increase NMDA receptor function have been suggested. A desired increase in NMDA receptor function may be obtained through an indirect modulation of NMDA receptors via metabotropic glutamate (mGlu) receptors, especially mGlu5 receptors.2 The mGlu5 receptor belongs to the Group I mGlu receptor subfamily, together with the mGlu1 receptor.5 They activate phospholipase C (PLC) via coupling to Gq proteins and are ubiquitously expressed on postsynaptic excitatory terminals in limbic brain regions that are involved in motivational, emotional, and cognitive processes.6 At GABA-ergic interneurons, mGlu5 and NMDA receptors are coex-pressed7 and are not only coupled via intracellular signaling pathways but also physically through scaffolding proteins.8 In summary, mGlu5 receptor-mediated activation of NMDA receptor functioning has recently emerged as a promising non-dopamine based approach for potential therapeutic intervention of schizophrenia.2
Taking into account the challenges associated with the development of orthosteric agonists for mGlu receptors (e.g., poor druglike properties, elusive selectivity, or potential tolerance development), current strategies have mainly focused on the identification of positive allosteric modulators (PAMs).9 This approach has already proven useful, and in vivo activity in different preclinical schizophrenia and cognition models has been reported for early prototypical mGlu5 PAMs such as 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide (CDPPB, 1)10,11 or S-(4-fluoro-phenyl)-{3-[3-(4-fluoro-phenyl)-[1,2,4]-oxadiazol-5-yl]-piperidin-1-yl}-methanone (ADX47273, 2)12 and more recently for novel structurally distinct chemical series including N-cyclobutyl-6-((3-fluorophenyl)ethynyl)-nicotinamide (VU0360172, 3),13N-(1-methylethyl)-5-(pyridin-4-ylethynyl)pyridine-2-carboxamide (LSN2463359, 4),14,15 4-butoxy-N-(2,4-difluor-ophenyl)benzamide (VU0357121, 5),16 2-(4-(2-(benzyloxy)acetyl)piperazin-1-yl)benzonitrile (VU0364289, 6),17 (1-(4-(2-chloro-4-fluorophenyl)piperazin-1-yl)-2-(pyridin-4-ylmethoxy)eth-anone (CPPZ, 7),18 and (7S)-3-tert-butyl-7-[3-(4-fluorophenyl)-1,2,4-oxadiazol-5-yl]-5,6,7,8-tetrahydro[1,2,4]triazolo[4,3-a]pyridine (LSN2814617, 8).15 These novel PAM classes represent a significant evolution from the first generation as they offer improved drug-likeliness (e.g., improved solubility, unbound fraction in brain or oral bioavailability and in vivo efficacy). Although the preclinical biological data clearly suggest the potential of mGlu5 receptor PAMs as a novel class of antipsychotic agents, the identification of a compound suitable for clinical proof-of-concept studies still remains a challenge and will be crucial to clarify the potential role of mGlu5 receptor PAMs as improved antipsychotics.
RESULTS
Given the promise offered by mGlu5 receptor PAMs as a potential novel treatment for schizophrenia, we performed an HTS campaign looking for for starting points for a medicinal chemistry program. The in vitro mGlu5 PAM calcium mobilization assay provides two measures of compound activity: pEC50, which is defined as the negative logarithm of the concentration at which half-maximal potentiation of glutamate is reached, and the maximal increase in observed glutamate response, the % Glu Max. Screening of the Janssen corporate library in a cell line expressing the human mGlu5 receptor with this functional calcium mobilization assay resulted in the identification of the subsequently reported chromanone 9,19 as a promising hit. Compound 9 possessed potent in vitro mGlu5 PAM activity (EC50 = 128 nM, Glu Max = 82%). As a first step, and since molecule 9 contains labile substructures such as the ester group, we verified that the products of the ester hydrolysis, benzoic acid and 6-hydroxychromanone, were not responsible for the mGlu5 PAM activity of the HTS sample of 9. Both compounds were found to be inactive under the same assay conditions (EC50 > 10 μM), while quality control of the sample 9 was good (LCMS purity > 95%), providing confidence in the HTS in vitro data. Furthermore, 9 showed satisfactory selectivity20 versus other representative members of the mGlu family (mGlu1,2,8 EC50 > 10 μM). Unfortunately, and as anticipated from the presence of the carboxylic ester functionality, 9 was found to be completely hydrolyzed in rat plasma, which precluded any further work for its in vivo pharmacological characterization. Nevertheless, compound 9 was considered as a good starting point for optimization due to its low molecular weight, moderate lipophilicity (clogP = 3.3), high ligand efficiency21 (LE = 0.34), and promising selectivity.
As seen in Figure 2, our strategy for the exploration of the chromanone 9 followed two parallel approaches, first fixing the chromanone scaffold while performing an extensive search for replacements for the labile ester group (A) and, in addition, seeking to replace and optimize the chromanone bicycle as it also contains a ketone moiety, a potential metabolic hot spot and biologically reactive group (B).
Figure 2.

Parallel simultaneous evolution from HTS hit 9 to optimized series 15 and 16.
Approach A involved the synthesis and evaluation of several libraries of diverse potential ester surrogates including, among others, alkyls, amides, amines, heterocycles, and ethers as alternate spacers between the chromanone core and the distal phenyl ring.22 Among this set of linkers, only previously published benzyloxy and phenoxymethyl derivatives 10 and 1119 retained significant mGlu5 receptor PAM activity (EC50 = 950 nM, 76% Glu Max and EC50 = 2.14 μM, 94% Glu Max respectively) and promising selectivity versus the other mGlu receptor subtypes (mGlu1–4,6–8 EC50 > 10 μM). Similar moieties have recently been reported for other mGlu5 PAM chemotypes.19,23
For the second prong of our strategy, chromanone replacements were sought using different approaches. It is now becoming clear in the literature that some of the most potent in vitro mGlu5 receptor PAMs have a common phenylacetylene moiety.9 For comparative purposes, we synthesized the corresponding phenylacetylene chromanone analogue 1219 which was also found to be an mGlu5 receptor PAM (EC50 = 682 nM, Glu Max 84%) although with an approximately 5-fold decrease in activity compared to the initial HTS hit 9. Following this result, we decided to keep the acetylenic spacer constant and combine it with different bicyclic systems with the underlying rationale that this could increase the ability to sample and identify chromanone alternatives. To prioritize among the multiple existing possibilities, our design principle focused on bicyclic scaffolds possessing hydrogen bond acceptors that could mimic those present in the chromanone core. This strategy proved fruitful as among the initial set of compounds synthesized, dihydrobenzothiazolone derivative 1324 emerged as a highly potent (~21 fold potency increase vs 12), efficacious and selective mGlu5 receptor PAM (EC50 = 33 nM, 71% Glu Max, mGlu1–4,6–8 > 10 μM). Despite its high potency and promising metabolic stability in human liver microsomes (55% remaining after 15 min incubation), 13 was found to be highly unstable in rat liver microsomes (2% remaining after 15 min incubation), likely due to the presence of the exposed keto-group. Furthermore, 13 was poorly soluble in aqueous buffer (30 μM, pH 7.4), and it could not be formulated in our standard carrier for in vivo testing [<0.25 mg/mL in 20% hydroxypropyl-β-cyclodextrin (HP-β-CD) formulation in water]. Therefore, we focused efforts on the identification of metabolically stable analogues of 13 bearing a carbonyl function less prone to metabolism. For this purpose, we considered the conversion of the dihydrobenzothiazolone of 13 into a dihydrothiazolopyridone scaffold. Despite an approximate 4-fold decrease in potency with respect to 13, compound 14a showed comparable mGlu5 potency (EC50 = 123 nM, 79% Glu Max) to HTS hit 9. In addition, 14a was selective versus the other mGlu receptors at the screening concentration of 10 μM. Importantly, 14a showed a remarkably reduced turnover in both human and rat liver microsomes (85% and 63% respectively remaining after 15 min incubation at 1 μM concentration). Furthermore, the amide functionality present in the dihydrothiazolopyridone core of 14a represents an attractive chemical handle amenable for further derivatization for structure–activity relationship (SAR) expansion purposes. A preliminary evaluation of tertiary amides showed that the introduction of alkyl substituents could lead to a comparable activity in the case of the methyl derivative 14b (EC50 = 193 nM, 57% Glu Max) or the methoxyethyl substituted example 14c (EC50 = 119 nM, 47% Glu Max) although with a drop of Glu Max in both examples.
At this point, having accomplished our initial goal of identifying separate replacements for the ester and keto functions, we decided to combine both approaches and target the synthesis of the corresponding series of benzyloxy- and phenoxymethyl-dihydrothiazolopyridone combinations 15 and 16, respectively (Table 1).25
Table 1.
Structures and Activities of Substituted Benzyloxy- and Phenoxymethyl-dihydrothiazolopyridones 15 and 16
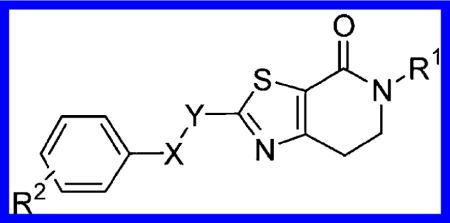
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Comp | X-Y | R1 | R2 | hmGlu5 PAM EC50a (nM)19 | hmGlu5 PAM pEC50a19 | Glu Maxa (%)19 | HLMb (%) | RLMb (%) | clogPc |
| 1 | -- | -- | -- | 176 | 6.75 (86.73-6.78) | 73 (70-76) | -- | -- | -- |
| 2 | -- | -- | -- | 199 | 6.70 (6.66-6.74) | 66 (63-70) | -- | -- | -- |
| 15b | CH2-O |

|
H | 1748 | 5.76 (5.43-6.08) | 76 (55-97) | n.t.d | n.t.d | 1.7 |
| 15c | CH2-O |
|
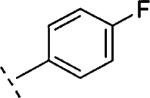
H | 1133 | 5.95 (5.76-6.13) | 67 (55-79) | 7 | 25 | 3.6 |
| 16a | O-CH2 |

|
H | 1195 | 5.92 (5.84-6.01) | 89 (79-99) | 22 | 68 | 1.3 |
| 16b | O-CH2 |
|
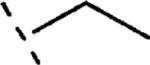
H | 696 | 6.16 (5.99-6.32) | 76 (67-85) | 72 | 99 | 1.8 |
| 16c | O-CH2 |
|
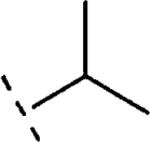
H | 1211 | 5.92 (5.60-6.24) | 42 (28-55) | n.t.d | n.t.d | 2.1 |
| 16d | O-CH2 |
|
H | 2314 | 5.64 (5.43-5.85) | 66 (49-83) | n.t.d | n.t.d | 1.6 |
| 16e | O-CH2 |
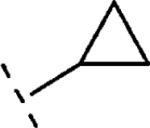
|
H | 1459 | 5.84 (5.61-6.06) | 66 (54-79) | n.t.d | n.t.d | 1.8 |
| 16f | O-CH2 |
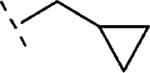
|
H | 250 | 6.60 (6.34-6.86) | 73 (55-91) | 46 | 99 | 2.3 |
| 16g | O-CH2 |
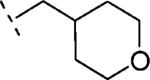
|
H | 851 | 6.07 (5.86-6.28) | 71 (60-81) | n.t.d | n.t.d | 1.5 |
| 16h | O-CH2 |
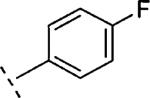
|
H | 96 | 7.02 (6.78-7.25) | 79 (63-95) | 3 | 44 | 3.2 |
| 16i | O-CH2 |
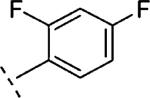
|
H | 66 | 7.18 (6.95-7.41) | 69 (54-83) | 14 | 43 | 3.2 |
| 16j | O-CH2 |
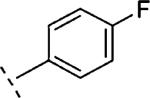
|
2-F | 553 | 6.26 (6.09-6.43) | 72 (64-80) | n.t.d | n.t.d | 3.3 |
| 16k | O-CH2 |
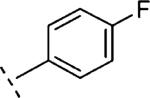
|
3-F | 77 | 7.12 (6.85-7.38) | 61 (40-83) | 18 | 48 | 3.5 |
| 16l | O-CH2 |
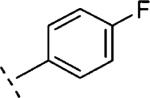
|
4-F | 739 | 6.13 (6.02-6.24) | 79 (73-85) | n.t.d | n.t.d | 3.5 |
| 16m | O-CH2 |
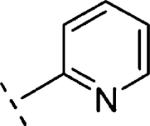
|
H | 1019 | 5.99 (5.82-6.16) | 78 (69-88) | 13 | 99 | 1.6 |
| 16n | O-CH2 |
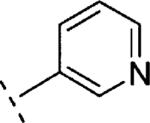
|
H | 1759 | 5.75 (5.59-5.91) | 77 (64-90) | n.t.d | n.t.d | 1.6 |
| 16o | O-CH2 |
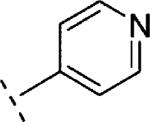
|
H | 2690e | 5.57e | 82e | n.t.d | n.t.d | 1.6 |
| 16p | O-CH2 |
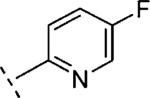
|
H | 621 | 6.21 (6.12-6.29) | 94 (89-99) | 9 | 29 | 1.8 |
| 16q | O-CH2 |
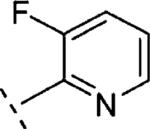
|
H | 791 | 6.10 (5.74-6.46) | 56 (41-70) | 33 | 97 | 1.6 |
Values were calculated from three independent experiments using a four-parameter logistic nonlinear regression model taking into account the heterogeneity between experiments and concentration—response curves for each compound.
HLM and RLM data refer to % of compound metabolized after incubation of tested compound with human and rat microsomes respectively, for 15 min at 1 μM concentration.
Calculated with Biobyte software.
Not tested.
Data obtained from a single experiment not replicated.
Chemistry
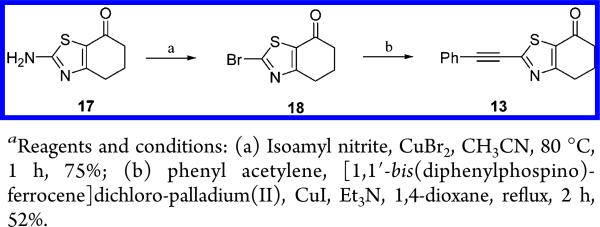
Compound 13 was readily prepared in two steps (39% overall yield), via Sandmeyer reaction and subsequent Sonogashira cross-coupling, from 2-amino-benzothiazolone 1726 (Scheme 1).
Scheme 1.
The synthesis of the targeted compounds 14a–c is shown in Scheme 2. Starting from commercially available Boc-protected 2,4-dioxopiperidine 19, bromination and condensation with thiourea in the presence of sodium hydrogen carbonate followed by Boc deprotection afforded key intermediate 20 (17% overall yield), which was transformed into the dihydrothiazolopyridone 14a with moderate yield (55%) following an analogous procedure to the one described for the synthesis of compound 13 in Scheme 1. Finally, reaction of 14a with the corresponding alkyl halides in the presence of cesium carbonate as base, yielded derivatives 14b,c (19 and 20% yield, respectively).
Scheme 2.

The synthetic methods used for the preparation of the final compounds 15b,c and 16a–q are outlined in Schemes 3–9. Preparation of most dihydrothiazolopyridones 15–16 followed a cyclization step performed with a piperidine-2,4-dione, which was commercially available or was prepared using a synthetic sequence shown in Scheme 3. Thus, commercially available β-amino esters 22a–e were reacted with ethyl malonyl chloride to afford the amides 23a–e in moderate to good yields (22–70%). Intramolecular cyclization reaction of 23a–e, promoted by sodium ethoxide, yielded the corresponding 3-carbethoxypiperidine-2,4-dione derivatives 24a–e. Acid mediated decarboxylation of 24a–e followed by regioselective bromination afforded the key intermediates 25a–e in good yields. These intermediates were found to be highly unstable and had to be used immediately after synthesis.
Scheme 3.

Scheme 9.

The benzyloxy-dihydrothiazolopiperidones 15b,c were prepared as shown in Scheme 4. Nucleophilic substitution of the bromothiazolopyridone 21 with benzyl alcohol yielded the target intermediate 15a, which was subsequently N-alkylated with methyl iodide affording compound 15b in 30% yield (equation (I), Scheme 4). Attempts to perform a copper catalyzed N-arylation reaction on 15a (CuI, K3PO4, DMF, microwave irradiation) were unsuccessful as compound 15c was found to be unstable under these reaction conditions, and the synthesis of compound 15c was carried out as shown in the equation (II) of Scheme 4. Thus, intermolecular cyclization of 25d with thiourea followed by Sandmeyer reaction led with low yield to the intermediate N-aryl bromothiazole derivative 26, which was treated with benzyl alcohol in the presence of sodium hydride to yield (63%) the target compound 15c.
Scheme 4.

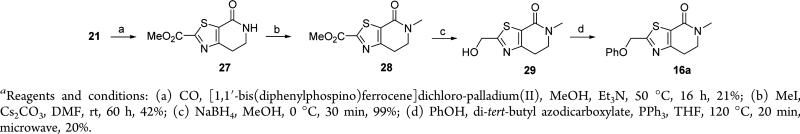
In the case of the phenoxymethyl-dihydrothiazolopyridones 16, five main synthesis routes (Schemes 5–9) were used to access the final compounds 16a–q. Starting from the bromothiazole derivative 21, palladium catalyzed carbonylation followed by alkylation with methyl iodide and reduction of the intermediate ester 28 led to the alcohol 29 which after Mitsunobu reaction with phenol afforded 16a (Scheme 5).
Scheme 5.
Key intermediate 31 was obtained in 26% yield from intermediate 27, in a similar way (Scheme 6). N-Alkylation of 31 with several primary alkyl halides 32 afforded the corresponding N-alkyl derivatives 16b,d,g in moderate yields (13–49%) (Scheme 6).
Scheme 6.

Alternatively, several compounds (16c,e,f,h,i,k) were prepared by intermolecular cyclization between the thioamides 33a,b and the corresponding 3-bromopiperidine-2,4-dione 25a–e (10–25%) (Scheme 7). Similarly, derivatives 16j,l were accessed by nucleophilic substitution on the chloromethyl thiazol 36, obtained via reaction of 25d with ethyl thiooxamate followed by ester reduction and subsequent treatment with thionyl chloride, with the corresponding phenols as shown in Scheme 8.
Scheme 7.

Scheme 8.

Finally, intermediate 31 was also used to prepare the N-pyridyl derivatives 16m-q by copper catalyzed coupling with the corresponding halopyridines 36 as shown in Scheme 9, in low to moderate yields (15–64%).
DISCUSSION
In the case of benzyloxy-dihydrothiazolopyridone derivatives 15, our SAR investigation started with the synthesis of the methyl and 4-fluorophenyl substituted analogues 15b (prepared by analogy to 14b) and 15c (selected because fluorine containing aromatic rings are common motifs as distal aromatic moieties for different mGlu5 PAM chemotypes).9,16 As seen in Table 1, the introduction of the 4-fluorophenyl and the methyl substituents resulted in compounds of similar potency. Furthermore, 15c showed good metabolic stability in human and rat fortified microsomes. Unfortunately, in the course of the aqueous solubility assessment of derivatives 15, these were found to be unstable in acidic media (20% HP-β-CD in water, pH = 4) leading to the corresponding 2-hydroxy-dihydrothiazolopyridone fragments. In light of this finding, we decided to stop the exploration of the benzyloxy-dihydrothiazolopyridone series 15. Instead we focused efforts on the phenoxymethyl subseries 16, which proved to be stable under the same acidic aqueous solubility conditions. As a first step, the alkyl substituent on the amide moiety was explored. Akin to the benzyloxy series, introduction of a methyl group in 16a resulted in a significant reduction (~6 fold) in mGlu5 PAM potency compared to the corresponding acetylene analogue 14b, or reference compounds 1 and 2, but still retained high efficacy. Replacement of the methyl group in 16a by other alkyl chains with increased size and diverse polarity either had a limited effect or was detrimental to activity. Thus, comparable activity was observed for the more lipophilic 16b (R1 = ethyl) and 16c (R1 = isopropyl), although a modest reduction in efficacy was also noted for the bulkier 16c. In the case of the methoxyethyl substituted 16d and in analogy with the acetylene containing series 14, the potency was again in the same range as for 16a. The introduction of a cyclopropyl group with pseudo aromatic character in 16e resulted as well in a similar potency when compared to 16a and its isopropyl congener 16c. Noteworthy, the introduction of a methylene spacer between the cyclopropyl group and the dihydrothiazolopyridone core (16f) delivered a remarkable increase in potency (~6 fold vs. 16a and 16e). Finally, ring expansion to the more polar tetrahidropyrane ring (16g) had almost negligible effect on both potency and efficacy compared to 16a. As observed with the benzyloxy series, better results were obtained with fluorophenyl tethered amides such as 16h or 16i with in vitro potencies below 100 nM (~2–3 fold more potent than reference compounds 1 and 2) and good efficacies. Taking 16h as a starting point, the introduction of fluorine substitution in the western aromatic ring was found to be preferred at position 3, with 16k showing comparable potency and minimally reduced efficacy to 16h. A ~7–10-fold decrease in potency was observed for the 2- and 4-substituted analogues 16j,l. In light of the promising in vitro results obtained with the aryl derivatives 16h–l, our SAR exploration concluded with the investigation of the effects of replacing the fluorophenyl amide substituents by more polar and weakly basic pyridine rings. From the data in Table 1, it can be concluded that the 2-substituted pyridine analogue 16m was preferred over the 3 and 4 regioisomers (16n and 16o). On the other hand, although 3-fluorine substitution (16q) did not induce any increase on mGlu5 potency, a significant improvement was accomplished by the introduction of the fluorine atom in the 5-position of the 2-substituted pyridine ring, with 16p showing similar potency to 16m and increased efficacy (94% vs. 79%).
In light of their good mGlu5 PAM activity and efficacy compounds 16a, 16b, 16f, 16h, and 16p were selected as representative examples for further characterization. Metabolic stability evaluation showed significant species differences, with lower turnovers in human vs. rat liver microsomes in all cases. Thus, despite low (16a, 16h, and 16p, > 75% remaining) to moderate-high (16b and 16f, 75–25% remaining) turnover in HLM for the five compounds, metabolic instability in RLM ranged from moderate (16p, ~30% remaining) to very high (16b and 16f, ~100% metabolized). As high metabolic instability in rodents would hamper further pharmacological evaluation, only compounds 16a, 16h, and 16p were selected for additional DMPK evaluation to confirm their potential utility for proof-of-concept in vivo studies for this series. Thus, 16a and 16b were evaluated in noncrossover in vivo pharmacokinetic (PK) studies in rats. Because of its poor solubility in aqueous buffer (<4 μM, pH 7.4) and in 20% HP-β-CD in water formulation (<0.20 mg/mL), 16h could only be dosed orally as a suspension (5 mg/mL) and was found to be undetectable in the brain after 2.0 h of administration and hence discarded for in vivo pharmacological evaluation. As a consequence of these findings, and because the improvement in kinetic solubility for 16p was minimal (10 μM, pH 7.4 and <1 mg/mL in 20% HP-β-CD in water) compared to 16h, 16p was discarded for further investigation. PK studies (1 mg/kg iv, 10 mg/kg p.o.) with the more soluble 16a (97 μM, aqueous buffer pH 7.4 and 4 mg/mL in 20% HP-β-CD in water), revealed a relatively high CLp of 50 mL/min/kg and a short half-life (T1/2) of 0.18 h. Examination of drug concentrations in portal vein, plasma, and brain at different time points (0.5, 1.0, 2.0, 4.0, and 7.0 h) showed a moderate first-pass clearance (Ehep 0.47) with a relatively low volume of distribution (Vss = 0.70 L/kg) and moderate systemic exposure (plasmaAUC = 2.9 μM-h). The relative CNS exposure of 16a was moderate as well (brainAUC = 2.4 μM·h) with a good distribution to the brain (brainAUC/plasmaAUC = 0.90). Additionally, a relatively high unbound fraction in brain (rat fu brain = 18%) contributed favorably to achieve good absolute brain levels (Cmax = 522 nM, Tmax = 0.5 h), which were considered adequate to warrant further pharmacological evaluation.
Subsequent in vitro examination of 16a in a low expressing rat receptor mGlu5 cell line27 resulted in a slightly reduced potency (~2 fold) and efficacy (EC50 = 3408 nM, 62% Glu Max) compared with the human receptor expressing cell line. 16a was found to be selective in an mGlu selectivity panel (mGlu1–4,6–8 EC50 > 10 μM)20 and also when tested at 10 μM concentration in a panel of 66 off-target class receptors, ion channels and transporters.28 Finally, positive allosteric modulator activity was further investigated by fold-shift experiments to determine the degree of cooperativity with glutamate of the compound. Thus, as shown in Figure 4, progressive fold-shift experiments in our cell line expressing the rat mGlu5 receptor revealed how the concentration response curve of (CRC) glutamate shifted to the left with increasing concentration of 16a (55 nM to 30 μM) with no significant effect change of the maximal response. A 5.4-fold leftward shift in the glutamate EC50 was observed in the presence of 30 μM 16a, and a predicted allosteric modulator affinity of approximately 10 μM (10069 nM) was calculated using the operational model of allosterism described by Gregory et al. (Figure 3).29 These results corroborate a positive allosteric interaction between glutamate and 16a and were confirmed in a cell line expressing the human mGlu5 receptor,27 where 10 μM 16a produced a 4.4-fold leftward shift in the glutamate CRC.
Figure 4.

Profile summary of 16a.
Figure 3.

Glutamate CRC in the presence of increasing concentrations of 16a.
Encouraged by its overall profile, 16a (Figure 4) was evaluated in a screen of potential antipsychotic efficacy, the reversal of amphetamine-induced hyperlocomotion in rats.10,11
As seen in Figure 5, when dosed orally in a 20% HP-β-CD in water formulation, 16a showed robust dose-dependent effects in reversal of hyperlocomotion with a lowest active oral dose of 10 mg/kg. The maximal effect (61%) was seen at the highest tested dose of 100 mg/kg, corresponding with an estimated average terminal unbound brain concentration of ~3.2 μM,30 which is well in line with the rat in vitro mGlu5 PAM EC50 of 16a and that, according with the progressive fold-shift experiments (Figure 3), would correlate with a left-ward shift in the glutamate CRC in the range of ~1.5–2.9 fold.
Figure 5.

Dose-dependent effect of 16a on the reversal of amphetamine-induced hyperlocomotion in rats.
The preliminary in vivo characterization of 16a concluded with the evaluation of balance and motor coordination and potential neurological side effects by means of rotarod and Irwin modified neurological battery tests respectively in rats. As it can be seen in Figure 6, when dosed orally at 30, 56.6, and 100 mg/kg (20% HP-β-CD in water), 16a had no statistically significant effect on general motor output as measured by performance on the rotarod (120 s cutoff) test in rats.31 Furthermore, 16a had no effect on any autonomic or somatomotor nervous system functions as measured using the modified Irwin neurological test battery in rats when tested at a single oral dose of 100 mg/kg (20% HP-β-CD in water).,32 These results highlight how 16a neither impacts motor behavior nor shows any overt neurological side effects in rats up to a dose of 100 mg/kg p.o., a dose that has been previously shown to produce maximal efficacy in the inhibition of the amphetamine-induced hyperlocomotion.
Figure 6.

Effects of 16a on rotarod performance in rats.
CONCLUSIONS
In summary, starting from a singleton phenyl ester chromanone HTS hit, two parallel approaches allowed the identification of the benzyloxy and phenoxymethyl moieties and the dihydrothiazolopyridone core as optimal replacements for the ester and the chromanone core respectively. While the benzyloxydihydrothiazolopyridone subseries was found susceptible to hydrolysis, the phenoxymethyl-dihydrothiazolopyridones 16 proved to be a promising mGlu5 PAM series. Preliminary focused SAR exploration of amide substituents revealed aromatic groups as preferred substituents for achieving high in vitro mGlu5 potentiation although their poor DMPK characteristics precluded further in vivo evaluation. On the other hand, the N-methyl analogue 16a presented a more balanced profile in terms of in vitro potency, selectivity and DMPK profile, which made it an attractive candidate for in vivo characterization in a model of antipsychotic activity. 16a showed robust, dose-dependent effects in the reversal of amphetamine induced hyperlocomotion, with a lowest active dose of 10 mg/kg p.o. and maximal efficacy at a dose of 100 mg/kg p.o. without showing any significant motor impairment or overt neurological side effects up to a dose of 100 mg/kg p.o. These results underscore the potential of 16a as an interesting tool compound suitable to unravel the in vivo pharmacology of mGlu5 positive allosteric modulation. Further investigation and evolution of this novel series of mGlu5 PAMs are underway and will be reported in due course.
EXPERIMENTAL SECTION
Chemistry
Unless otherwise noted, all reagents and solvents were obtained from commercial suppliers and used without further purification. Thin layer chromatography (TLC) was carried out on silica gel 60 F254 plates (Merck). Flash column chromatography was performed on silica gel, particle size 60 Å, mesh of 230–400 (Merck) under standard techniques. Microwave assisted reactions were performed in a single-mode reactor, Biotage Initiator Sixty microwave reactor (Biotage), or in a multimode reactor, MicroSYNTH Labstation (Milestone, Inc.). Nuclear magnetic resonance (NMR) spectra were recorded with a Bruker DPX-300 a Bruker DPX-400 or a Bruker AV-500 spectrometer (Bruker AG) with standard pulse sequences, operating at 300, 400, and 500 MHz, respectively, using CDCl3 and DMSO-d6 as solvents. Chemical shifts (δ) are reported in parts per million (ppm) downfield from tetramethylsilane (δ = 0). Coupling constants are reported in Hertz. Splitting patterns are defined by s (singlet), d (doublet), dd (double doublet), t (triplet), q (quartet), quin (quintet), sex (sextet), sep (septet), or m (multiplet). Liquid chromatography combined with mass spectrometry (LCMS) was performed on either a HP 1100 high-performance liquid chromatography (HPLC) system (Agilent Technologies) or Advanced Chromatography Technologies system composed of a quaternary or binary pump with degasser, an autosampler, a column oven, a diode array detector (DAD), and a column as specified in the respective methods below. Flow from the column was split to a MS spectrometer. The MS detector was configured with either an electrospray ionization source or an electrospray combined with atmospheric pressure chemical ionization (ESCI) dual ionization source. Nitrogen was used as the nebulizer gas. Data acquisition was performed with MassLynx- Openlynx software or with Chemstation-Agilent data browser software. More detailed information about the different LCMS methods employed can be found in the Supporting Information. Liquid chromatography combined with high resolution mass spectrometry (LC-HRMS) was performed using a Micromass (Waters) Q-Tof API-US calibrated and verified with sodium iodide. The samples were diluted with a 50:50 0.1% formic acid (in Milli-Q)/acetonitrile solution, directly infused using leucine-enkephalin ([M + H]+ = 556.2771) as a lockmass. Scan range was from 100 to 1000 Da, with a scan time of 1 s. For a number of compounds, melting points (Mpa) were determined with a WRS-2A melting point apparatus (Shanghai Precision and Scientific Instrument Co. Ltd.). Melting points were measured with a linear heating up rate of 0.2–5.0 °C/min (maximum temperature 300 °C), and the reported values are melt ranges. For another number of compounds, melting points (Mpb) were determined in open capillary tubes on a FP62 or on a FP81HTFP90 apparatus (Mettler). Melting points were measured with a temperature gradient of 10 °C/min (maximum temperature 300 °C), and the melting point was read from a digital display. Melting point values are peak values and were obtained with experimental uncertainties that are commonly associated with this analytical method.
Purities of all new compounds were determined by analytical reversed-phase high-performance liquid chromatography (RP-HPLC) using the area percentage method on the UV trace recorded at a wavelength of 254 nm, and compounds were found to have ≥95% purity unless otherwise specified.
2-Bromo-5,6-dihydro-4H-benzothiazol-7-one (18)
A mixture of 1727 (19 g, 112 mmol), CuBr2 (27 g, 120 mmol), and 3-methyl-1-nitrosooxy-butane (18 g, 153 mmol) in CH3CN (250 mL) was stirred at 80 °C for 1 h. The mixture was then cooled to rt and poured into a 10% solution of HCl. The mixture was extracted with CH2Cl2 and the organic layer was separated, dried (Na2SO4), and filtered, and the solvent was evaporated in vacuo to yield 19.6 g (75%) of 18 that was used in the next step without any further purification. 1H NMR (500 MHz, CDCl3) δ ppm 2.47–2.60 (m, 2 H), 3.07–3.15 (m, 1 H), 3.18–3.29 (m, 1 H), 4.64 (t, J = 3.9 Hz, 1 H).
2-Amino-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one hydrochloride salt (20)
To a solution of 19 (40 g, 187.58 mmol) in CCl4 (500 mL) was added NBS (33.38 g, 187.58 mmol) portionwise keeping the reaction temperature in the range of 10–15 °C. The mixture was further stirred at 10–15 °C for 2 h. The mixture was allowed to warm to rt, and the solvent was evaporated in vacuo. The residue thus obtained was dissolved in EtOAc and washed with H2O. The organic layer was separated, dried (Na2SO4), and filtered and the solvent was evaporated. The residue thus obtained was dissolved in EtOH (400 mL), and then thiourea (6.5 g, 85.6 mmol) and NaHCO3 (7.2 g, 85.6 mmol) were added. The resulting mixture was stirred at 80 °C for 2.5 h and then cooled to rt, and the solids were filtered off. The filtrate was evaporated in vacuo to give a residue that was crystallized in EtOH. The yellow crystals thus obtained were dissolved in a 4 M solution of HCl in 1,4-dioxane (100 mL), and the resulting mixture was stirred at rt for 30 min. The solvent was evaporated in vacuo to yield 10 g (88% pure) of 20 as a yellow powder which was used in the next step without any further purification. LCMS: m/z 170 [M + H]+, tR = 0.38 min.
2-Bromo-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one (21)
A mixture of 20 (8 g, 39.8 mmol), CuBr2 (10.43 g, 46.68 mmol), and 3-methyl-1-nitrosooxy-butane (6.8 g, 58.35 mmol) in CH3CN (100 mL) was stirred at rt for 1.5 h. The solvent was evaporated in vacuo. The residue thus obtained was dissolved in EtOAc and washed with H2O. The organic layer was separated, dried (Na2SO4), and filtered, and the solvent was evaporated in vacuo to yield 5 g (55%) of 21 that was used in the next step without any further purification. LCMS: m/z 233 [M + H]+, tR = 0.70 min.
N-(2-Ethoxycarbonyl-ethyl)-N-isopropyl-malonamic Acid Ethyl Ester (23a)
To a solution of 22a (13.5 g, 69 mmol) in CH2Cl2 (100 mL) was added Et3N (26 g, 255 mmol). Then ethyl malonyl chloride (14 g, 93 mmol) was added to the mixture at 0 °C, and the mixture was stirred at rt for 5 h. Then H2O was added and the organic layer was separated, dried (Na2SO4), and filtered and the solvent was evaporated in vacuo. The crude product was purified by flash column chromatography (silica gel, EtOAc in petroleum ether, 10/90 to 33/66). The desired fractions were collected and the solvents evaporated in vacuo to yield 5.1 g (87% pure) of 23a as an oil. LCMS: m/z 274 [M + H]+, tR = 1.25 min.
N-Cyclopropyl-N-(2-ethoxycarbonyl-ethyl)-malonamic Acid Ethyl Ester (23b)
Starting from 22b (40.3 g, 256 mmol) and following the procedure described for 23a, 23b was obtained as an oil (40 g, 60% pure). LCMS: m/z 272 [M + H]+, tR = 0.91 min.
N-Cyclopropylmethyl-N-(2-ethoxycarbonyl-ethyl)-malonamic Acid Ethyl Ester (23c)
Starting from 22c (31.3 g, 183 mmol) and following the procedure described for 23a, 23c was obtained as an oil (33 g, 93% pure). LCMS: m/z 286 [M + H]+, tR = 1.23 min.
N-(2-Ethoxycarbonyl-ethyl)-N-(4-fluoro-phenyl)-malonamic Acid Ethyl Ester (23d)
Starting from 22d (10 g, 47.3 mmol) and following the procedure described for 23a but using diisopropylethylamine as base, 23d was obtained as an orange oil (11 g, 73% pure). LCMS: m/z 326 [M + H]+, tR = 2.41 min.
N-(2-Ethoxycarbonyl-ethyl)-N-(2,4-difluoro-phenyl)-malonamic Acid Ethyl Ester (23e)
Starting from 22e (29 g, 127 mmol) and following the procedure described for 23a, 23e was obtained as an orange oil (35 g, 69% pure). LCMS: m/z 344 [M + H]+, tR = 1.21 min.
1-Isopropyl-2,4-dioxo-piperidine-3-carboxylic Acid Ethyl Ester (24a)
Sodium (0.86 g, 37.3 mmol) was added to EtOH (50 mL) at 0 °C. The mixture was stirred at rt for 1 h. Then 23a (5.1 g, 18.6 mmol) was added and the mixture was stirred at 85 °C for 16 h. The mixture was poured into ice H2O. The aqueous phase was neutralized by addition of a 2 N HCl solution and extracted with EtOAc. The organic layer was separated, dried (Na2SO4), and filtered, and the solvent was evaporated in vacuo to yield 3 g (91%) of 24a which was used in the next step without any further purification. LCMS: m/z 228 [M + H]+, tR = 0.95 min.
1-Cyclopropyl-2,4-dioxo-piperidine-3-carboxylic Acid Ethyl Ester (24b)
Starting from 23b (40 g, 147 mmol) and following the procedure described for 24a, 24b was obtained as an oil (20 g, 61%). LCMS: m/z 226 [M + H]+, tR = 0.91 min.
1-Cyclopropylmethyl-2,4-dioxo-piperidine-3-carboxylic Acid Ethyl Ester (24c)
Starting from 23c (20 g, 70.1 mmol) and following the procedure described for 24a, 24c was obtained as an oil (10 g, 60%). LCMS: m/z 240 [M + H]+, tR = 1.21 min.
1-(4-Fluoro-phenyl)-2,4-dioxo-piperidine-3-carboxylic Acid Ethyl Ester (24d)
A mixture of 23d (6.27 g, 19.27 mmol) in a 21% solution of sodium ethoxide in EtOH (14.39 mL, 38.55 mmol) was stirred at 85 °C for 16 h. The solvent was evaporated in vacuo and the residue was partitioned between EtOAc and H2O. The aqueous layer was separated, acidified by 1N HCl solution addition and extracted with CH2Cl2. The organic layer was separated, dried (Na2SO4), filtered and the solvent evaporated in vacuo to yield 5 g (66% pure) of 24d which was used in the next step without any further purification. LCMS: m/z 280 [M + H]+, tR = 0.73 min.
1-(2,4-Difluoro-phenyl)-2,4-dioxo-piperidine-3-carboxylic acid ethyl ester (24e)
Starting from 24e (4.8 g, 13.98 mmol) and following the procedure described for 24d, 24e was obtained as an oil (4.16 g, 55% pure). LCMS: m/z 298 [M + H]+, tR = 0.65 min.
3-Bromo-1-isopropyl-piperidine-2,4-dione (25a)
A solution of 24a (3 g, 13.2 mmol) in a mixture of acetic acid (10 mL) and H2O (90 mL) was stirred at 90 °C for 14 h. The mixture was cooled and extracted with CH2Cl2. The organic layer was separated, dried (Na2SO4), and filtered, and the solvent was evaporated in vacuo. The crude product was purified by flash column chromatography (silica gel; EtOAc in petroleum ether 10/90 to 33/66). The desired fractions were collected and the solvents evaporated in vacuo. The residue thus obtained was dissolved in CH2Cl2 (20 mL) and NBS (1.1 g, 6.44 mmol) was added. The mixture was stirred at rt for 4 h. Then H2O was added and the mixture was extracted with EtOAc. The organic layer was separated, dried (Na2SO4), and filtered, and the solvents were evaporated in vacuo to yield 0.6 g (53% pure) of 26a which was used in the next step without any further purification. LCMS: m/z 234 [M + H]+, tR = 0.35 min.
3-Bromo-1-cyclopropyl-piperidine-2,4-dione (25b)
Starting from 24b (20 g, 89 mmol) and following the procedure described for 25a, 25b was obtained as an oil (11 g, 73% pure). LCMS: m/z 232 [M + H]+, tR = 0.29 min.
3-Bromo-1-cyclopropylmethyl-piperidine-2,4-dione (25c)
Starting from 24c (10 g, 41.8 mmol) and following the procedure described for 25a, 25c was obtained as an oil (4 g, 66% pure). LCMS: m/z 246 [M + H]+, tR = 1.053 min.
3-Bromo-1-(4-fluoro-phenyl)-piperidine-2,4-dione (25d)
Starting from 24d (7.5 g, 26.86 mmol) and following the procedure described for 25a, 25d was obtained as an oil (7.7 g, 76% pure). LCMS: m/z 285 [M + H]+, tR = 0.42 min.
3-Bromo-1-(2,4-difluoro-phenyl)-piperidine-2,4-dione (25e)
Starting from 24e (18 g, 60 mmol) and following the procedure described for 25a, 25e was obtained as an oil (11 g, 38% pure). LCMS: m/z 304 [M + H]+, tR = 0.34 min.
2-Bromo-5-(4-fluoro-phenyl)-6,7-dihydro-5H-thiazolo[5,4-c]-pyridin-4-one (26)
A mixture of 25d (4.14 g, 14.48 mmol), thiourea (1.1 g, 14.48 mmol) and NaHCO3 (1.22 g, 14.48 mmol) in EtOH (60 mL) was stirred at 80 °C for 1 h. The mixture was then cooled to rt and the solids were filtered off. The filtrate was evaporated in vacuo. The residue thus obtained was dissolved in CH3CN (80 mL) and then CuBr2 (3.05 g, 13.67 mmol) and 3-methyl-1-nitrosooxy-butane (2.3 mL, 17.09 mmol) were added. The mixture was stirred at rt for 45 min and then the solvent was evaporated in vacuo. The residue thus obtained was partitioned between EtOAc and H2O. The organic layer was separated, dried (Na2SO4), filtered and the solvent evaporated in vacuo. The crude product was purified by flash column chromatography (silica gel, EtOAc in heptane, 0/100 to 30/70). The desired fractions were collected and the solvents evaporated in vacuo to yield 1.2 g (16%) of 26 as a white solid. LCMS: m/z 327 [M + H]+, tR = 2.51 min.
4-Oxo-4,5,6,7-tetrahydro-thiazolo[5,4-c]pyridine-2-carboxylic acid methyl ester (27)
A mixture of Et3N (17.2 g, 171 mmol) and [1,1′-bis(diphenylphosphino)ferrocene]dichloro-palladium(II) (2 g, 2.7 mmol) in THF (300 mL) was added to a solution of 21 (7.5 g, 23.6 mmol) in MeOH (300 mL). Then the mixture was stirred at 50 °C overnight under CO atmosphere (2.5 MPa). The mixture was cooled and filtered, and the solvents were evaporated in vacuo. The crude product was purified by flash column chromatography (silica gel, MeOH in CH2Cl2, 1/99). The desired fractions were collected and evaporated in vacuo to yield 4.5 g (21%) of 27 that crystallized from EtOAc as a yellow solid. Mpa 217.6–220.0 °C. 1H NMR (300 MHz, CDCl3) δ ppm 3.20 (t, J = 7.1 Hz, 2 H), 3.62–3.79 (m, 2 H), 4.02 (s, 3 H), 5.84 (br. s., 1 H). LCMS: m/z 213 [M + H]+, tR = 2.94 min.
5-Methyl-4-oxo-4,5,6,7-tetrahydro-thiazolo[5,4-c]pyridine-2-carboxylic acid methyl ester (28)
Iodomethane (4.4 mL, 70.7 mmol) was added to a suspension of 27 (10 g, 47.1 mmol) and Cs2CO3 (23 g, 70.68 mmol) in DMF (118 mL) under nitrogen. The mixture was stirred at rt for 60 h and then diluted with H2O and extracted with EtOAc. The organic layer was separated, dried (MgSO4), and filtered, and the solvent was evaporated in vacuo. The crude product was purified by flash column chromatography (silica gel, EtOAc in CH2Cl2, 0/100 to 50/50). The desired fractions were collected, and the solvents were evaporated in vacuo to yield 4.46 g (42%) of 28 as a pale brown oily solid. 1H NMR (400 MHz, CDCl3) δ ppm 3.13 (s, 3 H), 3.23 (t, J = 7.2 Hz, 2 H), 3.71 (t, J = 7.2 Hz, 2 H), 4.03 (s, 3 H). LCMS: m/z 227 [M + H]+, tR = 0.60 min.
2-Hydroxymethyl-5-methyl-6,7-dihydro-5H-thiazolo[5,4-c]-pyridin-4-one (29)
Sodium borohydride (0.15 g, 4.0 mmol) was added to a stirred solution of 28 (0.65 g, 2.87 mmol) in a mixture of THF (8.8 mL) and MeOH (8.8 mL). The mixture was stirred at 0 °C for 30 min in a sealed tube under nitrogen and then diluted with H2O and extracted with CH2Cl2. The organic layer was separated, dried (Na2SO4), and filtered and the solvents were evaporated in vacuo. The aqueous phase was acidified with 3 N HCl and extracted with CH2Cl2. The combined organic layers were dried (Na2SO4) and filtered, and the solvents were evaporated in vacuo. The crude product was purified by flash column chromatography (silica gel, MeOH in EtOAc, 0/100 to 20/80). The desired fractions were collected and the solvents were evaporated in vacuo to yield 0.59 g (99%) of 29 as a dark oil. 1H NMR (400 MHz, CDCl3) δ ppm 2.81 (t, J = 6.1 Hz, 1H), 3.10 (t, J = 7.2 Hz, 2 H), 3.10 (s, 3 H), 3.66 (t, J = 7.2 Hz, 2 H), 4.93 (d, J = 6.2 Hz, 2 H). LCMS: m/z 199 [M + H]+, tR = 0.28 min.
2-Hydroxymethyl-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one (30)
Starting from 27 (10 g, 47 mmol) and following the procedure described for 29, 30 was obtained as an oil (3.5 g, 40%). Mpa 212.1–230.9 °C. 1H NMR (400 MHz, DMSO-d6) δ ppm 2.92 (t, J = 7.0 Hz, 2 H), 3.47 (td, J = 7.0, 2.5 Hz, 2 H), 4.72 (s, 2 H), 6.24 (br. s., 1 H), 7.79 (br. s., 1 H). LCMS: m/z 185 [M + H]+, tR = 3.63 min.
2-Phenoxymethyl-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one (31)
Di-tert-butyl azodicarboxylate (3.0 g, 13.03 mmol) was added to a stirred solution of 30 (2 g, 10.86 mmol), phenol (1.23 g, 13.03 mmol), and PPh3 (3.42 g, 13.03 mmol) in THF (31 mL) under nitrogen. The mixture was stirred at 120 °C for 40 min under microwave irradiation. The solvent was evaporated in vacuo. The crude product was purified by flash column chromatography (silica gel, EtOAc in CH2Cl2, 0/100 to 100/0). The desired fractions were collected and evaporated in vacuo to yield 1.82 g (69% pure) of 31 as a white solid. LCMS: m/z 261 [M + H]+, tR = 1.50 min.
2-Phenoxy-thioacetamide (33a)
To a solution of phenoxyacetonitrile (38 g, 285 mmol) in DMF (380 mL), thioacetamide (42.8 g, 450 mmol) and a solution of 4 M HCl in 1.4-dioxane (380 mL) were added. The mixture was stirred at 100 °C for 16 h and then cooled to rt and poured into a saturated solution of NaHCO3. The solid formed was filtered off, washed with H2O, and dried in vacuo to yield 45 g (94%) of 33a as a beige solid which was used in the next step without any further purification. 1H NMR (500 MHz, CDCl3) δ ppm 4.88 (s, 2 H), 6.89–6.97 (m, 2 H), 7.04 (t, J = 7.4 Hz, 1 H), 7.29–7.36 (m, 2 H), 7.74 (br. s, 1 H), 7.97 (br. s, 1 H). LCMS: m/z 166 [M – H]–, tR = 1.18 min.
2-(3-Fluoro-phenoxy)-thioacetamide (33b)
Starting from (3-fluoro-phenoxy)-acetonitrile (3.39 g, 22.41 mmol) and thioacetamide (4.21 g, 56.02 mmol) and following the procedure described for 33a, 33b was obtained as a white solid (3.58 g, 86%). 1H NMR (400 MHz, DMSO-d6) δ ppm 4.78 (s, 2 H), 6.70–6.91 (m, 3 H), 7.25–7.42 (m, 1 H), 9.69 (m, 2 H). LCMS: m/z 184 [M – H]–, tR = 1.19 min.
5-(4-Fluoro-phenyl)-4-oxo-4,5,6,7-tetrahydro-thiazolo[5,4-c]-pyridine-2-carboxylic acid ethyl ester (34)
A mixture of 25d (11.8 g, 41.3 mmol), ethyl thiooxamate (5.5 g, 41.3 mmol), and NaHCO3 (8.7 g, 103.5 mmol) in EtOH (400 mL) was stirred at 80 °C for 2 h. The mixture was cooled and filtered, and the filtrate was evaporated in vacuo. The crude product was purified by flash column chromatography (silica gel, EtOAc in petroleum ether, 10/90 to 33/66). The desired fractions were collected and evaporated in vacuo to yield 2 g (68% pure) of 34. LCMS: m/z 321 [M + H]+, tR = 1.05 min.
2-Chloromethyl-5-(4-fluoro-phenyl)-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one (35)
Sodium borohydride (0.7 g, 18.7 mmol) was added to a solution of 34 (2 g, 6.3 mmol) in MeOH (50 mL) at 0 °C. The mixture was stirred at rt for 2 h, H2O was added, and the mixture was extracted with EtOAc. The organic layer was separated, dried (Na2SO4), and filtered, and the solvent was evaporated in vacuo. The crude product was purified by flash column chromatography (silica gel, EtOAc in petroleum ether, 20/80 to 66/33). The desired fractions were collected, and the solvents were evaporated in vacuo. The residue thus obtained was added to a mixture of thionyl chloride (10 mL) and CH2Cl2 (10 mL). The mixture was stirred at rt for 2 h. The solvents were then evaporated in vacuo to yield 1 g (87% pure) of 35 as a solid which was used in the next step without any further purification. LCMS: m/z 297 [M + H]+, tR = 1.03 min.
2-Phenylethynyl-5,6-dihydro-4H-benzothiazol-7-one (13)
To a solution of 18 (2.3 g, 9.86 mmol), phenylacetylene (2.01 g, 19.7 mmol), CuI (0.2 g, 1.05 mmol) and Et3N (2.98 g, 29.58 mmol) in 1,4-dioxane (50 mL) at rt was added [1,1′-bis(diphenylphosphino)-ferrocene]dichloro-palladium(II) (0.2 g, 0.25 mmol). The mixture was stirred at reflux for 2 h under nitrogen. The solvent was evaporated in vacuo. The crude product was purified by flash column chromatography (silica gel, EtOAc in petroleum ether, 66/33). The desired fractions were collected, and the solvent was evaporated in vacuo to yield 1.2 g (52%) of 13 as a solid. Mpa 117.1–119.9 °C. 1H NMR (400 MHz, CDCl3) δ ppm 2.28 (quin, J = 6.4 Hz, 2 H), 2.70 (t, J = 6.3 Hz, 2 H), 3.13 (t, J = 6.1 Hz, 2 H), 7.39 − 7.53 (m, 3 H), 7.60–7.70 (m, 2 H). LCMS: m/z 254 [M + H]+, tR = 5.49 min.
2-Phenylethynyl-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one (14a)
To a solution of 21 (2.33 g, 10 mmol), phenylacetylene (2.0 g, 20 mmol) and Et3N (4.5 g, 45 mmol) in 1,4-dioxane (50 mL) at rt under nitrogen were added [1,1′-bis(diphenylphosphino)ferrocene]-dichloro-palladium(II) (0.73 g, 1 mmol) and CuI (0.75 g, 4 mmol). The mixture was stirred at 80 °C for 2 h and then cooled to rt, and the solvent was evaporated in vacuo. The resulting residue was dissolved in EtOAc and washed with H2O. The organic layer was separated, dried (Na2SO4), and filtered, and the solvent was evaporated in vacuo. The crude product was purified by flash column chromatography (silica gel, EtOAc in petroleum ether, 10/90 to 50/50). The desired fractions were collected, and the solvent was evaporated in vacuo to yield 0.7 g (30%) of 14a as a brown solid. Mpa 232.1–235.1 °C. 1H NMR (400 MHz, DMSO-d6) δ ppm 3.00 (t, J = 7.0 Hz, 2 H), 3.50 (td, J = 7.0, 2.5 Hz, 2 H), 7.44–7.57 (m, 3 H), 7.63–7.72 (m, 2 H), 8.07 (br. s, 1 H). LCMS: m/z 255 [M + H]+, tR = 5.00 min.
5-Methyl-2-phenylethynyl-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one (14b)
A mixture of 14a (0.25 g, 0.98 mmol), methyl iodide (0.84 g, 5.9 mmol), and Cs2CO3 (1.9 g, 5.9 mmol) in CH3CN (50 mL) was stirred at 80 °C for 16 h. The mixture was then cooled to rt and concentrated in vacuo. The crude product was purified by reverse phase HPLC (0.1% TFA in CH3CN/0.1% TFA in H2O). The desired fractions were collected, washed with a saturated solution of NaHCO3 and extracted with EtOAc. The organic layer was separated, dried (Na2SO4), and filtered, and the solvent was evaporated in vacuo to yield 50 mg (19%) of 14b. Mpa 146.1–147.2 °C. 1H NMR (400 MHz, DMSO-d6) δ ppm 2.97 (s, 3 H), 3.09 (t, J = 7.2 Hz, 2 H), 3.67 (t, J = 7.2 Hz, 2 H), 7.43–7.58 (m, 3 H), 7.67 (d, J = 6.8 Hz, 2 H). LCMS: m/z 269 [M + H]+, tR = 5.29 min.
5-(2-Methoxy-ethyl)-2-phenylethynyl-6,7-dihydro-5H-thiazolo-[5,4-c]pyridin-4-one (14c)
Starting from 14a (0.7 g, 2.9 mmol) and 2-bromoethyl methyl ether and following the procedure described for 14b, 14c was obtained as a yellow solid (150 mg, 20%). Mpa 144.5–148.2 °C. 1H NMR (300 MHz, CDCl3) δ ppm 3.06 (t, J = 7.0 Hz, 2 H), 3.30 (s, 3 H), 3.49–3.59 (m, 2 H), 3.59–3.68 (m, 2 H), 3.74 (t, J = 7.0 Hz, 2 H), 7.26–7.43 (m, 3 H), 7.45–7.62 (m, 2 H). LCMS: m/z 313 [M + H]+, tR = 6.01 min.
2-Benzyloxy-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one (15a)
Benzyl alcohol (0.145 mL, 1.42 mmol) was added dropwise to a suspension of NaH (0.067 g, 1.67 mmol, 60% in mineral oils) in THF (6 mL) under nitrogen. The mixture was stirred at rt for 15 min and then 21 (0.3 g, 1.29 mmol) was added. The mixture was stirred at 120 °C for 15 min under microwave irradiation in a sealed tube. The solvent was removed with a nitrogen flow to yield 0.340 g (70% pure) of 15a. LCMS: m/z 261 [M + H]+, tR = 2.15 min.
2-Benzyloxy-5-methyl-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one (15b)
15a (0.34 g, 1.29 mmol) was added dropwise to a suspension of NaH (0.067 g, 1.67 mmol, 60% in mineral oils) in THF (2 mL) under nitrogen. The mixture was stirred for 15 min then methyl iodide (0.12 mL, 1.68 mmol). The mixture was stirred at 120 °C for 15 min under microwave irradiation in a sealed tube. The mixture was diluted with EtOAc and washed with a saturated solution of NH4Cl. The organic layer was separated, dried (Na2SO4), and filtered, and the solvent was evaporated in vacuo. The crude product was purified by flash column chromatography (silica gel, EtOAc in CH2Cl2, 0/100 to 10/90). The desired fractions were collected and the solvents evaporated in vacuo. The solid obtained was triturated with heptane/Et2O to yield 0.190 g (54%) of 15b. Mpb 106.4 °C. 1H NMR (500 MHz, CDCl3) δ ppm 2.96 (t, J = 7.2 Hz, 2 H), 3.05 (s, 3 H), 3.60 (t, J = 7.2 Hz, 2 H), 5.45 (s, 2 H), 7.34–7.50 (m, 5 H). LCMS: m/z 275 [M + H]+, tR = 1.68 min.
2-Benzyloxy-5-(4-fluoro-phenyl)-6,7-dihydro-5H-thiazolo[5,4-c]-pyridin-4-one (15c)
Benzyl alcohol (0.38 mL, 3.67 mmol) was added dropwise to a suspension of NaH (0.183 g, 4.58 mmol, 60% in mineral oils) in THF (12 mL) under nitrogen. The mixture was stirred at rt for 15 min and then 26 (1 g, 3.06 mmol) was added. The mixture was stirred at 120 °C for 25 min under microwave irradiation in a sealed tube. The mixture was partitioned between CH2Cl2 and H2O. The organic layer was separated, dried (MgSO4), and filtered, and the solvent was evaporated in vacuo. The crude product was purified by flash column chromatography (silica gel, EtOAc in CH2Cl2 in heptane, 0/0/100 to 10/10/80). The desired fractions were collected and evaporated in vacuo to yield 0.68 g (63%) of 15c as a white solid. Mpb 130.0 °C. 1H NMR (400 MHz, CDCl3) δ ppm 3.08 (t, J = 6.9 Hz, 2 H), 4.01 (t, J = 6.9 Hz, 2 H), 5.49 (s, 2 H), 7.08 (t, J = 8.7 Hz, 2 H), 7.29 (dd, J = 9.0, 4.9 Hz, 2 H), 7.34–7.54 (m, 5 H). 13C NMR (126 MHz, CDCl3) δ ppm 26.56 (s, 1 C) 49.57 (s, 1 C) 73.78 (s, 1 C) 115.68 (d, J = 22.00 Hz, 2 C) 119.25 (s, 1 C) 127.02 (d, J = 8.25 Hz, 2 C) 128.32 (s, 2 C) 128.64 (s, 2 C) 128.78 (s, 1 C) 134.65 (s, 1 C) 138.24 (d, J = 3.67 Hz, 1 C) 155.14 (s, 1 C) 160.62 (d, J = 245.61 Hz, 1 C) 160.68 (s, 1 C) 177.92 (s, 1 C). LCMS: m/z 355 [M+H]+, tR = 2.44 min. HRMS (ES+, M + H) calcd. for C19H16N2O2SF: 355.0917, found 355.0914.
5-Methyl-2-phenoxymethyl-6,7-dihydro-5H-thiazolo[5,4-c]-pyridin-4-one (16a)
Di-tert-butyl azodicarboxylate (1.55 g, 5.90 mmol) was added to a stirred solution of 29 (0.90 g, 4.54 mmol), phenol (0.534 g, 5.68 mmol) and PPh3 (1.55 g, 5.90 mmol) in THF (15 mL) under nitrogen. The mixture was stirred at 120 °C for 20 min under microwave irradiation. The solvent was evaporated in vacuo. The crude product was purified by flash column chromatography (silica gel, EtOAc in CH2Cl2, 0/100 to 50/50). The desired fractions were collected and evaporated in vacuo, and the residue was triturated with diisopropyl ether to yield 0.253 g (20%) of 16a as a white solid. Mpa 141.5–142.4 °C. 1H NMR (500 MHz, CDCl3) δ ppm 3.10 (s, 3 H), 3.14 (t, J = 7.2 Hz, 2 H), 3.67 (t, J = 7.1 Hz, 2 H), 5.33 (s, 2 H), 6.96–7.05 (m, 3 H), 7.28–7.35 (m, 2 H). 13C NMR (126 MHz, CDCl3) δ ppm 25.64 (s, 1 C) 34.14 (s, 1 C) 48.56 (s, 1 C) 67.30 (s, 1 C) 114.85 (s, 2 C) 121.89 (s, 1 C) 127.04 (s, 1 C) 129.56 (s, 2 C) 157.41–157.65 (m, 1 C) 157.69–157.96 (m, 1 C) 160.76–161.07 (m, 1 C) 171.74 (s, 1 C). LCMS: m/z 275 [M + H]+ tR = 1.76 min. HRMS (ES+, M + H) calcd. for C14H15N2O2S: 275.0854, found 275.0853.
5-Ethyl-2-phenoxymethyl-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one (16b)
Iodoethane (0.069 mL, 0.86 mmol) was added to a suspension of 31 (150 mg, 0.058 mmol) and Cs2CO3 (281 mg, 0.86 mmol) in anhydrous DMF (2.5 mL). The mixture was stirred at rt for 16 h under nitrogen then at 100 °C for 1 h. The mixture was diluted with H2O and extracted with EtOAc. The organic layer was separated, washed with brine, dried (Na2SO4), and filtered, and the solvents were evaporated in vacuo. The crude product was purified by flash column chromatography (silica gel, EtOAc in CH2Cl2, 0/100 to 50/50). The desired fractions were collected and the solvents evaporated in vacuo. The impure product was triturated with diisopropyl ether and purified by reverse phase HPLC (gradient elution: 80% 0.1% NH4CO3H/NH4OH pH 9 solution in H2O, 20% CH3CN to 0% 0.1% NH4CO3H/ NH4OH pH 9 solution in H2O, 100% CH3CN). The desired fractions were collected and evaporated in vacuo to yield 62 mg (37%) of 16b as a yellow solid. Mpb 116.0 °C. 1H NMR (400 MHz, CDCl3) δ ppm 1.21 (t, J = 7.2 Hz, 3 H), 3.12 (t, J = 7.1 Hz, 2 H), 3.57 (q, J = 7.2 Hz, 2 H), 3.67 (t, J = 7.2 Hz, 2 H), 5.33 (s, 2 H), 6.95 - 7.07 (m, 3 H), 7.28 - 7.36 (m, 2 H). 13C NMR (101 MHz, CDCl3) δ ppm 12.79 (s, 1 C) 25.90 (s, 1 C) 41.35 (s, 1 C) 45.97 (s, 1 C) 67.30 (s, 1 C) 114.87 (s, 2 C) 121.92 (s, 1 C) 127.46 (s, 1 C) 129.61 (s, 2 C) 157.54 (s, 1 C) 157.82 (s, 1 C) 160.31 (s, 1 C) 171.75 (s, 1 C). LCMS: m/z 289 [M + H]+, tR = 2.00 min. HRMS (ES+, M + H) calcd. for C15H17N2O2S: 289.1011, found 289.1013.
5-Isopropyl-2-phenoxymethyl-6,7-dihydro-5H-thiazolo[5,4-c]-pyridin-4-one (16c)
A mixture of 25a (0.23 g, 1.29 mmol) and 33a (0.24 g, 1.4 mmol) in EtOH (10 mL) was stirred at rt for 15 min before NaHCO3 (0.4 g, 3.9 mmol) was added. Then the mixture was stirred at 70 °C for 15 min, diluted with EtOAc, and washed with H2O. The organic layer was separated, washed with brine, dried (Na2SO4), and filtered, and the solvents were evaporated in vacuo. The crude product was purified by reverse phase HPLC (gradient elution: 0.1% TFA in CH3CN/0.1% TFA in H2O). The desired fractions were collected and washed with a saturated solution of NaHCO3 and extracted with EtOAc (2 × 100 mL). The organic layer was separated, washed with brine, dried (Na2SO4), filtered and the solvents evaporated in vacuo to yield 81 mg (21%) of 16c as a white solid. Mpa 104.1–106.0 °C. 1H NMR (400 MHz, CDCl3) δ ppm 1.13 (d, J = 6.8 Hz, 6 H), 3.00 (t, J = 7.0 Hz, 2 H), 3.49 (t, J = 7.0 Hz, 2 H), 4.87 (spt, J = 6.8 Hz, 1 H), 5.26 (s, 2 H), 6.86–7.01 (m, 3 H), 7.21– 7.31 (m, 2 H). LCMS: m/z 303 [M + H]+, tR = 5.42 min.
5-(2-Methoxy-ethyl)-2-phenoxymethyl-6,7-dihydro-5H-thiazolo-[5,4-c]pyridin-4-one (16d)
Starting from 31 (150 mg, 0.58 mmol) and 2-bromoethyl methyl ether and following the procedure described for 16b, 16d was obtained as a yellow solid (90 mg, 49%). Mpb 82.6 °C. 1H NMR (500 MHz, CDCl3) δ ppm 3.10 (t, J = 7.1 Hz, 2 H), 3.36 (s, 3 H), 3.60 (t, J = 5.2 Hz, 2 H), 3.69 (t, J = 5.2 Hz, 2 H), 3.78 (t, J = 7.2 Hz, 2 H), 5.33 (s, 2 H), 6.97–7.04 (m, 3 H), 7.28–7.35 (m, 2 H). LCMS: m/z 319 [M + H]+, tR = 1.90 min.
5-Cyclopropyl-2-phenoxymethyl-6,7-dihydro-5H-thiazolo[5,4-c]-pyridin-4-one (16e)
Starting from 25b (0.28 g, 1.2 mmol) and 33a (0.23 g, 1.4 mmol) and following the procedure described for 16c, 16e was obtained as a white solid (72 mg, 20%). Mpa 252.1–255.3 °C. 1H NMR (400 MHz, CDCl3) δ ppm 0.62 - 0.69 (m, 2 H), 0.80 - 0.88 (m, 2 H), 2.65 (tt, J = 7.2, 3.7 Hz, 1 H), 2.99 (t, J = 7.0 Hz, 2 H), 3.63 (t, J = 7.0 Hz, 2 H), 5.25 (s, 2 H), 6.89–6.99 (m, 3 H), 7.21–7.29 (m, 2 H). LCMS: m/z 301 [M + H]+, tR = 4.86 min.
5-Cyclopropylmethyl-2-phenoxymethyl-6,7-dihydro-5H-thiazolo-[5,4-c]pyridin-4-one (16f)
Starting from 25c (250 mg, 1.01 mmol) and 33a (0.2 g, 1.22 mmol) and following the procedure described for 16c, 16f was obtained as a white solid (78 mg, 25%). Mpa 292.6–302.9 °C. 1H NMR (400 MHz, CDCl3) δ ppm 0.23 (q, J = 4.9 Hz, 2 H), 0.40 - 0.56 (m, 2 H), 0.87 - 1.06 (m, 1 H), 3.08 (t, J = 7.2 Hz, 2 H), 3.35 (d, J = 7.0 Hz, 2 H), 3.72 (t, J = 7.2 Hz, 2 H), 5.28 (s, 2 H), 6.86 - 7.02 (m, 3 H), 7.21 - 7.33 (m, 2 H). 13C NMR (101 MHz, CDCl3) δ ppm 3.49 (s, 2 C) 9.53 (s, 1 C) 25.89 (s, 1 C) 46.52 (s, 1 C) 50.68 (s, 1 C) 67.32 (s, 1 C) 114.89 (s, 2 C) 121.93 (s, 1 C) 127.46 (s, 1 C) 129.62 (s, 2 C) 157.55 (s, 1 C) 157.85 (s, 1 C) 160.56 (s, 1 C) 171.80 (s, 1 C). LCMS: m/z 303 [M + H]+, tR = 5.32 min. HRMS (ES+, M + H) calcd. for C17H19N2O2S: 315.1167, found 315.1169.
2-Phenoxymethyl-5-(tetrahydro-pyran-4-ylmethyl)-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one (16g)
Starting from 31 (150 mg, 0.58 mmol) and 4-(bromomethyl)tetrahydropyran and following the procedure described for 16a, 16g was obtained as a white solid (27 mg, 13%). 1H NMR (400 MHz, CDCl3) δ ppm 1.34–1.48 (m, 2 H), 1.59–1.68 (m, 2 H), 1.90–2.04 (m, 1 H), 3.11 (t, J = 7.0 Hz, 2 H), 3.32–3.42 (m, 2 H), 3.40 (d, J = 7.4 Hz, 2 H), 3.69 (t, J = 7.0 Hz, 2 H), 3.94–4.03 (m, 2 H), 5.33 (s, 2 H), 6.97–7.05 (m, 3 H), 7.28–7.35 (m, 2 H). LCMS: m/z 359 [M + H]+, tR = 2.10 min.
5-(4-Fluoro-phenyl)-2-phenoxymethyl-6,7-dihydro-5H-thiazolo-[5,4-c]pyridin-4-one (16h)
A mixture of 25d (0.41 g, 1.45 mmol) and 33a (0.22 g, 1.3 mmol) in DMF (5 mL) was stirred at rt for 15 min before NaHCO3 (0.19 g, 2.3 mmol) was added. Then the mixture was stirred at 100 °C for 30 min, diluted with EtOAc and washed with H2O. The organic layer was separated, dried (MgSO4), and filtered, and the solvents were evaporated in vacuo. The crude product was purified by flash column chromatography (silica gel, EtOAc in CH2Cl2, 0/100 to 2/98). The desired fractions were collected, and the solvents were evaporated in vacuo to yield 0.084 g (16%) of 16h as a white solid. Mpb 163.3 °C. 1H NMR (500 MHz, CDCl3) δ ppm 3.26 (t, J = 6.9 Hz, 2 H), 4.08 (t, J = 6.9 Hz, 2 H), 5.36 (s, 2 H), 6.98–7.06 (m, 3 H), 7.07–7.13 (m, 2 H), 7.28–7.36 (m, 4 H). 13C NMR (126 MHz, CDCl3) δ ppm 26.32 (s, 1 C) 50.16 (s, 1 C) 67.37 (s, 1 C) 114.87 (s, 2 C) 115.82 (d, J = 22.91 Hz, 2 C) 122.01 (s, 1 C) 127.15 (d, J = 8.25 Hz, 2 C) 127.38 (s, 1 C) 129.64 (s, 2 C) 137.93 (d, J = 3.67 Hz, 1 C) 157.50 (s, 1 C) 158.79 (s, 1 C) 160.79 (d, J = 246.52 Hz, 1 C) 160.36 (s, 1 C) 172.95 (s, 1 C). LCMS: m/z 355 [M + H]+, tR = 6.57 min. HRMS (ES+, M + H) calcd. for C19H16N2O2SF: 355.0917, found 355.0916.
5-(2,4-Difluoro-phenyl)-2-phenoxymethyl-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one (16i)
Starting from 25e (300 mg, 0.99 mmol) and 33a (148 mg, 0.89 mmol) and following the procedure described for 16h, 16i was obtained as a yellow solid (37 mg, 10%). Mpa 268.9–284.1 °C. 1H NMR (400 MHz, CDCl3) δ ppm 3.28 (t, J = 6.9 Hz, 2 H), 4.01 (t, J = 6.8 Hz, 2 H), 5.37 (s, 2 H), 6.89–6.99 (m, 2 H), 6.99–7.07 (m, 3 H), 7.29–7.39 (m, 3 H). LCMS: m/z 373 [M + H]+, tR = 5.63 min.
2-(2-Fluoro-phenoxymethyl)-5-(4-fluoro-phenyl)-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one (16j)
A mixture of 35 (0.3 g, 1.01 mmol), 2-fluorophenol (0.15 g, 1.3 mmol) and K2CO3 (0.4 g, 3 mmol) in DMF (40 mL) was stirred at rt for 1 h. The mixture was filtered and the solvent evaporated in vacuo. The crude product was purified by reverse phase HPLC (gradient elution: 0.1% TFA in CH3CN/0.1% TFA in H2O). The desired fractions were collected and washed with a saturated solution of NaHCO3 and extracted with EtOAc (2 × 100 mL). The organic layer was separated, dried (Na2SO4), filtered and the solvent evaporated in vacuo to yield 31 mg (8%) of 16j as a solid. Mpa > 280 °C. 1H NMR (300 MHz, CDCl3) δ ppm 3.25 (t, J = 6.9 Hz, 2 H), 4.07 (t, J = 6.8 Hz, 2 H), 5.41 (s, 2 H), 6.92–7.19 (m, 6 H), 7.19–7.42 (m, 2 H). LCMS: m/z 373 [M + H]+, tR = 4.58 min.
2-(3-Fluoro-phenoxymethyl)-5-(4-fluoro-phenyl)-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one (16k)
Starting from 25d (0.39 g, 1.38 mmol) and 33b (230 mg, 1.24 mmol) and following the procedure described for 16h, 16k was obtained as a white solid (120 mg, 23%). Mpb 152.8 °C. 1H NMR (500 MHz, CDCl3) δ ppm 3.27 (t, J = 6.9 Hz, 2 H), 4.09 (t, J = 6.9 Hz, 2 H), 5.35 (s, 2 H), 6.69–6.84 (m, 2 H), 7.07–7.16 (m, 2 H), 7.23–7.35 (m, 4 H). LCMS: m/z 372 [M + H]+, tR = 2.88 min.
2-(4-Fluoro-phenoxymethyl)-5-(4-fluoro-phenyl)-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one (16l)
Starting from 35 (0.3 g, 1.01 mmol) and 4-fluorophenol (0.15 g, 1.3 mmol) and following the procedure described for 16j, 16l was obtained as a solid (74 mg, 20%). Mpa > 280 °C. 1H NMR (300 MHz, CDCl3) δ ppm 3.25 (t, J = 6.7 Hz, 2 H), 4.07 (t, J = 6.8 Hz, 2 H), 5.31 (s, 2 H), 6.85–7.05 (m, 4 H), 7.09 (br. t, J = 8.4, 8.4 Hz, 2 H), 7.26–7.35 (m, 2 H). LCMS: m/z 373 [M + H]+, tR = 4.62 min.
2-Phenoxymethyl-5-pyridin-2-yl-6,7-dihydro-5H-thiazolo[5,4-c]-pyridin-4-one (16m)
K2CO3 (111 mg, 0.81 mmol) was added to a stirred suspension of 31 (150 mg, 0.4 mmol), 2-iodopyridine (0.043 mL, 0.4 mmol), CuI (0.015 g, 0.081 mmol) and N,N′-dimethylethylenediamine (0.026 mL, 0.24 mmol) in toluene (3 mL) in a sealed tube and under nitrogen. The mixture was stirred at 120 °C for 16 h, filtered through a Celite pad, and washed with EtOAc and the filtrate was evaporated in vacuo. The crude product was purified by flash column chromatography (silica gel, EtOAc in CH2Cl2, 0/100 to 100/0). The desired fractions were collected and the solvents were evaporated in vacuo. The solid was triturated with diisopropyl ether to yield 57 mg (57%) of 16m as a white solid. Mpb 143.9 °C. 1H NMR (400 MHz, CDCl3) δ ppm 3.23 (t, J = 6.8 Hz, 2 H), 4.47 (t, J = 6.8 Hz, 2 H), 5.37 (s, 2 H), 7.00–7.06 (m, 3 H), 7.07–7.13 (m, 1 H), 7.29–7.37 (m, 2 H), 7.71 (ddd, J = 8.6, 7.1, 1.8 Hz, 1 H), 7.89 (d, J = 8.3 Hz, 1 H), 8.44 (br. s, 1 H). LCMS: m/z 338 [M + H]+, tR = 2.42 min.
2-Phenoxymethyl-5-pyridin-3-yl-6,7-dihydro-5H-thiazolo[5,4-c]-pyridin-4-one (16n)
Starting from 31 (150 mg, 0.58 mmol) and 3-bromopyridine and following the procedure described for 16m, 16n was obtained as a white solid (29 mg, 15%). 1H NMR (400 MHz, CDCl3) δ ppm 3.30 (t, J = 6.8 Hz, 2 H), 4.17 (t, J = 6.8 Hz, 2 H), 5.37 (s, 2 H), 6.96–7.09 (m, 3 H), 7.29–7.41 (m, 3 H), 7.71–7.80 (m, 1 H), 8.50 (dd, J = 4.7, 1.3 Hz, 1 H), 8.65 (d, J = 2.3 Hz, 1 H). LCMS: m/z 338 [M + H]+, tR = 2.00 min.
2-Phenoxymethyl-5-pyridin-4-yl-6,7-dihydro-5H-thiazolo[5,4-c]-pyridin-4-one (16o)
Starting from 31 (150 mg, 0.58 mmol) and 4-iodopyridine and following the procedure described for 16m, 16o was obtained as a white solid (5 mg, 2.5%). 1H NMR (500 MHz, CDCl3) δ ppm 3.29 (t, J = 6.8 Hz, 2 H), 4.19 (t, J = 6.8 Hz, 2 H), 5.37 (s, 2 H), 6.90–7.12 (m, 3 H), 7.29 - 7.41 (m, 4 H), 8.62 (d, J = 6.4 Hz, 2 H). LCMS: m/z 338 [M + H]+, tR = 2.09 min.
5-(5-Fluoro-pyridin-2-yl)-2-phenoxymethyl-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one (16p)
K3PO4 (1.6 g, 7.49 mmol) was added to a stirred suspension of 31 (650 mg, 2.5 mmol), 2-bromo-5-fluoropyridine (879 mg, 4.99 mmol), CuI (475 mg, 2.5 mmol) and N,N′-dimethylethylenediamine (0.81 mL, 7.49 mmol) in toluene (12 mL) in a sealed tube and under nitrogen. The mixture was stirred at 140 °C for 16 h and then filtered through a Celite pad. The filtrate was evaporated in vacuo. The crude product was purified by flash column chromatography (silica gel, EtOAc in CH2Cl2, 0/100 to 30/70). The desired fractions were collected and the solvents evaporated in vacuo. The solid was triturated with diisopropyl ether to yield 570 mg (64%) of 16p as a white solid. Mpa 131.7–132.8 °C. 1H NMR (500 MHz, CDCl3) δ ppm 3.23 (t, J = 6.8 Hz, 2 H), 4.42 (t, J = 6.9 Hz, 2 H), 5.37 (s, 2 H), 6.86–7.12 (m, 3 H), 7.29–7.38 (m, 2 H), 7.39 - 7.50 (m, 1 H), 7.89 (dd, J = 9.2, 4.0 Hz, 1 H), 8.27 (d, J = 3.2 Hz, 1 H). 13C NMR (101 MHz, CDCl3) δ ppm 26.15 (s, 1 C) 46.39 (s, 1 C) 67.41 (s, 1 C) 114.83 (s, 2 C) 120.72 (d, J = 4.24 Hz, 1 C) 122.02 (s, 2 C) 124.21 (d, J = 19.78 Hz, 1 C) 127.41 (s, 1 C) 129.65 (s, 2 C) 134.92 (d, J = 25.43 Hz, 1 C) 149.26 (d, J = 2.12 Hz, 1 C) 158.76 (d, J = 259.96 Hz, 1 C) 157.89 (s, 1 C) 160.67 (s, 1 C) 173.65 (s, 1 C). LCMS: m/z 356 [M + H]+, tR = 5.99 min. HRMS (ES+, M + H) calcd. for C18H15N3O2SF: 356.0869, found 356.0870.
5-(3-Fluoro-pyridin-2-yl)-2-phenoxymethyl-6,7-dihydro-5H-thiazolo[5,4-c]pyridin-4-one (16q)
Starting from 31 (150 mg, 0.58 mmol) and 2-chloro-3-fluoropyridine and following the procedure described for 16m, 16q was obtained as a white solid (22 mg, 15%). 1H NMR (400 MHz, CDCl3) δ ppm 3.29 (t, J = 6.8 Hz, 2 H), 4.25 (t, J = 6.8 Hz, 2 H), 5.38 (s, 2 H), 6.98–7.06 (m, 3 H), 7.23–7.29 (m, 1 H), 7.30 - 7.40 (m, 2 H), 7.51 (ddd, J = 9.7, 8.1, 1.6 Hz, 1 H), 8.31 (dt, J = 4.8, 1.2 Hz, 1 H). LCMS: m/z 356 [M + H]+, tR = 2.37 min.
Biology
Cell Culture
Human embryonic kidney (HEK) 293 cells were stably transfected with human mGluR5a cDNA in expression vector pcDNA4/TO. hmGlu5 receptor-expressing HEK293 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat inactivated dialyzed fetal bovine serum and antibiotics (88 IU/mL penicillin G and 88 μg/mL streptomycin sulfate). Cells were detached with PBS w/o Ca2+/Mg2+ containing 0.04% EDTA and 0.005% trypsin. The cells were grown in 175 cm2 culture flasks at 37 °C in a 5% CO2 environment and hold under permanent selection with 200 μg/mL zeocin and 5 μg/mL blasticidin. Cells were cultured at 40 000 cells/cm2 or 20 000 cells/cm2 for respectively 3 or 4 days of growth in a F175 flask.
Calcium Mobilization Assay
Cells were seeded at 40 000 cells/well in PDL-coated MW384, and a day later, medium was removed before addition of a fluo-4-AM solution (HBSS, 2.5 mM probenecid and ~2 μM fluo-4-AM, pH 7.4). Measurements were performed 90 min later (while incubation at 37 °C and 5% CO2) using the FDSS. Two additions were made: first, compound alone was added at 6 s to detect any agonist activity. Next, a suboptimal concentration (nominally 20% of the maximum response, EC20) of glutamate was added at 157 s to detect any potentiation of the glutamate response. For each interval, Ca2+ fluorescence of the Ca2+-bound dye was monitored every second, at an excitation wavelength of 480 nm and emission at 540 nm. The ratio of peak versus basal fluorescent signals was used for data analysis.
Data Analysis
Data were normalized to the control agonist response and sigmoid concentration–response curves plotting these percentages versus the log concentration of the test compound were analyzed using a 4 parameter logistic nonlinear regression model programmed in R (R Development Core Team, 2010. R: A language and environment for statistical computing. R Foundation for Statistical Computing,Vienna, Austria. ISBN 3–900051–07–0, URL http://www.R-project.org/.). The model was determined by the maximal effect of each compound (Emax), its EC50 and the response in the absence of compound. The fourth parameter determining the slope was fixed to 1. In addition, the heterogeneity between experiments and concentration–response curves for each compound was taken into account by the inclusion of a random effect in both ‘E0’ (response in the absence of compound) and Emax of the model
In Vivo Pharmacology
(a) Animal Husbandry
Animals were housed in the animal care facility certified by the American Association for the Accreditation of Laboratory Animal Care (AAALAC) under a 12-h light/dark cycle (lights on: 7 a.m.; lights off: 7 p.m.) and had free access to food and water. The animals used in these experiments were food-deprived the evening before experimentation for oral administration of test compound. The experimental protocols performed during the light cycle were approved by the Institutional Animals Care and Use Committee of Vanderbilt University and conformed to the guidelines established by the National Research Council Guide for the Care and Use of Laboratory Animals.
(b) Preparation of Test Article
16a was formulated in volumes specific to the number of animals dosed each day. The solutions were formulated so that animals were injected with a maximal dosing volume of 10 mL/kg. The appropriate amount according to the dosage was mixed into a 20% 2-(hydroxypropyl)-β-cyclodextrin in sterile water (HP-β-CD; Sigma, Cat. No. C0926-10G) solution. Each mixture was ultrahomogenized on ice for 2–3 min using a hand-held tissue homogenizer. Next, the pH of all solutions was checked using 0–14 EMD pH strips and adjusted to a pH of 6–7 if necessary with 1N NaOH. The mixtures were then vortexed, and stored in a sonication bath at 40 °C until time of injection. d-Amphetamine hemisulfate (AMP) was obtained from Sigma (Cat. No. A5880-1G; St. Louis, MO). Salt-correction was used to determine the correct amount of the d-amphetamine hemisulfate form in mg to add to sterile water in order to yield a 1 mg/mL solution; injected with a maximal dosing volume of 10 mL/kg.
Reversal of Amphetamine-Induced Hyperlocomotion
Studies were conducted using Smart Open Field activity chambers (27 × 27 × 20 cm) (Kinder Scientific, Poway, CA) equipped with 16 horizontal (x- and y-axes) infrared photobeams. Changes in locomotor activity were measured as the number of photobeam breaks over time and were recorded with a Pentium I computer equipped with rat activity monitoring system software (Motor Monitor, Kinder Scientific, Poway, CA). Male Harlan Sprague–Dawley rats (Harlan Laboratories, Indianapolis, IN) weighing 250–375 g were used. Animals were habituated in the locomotor activity test chambers for 30 min. Animals were next pretreated for an additional 30 min with either vehicle or a dose of 16a p.o., followed by a subcutaneous injection of 1 mg/kg amphetamine or vehicle and monitored for an additional 60 min.
Treatment groups
Dose group 1: VAMP = 20% β-CD (16a vehicle), p.o. +1 mg/kg AMP, sc (n = 8). Dose group 2: 3AMP = 3 mg/kg 16a, p.o. +1 mg/kg AMP, sc (n = 6). Dose group 3: 10AMP = 10 mg/kg 16a, po +1 mg/kg AMP, sc (n = 7). Dose group 4: 30AMP = 30 mg/kg 16a, p.o. +1 mg/kg AMP, sc (n = 8). Dose group 5: 56.6AMP = 56.6 mg/kg 16a, p.o. + 1 mg/kg AMP, sc (n = 8). Dose group 6: 100AMP = 100 mg/kg 16a, p.o. +1 mg/kg AMP, sc (n = 8). Dose group 7: 100 V = 100 mg/kg 16a, p.o. + sterile water (AMP vehicle), sc (n = 8). Changes in locomotor activity were recorded for a total of 120 min. Data were expressed as changes in ambulation defined as the total number of photobeam breaks per 5 min interval. At the end of this behavioral study, each animal was euthanized, then decapitated, and the plasma and brain tissues were collected for the evaluation of exposure levels of 16a. Data Analysis. Behavioral data were analyzed using a one-way ANOVA with main effects of treatment and time. Post hoc analyses were performed using a Dunnett's t-test with all treatment groups compared to the VAMP group using JMP 8.0 (SAS Institute, Cary, NC) statistical software. Data were graphed using SigmaPlot for Windows Version 11.0 (Saugua, MA). A probability of p ≤ 0.05 was taken as the level of statistical significance. Percent Effect and Reversal calculations for the 3AMP, 10AMP, 30AMP, 56.6AMP, and 100AMP treatment groups were performed with the following formula relative to the VAMP treatment group: (1) Total number of photobeam breaks in the time interval from t = 60 to t = 120 was calculated for each rat in each treatment group; (2) Mean total number of photobeam breaks in the time interval from t = 60 to t = 120 was calculated for the VAMP group; (3) Percent effect = ratio of the total number of photobeam breaks for each rat in each treatment group divided by the mean total number of photobeam breaks in the time interval from t = 60 to t = 120 of the VAMP group multiplied by 100; (4) Percent reversal = 100-the percent effect for each rat in each treatment group; (5) finally the mean ± SE percent reversal for each treatment group was calculated from the individual percent reversal values. Percent effect and change calculations for the VAMP and 100 V treatment groups relative to the VV treatment group were also performed with the following formula: (1) Total number of photobeam breaks in the time interval from t = 60 to t = 120 was calculated for each rat in each treatment group; (2) Mean total number of photobeam breaks in the time interval from t = 60 to t = 120 was calculated for the VV group; (3) Percent Effect = Ratio of the total number of photobeam breaks for each rat in each treatment group divided by the mean total number of photobeam breaks in the time interval from t = 60 to t = 120 of the VV group multiplied by 100; (4) percent change = the percent effect for each rat in each treatment group –100; (5) finally the Mean ± SE percent change for each treatment group was calculated from the individual percent change values. LC-MS/MS Sample Preparation and Analysis Methodology. Brain samples were homogenized in 3 mL of 70:30 isopropanol/water and then centrifuged at 3500 rpm for 5 min. Resulting brain sample supernatant and plasma samples were transferred into a 96-well plate containing plasma blanks, plasma double blank, a standard curve of the test article, and quality control samples for subsequent LC-MS/MS analysis. Each sample was diluted with acetonitrile containing 50 nM carbamazepine (internal standard), centrifuged at 3500 rpm, and the resulting supernatant was transferred to a new 96-well plate. After an equal volume of water was added to each sample (to provide 50:50 acetonitrile/water solution), the plate was analyzed via ESI on a triple-quadrupole mass spectrometer (AB Sciex API-4000) coupled with an autosampling liquid chromatography system (Shimadzu LC-10AD pumps, Leap Technologies CTC PAL autosampler). Analyte (test article) was separated by gradient elution using a C18 3.0 × 50 mm, 3 μm column (Fortis Technologies) thermostatted at 40 °C. HPLC mobile phase A was 0.1% formic acid in water (pH unadjusted), mobile phase B was 0.1% formic acid in acetonitrile (pH unadjusted), and the gradient used was 30–90% mobile phase B with 0.2 min hold at 30% B, linear increase to 90% B over 0.8 min, hold at 90% B for 0.6 min, and a return to 30% B over 0.1 min followed by a re-equilibration (0.5 min) to provide a 2.0 min total run time per sample. HPLC flow rate was 0.5 mL/min, source temperature was 500 °C, and mass spectral analyses were performed using the following MRM transitions: 353.5 → 259.2 and 353.5 → 123.2 m/z. ESI was achieved by a Turbo-Ionspray source in positive ionization mode (5.0 kV spray voltage). The raw plasma and brain concentration data obtained by LC-MS/MS were analyzed by Analyst software (AB Sciex, version 1.5.1), which used the ratio of peak area responses of drug relative to internal standard to construct a standard curve with a dynamic range covering the concentrations found in the samples. Free plasma and brain concentrations were calculated by multiplication of the total concentration values by fu plasma and fu brain, respectively, based on data from in vitro rat plasma protein and rat brain homogenate binding experiments.
Rotarod
The effects of 16a on motor performance were evaluated using an automated rotarod setup with a rotarod (7.0 cm in diameter) rotating at a constant speed of 20 rotations/min (MedAssociates, Inc., St Albans, CA). Male Harlan Sprague–Dawley rats (Harlan Laboratories, Indianapolis, IN) weighing 250–300 g were used. Animals were given two training trials of 120 s on the rotarod with a 10 min interval between trials, followed by baseline assessment of performance with a 120 s trial, any animals that did not reach a performance criteria of 85 s were excluded from the study. Animals were next pretreated for 30 min with vehicle (20% HP-β-CD) or a dose of 16a (n = 6 each group) and then tested on the rotarod using a 120 s trial. The amount of time in s that each animal remained on the rotarod was recorded; animals not falling off of the rotarod were given a maximal score of 120 s. Data were expressed as the mean latency to fall off the rotarod in seconds for each treatment group. Data Analysis. Behavioral data were analyzed using a one-way ANOVA with main effects of treatment. Post hoc analyses were performed using a Dunnett's t test with all treatment groups compared to the vehicle group using JMP 8.0 (SAS Institute, Cary, NC) statistical software. Data were graphed using SigmaPlot for Windows Version 11.0 (Saugua, MA). A probability of p ≤ 0.05 was taken as the level of statistical significance.
Modified Irwin Neurological Test Battery
Male Harlan Sprague–Dawley rats (Harlan Laboratories, Indianapolis, IN) weighing 250– 300 g were used. Animals were evaluated in the modified Irwin neurological test battery at t = 0 to provide a baseline measurement across each functional end point. Animals were next administered either vehicle (20% HP-β-CD) or a 100 mg/kg p.o. dose of 16a (n = 6 each group) and then assessed after a 30 min, 1 h, and 4 h pretreatment interval in the modified Irwin neurological test battery. Changes in the different functional end points of the Irwin test battery were given a score of 0, 1 or 2; with 0 = no effect, 1 = moderate effect, and 2 = robust full effect. All data were collected blinded to treatment for each animal. Following functional end points were scored. Autonomic nervous system functions: ptosis, exophthalmos, miosis, mydriasis, corneal reflex, pinna reflex, piloerection, respiratory rate, writhing, tail erection, lacrimation, salivation, vasodilation, skin color, irritability and rectal temperature; Somatomotor nervous system functions: motor activity, ataxia, arch/roll, tremors, leg weakness, rigid stance, spraddle, placing loss, grasping loss, righting loss, catalepsy, tail pinch reaction, escape loss and physical appearance. Data Analysis. Mean change values for each functional end point in the Irwin test battery of each treatment group were calculated using Microsoft Excel. Behavioral data were then analyzed using a one-way ANOVA with main effect of treatment. Post hoc analyses were performed using a Dunnett's t test with all treatment groups compared to the vehicle group using JMP 8.0 (SAS Institute, Cary, NC) statistical software. Data were graphed using SigmaPlot for Windows Version 11.0 (Saugua, MA). A probability of p ≤ 0.05 was taken as the level of statistical significance.
Supplementary Material
Figure 1.

mGlu5 receptor PAMs with reported efficacy in preclinical models of schizophrenia and cognition.
ACKNOWLEDGMENTS
Vanderbilt Center for Neuroscience Drug Discovery (VCNDD) research was supported by grants from Janssen Pharmaceutical Companies of Johnson & Johnson and in part by the NIH (NS031373, MH062646). The authors would like to thank Dr. Lieve Heylen and Dr. Tom Jacobs for their help with data management and interpretation, Dr. Han Min Tong for her assistance in the preparation of this manuscript, and the members of the purification and analysis group and the biology and ADME/Tox teams from Janssen R&D for their experimental contribution.
ABBREVIATIONS USED
- AMP
d-amphetamine hemisulfate
- AUC
area under the curve
- b/p
brain to plasma ratio
- chromanone
2,3-dihydro-4H-1-benzopyran-4-one
- CLp
plasma clearance
- Cmax
peak plasma concentration
- CNS
central nervous system
- CRC
concentration response curve
- D2
dopamine 2
- DAD
diode array detector
- dihydrobenzothiazolone
5,6-dihydro-7(4H)-benzothiazolone
- dihydrothiazolopyridone
6,7-dihydro-thiazolo[5,4-c]pyridin-4(5H)-one
- DMPK
drug metabolism and pharmacokinetic
- Ehep
hepatic extraction ratio
- EPS
extrapyramidal symptoms
- ESCI
electrospray combined with atmospheric pressure chemical ionization
- fu
fraction unbound
- GABAergic
γ-aminobutiric acidergic
- HEK
human embryonic kidney
- HLM
human liver microsome
- HP-β-CD
2-hydroxylpropyl-β-cyclodextrin
- HPLC
high-performance liquid chromatography
- HTS
high throughput screening
- iv
intravenous
- LCHRMS
liquid chromatography combined with high resolution mass spectrometry
- LCMS
liquid chromatography combined with mass spectrometry
- LE
ligand efficiency
- Mp
melting point
- NMDA
N-methyl-d-aspartate
- mGlu
metabotropic glutamate
- NMR
nuclear magnetic resonance (NMR)
- mGlu5
metabotropic glutamate 5
- PAM
positive allosteric modulator
- PK
pharmacokinetic
- PKC
protein kinase C
- ppm
parts per million
- p.o.
oral
- RLM
rat liver microsome
- RPHPLC
reversed-phase high-performance liquid chromatography
- SAR
structure activity relationship
- sc
subcutaneous
- SPR
structure properties relationship
- T1/2
half-life
- Tmax
tR
- retention time
time to reach peak concentration
- TLC
thin layer chromatography
- VCNDD
Vanderbilt Center for Neuroscience Drug Discovery
Footnotes
Supporting Information
The different LCMS methods used for the characterization of intermediate and final compounds. Mean scores of 16a effects on autonomic and somatomotor nervous system functions in rats. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.Tandon R, Nasrallah HA, Keshavan MS. Schizophrenia, “Just the Facts” 4. Clinical Features and Conceptualization. Schizophr. Res. 2009;110:1–23. doi: 10.1016/j.schres.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 2.Conn PJ, Lindsley CW, Jones CK. Activation of Metabotropic Glutamate Receptors as a Novel Approach for the Treatment of Schizophrenia. Trends Pharmacol. Sci. 2009;30:148–155. doi: 10.1016/j.tips.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Macdonald GJ, Bartolome JM. A Decade of Progress in the Discovery and Development of ‘Atypical’ Antipsychotics. Prog. Med. Chem. 2010;49:37–80. doi: 10.1016/S0079-6468(10)49002-5. [DOI] [PubMed] [Google Scholar]
- 4.Olney JW, Farber NB. NMDA Antagonists as Neuro-therapeutic Drugs, Psychotogens, Neurotoxins, and Research Tools for Studying Schizophrenia. Arch. Gen. Psychiatry. 1995;52:998–1007. doi: 10.1016/0893-133X(95)00079-S. [DOI] [PubMed] [Google Scholar]
- 5.Nakanishi S. Molecular Diversity of Glutamate Receptors and Implications for Brain Functions. Science. 1992;258:597–603. doi: 10.1126/science.1329206. [DOI] [PubMed] [Google Scholar]
- 6.Shigemoto R, Mizuno N. Metabotropic Glutamate Receptors − Immunocytochemical and In Situ Hybridization Analysis. In: Ottersen OP, Storm-Mathisen J, editors. Handbook of Chemical Neuroanatomy. Elsevier; Amsterdam: 2000. pp. 63–98. [Google Scholar]
- 7.Cauli B, Porter JT, Tsuzuki K, Lambolez B, Rossier J, Quenet B, Audinat E. Classification of Fusiform Neocortical Interneurons Based on Unsupervised Clustering. Proc. Natl. Acad. Sci. U. S. A. 2000;97:6144–6149. doi: 10.1073/pnas.97.11.6144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Perroy J, Raynaud F, Homburger V, Rousset M-C, Telley L, Bockaert J, Fagni L. Direct Interaction Enables Cross-talk between Ionotropic and Group I Metabotropic Glutamate Receptors. J. Biol. Chem. 2008;283:6799–6805. doi: 10.1074/jbc.M705661200. [DOI] [PubMed] [Google Scholar]
- 9.Stauffer SR. Progress toward Positive Allosteric Modulators of the Metabotropic Glutamate Receptor Subtype 5 (mGluR5). ACS Chem. Neurosci. 2011;2:450–470. doi: 10.1021/cn2000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lindsley CW, Wisnoski DD, Leister WH, O'Brien JA, Lemiare W, Williams DL, Jr., Burno M, Sur C, Kinney GG, Pettibone DJ, Miller PR, Smith S, Duggan ME, Hartman GD, Conn PJ, Huff JR. Discovery of Positive Allosteric Modulators for the Metabotropic Glutamate Receptor Subtype 5 from a Series of N-(1,3-Diphenyl-1H-pyrazol-5-yl)benzamides That Potentiate Receptor Function In Vivo. J. Med. Chem. 2004;47:5825–5828. doi: 10.1021/jm049400d. [DOI] [PubMed] [Google Scholar]
- 11.Kinney GG, O'Brien JA, Lemaire W, Burno M, Bickel DJ, Clements MK, Wisnoski DD, Lindsley CW, Tiller PR, Smith S, Jacobson MA, Sur C, Duggan ME, Pettibone DJ, Williams DW., Jr. A Novel Selective Positive Allosteric Modulator of Metabotropic Glutamate Receptor Subtype 5 Has In Vivo Activity and Antipsychotic-like Effects in Rat Behavioral Models. J. Pharmacol. Exp. Ther. 2005;313:199–206. doi: 10.1124/jpet.104.079244. [DOI] [PubMed] [Google Scholar]
- 12.Liu F, Grauer S, Kelley C, Navarra R, Graf R, Zhang G, Atkinson PJ, Popiolek M, Wantuch C, Khawaja X, Smith D, Olsen M, Kouranova E, Lai M, Pruthi F, Pulcicchio C, Day M, Gilbert A, Pausch MH, Brandon NJ, Beyer CE, Comery TA, Logue S, Rosenzweig-Lipson S, Marquis KL. ADX47273 [S-(4-Fluoro-phenyl)-{3-[3-(4-fluoro-phenyl)-[1,2,4]oxadiazol-5-yl]-piperidin-1-yl}-methanone]: A Novel Metabotropic Glutamate Receptor 5-Selective Positive Allosteric Modulator with Preclinical Antipsychotic-like and Procognitive Activities. J. Pharmacol. Exp. Ther. 2008;327:827–839. doi: 10.1124/jpet.108.136580. [DOI] [PubMed] [Google Scholar]
- 13.Rodriguez AL, Grier MD, Jones CK, Herman EJ, Kane AS, Smith RL, Williams R, Zhou Y, Marlo JE, Days EL, Blatt TN, Jadhav S, Menon U, Vinson PN, Rook JM, Stauffer SR, Niswender CM, Lindsley CW, Weaver CD, Conn PJ. Discovery of Novel Allosteric Modulators of Metabotropic Glutamate Receptor Subtype 5 Reveals Chemical and Functional Diversity and In Vivo Activity in Rat Behavioral Models of Anxiolytic and Antipsychotic Activity. Mol. Pharmacol. 2010;78:1105–1123. doi: 10.1124/mol.110.067207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a Gastambide F, Cotel MC, Gilmour G, ÓNeill MJ, Robbins TW, Tricklebank MD. Selective Remediation of Reversal Learning Deficits in the Neurodevelopmental MAM Model of Schizophrenia by a Novel mGlu5 Positive Allosteric Modulator. Neuropsychopharmacology. 2012;37:1057–1066. doi: 10.1038/npp.2011.298. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gastambide F, Gilmour G, Robbins TW, Tricklebank MD. The mGlu5 Positive Allosteric Modulator LSN2463359 Differentially Modulates Motor, Instrumental and Cognitive Effects of NMDA Receptor Antagonists in the Rat. Neuropharmacology. 2013;64:240–247. doi: 10.1016/j.neuropharm.2012.07.039. [DOI] [PubMed] [Google Scholar]
- 15.Gilmour G, Broad LM, Wafford KA, Britton T, Colvin EM, Fivush A, Gastambide F, Getman B, Heinz BA, McCarthy AP, Prieto L, Shanks E, Smith JW, Taboada L, Edgard DM, Tricklebank MD. In Vitro Characterisation of the Novel Positive Allosteric Modulators of the mGlu5 Receptor, LSN2463359 and LSN2814617, and Their Effects on Sleep architecture and Operant Responding in the Rat. Neuropharmacology. 2013;64:224–239. doi: 10.1016/j.neuropharm.2012.07.030. [DOI] [PubMed] [Google Scholar]
- 16.Hammond AS, Rodriguez AL, Townsend SD, Niswender CM, Jones CK, Linsdley CW, Conn PJ. Discovery of a Novel Chemical Class of mGlu5 Allosteric Ligands with Distinct Modes of Pharmacology. ACS Chem. Neurosci. 2010;1:702–716. doi: 10.1021/cn100051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou Y, Manka J, Rodriguez AL, Weaver CD, Jones CK, Conn PJ, Lindsley CW, Stauffer SR. Discovery of N-Aryl Piperazines as Selective mGluR5 Potentiators with Improved In Vivo Utility. ACS Med. Chem. Lett. 2010;1:433–438. doi: 10.1021/ml100181a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spear N, Gadient RA, Wilkins DE, Do ML, Smith JS, Zeller KL, Schroeder P, Zhang M, Arora J, Chhajlani V. Preclinical Profile of a Novel Metabotropic Glutamate Receptor 5 Positive Allosteric Modulator. Eur. J. Pharmacol. 2011;659:146–154. doi: 10.1016/j.ejphar.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 19.Chromanone derivatives 9, 10, 11 and 12 have recently also been described as mGlu5 receptor PAMs by Varnes et al. confirming our findings. Varnes JG, Marcus AP, Mauger RC, Throner SR, Hoesch V, King MM, Wang X, Sygowski LA, Spear N, Gadient R, Brown DG, Campbell JB. Discovery of Novel Positive Allosteric Modulators of the Metabotropic Glutamate Receptor 5 (mGlu5). Bioorg. Med. Chem. Lett. 2011;21:1402–1406. doi: 10.1016/j.bmcl.2011.01.027.
- 20.Compounds were tested for agonist, PAM, or antagonist activity on mGlu receptors in fluorescent Ca2+ assays using HEK293 cells expressing human mGlu1, mGlu2, mGlu3, mGlu7, or mGlu8 receptors. Effects on the human mGlu4 receptor were tested in Ca2+ or [35S]-GTPγS functional assays (receptor either expressed in HEK293 or L929sA cells). Evaluation of effects on the rat mGlu6 receptors expressed in CHO cells were assessed in [35S]-GTPγS functional assays.
- 21.Hopkins AL, Groom CR, Alex A. Ligand Efficiency: a Useful Metric for Lead Selection. Drug Discovery Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 22.Data not shown [Google Scholar]
- 23.Manka JT, Vinson PN, Gregory KJ, Zhou Y, Williams R, Gogi K, Days E, Jadhav S, Herman EJ, Lavreysen H, Mackie C, Bartolomé JM, Macdonald GJ, Steckler T, Daniels JS, Weaver CD, Niswender CM, Jones CK, Conn PJ, Lindsley CW, Stauffer SR. Optimization of an Ether Series of mGlu5 Positive Allosteric Modulators: Molecular Determinants of MPEP-site Interaction Crossover. Bioorg. Med. Chem. Lett. 2012;22:6481–6485. doi: 10.1016/j.bmcl.2012.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Macdonald GJ, Tresadern GJ, Trabanco-Suarez AA, Pastor-Fernandez J. Preparation of Bicyclic Thiazoles as Allosteric Modulators of mGluR5 Receptors. 2011 WO 2011073347 A1.
- 25.Macdonald GJ, Trabanco-Suarez AA, Conde-Ceide S, Tresadern GJ, Bartolome-Nebreda JM, Pastor-Fernandez J. Preparation of Bicyclic Thiazoles as Allosteric Modulators of mGluR5 Receptors. 2011 WO 2011073339 A1.
- 26.Brandl T, Maier U, Hoffmann M, Scheuerer S, Joergensen AT, Pautsch A, Breitfelder S, Grauert M, Hoenke C, Erb K, Pieper M, Pragst I. Preparation of Dihydrothiazoloquinazolines as PI-3 Kinase Inhibitors. 2007 WO 2007115929 A1.
- 27.Noetzel MJ, Rook JM, Vinson PN, Cho HP, Days E, Zhou Y, Rodriguez AL, Lavreysen H, Stauffer SR, Niswender CM, Xiang Z, Daniels JS, Lindsley CW, Weaver CD, Conn PJ. Functional Impact of Allosteric Agonist Activity of Selective Positive Allosteric Modulators of Metabotropic Glutamate Receptor Subtype 5 in Regulating Central Nervous System Function. Mol. Pharmacol. 2012;81:120–133. doi: 10.1124/mol.111.075184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.LeadProfilingScreen. Ricerca Biosciences. Screen performed on the following targets: A1, A2A, A3, α1A, α1B, α1D, α2A, β1, β2, AR, B1, B2, CaCH L-Type (benzothiazepine), CaCH L-Type (dihydropyridine), CaCH N-Type, CB1, D1, D2S, D3, D4.2, ETA, ETB, EGF, ERα, GABAA (flunitrazepam), GABAA (muscinol), GABAB1A, GR, kainate, NMDA (agonism), NMDA (glycine), NMDA (phencyclidine), H1, H2, H3, IL-1, CysLT1, MT1, M1, M2, M3, NPY1, NPY2, acetylcholine, acetylcholine α1 (bungarotoxin), OP1, OP2, OP3, phorbol ester, PAF, KCHATP, hERG, EP4, P2X, P2Y, rolipram, 5HT1A, 5HT2B, 5HT3, σ1, NK1, thyroid hormone, DAT, GABA transporter, NET, SERT.
- 29.Gregory KJ, Noetzel MJ, Rook JM, Vinson PN, Stauffer SR, Rodriguez AL, Emmitte KA, Zhou Y, Chun AC, Felts AS, Chauder BA, Lindsley CW, Niswender CM, Conn PJ. Investigating Metabotropic Glutamate Receptor 5 Allosteric Modulator Cooperativity, Affinity and Agonism: Enriching Structure-function Relationships. Mol. Pharmacol. 2012;82:860–875. doi: 10.1124/mol.112.080531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Based upon rat brain homogenate binding fraction unbound (18%) and total brain levels ([brain]90min = 17.5 ± 13.4 μM). Estimated average terminal unbound brain concentrations of 16a after the 3, 10, 30, and 56.6 p.o. doses were 14 ± 11, 15 ± 13, 551 ± 449, and 908 ± 1029 nM respectively.
- 31.Carter RJ, Morton J, Dunnett SB. Motor Coordination and Balance in Rodents. Curr. Protocols Neurosci. 2001:8.12.1–8.12.14. doi: 10.1002/0471142301.ns0812s15. [DOI] [PubMed] [Google Scholar]
- 32.Measures included scoring for ptosis, exophthalmos, miosis, mydriasis, corneal reflex, pinna reflex, piloerection, respiratory rate, writhing, tail erection, lacrimation, salivation, vasodilation, skin color, irritability, rectal temperature, motor activity, ataxia, arch/roll, tremors, leg weakness, rigid stance, spraddle, placing loss, grasping loss, righting loss, catalepsy, tail pinch reaction, escape loss and physical appearance.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.