

Abstract
Tumor microenvironment (TME) refers to the dynamic cellular and extra-cellular components surrounding tumor cells at each stage of the carcinogenesis. TME has now emerged as an integral and inseparable part of the carcinogenesis that plays a critical role in tumor growth, angiogenesis, epithelial to mesenchymal transition (EMT), invasion, migration and metastasis. Besides its vital role in carcinogenesis, TME is also a better drug target because of its relative genetic stability with lesser probability for the development of drug-resistance. Several drugs targeting the TME (endothelial cells, macrophages, cancer-associated fibroblasts, or extra-cellular matrix) have either been approved or are in clinical trials. Recently, non-steroidal anti-inflammatory drugs targeting inflammation were reported to also prevent several cancers. These exciting developments suggest that cancer chemopreventive strategies targeting both tumor and TME would be better and effective towards preventing, retarding or reversing the process of carcinogenesis. Here, we have reviewed the effect of a well established hepatoprotective and chemopreventive agent silibinin on cellular (endothelial, fibroblast and immune cells) and non-cellular components (cytokines, growth factors, proteinases etc.) of the TME. Silibinin targets TME constituents as well as their interaction with cancer cells, thereby inhibiting tumor growth, angiogenesis, inflammation, EMT, and metastasis. Silibinin is already in clinical trials, and based upon completed studies we suggest that its chemopreventive effectiveness should be verified through its effect on biological end points in both tumor and TME. Overall, we believe that the chemopreventive strategies targeting both tumor and TME have practical and translational utility in lowering the cancer burden.
Keywords: Angiogenesis, Chemoprevention, Inflammation, Metastasis, Silibinin, Tumor microenvironment
Tumor microenvironment
Tumor microenvironment (TME) refers to the dynamic cellular and extra-cellular components surrounding tumor cells at each stage of the carcinogenesis that plays a critical role in tumor growth, angiogenesis, epithelial to mesenchymal transition (EMT), invasion, migration and metastasis. TME encompasses several distinct cell types, including cancer-associated fibroblasts (CAFs), endothelial cells, pericytes, smooth muscle cells, neutrophils, macrophages, adipocytes, dendritic cells, natural killer cells, lymphocytes etc. Besides, TME is also rich in non-cellular components such as cytokines, growth factors, hormones, and components of the extracellular matrix (ECM). The TME is not merely a passive response to transformed or growing tumor cells; instead, TME should be viewed as an equal and active partner in cancer growth and progression. In several instances, the TME determines whether dysfunctional epithelial cells will continue to grow and invade in particular milieu or will become dormant or even be removed [1]. Therefore, the tumor microenvironment is an essential, integrated and inseparable component of carcinogenesis. Few tumor microenvironment components, including their roles in carcinogenesis and the opportunities they offer for drug development, are described next.
CAFs or myofibroblasts are large spindle-shaped cells present in the microenvironment or stroma of several cancer types such as prostate, breast, ovarian, and lung cancer [2–6]. Cytokines secreted by cancer cells promote differentiation of normal fibroblasts into CAFs [2–5]. These fibroblasts are highly proliferative and express typical biomarkers such as α-smooth muscle actin (SMA), fibroblast activation protein (FAP), vimentin, and desmin. CAFs play an important role in ECM remodeling, angiogenesis, and the recruitment of inflammatory cells to the TME [1, 3, 4, 6]. For example, CAFs produce CXCL12, a chemokine ligand for the chemokine receptor CXCR4, thereby promoting the recruitment of bone marrow-derived precursors that contribute to vessel development in tumors [1]. CAFs have also been reported to secrete pro-migratory ECM components such as tenascin [6, 7]. Giannoni et al. recently reported the reciprocal interactions between prostate cancer cells and fibroblasts, where prostate cancer cells-derived IL6 activated the fibroblasts, which then promoted EMT, growth, metastases, and stemness in prostate cancer cells [8]. Considering the important role of CAFs in tumor growth and progression, efforts have been made to target CAFs for cancer treatment. Humanized monoclonal antibody (Sibrotuzumab) directed against FAP has been successfully tested clinically (Phase I/II) in patients with colorectal carcinoma and non-small cell lung cancer [9–11].
Tumor vasculature comprising endothelial cells and pericytes is the most important element of the TME as the development of vasculature (known as ‘neo-angiogenesis’) is the critical step for tumor growth as well as progression from a pre-malignant to invasive and malignant phenotype. Beyond a critical size of 1–2 mm, oxygen and nutrients have difficulty diffusing to the core cells of the tumor, causing a state of cellular hypoxia. Under hypoxic conditions, cancer cells secrete several pro-angiogenic factors such as VEGF and bFGF, which recruit endothelial cells from the neighboring blood vessels [12, 13]. Beside cancer cells, other components of the TME (such as CAFs or immune cells) also promote angiogenesis via ECM remodeling and secretion of pro-angiogenic factors [1, 3, 4, 6]. For example, the secretion of cytokines in the TME (such as CXCL12 by CAFs, as mentioned above) attracts circulating bone marrow-derived endothelial progenitor cells (EPCs), which contribute to the blood vessels formation, a process known as ‘vasculogenesis’ [1, 14]. The continuous and excessive presence of pro-angiogenic stimuli in the TME interferes with the normal maturation of the vessel network, and as a result, vessels in the tumor area show abnormal morphology and physiology and are usually dilated, leaky, and tortuous [15]. The process of vasculature formation also includes the interaction between endothelial cells and surrounding pericytes involving several growth factors and signaling pathways such as PDGF/PDGFR, VEGF/VEGFR2, and the angiopoietin/Tie system [4]. In this interaction, pericytes provide the survival and structural support to endothelial cells; therefore, pericytes are also considered a drug target. About four decades ago, Judah Folkman first predicted a potential role for anti-angiogenic inhibitors against solid cancers, and to date, numerous angiogenesis inhibitors have been tested against several malignancies [16–19]. Many of these inhibitors have already been FDA approved for their use either alone or in combination with other cancer chemotherapeutic drugs [17, 19, 20]. For example, humanized VEGF antibody (‘Avastin’) has been approved against colorectal, brain, lung, and renal cancers [20]. Similarly, the tyrosine kinase inhibitor SU11248 (‘Sunitinib’), which targets VEGFR and PDGFR activity, has been approved for use against advanced renal cell carcinoma and gastrointestinal stromal tumors [20–22].
Besides angiogenesis, inflammation is another phenomenon that is intricately linked to carcinogenesis [1, 3, 4]. Tumor cells over-express inflammatory cytokines that help to recruit hematopoietic cells such as lymphocytes, monocytes and neutrophils into the TME. For example, colony stimulating factor-1 (CSF1), secreted by several tumors, plays an important role in the growth and differentiation of macrophages [3]. In CSF1-null mice, the loss of CSF1 prevented the accumulation of macrophages in the tumor vicinity and delayed the development of invasive and metastatic carcinoma confirming the importance of the presence of macrophages in the TME [23]. In fact, macrophages are one of the most studied inflammatory cells for their role in tumorigenesis. Macrophages possess phenotypic plasticity that can be classified into two types, M1 (type I) and M2 (type II) polarized macrophages. In general, M1 macrophages are known as classically activated macrophages and play diverse roles in the immune system including defending against tumor cells. M2 macrophages are known as alternatively activated macrophages and are better adapted to scavenge debris and secrete growth factors that promote angiogenesis [24]. Pre-invasive tumor cells release chemotactic factors (e.g. CSF-1, CCL2, CCL3, CCL4, CCL5, CCL8, and MCP-1) that attract circulating monocytes into tumor stroma [1, 4, 23–26]. In the tumor stroma, presence of tumor cells as well as additional tumor-derived factors promotes macrophage differentiation into tumor-associated macrophages (TAMs) with characteristics akin to those of the M2 macrophages. This is advantageous to the tumor as TAMs support growth, angiogenesis, invasion, migration, intravasation and extravasation during metastatic spread [4, 24–26]. In addition, TAMs induce local immunosuppression that helps cancer cells evade detection by immune cells; thereby tumor cell travels unharmed in the circulation and extravasate at distant sites [24, 25]. It has been estimated that in majority of human malignancies, increased TAMs density correlates with poor prognosis [24, 27]. Considering the significant role of the inflammatory milieu in cancer development, in recent times, NSAIDs (non-steroidal anti-inflammatory drugs) have been extensively tested for their potential to prevent and treat various cancers [1, 28–30]. COX2 inhibitor, Celecoxib has been approved to reduce polyp growth in people with a rare genetic disorder (adenomatous polyposis). Furthermore, bisphosphonates, which are widely used clinically to target osteoclasts, are being tested for their potential to target TAMs in the TME [24, 31, 32].
One major advantage of therapies targeting the TME is that non-tumor cells are relatively genetically stable and thus the chances of them developing drug resistance are presumably remote [3, 15, 33]. Furthermore, whereas cancer cells exhibit an enormous variety of genetic and epigenetic changes during tumorigenesis, changes in the cancer microenvironment are usually common among many tumor types, raising the hope that therapeutic targeting of these events could be generally applicable [4]. For example, NSAIDs are effective against several cancers (colorectal, breast, prostate etc.). Similarly, the antiangiogenic drug Avastin has been approved for the treatment of several cancers (colorectal cancer, non-small cell lung cancer, kidney cancer, and glioblastoma). Moreover, the better knowledge of the TME has helped to devise novel and improved multi-targeted therapeutic approaches, in which both tumor cells and microenvironment components are simultaneously inhibited. Currently, the agents targeting the TME (Avastin, Suramin, NSAIDs, humanized monoclonal antibodies against integrins etc.) are increasingly employed along with chemotherapeutic drugs with the goal to better treat cancer. Furthermore, targeting the TME at metastatic sites has also shown promising results [34, 35]. For example, osteoclasts play an important role in bone metastatic cancers and RANKL is the critical regulator of osteoclast differentiation and activity. Humanized monoclonal antibody (‘Denosumab’) targeting RANKL has been reported to prevent skeletal metastasis and was recently approved by the FDA for the use in prostate and breast cancer patients [34, 35]. Overall, an understanding of the TME has helped tremendously to better comprehend the complexity of the carcinogenesis process and has also offered several new opportunities to control cancer.
Translational chemopreventive opportunities in targeting tumor microenvironment
Cancer chemoprevention refers to preventing, inhibiting, or reversing the process of carcinogenesis through dietary or pharmacological interventions. Cancer chemoprevention is based upon the rationale that the common epithelial cancers (prostate, breast, lung, pancreas, colorectal, etc.) which account for most of the mortality, have a long latency period and often take 2–3 decades to become malignant. Therefore, the life style changes (dietary habits, exercise etc.) and/or non-toxic chemopreventive agents could be effectively employed to arrest or reverse the progression of pre-malignant cells towards full malignancy [30]. Similar to the oncology field in general, cancer chemoprevention strategies in the past have been solely directed towards initiated or mutated cancer cells. Carcinogenesis has long been understood as a multistep process during which cancer cells accumulate multiple and consecutive genetic alterations and increasingly become proliferative, invasive and metastatic (Figure 1). But in the past few decades, numerous reports in the literature (as described above) have confirmed that cancer is not just a homogeneous mass of proliferative cells but essentially includes the TME, which consist of several interacting cellular and non-cellular components surrounding cancer cells at each stage of the carcinogenesis (Figure 1). In light of the critical importance of the TME in carcinogenesis, it is pertinent to identify and establish chemopreventive targets in the complex and heterogeneous TME along with the tumor (Figure 1). There is already ample pre-clinical, clinical and epidemiological evidence that advocates the TME to be a prime target in any translational cancer chemoprevention strategy [1, 28, 30, 36].
Figure 1.

The classical and current view of carcinogenesis and chemoprevention targets. Classically, tumor has been viewed as a homogenous mass of rapidly growing cells, though could be of different sizes; while the current view considers tumor as a complex community of interacting heterogeneous population of cells.
There are several instances where inflammatory lesions precede neoplasm and helps to create an environment favorable for cancer development [37]. For example, ulcerative colitis and chronic reflux esophagitis are associated with enhanced risk of developing colorectal and esophageal cancer, respectively. Furthermore, several infectious diseases causing chronic inflammation have also been linked with cancer development such as Human Papillomavirus (HPV) infection with cervical and other cancers, Helicobacter pylori infection with gastric cancer, Hepatitis C with hepatic cancer, urinary form of Schistosomiasis with bladder cancer. The inflammatory cells in the TME produce prostaglandins, reactive oxygen species, reactive nitrogen species etc. that promote genetic instability, cell proliferation, survival, motility as well as angiogenesis [38, 39]. Therefore, the use of anti-inflammatory drugs (such as COX2 inhibitors) is gaining popularity for preventing or treating cancer. The chronic use of NSAIDs has been reported to reduce the risk of colon cancer, and the new evidence suggests that this could probably also be true for other cancers such as breast, prostate, and ovarian [28, 30, 40, 41]. In a recent study, Lyon et al. showed that inhibiting the COX2 in the involuting mammary gland reduced the collagen fibrillogenesis associated with involution and inhibited breast tumor growth and infiltration to the lung [36]. This study suggested that women at high risk for postpartum breast cancer would benefit from treatment with NSAIDs during postpartum involution [36]. These findings are significant as there are millions of women that could be potentially protected from developing breast cancer simply by using NSAIDs. Therefore, several cancers could be prevented or delayed through chemopreventive approaches targeted against an inflammatory TME.
Angiogenesis inhibitors were originally envisioned for preventing the progression of non-angiogenic, localized indolent tumors to aggressive and invasive carcinoma. However, most of the clinical anti-angiogenic agents are employed at a terminal or late-stage of the cancer in combination with chemotherapeutic agents. Considering their exorbitant cost as well as toxicity, the use of anti-angiogenic inhibitors in cancer prevention seems implausible. Several chemopreventive agents (silibinin, GSE, isothiocyanate, EGCG, triterpenoids etc.) have shown remarkable angiopreventive efficacy in pre-clinical models [42–47] and these chemopreventive agents could be expediently tested and judiciously employed for angioprevention in humans. We have learnt hard-lessons from earlier failed clinical trials with chemopreventive agents (β-carotene, selenium, and tocopherol) [30]; therefore, only agents that have been tested extensively and exhibiting high efficacy in animal models should move forward and be tested in high-risk populations for angiopreventive efficacy.
Epidemiological and pre-clinical studies have suggested that excessive adiposity, decreased physical activity, and unhealthy diets are few of the important players in the pathogenesis of common cancers [48]. These conditions result in chronically elevated levels of insulin, IGF-1, hormones, and inflammatory cytokines that promote genomic instability, proliferation, and inhibit apoptosis, thereby accelerate carcinogenesis [49, 50]. Cancer chemopreventive agents could lower the risk of cancer by reducing the levels of anabolic hormones, growth factors, inflammatory cytokines, and metabolites. For example, Statins, widely used to lower cholesterol level, have been reported to reduce the risk of several cancers and the possible mechanisms for this effect include the suppression of inflammation and angiogenesis in the TME [1]. Therefore, efficacy of established cancer chemopreventive agents/practices (phytochemicals, energy restriction, low fat diets etc.) could be partly through targeting the adverse TME components and this aspect needs further scrutiny.
Cancer chemoprevention approaches should emphasize identifying and targeting the molecular signatures specific to the TME, because a generalized targeting of the microenvironment might also adversely affect normal tissue homeostasis and cause undesirable effects. For example, the chronic use of NSAIDs causes serious gastrointestinal complications, and COX2 inhibitor use could enhance the risk of cardiovascular diseases (heart-attack, stroke). Lately, focus has also been on the ‘normalization of the TME’ instead of inhibiting or wiping out the TME components [3, 4, 51, 52]. The normalization of tumor vasculature by anti-angiogenic agents has shown promising results in terms of improving the sensitivity of cancer cells towards drugs/radiations [53, 54]. Similarly, in a recent study, Coscia et al. reported the effect of Zoledronic acid treatment on the normalization of immune cells in a murine model of mammary carcinoma, where Zoledronic acid reversed the polarity of TAM from M2 to M1 type [55]. These results are important as M1 macrophages are considered tumoricidal, and Zoledronic acid increased the tumor-free survival, reduced the in tumor growth rate and tumor multiplicity as well lung metastasis [55]. These results also confirm the bipolar action of TME components i.e. in normal homeostasis, microenvironment components (immune cells, fibroblasts etc.) seek to prevent tumor growth, but once educated by tumor cells they promote tumor growth and progression. Therefore, normalization of the TME through cancer chemopreventive strategies should prevent/delay/retard carcinogenesis via a non-conducive environment for growth. An in-depth knowledge of the early interactions in the TME would be critical in arresting cancer growth at early stages. Overall, there are many avenues to target both tumor and TME using chemopreventive agents. Next, we discuss in detail the effect of an established cancer chemopreventive agent Silibinin (Figure 2) on various components of the TME.
Figure 2.
Chemical structure of silibinin.
Silibinin and tumor microenvironment
Silibinin (C25H22O10, molecular weight, 482.44) is isolated from the seeds of Silybum marianum (L.) Gaertn (Family Asteraceae), and is one of the most-widely consumed dietary supplements for its hepatoprotective efficacy [56, 57]. Silibinin source, metabolism, bioavailability and anti-cancer efficacy have been reviwed extensively earlier [58–61]; therefore here we have focused mainly on silibinin’s effect on the TME components.
Silibinin and angiogenesis
As mentioned earlier, neo-angiogenesis is an essential component of the TME in solid tumors. Clinical and experimental evidence have suggested that human tumors can persist for years as microscopic lesions in a state of dormancy and their further growth is critically dependent upon attaining an ‘angiogenic phenotype’[62–65]. The formation of vasculature is necessary to provide nutrients and oxygen to the growing tumors and also to remove waste products. Furthermore, angiogenesis in tumors provides tumor cells the route to metastasize at secondary sites. Therefore, preventing the onset of angiogenesis in indolent tumors (referred to as ‘angioprevention’) has been suggested as a novel and rational approach to control cancer growth, malignant progression and metastasis to secondary sites. Now, there is a plethora of reports suggesting the strong angiopreventive efficacy of silibinin in several cancer models [43, 44, 66–73].
We have first reported that silibinin feeding inhibits micro-vessel density (MVD) in growing prostate carcinoma DU145 tumors in athymic nude mice, which was associated with a decrease in VEGF expression in the tumors [74]. Similar anti-angiogenic efficacy of silibinin was also observed in prostate tumors growing in the prostate microenvironment in nude mice [70]. Extensive studies in TRAMP (transgenic adenocarcinoma of the mouse prostate) revealed that silibinin targets the onset of ‘angiogenic switch’ in prostate tumors [44, 73]. Huss et al. have shown that angiogenic switch in TRAMP involves an increase in VEGF, VEGFR2 and HIF-1α expression accompanied with increased intra-ductal microvessels, and disease progression from low-grade prostate intraepithelial neoplasia (LGPIN) stage to high-grade PIN (HGPIN), adenocarcinoma and metastasis [13]. Our completed studies showed that silibinin feeding decreased the VEGFR2, VEGF, and HIF-1α expression, and strongly inhibited the MVD in TRAMP prostate tissues [44]. Silibinin treatment also decreased the levels of circulating angiogenic factors VEGF and bFGF in TRAMP mice [73]. These anti-angiogenic effects of silibinin were associated with a potent inhibition of tumor grade as well as metastasis confirming its strong angiopreventive efficacy against prostate cancer [44, 73].
Silibinin has also been extensively tested for its anti-angiogenic efficacy in several animal models of colorectal cancer [69, 71, 72, 75]. We have reported that silibinin feeding inhibited the angiogenesis in HT29 tumor via down-regulating iNOS (inducible nitric oxide synthase), COX2, HIF-1α, and VEGF expression [71]. Silibinin also exerted sustained growth suppressive effects in human colorectal cancer SW480 tumors via decreasing MVD, VEGF and iNOS expression [75]. In transgenic APCmin+ mice, silibinin feeding inhibited the nestin-positive microvessels selectively in the small intestinal polyps by down-regulating the expression of HIF-1α, VEGF, and eNOS (endothelial nitric oxide synthase) in the polyps [69]. Importantly, silibinin feeding did not affect the expression of these molecules in the crypt-villus region in the small intestine of APCmin+ and wild type C57BL/6J mice confirming the polyp specific effect of silibinin [69]. These angiopreventive effects of silibinin were associated with the prevention of spontaneous intestinal polyposis in APCmin+ mice and we observed a decrease in both the number of polyps as well as size of the polyps formed [69]. Similarly, silibinin treatment inhibited VEGF and iNOS expression in azoxymethane-induced colon tumors in A/J mice [76]. Earlier, Yang et al. reported that silibinin treatment inhibits the vascular density index induced by human colorectal cancer LoVo cells in CAM (chicken chorioallantoic membrane) assay through inhibiting VEGF expression [77]. Overall, silibinin has shown strong angiopreventive efficacy against colorectal cancer in xenografts, chemical carcinogenesis, CAM and transgenic models.
Sustained NO (nitric oxide) generation positively correlates with lung cancer development and progression; and our completed studies in lung tumorigenesis models suggest that silibinin’s chemopreventive and angiopreventive effects could be through targeting iNOS expression [43, 68, 78]. In urethane-induced lung tumorigenesis model, silibinin feeding strongly decreased the MVD, VEGF and iNOS levels in lung tumors [43]. The angiopreventive effects of silibinin resulted in a significant decrease in the lung tumor multiplicity as well as tumor size [43]. Using iNOS−/− mice we confirmed that silibinin exerts its chemopreventive and angiopreventive effects against lung tumorigenesis via inhibiting iNOS expression [78]. Our in vitro mechanistic studies also showed that silibinin targets multiple signaling molecules [STATs (1 and 3), AP1, NF-κB, MAPKs and HIF-1α], and that it inhibits cytokine mixture (IFN-γ + IL-1β + TNF-α)-induced iNOS expression in human lung epithelial carcinoma A549 cells [79]. Lung tumor analyses supported these in vitro observations and silibinin was found to inhibit the expression of IFNγ, interleukins, and TNFα in lung tumors [68]. Silibinin also decreased HIF-1α, NF-κB and phosphorylated STAT3 expression in lung tumors [68]. These results suggested that silibinin targets multiple signaling pathways regulating iNOS expression in lung tumor cells, and thereby, it inhibits the angiogenesis and overall tumor progression in lung tumors.
The anti-angiogenic effects of silibinin have also been observed in other cancers such as skin cancer and bladder cancer [80, 81] suggesting the broad-spectrum angiopreventive efficacy of silibinin. Additionally, several in vitro studies have supported the anti-angiogenic effects of silibinin and provided detailed insight into the mechanisms for silibinin’s angiopreventive action. Silibinin was reported to inhibit the growth of HUVEC (human umbilical vein endothelial cells) and HMVEC (human microvascular endothelial cells) at pharmacologically achievable doses in cell culture [67]. Silibinin also inhibited capillary tube formation on matrigel as well as inhibited HUVEC invasion and migration [67]. Molecular analyses revealed that silibinin induces G1 arrest in endothelial cells via promoting the expression of CDKIs (cyclin dependent kinase inhibitors) and p53 [67]. Furthermore, silibinin treatment induced apoptotic death involving both caspases-dependent and –independent mechanisms [67]. Silibinin also targeted Akt, NF-κB and survivin as well as MMP-2 activity (Figure 3) [67]. Yang et al. have reported similar effects of silibinin on the growth as well as the differentiation of endothelial EA.hy 926 cells [82]. Silibinin also inhibited the chemotactic migration of EA.hy 926 cells towards LoVo colon cancer cells [82]. Yoo et al. have also shown that silibinin suppresses growth and induces apoptotic death in human endothelial ECV304 cells by modulating NF-κB, Bcl-2 family members and caspases [83].
Figure 3.

Silibinin exhibits angiopreventive efficacy through targeting the secretion of pro-angiogenic factors by cancer cells (blue arrow); by reducing the recruitment of macrophages in the tumor microenvironment (yellow arrow); and through inhibiting various signaling molecules in endothelial cells, compromising their survival as well as chemotactic movement (purple arrow) towards pro-angiogenic stimuli.
Along with targeting endothelial cells, silibinin has also been reported to target cancer cells towards inhibiting the secretion of pro-angiogenic factors (Figure 3). HIF-1α has emerged as a master regulator of angiogenesis, tumor metabolism and metastasis [84]. Together with other regulators (such as ERK, NF-κB, STATs), HIF-1α controls the expression and secretion of several pro-angiogenic growth factors (Figure 3), that promote chemotactic movement, survival, proliferation and differentiation of endothelial cells. Garcia-Maceira et al. showed that silibinin strongly inhibits hypoxia-induced HIF-1α accumulation and VEGF release in human cervical HeLa and hepatoma Hep3B cells [85]. This effect was correlated with silibinin’s inhibitory effect on the HIF-1α translation through targeting the mTOR-p70S6K and 4E-BP1 pathways [85]. Similarly, Jung et al. have reported that silibinin inhibits HIF-1α protein expression via targeting its synthesis in human prostate cancer cells [86]. This study also showed that silibinin inhibits global protein synthesis via decreasing the levels of eIF4E-associated with eIF4F complex, increasing the levels of eIF4E associated with 4E-BP1 and promoting the eIF2α phosphorylation [86]. Kim et al. (2009) have shown that silibinin treatment inhibits 12-O-tetradecanoyl phorbol-13-acetate (TPA)-induced MMP9 and VEGF expression via suppressing the RAF/MEK/ERK pathway in MCF-7 breast cancer cells [87]. Overall, it is clear that silibinin targets both cancer and endothelial cells to effectively inhibit angiogenesis. Specifically, on the one hand silibinin targets multiple signaling cascades in cancer cells to inhibit the secretion of pro-angiogenic factors in the microenvironment, and on the other hand it targets endothelial cell responses (motility, proliferation, survival, and differentiation) to pro-angiogenic stimuli (Figure 3).
Silibinin and metastasis
Metastasis is an extremely complex, multi-step and multi-functional biological event that is responsible for high mortality and morbidity in cancer patients [58, 88–90]. Successful metastasis is dependent on the cumulative ability of cancer cells to suitably respond to the distinct microenvironment at each step in the metastatic cascade starting from primary tumor growth to final metastatic site [58]. Over a century ago, Stephen Paget first reported a non-random pattern of metastasis of cancer cells to certain organs [91, 92]. He proposed “seed and soil hypothesis”, in which he compared the metastasis of cancer cells to the dispersal of seeds by plants. He postulated that seeds (‘cancer cells’) could grow only in a congenial soil (‘specific microenvironment’). For example, osteotropic cancer cells possess certain intrinsic properties that enable them to grow in the bone; and the bone microenvironment provides a fertile soil for their growth. This theory, which placed main emphasis on the compatibility between metastatic cancer cells and their microenvironment, is still relevant, and the metastatic microenvironment has now become an important drug target to treat or prevent metastasis. There are several reports that suggest that silibinin targets the multiple interactions between tumor cells and their microenvironment and prevents/inhibits metastasis [58].
The extracellular matrix (ECM) and integrins interact to regulate a variety of cellular functions including adhesion, survival, and motility [3, 93, 94]. Fibronectin, a matrix glycoprotein, is one such ECM component, that has been reported to be up-regulated in several malignant tumors and its expression positively correlates with an invasive and metastatic phenotype [94–96]. We have reported that fibronectin expression increases with tumor progression in prostate tumors in TRAMP mice, and that fibronectin expression was significantly decreased by silibinin treatment [44]. Fibronectin-integrin interaction activates several signaling pathways (FAK, Src, Akt, and GTPase) involved in cell survival and actin-remodeling. In our unpublished studies, we have observed that silibinin targets the fibronectin-prostate cancer cell interaction and inhibits integrins expression as well as down-stream signaling involved in actin-remodeling; thereby inhibits the formation of motile structures. Besides fibronectin, silibinin has been reported to significantly decrease the adhesion of prostate cancer cells to type I collagen [97]. It is important to highlight here that bones are rich in type I collagen and prostate cancer cells generally metastasize to bones. Silibinin treatment also inhibited the adhesion of human prostate cancer PC3M cells with ECM proteins hyaluronan and fibronectin by targeting the expression of transmembrane protein CD44 and its variant form CD44v7-10 [98]. Furthermore, silibinin treatment inhibited the adhesive capability of human osteosarcoma MG-63 cells towards type IV collagen [99]. These studies suggest that silibinin treatment significantly attenuates the interaction of cancer cells with their ECM components, which could adversely affect their motility and invasiveness.
Proteinases have been implicated in many cancer-related biological activities (angiogenesis, metastasis etc.), mainly because of their ability to break down components of the ECM, allowing cancer and other cells to migrate [58]. Silibinin treatment has been shown to significantly inhibit the expression of MMPs (matrix metalloproteinases) and to increase expression of TIMP-2 (tissue inhibitors of metalloproteinases-2) in vitro in a wide variety of cancer cells [58, 100–104]. In vivo, we have observed in TRAMP mice that silibinin feeding significantly decreased the expression of MMP-2, MMP-3 and MMP-9, but increased the TIMP-2 expression in prostate tumor tissue [44, 73]. Furthermore, silibinin treatment has been reported to inhibit serine protease uPA and its receptor uPAR expression in several cancer cell lines in vitro and in vivo [58, 99, 103–105]. Silibinin has also been reported to decrease the expression of cysteine proteinases cathepsin B in highly invasive human glioma cells [105].
During metastasis, several cancer cells undergo a phenomenon known as ‘epithelial to mesenchymal transition’ (EMT). EMT refers to a dynamic, multistep, and highly coordinated process that includes the loss of inter-cellular junctions, disruption of the tumor basement membrane, activation and rearrangement of cytoskeleton elements resulting in increased motility and invasiveness, and the release of cells from parent epithelial tissue [58, 106, 107]. EMT is regulated by a multitude of factors located in the TME. For example, CAFs have been reported to promote EMT by secreting MMPs [8]. In our unpublished studies we have also observed that CAFs promote the invasiveness of human prostate cancer LNCaP cells, which is associated with increased vimentin and Akt phosphorylation; and silibinin treatment inhibits the CAFs-induced invasiveness of LNCaP cells as well as strongly decreases the Akt phosphorylation and vimentin expression. Silibinin has also been reported to inhibit EMT through promoting the E-cadherin expression and inhibiting the expression of EMT transcriptional regulators [58, 108]. These results have been accompanied with a strong decrease in the migratory, invasive and metastasis properties of cancer cells both in vitro and in vivo [44, 58, 73, 108]. We have reported that silibinin feeding strongly inhibits the local invasion of prostate cancer cells to the seminal vesicle as well as distant metastasis to liver, lung and kidney in TRAMP mouse model [44, 73].
Silibinin has also been reported to affect the TME at metastatic site in prostate cancer model. Prostate cancer cells have a high propensity to metastasize to bones [58, 109]. During bone metastasis, prostate cancer cells even express genes like osteocalcin, bone sialoprotein, osteopontin, RANKL, whose expression is normally restricted to bone cells [58, 91, 110]. This phenomenon is termed ‘osteomimicry’ and is considered as an effort by cancer cells to adapt to their microenvironment, helping cancer cells to settle in the bones. Our unpublished data has shown that silibinin inhibits the expression of many osteomimicry related proteins such as RANKL, PTHrP, osteocalcin, and RunX2 in prostate cancer cells both in vitro and in vivo. Once settled in the bones, prostate cancer cells alter the delicate balance of bone remodeling orchestrated by two types of bone cells namely osetoclasts (involved in bone degradation) and osteoblasts (involved in bone formation) [58, 91, 110, 111]. Prostate cancer cells secrete factors that are involved in osetoclast maturation and activation, thereby promoting bone mineralization and the liberation of various growth factors [58, 91, 110, 111]. Bone degradation provides prostate cancer cells the initial space to expand, and the released growth factors promote prostate cancer cell survival and proliferation. These growth factors secreted by bone degradation and those secreted by prostate cancer cells like endothelin-1, BMPs (bone morphogenetic proteins), Wnts, promote osteoblasts maturation and formation of new bone [58, 91, 110, 111]. Mature osteoblasts also secrete growth factors which further promote prostate cancer cell growth in bone [91, 110]. Overall, this vicious cycle involving prostate cancer cells, osteoclasts and osteoblasts promotes bone degradation as well as deposition of new ‘woven type bone’ (uneven/immature/embryonic), and thereby compromises bone health and leads to bone complications in prostate cancer patients. Silibinin has been reported to affect both the components (osteoclasts and osteoblasts) of the bone microenvironment [112, 113]. Kim et al. have reported that silibinin treatment inhibited the formation of TRAP-positive multinuclear osteoclasts in bone marrow-derived macrophage cells cultured in the presence of M-CSF and RANKL or with osteoblasts and 1,25(OH)2D3 [113]. Silibinin also inhibited osteoclast differentiation in murine monocyte/macrophage cell line RAW264.7 stimulated with RANKL [113]. However, silibinin treatment did not affect osteoclast function when mature osteoclasts were treated with silibinin [113]. Silibinin also inhibited TNFα-induced osteoclastogenesis in bone marrow-derived macrophage cells treated with M-CSF and TGF-β [113]. Silibinin effect on osteoclast differentiation seems to be through targeting the fusion of TRAP+ mononuclear pre-osteoclasts forming TRAP+ multinucleate mature osteoclasts [113]. In our unpublished studies, we have observed that prostate cancer cells promote osteoclast activation in RAW264.7 cells, while silibinin treatment inhibits the prostate cancer cells potential to induce osteoclastogenesis. Silibinin seems to target multiple RANKL-induced signaling pathways such as NF-κB, MAPKs, Akt, AP1, and NFATc1 and also to inhibit NFATc1 regulated genes (TRAP, OSCAR and Cathepsin K) (our unpublished results) that are important in osteoclastogenesis (Figure 4) [114, 115]. In another study, the bone-forming and osteoprotective effects of silibinin were studied in cell culture in murine osteoblastic MC3T3-E1 cells [112]. Silibinin treatment increased the bone nodule formation by enhancing calcium deposits [112]. Silibinin also increased the induction of osteoblastogenic biomarkers alkaline phosphatase (ALP), collagen type 1, connective tissue growth factor (CTGF), and BMP-2 (Figure 4) [112]. Silibinin treatment also inhibited RANKL secretion by differentiated MC3T3-E1 cells (Figure 4) [112]. But the effect of silibinin on cancer cell-induced osteoblastogenesis remains unknown (Figure 4). Overall, these studies confirmed that silibinin targets several TME components towards lowering the metastatic growth of cancer cells.
Figure 4.

Silibinin targets prostate cancer cell, osteoblast, and osteoclast interaction towards inhibiting the osteoclast activation and differentiation.
Silibinin and inflammation
Chronic inflammation is a frequent cause of cancer. Even in cancers that are not necessarily the outcome of chronic inflammation, invariably, there are inflammatory components in their microenvironment. In fact, the constant disruption of homeostasis by proliferating transformed cells produces a local chronic inflammatory environment, which is considered an attempt by the body to re-establish normal homeostasis [1]. However, in the presence of cancer cells, immune cells react paradoxically and promote the survival and proliferation of cancer cells [1]. As a result tumors have been characterized as ‘wounds that do not heal’. These observations clearly suggest that reducing inflammation in the TME should inhibit or prevent cancer growth. There are plenty of evidences now suggesting that silibinin could modify inflammatory or immune components towards preventing carcinogenesis [69, 72, 76, 116–119]. Provinciali et al. have reported that silibinin administration delayed the development of spontaneous mammary tumors, reduced the number and size of tumors and diminished lung metastasis in HER-2/neu transgenic mice [116]. Silibinin treatment affected the leukocytes infiltration into the tumors and there were increased numbers of neutrophils, CD4+ and CD+8 lymphocytes but there was a slight decrease in macrophage number [116]. We have also reported that silibinin treatment inhibits TAMs in the TME, which was correlated with angiopreventive effects of silibinin against lung tumorigenesis (Figure 3) [68]. Meeran et al. have reported that silibinin treatment inhibited UVB-induced local and systemic immuno-suppression [118]. In trinitrobenzene sulfonic acid (TNBS)-induced colitis model, silibinin treatment significantly reduced several components of inflammatory colitis such as NF-κB activity, levels of IL-1β, TNFα, thiobarbituric acid reactive substances (TBARS), protein carbonyl, myeloperoxidase activity, and an improvement in antioxidant capability of the colon tissue [119].
The arachidonic acid pathway is at the core of inflammatory response. In this pathway, COX enzymes are responsible for the formation of prostaglandins (PGE2, PGF2α, and PGD2), prostacyclin and thromboxane, while lipoxygenase generates 5-HPETE which is converted to leukotrienes. COX2 is over-expressed in several cancers and considered an attractive drug target [120]. We have reported that chronic exposure to a physiological dose of UVB strongly increased the COX2 levels in the skin and skin tumors [80]. Pre- or post-topical treatment or dietary feeding of silibinin was reported to strongly inhibit UVB-induced COX2 levels [80]. Silibinin has also been reported to inhibit COX2 expression in colorectal cancer in xenografts, transgenic and chemically-induced colorectal cancer models [69, 71, 72, 76]. Silibinin treatment has been shown to inhibit the formation of cyclooxygenase pathway metabolites (PGE2, prostacyclin, and thromoxanes) by human mononuclear cells, platelets, and endothelial cells stimulated with LPS or A23187 [117]. Silibinin treatment also strongly inhibited the formation of 5-lipoxygenase metabolites by human granulocytes (leukotrienes LTB4, LTC4/D4/E4/F4) when stimulated with A23187, FMLP, or opsonized zymosan. Silibinin was also reported to be a strong scavenger of HOCl (IC50 7 μM) produced by human granulocytes [117]. Together, these studies suggest a strong inhibitory effect of silibinin on arachidonic acid pathway.
Transcriptional factors (AP1, NF-κB, STATs etc.) regulate the expression of several pro-inflammatory cytokines [79, 121–124]. Recently, we reported that silibinin targets several signaling pathways (ERK1/2, STAT1/3, NF-κB, and EGFR) towards inhibiting the TNFα and IFNγ induced expression of pro-inflammatory enzymes COX2 and iNOS [123]. Silibinin has been reported to inhibit TNFα-induced NF-κB activation in prostate cancer and colorectal cancer cells [124, 125]. Silibinin treatment also strongly inhibited the UVB-induced activation of STAT3 and NF-κB in skin and skin tumors in SKH-1 hairless mice [80]. Overall, silibinin targets multiple signaling pathways towards inhibiting the secretion of pro-inflammatory cytokines.
In general, published literature shows that silibinin targets many cellular as well as non-cellular components of the TME (Figure 5) towards inhibiting angiogenesis, metastasis and inflammation. There is also evidence now that silibinin targets the abnormal tumor metabolism as well as insulin signaling pathways (IGF/IGFBP3) [74, 126, 127]. Silibinin is already been tested clinically for its efficacy against several cancers including prostate and colon cancer [58, 128–130]. Considering the important role of TME in carcinogenesis, it is important that we focus on its chemopreventive efficacy not only in terms of effect on cancer cells but also the biomarkers in the TME (such as macrophages, CAFs, angiogenesis, and cytokines).
Figure 5.
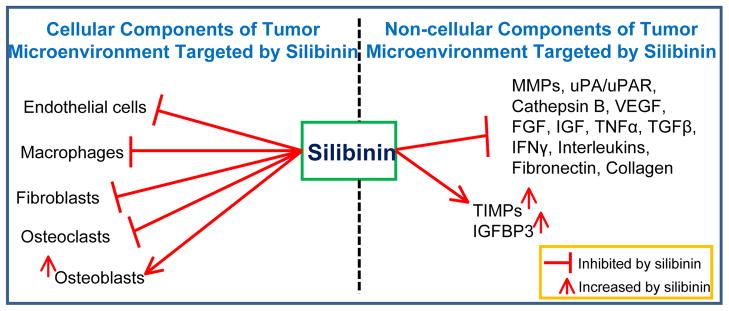
Silibinin targets several cellular and non-cellular components in the tumor microenvironment.
Conclusions and future directions
The essential role of the TME in carcinogenesis is now established beyond any doubt, and several drugs targeting the TME components are already in clinical use. But, whether the TME could also be exploited for translational cancer chemopreventive purposes is a relatively new concept. In the past, the main focus in cancer chemoprevention has been to prevent the promotion and progression of proliferating mutated cancer cells. But in recent times, there have been encouraging pre-clinical, clinical and epidemiological evidences that warrant the consideration of the TME as a prime target in cancer chemoprevention. To fully exploit the cancer chemoprevention opportunities in the TME, it is essential to understand the complexity of the TME especially at the early stages of carcinogenesis. We need to develop novel pre-clinical models that more closely mimic the conditions of the human TME and these models should be exploited to rapidly screen the usefulness of cancer chemopreventive agents. Further, we need to more frequently employ computational and system biology tools coupled with “omic” approaches (genomics, proteomics, and metabolomics) and laser capture microdissection techniques to better understand the complex interactions between tumor and TME constituents. As cancer chemopreventive agents are mostly intended for the normal healthy individuals, high risk populations, or early stage cancer patients; it is mandatory that these agents have no or minimal side effects. Also, in terms of their effect on the TME, it is desirable that cancer chemopreventive agents force a normalization of the TME. Similarly, it is important to identify and target the molecular signature/s specific to the TME to reduce the adverse effects on normal tissue homeostasis. Overall, there are tremendous translational cancer chemoprevention opportunities in the TME and they should be essentially targeted along with the tumor for effective cancer control.
Acknowledgments
This work was supported by NCI RO1 grants CA102514 and CA112304.
References
- 1.Albini A, Sporn MB. The tumour microenvironment as a target for chemoprevention. Nat Rev Cancer. 2007;7(2):139–147. doi: 10.1038/nrc2067. [DOI] [PubMed] [Google Scholar]
- 2.Bremnes RM, Donnem T, Al-Saad S, Al-Shibli K, Andersen S, Sirera R, Camps C, Marinez I, Busund LT. The role of tumor stroma in cancer progression and prognosis: emphasis on carcinoma-associated fibroblasts and non-small cell lung cancer. J Thorac Oncol. 2011;6(1):209–217. doi: 10.1097/JTO.0b013e3181f8a1bd. [DOI] [PubMed] [Google Scholar]
- 3.Mueller MM, Fusenig NE. Friends or foes - bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004;4(11):839–849. doi: 10.1038/nrc1477. [DOI] [PubMed] [Google Scholar]
- 4.Joyce JA. Therapeutic targeting of the tumor microenvironment. Cancer Cell. 2005;7(6):513–520. doi: 10.1016/j.ccr.2005.05.024. [DOI] [PubMed] [Google Scholar]
- 5.Schauer IG, Sood AK, Mok S, Liu J. Cancer-associated fibroblasts and their putative role in potentiating the initiation and development of epithelial ovarian cancer. Neoplasia. 2011;13(5):393–405. doi: 10.1593/neo.101720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allen M, Louise Jones J. Jekyll and Hyde: the role of the microenvironment on the progression of cancer. J Pathol. 2011;223(2):162–176. doi: 10.1002/path.2803. [DOI] [PubMed] [Google Scholar]
- 7.Adam B, Toth L, Pasti G, Balazs M, Adany R. Contact stimulation of fibroblasts for tenascin production by melanoma cells. Melanoma Res. 2006;16(5):385–391. doi: 10.1097/01.cmr.0000205022.25397.86. [DOI] [PubMed] [Google Scholar]
- 8.Giannoni E, Bianchini F, Masieri L, Serni S, Torre E, Calorini L, Chiarugi P. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 2010;70(17):6945–6956. doi: 10.1158/0008-5472.CAN-10-0785. [DOI] [PubMed] [Google Scholar]
- 9.Kloft C, Graefe EU, Tanswell P, Scott AM, Hofheinz R, Amelsberg A, Karlsson MO. Population pharmacokinetics of sibrotuzumab, a novel therapeutic monoclonal antibody, in cancer patients. Invest New Drugs. 2004;22(1):39–52. doi: 10.1023/b:drug.0000006173.72210.1c. [DOI] [PubMed] [Google Scholar]
- 10.Scott AM, Wiseman G, Welt S, Adjei A, Lee FT, Hopkins W, Divgi CR, Hanson LH, Mitchell P, Gansen DN, Larson SM, Ingle JN, Hoffman EW, Tanswell P, Ritter G, Cohen LS, Bette P, Arvay L, Amelsberg A, Vlock D, Rettig WJ, Old LJ. A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin Cancer Res. 2003;9(5):1639–1647. [PubMed] [Google Scholar]
- 11.Hofheinz RD, al-Batran SE, Hartmann F, Hartung G, Jager D, Renner C, Tanswell P, Kunz U, Amelsberg A, Kuthan H, Stehle G. Stromal antigen targeting by a humanised monoclonal antibody: an early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie. 2003;26(1):44–48. doi: 10.1159/000069863. [DOI] [PubMed] [Google Scholar]
- 12.Bicknell R, Harris AL. Novel angiogenic signaling pathways and vascular targets. Annu Rev Pharmacol Toxicol. 2004;44:219–238. doi: 10.1146/annurev.pharmtox.44.101802.121650. [DOI] [PubMed] [Google Scholar]
- 13.Huss WJ, Hanrahan CF, Barrios RJ, Simons JW, Greenberg NM. Angiogenesis and prostate cancer: identification of a molecular progression switch. Cancer Res. 2001;61(6):2736–2743. [PubMed] [Google Scholar]
- 14.Georgiou HD, Namdarian B, Corcoran NM, Costello AJ, Hovens CM. Circulating endothelial cells as biomarkers of prostate cancer. Nat Clin Pract Urol. 2008;5(8):445–454. doi: 10.1038/ncpuro1188. [DOI] [PubMed] [Google Scholar]
- 15.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407(6801):249–257. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 16.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285(21):1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 17.Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6(4):273–286. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 18.Folkman J. Angiogenesis. Annu Rev Med. 2006;57:1–18. doi: 10.1146/annurev.med.57.121304.131306. [DOI] [PubMed] [Google Scholar]
- 19.Cook KM, Figg WD. Angiogenesis inhibitors: current strategies and future prospects. CA Cancer J Clin. 2010;60(4):222–243. doi: 10.3322/caac.20075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Samant RS, Shevde LA. Recent advances in anti-angiogenic therapy of cancer. Oncotarget. 2011;2(3):122–134. doi: 10.18632/oncotarget.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim EJ, Zalupski MM. Systemic therapy for advanced gastrointestinal stromal tumors: beyond imatinib. J Surg Oncol. 2011;104(8):901–906. doi: 10.1002/jso.21872. [DOI] [PubMed] [Google Scholar]
- 22.Purmonen TT. Cost-effectiveness of sunitinib in metastatic renal cell carcinoma. Expert Rev Pharmacoecon Outcomes Res. 2011;11(4):383–393. doi: 10.1586/erp.11.33. [DOI] [PubMed] [Google Scholar]
- 23.Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193(6):727–740. doi: 10.1084/jem.193.6.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rogers TL, Holen I. Tumour macrophages as potential targets of bisphosphonates. J Transl Med. 2011;9:177. doi: 10.1186/1479-5876-9-177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages. Crit Rev Oncol Hematol. 2008;66(1):1–9. doi: 10.1016/j.critrevonc.2007.07.004. [DOI] [PubMed] [Google Scholar]
- 26.Sica A, Rubino L, Mancino A, Larghi P, Porta C, Rimoldi M, Solinas G, Locati M, Allavena P, Mantovani A. Targeting tumour-associated macrophages. Expert Opin Ther Targets. 2007;11(9):1219–1229. doi: 10.1517/14728222.11.9.1219. [DOI] [PubMed] [Google Scholar]
- 27.Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 2002;196(3):254–265. doi: 10.1002/path.1027. [DOI] [PubMed] [Google Scholar]
- 28.Langley RE, Burdett S, Tierney JF, Cafferty F, Parmar MK, Venning G. Aspirin and cancer: has aspirin been overlooked as an adjuvant therapy? Br J Cancer. 2011;105(8):1107–1113. doi: 10.1038/bjc.2011.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meyerhardt JA. Beyond standard adjuvant therapy for colon cancer: role of nonstandard interventions. Semin Oncol. 2011;38(4):533–541. doi: 10.1053/j.seminoncol.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sporn MB. Perspective: The big C - for Chemoprevention. Nature. 2011;471(7339):S10–11. doi: 10.1038/471S10a. [DOI] [PubMed] [Google Scholar]
- 31.Miselis NR, Wu ZJ, Van Rooijen N, Kane AB. Targeting tumor-associated macrophages in an orthotopic murine model of diffuse malignant mesothelioma. Mol Cancer Ther. 2008;7(4):788–799. doi: 10.1158/1535-7163.MCT-07-0579. [DOI] [PubMed] [Google Scholar]
- 32.Veltman JD, Lambers ME, van Nimwegen M, Hendriks RW, Hoogsteden HC, Hegmans JP, Aerts JG. Zoledronic acid impairs myeloid differentiation to tumour-associated macrophages in mesothelioma. Br J Cancer. 2010;103(5):629–641. doi: 10.1038/sj.bjc.6605814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boehm T, Folkman J, Browder T, O’Reilly MS. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature. 1997;390(6658):404–407. doi: 10.1038/37126. [DOI] [PubMed] [Google Scholar]
- 34.Smith MR, Saad F, Coleman R, Shore N, Fizazi K, Tombal B, Miller K, Sieber P, Karsh L, Damiao R, Tammela TL, Egerdie B, Van Poppel H, Chin J, Morote J, Gomez-Veiga F, Borkowski T, Ye Z, Kupic A, Dansey R, Goessl C. Denosumab and bone-metastasis-free survival in men with castration-resistant prostate cancer: results of a phase 3, randomised, placebo-controlled trial. Lancet. 2012;379(9810):39–46. doi: 10.1016/S0140-6736(11)61226-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fizazi K, Carducci M, Smith M, Damiao R, Brown J, Karsh L, Milecki P, Shore N, Rader M, Wang H, Jiang Q, Tadros S, Dansey R, Goessl C. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: a randomised, double-blind study. Lancet. 2011;377(9768):813–822. doi: 10.1016/S0140-6736(10)62344-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lyons TR, O’Brien J, Borges VF, Conklin MW, Keely PJ, Eliceiri KW, Marusyk A, Tan AC, Schedin P. Postpartum mammary gland involution drives progression of ductal carcinoma in situ through collagen and COX-2. Nat Med. 2011;17(9):1109–1115. doi: 10.1038/nm.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lorusso G, Ruegg C. The tumor microenvironment and its contribution to tumor evolution toward metastasis. Histochem Cell Biol. 2008;130(6):1091–1103. doi: 10.1007/s00418-008-0530-8. [DOI] [PubMed] [Google Scholar]
- 38.Hussain SP, Hofseth LJ, Harris CC. Radical causes of cancer. Nat Rev Cancer. 2003;3(4):276–285. doi: 10.1038/nrc1046. [DOI] [PubMed] [Google Scholar]
- 39.Maeda H, Akaike T. Nitric oxide and oxygen radicals in infection, inflammation, and cancer. Biochemistry (Mosc) 1998;63(7):854–865. [PubMed] [Google Scholar]
- 40.Brasky TM, Potter JD, Kristal AR, Patterson RE, Peters U, Asgari MM, Thornquist MD, White E. Non-steroidal anti-inflammatory drugs and cancer incidence by sex in the VITamins And Lifestyle (VITAL) cohort. Cancer Causes Control. 2012 doi: 10.1007/s10552-011-9891-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lo-Ciganic WH, Zgibor JC, Bunker CH, Moysich KB, Edwards RP, Ness RB. Aspirin, Nonaspirin Nonsteroidal Anti-inflammatory Drugs, or Acetaminophen and Risk of Ovarian Cancer. Epidemiology. 2012 doi: 10.1097/EDE.0b013e3182456ad3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tosetti F, Ferrari N, De Flora S, Albini A. Angioprevention’: angiogenesis is a common and key target for cancer chemopreventive agents. FASEB J. 2002;16(1):2–14. doi: 10.1096/fj.01-0300rev. [DOI] [PubMed] [Google Scholar]
- 43.Singh RP, Deep G, Chittezhath M, Kaur M, Dwyer-Nield LD, Malkinson AM, Agarwal R. Effect of silibinin on the growth and progression of primary lung tumors in mice. J Natl Cancer Inst. 2006;98(12):846–855. doi: 10.1093/jnci/djj231. [DOI] [PubMed] [Google Scholar]
- 44.Raina K, Rajamanickam S, Singh RP, Deep G, Chittezhath M, Agarwal R. Stage-specific inhibitory effects and associated mechanisms of silibinin on tumor progression and metastasis in transgenic adenocarcinoma of the mouse prostate model. Cancer Res. 2008;68(16):6822–6830. doi: 10.1158/0008-5472.CAN-08-1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kavitha CV, Agarwal C, Agarwal R, Deep G. Asiatic Acid inhibits pro-angiogenic effects of VEGF and human gliomas in endothelial cell culture models. PLoS One. 2011;6(8):e22745. doi: 10.1371/journal.pone.0022745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cavell BE, Syed Alwi SS, Donlevy A, Packham G. Anti-angiogenic effects of dietary isothiocyanates: mechanisms of action and implications for human health. Biochem Pharmacol. 2011;81(3):327–336. doi: 10.1016/j.bcp.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 47.Pfeffer U, Ferrari N, Morini M, Benelli R, Noonan DM, Albini A. Antiangiogenic activity of chemopreventive drugs. Int J Biol Markers. 2003;18(1):70–74. doi: 10.1177/172460080301800113. [DOI] [PubMed] [Google Scholar]
- 48.Longo VD, Fontana L. Calorie restriction and cancer prevention: metabolic and molecular mechanisms. Trends Pharmacol Sci. 2010;31(2):89–98. doi: 10.1016/j.tips.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fontana L, Klein S. Aging, adiposity, and calorie restriction. JAMA. 2007;297(9):986–994. doi: 10.1001/jama.297.9.986. [DOI] [PubMed] [Google Scholar]
- 50.Hursting SD, Lavigne JA, Berrigan D, Perkins SN, Barrett JC. Calorie restriction, aging, and cancer prevention: mechanisms of action and applicability to humans. Annu Rev Med. 2003;54:131–152. doi: 10.1146/annurev.med.54.101601.152156. [DOI] [PubMed] [Google Scholar]
- 51.Jain RK. Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med. 2001;7(9):987–989. doi: 10.1038/nm0901-987. [DOI] [PubMed] [Google Scholar]
- 52.Fukumura D, Jain RK. Tumor microenvironment abnormalities: causes, consequences, and strategies to normalize. J Cell Biochem. 2007;101(4):937–949. doi: 10.1002/jcb.21187. [DOI] [PubMed] [Google Scholar]
- 53.Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK. Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev. 2011;91(3):1071–1121. doi: 10.1152/physrev.00038.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sorensen AG, Emblem KE, Polaskova P, Jennings D, Kim H, Ancukiewicz M, Wang M, Wen PY, Ivy P, Batchelor TT, Jain RK. Increased Survival of Glioblastoma Patients Who Respond to Antiangiogenic Therapy with Elevated Blood Perfusion. Cancer Res. 2012;72(2):402–407. doi: 10.1158/0008-5472.CAN-11-2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Coscia M, Quaglino E, Iezzi M, Curcio C, Pantaleoni F, Riganti C, Holen I, Monkkonen H, Boccadoro M, Forni G, Musiani P, Bosia A, Cavallo F, Massaia M. Zoledronic acid repolarizes tumour-associated macrophages and inhibits mammary carcinogenesis by targeting the mevalonate pathway. J Cell Mol Med. 2010;14(12):2803–2815. doi: 10.1111/j.1582-4934.2009.00926.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Agarwal R, Agarwal C, Ichikawa H, Singh RP, Aggarwal BB. Anticancer potential of silymarin: from bench to bed side. Anticancer Res. 2006;26(6B):4457–4498. [PubMed] [Google Scholar]
- 57.Pradhan SC, Girish C. Hepatoprotective herbal drug, silymarin from experimental pharmacology to clinical medicine. Indian J Med Res. 2006;124(5):491–504. [PubMed] [Google Scholar]
- 58.Deep G, Agarwal R. Antimetastatic efficacy of silibinin: molecular mechanisms and therapeutic potential against cancer. Cancer Metastasis Rev. 2010;29(3):447–463. doi: 10.1007/s10555-010-9237-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Singh RP, Agarwal R. A cancer chemopreventive agent silibinin, targets mitogenic and survival signaling in prostate cancer. Mutat Res. 2004;555(1–2):21–32. doi: 10.1016/j.mrfmmm.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 60.Singh RP, Agarwal R. Mechanisms and preclinical efficacy of silibinin in preventing skin cancer. Eur J Cancer. 2005;41(13):1969–1979. doi: 10.1016/j.ejca.2005.03.033. [DOI] [PubMed] [Google Scholar]
- 61.Singh RP, Agarwal R. Prostate cancer chemoprevention by silibinin: bench to bedside. Mol Carcinog. 2006;45(6):436–442. doi: 10.1002/mc.20223. [DOI] [PubMed] [Google Scholar]
- 62.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86(3):353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 63.Naumov GN, Akslen LA, Folkman J. Role of angiogenesis in human tumor dormancy: animal models of the angiogenic switch. Cell Cycle. 2006;5(16):1779–1787. doi: 10.4161/cc.5.16.3018. [DOI] [PubMed] [Google Scholar]
- 64.Menakuru SR, Brown NJ, Staton CA, Reed MW. Angiogenesis in pre-malignant conditions. Br J Cancer. 2008;99(12):1961–1966. doi: 10.1038/sj.bjc.6604733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bergers G, Benjamin LE. Tumorigenesis and the angiogenic switch. Nat Rev Cancer. 2003;3(6):401–410. doi: 10.1038/nrc1093. [DOI] [PubMed] [Google Scholar]
- 66.Deep G, Raina K, Singh RP, Oberlies NH, Kroll DJ, Agarwal R. Isosilibinin inhibits advanced human prostate cancer growth in athymic nude mice: comparison with silymarin and silibinin. Int J Cancer. 2008;123(12):2750–2758. doi: 10.1002/ijc.23879. [DOI] [PubMed] [Google Scholar]
- 67.Singh RP, Dhanalakshmi S, Agarwal C, Agarwal R. Silibinin strongly inhibits growth and survival of human endothelial cells via cell cycle arrest and downregulation of survivin, Akt and NF-kappaB: implications for angioprevention and antiangiogenic therapy. Oncogene. 2005;24(7):1188–1202. doi: 10.1038/sj.onc.1208276. [DOI] [PubMed] [Google Scholar]
- 68.Tyagi A, Singh RP, Ramasamy K, Raina K, Redente EF, Dwyer-Nield LD, Radcliffe RA, Malkinson AM, Agarwal R. Growth inhibition and regression of lung tumors by silibinin: modulation of angiogenesis by macrophage-associated cytokines and nuclear factor-kappaB and signal transducers and activators of transcription 3. Cancer Prev Res (Phila Pa) 2009;2(1):74–83. doi: 10.1158/1940-6207.CAPR-08-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rajamanickam S, Velmurugan B, Kaur M, Singh RP, Agarwal R. Chemoprevention of intestinal tumorigenesis in APCmin/+ mice by silibinin. Cancer Res. 2010;70(6):2368–2378. doi: 10.1158/0008-5472.CAN-09-3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Singh RP, Raina K, Deep G, Chan D, Agarwal R. Silibinin suppresses growth of human prostate carcinoma PC-3 orthotopic xenograft via activation of extracellular signal-regulated kinase 1/2 and inhibition of signal transducers and activators of transcription signaling. Clin Cancer Res. 2009;15(2):613–621. doi: 10.1158/1078-0432.CCR-08-1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Singh RP, Gu M, Agarwal R. Silibinin inhibits colorectal cancer growth by inhibiting tumor cell proliferation and angiogenesis. Cancer Res. 2008;68(6):2043–2050. doi: 10.1158/0008-5472.CAN-07-6247. [DOI] [PubMed] [Google Scholar]
- 72.Velmurugan B, Singh RP, Tyagi A, Agarwal R. Inhibition of azoxymethane-induced colonic aberrant crypt foci formation by silibinin in male Fisher 344 rats. Cancer Prev Res (Phila Pa) 2008;1(5):376–384. doi: 10.1158/1940-6207.CAPR-08-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Singh RP, Raina K, Sharma G, Agarwal R. Silibinin inhibits established prostate tumor growth, progression, invasion, and metastasis and suppresses tumor angiogenesis and epithelial-mesenchymal transition in transgenic adenocarcinoma of the mouse prostate model mice. Clin Cancer Res. 2008;14(23):7773–7780. doi: 10.1158/1078-0432.CCR-08-1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Singh RP, Dhanalakshmi S, Tyagi AK, Chan DC, Agarwal C, Agarwal R. Dietary feeding of silibinin inhibits advance human prostate carcinoma growth in athymic nude mice and increases plasma insulin-like growth factor-binding protein-3 levels. Cancer Res. 2002;62(11):3063–3069. [PubMed] [Google Scholar]
- 75.Velmurugan B, Gangar SC, Kaur M, Tyagi A, Deep G, Agarwal R. Silibinin exerts sustained growth suppressive effect against human colon carcinoma SW480 xenograft by targeting multiple signaling molecules. Pharm Res. 2010;27(10):2085–2097. doi: 10.1007/s11095-010-0207-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ravichandran K, Velmurugan B, Gu M, Singh RP, Agarwal R. Inhibitory effect of silibinin against azoxymethane-induced colon tumorigenesis in A/J mice. Clin Cancer Res. 2010;16(18):4595–4606. doi: 10.1158/1078-0432.CCR-10-1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yang SH, Lin JK, Huang CJ, Chen WS, Li SY, Chiu JH. Silibinin inhibits angiogenesis via Flt-1, but not KDR, receptor up-regulation. J Surg Res. 2005;128(1):140–146. doi: 10.1016/j.jss.2005.04.042. [DOI] [PubMed] [Google Scholar]
- 78.Ramasamy K, Dwyer-Nield LD, Serkova NJ, Hasebroock KM, Tyagi A, Raina K, Singh RP, Malkinson AM, Agarwal R. Silibinin prevents lung tumorigenesis in wild-type but not in iNOS−/− mice: potential of real-time micro-CT in lung cancer chemoprevention studies. Clin Cancer Res. 2011;17(4):753–761. doi: 10.1158/1078-0432.CCR-10-2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chittezhath M, Deep G, Singh RP, Agarwal C, Agarwal R. Silibinin inhibits cytokine-induced signaling cascades and down-regulates inducible nitric oxide synthase in human lung carcinoma A549 cells. Mol Cancer Ther. 2008;7(7):1817–1826. doi: 10.1158/1535-7163.MCT-08-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gu M, Singh RP, Dhanalakshmi S, Agarwal C, Agarwal R. Silibinin inhibits inflammatory and angiogenic attributes in photocarcinogenesis in SKH-1 hairless mice. Cancer Res. 2007;67(7):3483–3491. doi: 10.1158/0008-5472.CAN-06-3955. [DOI] [PubMed] [Google Scholar]
- 81.Singh RP, Tyagi A, Sharma G, Mohan S, Agarwal R. Oral silibinin inhibits in vivo human bladder tumor xenograft growth involving down-regulation of survivin. Clin Cancer Res. 2008;14(1):300–308. doi: 10.1158/1078-0432.CCR-07-1565. [DOI] [PubMed] [Google Scholar]
- 82.Yang SH, Lin JK, Chen WS, Chiu JH. Anti-angiogenic effect of silymarin on colon cancer LoVo cell line. J Surg Res. 2003;113(1):133–138. doi: 10.1016/s0022-4804(03)00229-4. [DOI] [PubMed] [Google Scholar]
- 83.Yoo HG, Jung SN, Hwang YS, Park JS, Kim MH, Jeong M, Ahn SJ, Ahn BW, Shin BA, Park RK, Jung YD. Involvement of NF-kappaB and caspases in silibinin-induced apoptosis of endothelial cells. Int J Mol Med. 2004;13(1):81–86. [PubMed] [Google Scholar]
- 84.Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006;441(7092):437–443. doi: 10.1038/nature04871. [DOI] [PubMed] [Google Scholar]
- 85.Garcia-Maceira P, Mateo J. Silibinin inhibits hypoxia-inducible factor-1alpha and mTOR/p70S6K/4E-BP1 signalling pathway in human cervical and hepatoma cancer cells: implications for anticancer therapy. Oncogene. 2009;28(3):313–324. doi: 10.1038/onc.2008.398. [DOI] [PubMed] [Google Scholar]
- 86.Jung HJ, Park JW, Lee JS, Lee SR, Jang BC, Suh SI, Suh MH, Baek WK. Silibinin inhibits expression of HIF-1alpha through suppression of protein translation in prostate cancer cells. Biochem Biophys Res Commun. 2009;390(1):71–76. doi: 10.1016/j.bbrc.2009.09.068. [DOI] [PubMed] [Google Scholar]
- 87.Kim S, Choi JH, Lim HI, Lee SK, Kim WW, Kim JS, Kim JH, Choe JH, Yang JH, Nam SJ, Lee JE. Silibinin prevents TPA-induced MMP-9 expression and VEGF secretion by inactivation of the Raf/MEK/ERK pathway in MCF-7 human breast cancer cells. Phytomedicine. 2009;16(6–7):573–580. doi: 10.1016/j.phymed.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 88.Mehlen P, Puisieux A. Metastasis: a question of life or death. Nat Rev Cancer. 2006;6(6):449–458. doi: 10.1038/nrc1886. [DOI] [PubMed] [Google Scholar]
- 89.Gupta GP, Minn AJ, Kang Y, Siegel PM, Serganova I, Cordon-Cardo C, Olshen AB, Gerald WL, Massague J. Identifying site-specific metastasis genes and functions. Cold Spring Harb Symp Quant Biol. 2005;70:149–158. doi: 10.1101/sqb.2005.70.018. [DOI] [PubMed] [Google Scholar]
- 90.Nguyen DX, Massague J. Genetic determinants of cancer metastasis. Nat Rev Genet. 2007;8(5):341–352. doi: 10.1038/nrg2101. [DOI] [PubMed] [Google Scholar]
- 91.Buijs JT, van der Pluijm G. Osteotropic cancers: from primary tumor to bone. Cancer Lett. 2009;273(2):177–193. doi: 10.1016/j.canlet.2008.05.044. [DOI] [PubMed] [Google Scholar]
- 92.Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3(6):453–458. doi: 10.1038/nrc1098. [DOI] [PubMed] [Google Scholar]
- 93.Boudreau NJ, Jones PL. Extracellular matrix and integrin signalling: the shape of things to come. Biochem J. 1999;339(Pt 3):481–488. [PMC free article] [PubMed] [Google Scholar]
- 94.Mitra AK, Sawada K, Tiwari P, Mui K, Gwin K, Lengyel E. Ligand-independent activation of c-Met by fibronectin and alpha(5)beta(1)-integrin regulates ovarian cancer invasion and metastasis. Oncogene. 2011;30(13):1566–1576. doi: 10.1038/onc.2010.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lee M, Park JJ, Lee YS. Adhesion of ST6Gal I-mediated human colon cancer cells to fibronectin contributes to cell survival by integrin beta1-mediated paxillin and AKT activation. Oncol Rep. 2010;23(3):757–761. [PubMed] [Google Scholar]
- 96.Yang Z, Zhang X, Gang H, Li X, Li Z, Wang T, Han J, Luo T, Wen F, Wu X. Up-regulation of gastric cancer cell invasion by Twist is accompanied by N-cadherin and fibronectin expression. Biochem Biophys Res Commun. 2007;358(3):925–930. doi: 10.1016/j.bbrc.2007.05.023. [DOI] [PubMed] [Google Scholar]
- 97.Mokhtari MJ, Motamed N, Shokrgozar MA. Evaluation of silibinin on the viability, migration and adhesion of the human prostate adenocarcinoma (PC-3) cell line. Cell Biol Int. 2008;32(8):888–892. doi: 10.1016/j.cellbi.2008.03.019. [DOI] [PubMed] [Google Scholar]
- 98.Handorean AM, Yang K, Robbins EW, Flaig TW, Iczkowski KA. Silibinin suppresses CD44 expression in prostate cancer cells. Am J Transl Res. 2009;1(1):80–86. [PMC free article] [PubMed] [Google Scholar]
- 99.Hsieh YS, Chu SC, Yang SF, Chen PN, Liu YC, Lu KH. Silibinin suppresses human osteosarcoma MG-63 cell invasion by inhibiting the ERK-dependent c-Jun/AP-1 induction of MMP-2. Carcinogenesis. 2007;28(5):977–987. doi: 10.1093/carcin/bgl221. [DOI] [PubMed] [Google Scholar]
- 100.Chen PN, Hsieh YS, Chiou HL, Chu SC. Silibinin inhibits cell invasion through inactivation of both PI3K-Akt and MAPK signaling pathways. Chem Biol Interact. 2005;156(2–3):141–150. doi: 10.1016/j.cbi.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 101.Wu KJ, Zeng J, Zhu GD, Zhang LL, Zhang D, Li L, Fan JH, Wang XY, He DL. Silibinin inhibits prostate cancer invasion, motility and migration by suppressing vimentin and MMP-2 expression. Acta Pharmacol Sin. 2009;30(8):1162–1168. doi: 10.1038/aps.2009.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lee SO, Jeong YJ, Im HG, Kim CH, Chang YC, Lee IS. Silibinin suppresses PMA-induced MMP-9 expression by blocking the AP-1 activation via MAPK signaling pathways in MCF-7 human breast carcinoma cells. Biochem Biophys Res Commun. 2007;354(1):165–171. doi: 10.1016/j.bbrc.2006.12.181. [DOI] [PubMed] [Google Scholar]
- 103.Chu SC, Chiou HL, Chen PN, Yang SF, Hsieh YS. Silibinin inhibits the invasion of human lung cancer cells via decreased productions of urokinase-plasminogen activator and matrix metalloproteinase-2. Mol Carcinog. 2004;40(3):143–149. doi: 10.1002/mc.20018. [DOI] [PubMed] [Google Scholar]
- 104.Chen PN, Hsieh YS, Chiang CL, Chiou HL, Yang SF, Chu SC. Silibinin inhibits invasion of oral cancer cells by suppressing the MAPK pathway. J Dent Res. 2006;85(3):220–225. doi: 10.1177/154405910608500303. [DOI] [PubMed] [Google Scholar]
- 105.Momeny M, Malehmir M, Zakidizaji M, Ghasemi R, Ghadimi H, Shokrgozar MA, Emami AH, Nafissi S, Ghavamzadeh A, Ghaffari SH. Silibinin inhibits invasive properties of human glioblastoma U87MG cells through suppression of cathepsin B and nuclear factor kappa B-mediated induction of matrix metalloproteinase 9. Anticancer Drugs. 21(3):252–260. doi: 10.1097/cad.0b013e3283340cd7. [DOI] [PubMed] [Google Scholar]
- 106.Christiansen JJ, Rajasekaran AK. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res. 2006;66(17):8319–8326. doi: 10.1158/0008-5472.CAN-06-0410. [DOI] [PubMed] [Google Scholar]
- 107.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9(4):265–273. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 108.Deep G, Gangar SC, Agarwal C, Agarwal R. Role of E-cadherin in antimigratory and antiinvasive efficacy of silibinin in prostate cancer cells. Cancer Prev Res (Phila) 2011;4(8):1222–1232. doi: 10.1158/1940-6207.CAPR-10-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tantivejkul K, Kalikin LM, Pienta KJ. Dynamic process of prostate cancer metastasis to bone. J Cell Biochem. 2004;91(4):706–717. doi: 10.1002/jcb.10664. [DOI] [PubMed] [Google Scholar]
- 110.Msaouel P, Pissimissis N, Halapas A, Koutsilieris M. Mechanisms of bone metastasis in prostate cancer: clinical implications. Best Pract Res Clin Endocrinol Metab. 2008;22(2):341–355. doi: 10.1016/j.beem.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 111.Clarke NW, Hart CA, Brown MD. Molecular mechanisms of metastasis in prostate cancer. Asian J Androl. 2009;11(1):57–67. doi: 10.1038/aja.2008.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kim JL, Kang SW, Kang MK, Gong JH, Lee ES, Han SJ, Kang YH. Osteoblastogenesis and osteoprotection enhanced by flavonolignan silibinin in osteoblasts and osteoclasts. J Cell Biochem. 2012;113(1):247–259. doi: 10.1002/jcb.23351. [DOI] [PubMed] [Google Scholar]
- 113.Kim JH, Kim K, Jin HM, Song I, Youn BU, Lee J, Kim N. Silibinin inhibits osteoclast differentiation mediated by TNF family members. Mol Cells. 2009 doi: 10.1007/s10059-009-0123-y. [DOI] [PubMed] [Google Scholar]
- 114.Asagiri M, Takayanagi H. The molecular understanding of osteoclast differentiation. Bone. 2007;40(2):251–264. doi: 10.1016/j.bone.2006.09.023. [DOI] [PubMed] [Google Scholar]
- 115.Zhao Q, Wang X, Liu Y, He A, Jia R. NFATc1: functions in osteoclasts. Int J Biochem Cell Biol. 2010;42(5):576–579. doi: 10.1016/j.biocel.2009.12.018. [DOI] [PubMed] [Google Scholar]
- 116.Provinciali M, Papalini F, Orlando F, Pierpaoli S, Donnini A, Morazzoni P, Riva A, Smorlesi A. Effect of the silybin-phosphatidylcholine complex (IdB 1016) on the development of mammary tumors in HER-2/neu transgenic mice. Cancer Res. 2007;67(5):2022–2029. doi: 10.1158/0008-5472.CAN-06-2601. [DOI] [PubMed] [Google Scholar]
- 117.Dehmlow C, Murawski N, de Groot H. Scavenging of reactive oxygen species and inhibition of arachidonic acid metabolism by silibinin in human cells. Life Sci. 1996;58(18):1591–1600. doi: 10.1016/0024-3205(96)00134-8. [DOI] [PubMed] [Google Scholar]
- 118.Meeran SM, Katiyar S, Elmets CA, Katiyar SK. Silymarin inhibits UV radiation-induced immunosuppression through augmentation of interleukin-12 in mice. Mol Cancer Ther. 2006;5(7):1660–1668. doi: 10.1158/1535-7163.MCT-06-0095. [DOI] [PubMed] [Google Scholar]
- 119.Esmaily H, Vaziri-Bami A, Miroliaee AE, Baeeri M, Abdollahi M. The correlation between NF-kappaB inhibition and disease activity by coadministration of silibinin and ursodeoxycholic acid in experimental colitis. Fundam Clin Pharmacol. 2011;25(6):723–733. doi: 10.1111/j.1472-8206.2010.00893.x. [DOI] [PubMed] [Google Scholar]
- 120.Gasparini G, Longo R, Sarmiento R, Morabito A. Inhibitors of cyclo-oxygenase 2: a new class of anticancer agents? Lancet Oncol. 2003;4(10):605–615. doi: 10.1016/s1470-2045(03)01220-8. [DOI] [PubMed] [Google Scholar]
- 121.Ravichandran K, Tyagi A, Deep G, Agarwal C, Agarwal R. Interleukin-1beta-induced iNOS expression in human lung carcinoma A549 cells: involvement of STAT and MAPK pathways. Indian J Exp Biol. 2011;49(11):840–847. [PubMed] [Google Scholar]
- 122.Nickoloff BJ, Ben-Neriah Y, Pikarsky E. Inflammation and cancer: is the link as simple as we think? J Invest Dermatol. 2005;124(6):x–xiv. doi: 10.1111/j.0022-202X.2005.23724.x. [DOI] [PubMed] [Google Scholar]
- 123.Tyagi A, Agarwal C, Dwyer-Nield LD, Singh RP, Malkinson AM, Agarwal R. Silibinin modulates TNF-alpha and IFN-gamma mediated signaling to regulate COX2 and iNOS expression in tumorigenic mouse lung epithelial LM2 cells. Mol Carcinog. 2011 doi: 10.1002/mc.20851. [DOI] [PubMed] [Google Scholar]
- 124.Raina K, Agarwal C, Agarwal R. Effect of silibinin in human colorectal cancer cells: Targeting the activation of NF-kappaB signaling. Mol Carcinog. 2011 doi: 10.1002/mc.21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Dhanalakshmi S, Singh RP, Agarwal C, Agarwal R. Silibinin inhibits constitutive and TNFalpha-induced activation of NF-kappaB and sensitizes human prostate carcinoma DU145 cells to TNFalpha-induced apoptosis. Oncogene. 2002;21(11):1759–1767. doi: 10.1038/sj.onc.1205240. [DOI] [PubMed] [Google Scholar]
- 126.Raina K, Serkova NJ, Agarwal R. Silibinin feeding alters the metabolic profile in TRAMP prostatic tumors: 1H-NMRS-based metabolomics study. Cancer Res. 2009;69(9):3731–3735. doi: 10.1158/0008-5472.CAN-09-0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zi X, Zhang J, Agarwal R, Pollak M. Silibinin up-regulates insulin-like growth factor-binding protein 3 expression and inhibits proliferation of androgen-independent prostate cancer cells. Cancer Res. 2000;60(20):5617–5620. [PubMed] [Google Scholar]
- 128.Flaig TW, Glode M, Gustafson D, van Bokhoven A, Tao Y, Wilson S, Su LJ, Li Y, Harrison G, Agarwal R, Crawford ED, Lucia MS, Pollak M. A study of high-dose oral silybin-phytosome followed by prostatectomy in patients with localized prostate cancer. Prostate. 2010;70(8):848–855. doi: 10.1002/pros.21118. [DOI] [PubMed] [Google Scholar]
- 129.Flaig TW, Gustafson DL, Su LJ, Zirrolli JA, Crighton F, Harrison GS, Pierson AS, Agarwal R, Glode LM. A phase I and pharmacokinetic study of silybin-phytosome in prostate cancer patients. Invest New Drugs. 2007;25(2):139–146. doi: 10.1007/s10637-006-9019-2. [DOI] [PubMed] [Google Scholar]
- 130.Hoh C, Boocock D, Marczylo T, Singh R, Berry DP, Dennison AR, Hemingway D, Miller A, West K, Euden S, Garcea G, Farmer PB, Steward WP, Gescher AJ. Pilot study of oral silibinin, a putative chemopreventive agent, in colorectal cancer patients: silibinin levels in plasma, colorectum, and liver and their pharmacodynamic consequences. Clin Cancer Res. 2006;12(9):2944–2950. doi: 10.1158/1078-0432.CCR-05-2724. [DOI] [PubMed] [Google Scholar]