

Abstract
The fundamental theory of Förster resonance energy transfer (FRET) was established in the 1940's. Its great power was only realized in the past 20 years after different techniques were developed and applied to biological experiments. This success was made possible by the availability of suitable fluorescent probes, advanced optics, detectors, microscopy instrumentation and analytical tools. Combined with state-of-the-art microscopy and spectroscopy, FRET imaging allows scientists to study a variety of phenomena that produce changes in molecular proximity, thereby leading to many significant findings in the life sciences. In this review, we outline various FRET imaging techniques and their strengths and limitations; we also provide a biological model to demonstrate how to investigate protein-protein interactions in living cells using both intensity- and fluorescence lifetime-based FRET microscopy methods.
Keywords: FRET, FLIM, FLIM-FRET, protein-protein interactions
Introduction
Förster resonance energy transfer (FRET) is a non-radiative energy transfer process from an excited molecule (the donor) to another nearby molecule (the acceptor), via a long-range dipole-dipole coupling mechanism. The efficiency of energy transfer (E) from the donor to the acceptor is dependent upon the inverse of the sixth power of the distance (d) separating them, subject to the Förster distance (Ro) at which E is equal to 50% (Equation 1 and Figure 1A) (1–4). As shown in Equation 2, Ro (in Ångstrom) of a FRET pair depends upon: (i) the relative orientation between the dipoles of the donor emission and the acceptor absorption - κ2 ranging from 0 to 4; (ii) the refractive index of the medium - n; (iii) the donor quantum yield - QYD; (iv) the acceptor extinction coefficient - εA; and (v) the overlap integral between the donor emission and acceptor absorption spectra. For the majority of FRET pairs, Ro values are in the order of a few nanometers (3~7 nm). Therefore, FRET is typically limited to within 10 nm, providing a sensitive tool for studying a variety of phenomena that produce changes in molecular proximity.
| (1) |
| (2) |
FRET imaging is a powerful tool in life-science research (5–14). FRET measurements have been made between single-molecules (15), in tissues and even in whole animals (16,17). In flow cytometry, both in vitro and in vivo, FRET assays have been widely used to characterize DNA-protein, lipid-protein and protein-protein interactions (8,18–22). For example, Dye et al. (20) applied a FRET-based flow cytometric analysis of fusion proteins in live yeast cells to study the dimerization of the wild-type and the mutated Tom70p N-terminal transmembrane domain, and demonstrated that flow cytometry combined with FRET is a powerful tool for studying protein-protein interactions in a large number of individual cells. FRET microscopy imaging can provide spatial and temporal information of protein interactions in living cells under physiological conditions to address fundamental biological questions (23–32). Confocal FRET microscopy was employed to track the internalization of transferrin receptor-ligand complexes in live cells (27,33). FRET imaging has also been used to investigate the causes along with potential diagnostic tools and treatments for diseases (34–36). In the Alzheimer's disease study, advanced fluorescence lifetime-based FRET imaging enabled the detection of the spatial abnormalities of tau molecules as pathogenic markers in tissue sections (36). In pharmaceutical research, FRET imaging is used extensively in high throughput or content screening platforms for compound or drug screening (37–39). A FRET assay was designed for the screening of inhibitors of ADAMTS1, which plays a crucial role in inflammatory joint diseases; the results demonstrated the FRET assay to be an excellent tool not only for measuring ADAMTS1 activity, but also identifying new potential inhibitors (39).
Figure 1. The basic concepts of measuring FRET.

(A) The energy transfer efficiency (E) is plotted as a function of the donor-acceptor separation distance (d), expressed in units of the Förster distance (Ro) of the FRET pair, typically in the order of a few nanometers; the sharp decrease from 0.5Ro to 1.5Ro demonstrates the power of FRET for investigating phenomena that produce changes in molecular proximity. (B, C, D) FRET imaging methods are generally categorized into (B) fluorescence intensity-based, (C) fluorescence lifetime-based and (D) fluorescence anisotropy-based measurements (see text for more details).
In this review, we first provide some basic guidelines for choosing a FRET pair, and then describe various FRET microscopy and spectroscopy imaging techniques – their strengths and limitations, and avoiding pitfalls for using them are also emphasized. Through our annual FRET workshops (www.kcci.virginia.edu/workshop) and users of our imaging core, we have learned that interpreting FRET signals and energy transfer efficiency data with respect to their significance in a complex biological system presents the biggest challenge for most biologists. Using a real biological model as a showcase, we demonstrate how to employ both intensity- and fluorescence lifetime-based FRET imaging methods to investigate protein-protein interactions in living cells, and how to interpret the scattered FRET data points using quantitative FRET data analysis strategies.
FRET Pairs
Choosing a suitable FRET pair is the first step for successful FRET imaging. We are fortunate to have many choices: various fluorescent proteins have been developed and employed for FRET imaging to visualize dynamic protein interactions under physiological conditions (26,28,29,40–55). The development of organic dyes with improved photo-stability and excellent spectral characteristics provides additional choices for FRET imaging (27,33,56–60), as well as the utility of the emerging field of quantum dots (61,62). The selection of a correct FRET pair depends on the actual biological question to be investigated, the type of biological specimen to be imaged, the available instrumentation and the technique applied to measure FRET. A few selected fluorescent probes that have been used extensively for FRET imaging are given in Table 1.
Table 1.
Selected FRET Pairs in Biology
| FRET Pairs(1) | Ex / Em (nm)(2) | Comments | Ref |
|---|---|---|---|
|
| |||
| Fluorescent proteins (FPs) | |||
| BFP-GFP | BFP: 383/448 | BFP was one of the early FPs used in FRET experiments; however, it has a low quantum yield (0.31) and typically requires UV excitation. | (26,28) |
| BFP-YFP | GFP: 488/507 | ||
| YFP: 514/527 | |||
| CFP-YFP (49.14 Å) | CFP: 439/476 | CFP-YFP has been a widely used FRET pair. Cerulean, mCerulean3, mTurquoise - Venus are better substitutes with improved quantum yield, brightness and photostability. | (28,40–43,45,54,55) |
| Cerulean-Venus (53.76 Å) | Cerulean: 433/475 | ||
| mCerulean3-Venus (56.88 Å) | mCerulean3: 433/475 | ||
| mTurquoise-Venus (56.55 Å) | mTurquoise: 434/474 | ||
| Venus: 515/528 | |||
| mTFP1-Venus (58.77 Å) | mTFP1: 462/492 | mTFP1 has a high quantum yield (0.85) and is photostable. | (44,47,51) |
| mTFP1-mKO2 (54.74 Å) | mKO2: 551/565 | ||
| mTFP1-tdTomato (64.16 Å) | tdTomato: 554/581 | ||
| GFP-mRFP1 (52.45 Å) | GFP: 488/507 | GFP can be a good FRET donor to pair with a red FP acceptor. | (45,52,53) |
| GFP-mCherry (52.97 Å) | mRFP1: 584/607 | ||
| GFP-tdTomato (63.52 Å) | mCherry: 587/610 | ||
| Small molecules | |||
| Tryptophan-Dansyl (21 Å) | Tryptophan: 288/348 | Tryptophan naturally exists in animal and plant cells. | (90) |
| Dansyl: 331/535 | |||
| Fluorescein-Rhodamine (52.5Å, see Ref. 56) | Fluorescein: 492/520 | Many combinations of FRET pairs can be made from Alexa and Cy dyes with higher quantum yields, and better pH and photo stability, compared to the fluorescein (FITC), TRITC, Rhodamine and Texas Red dyes. | (27,33,56–60) |
| Rhodamine: 542/564 | |||
| Fluorescein-Cy3 | Alexa488: 496/519 | ||
| Alexa488-Alexa555 (67.5Å, see Ref. 27) | Alexa555: 555/565 | ||
| Alexa568: 578/603 | |||
| Alexa488-Cy3 | Alexa633: 632/647 | ||
| Alexa568-Alexa633 | Alexa647: 650/665 | ||
| Alexa568-Alexa647 | Cy3: 550/570 | ||
| Cy3-Cy5 (60Å, see Ref. 58) | Cy5: 650/670 | ||
| Cy5-Cy5.5 (73Å, see Ref. 58) | Cy5.5: 675/695 | ||
The Förster distance of the FRET pair in Angstrom (Å) in parentheses. The Förster distance of a FP FRET pair is calculated by an in-house developed numerical program based on Equation 2, where k2 and n are assumed to be 2/3 and 1.33, respectively (see Ref. (51). For small molecules, the Förster distances are obtained from the corresponding references.
The approximate peak excitation (Ex) and emission (Em) wavelengths (within a range of 250 to 800 nm) of a fluorophore.
A FRET pair with a large Ro value is generally favored, because of increased likelihood of FRET occurrence. The Ro value of a FRET pair can be estimated from their photophysical properties based on Equation 2 as described above. A sufficient overlap (> 30%) between the donor emission and acceptor absorption spectra is critical for FRET to occur. However, this overlap often causes the spectral bleedthrough problem for quantifying FRET based on acceptor sensitized emission (described below). However, the spectral bleedthrough is usually not an issue for measuring FRET based on the donor signals only, such as in acceptor photobleaching and fluorescence lifetime-based FRET measurements (described below). The donor quantum yield is also an important factor to achieve robust FRET efficiencies. Comparing the Ro values of CFP-YFP (49 Å), mCerulean-YFP (53 Å), and mCerulean3-YFP (56 Å) indicates that the differences are mainly caused by the different quantum yields of CFP (0.4) vs. mCerulean (0.62) and vs. mCerulean3 (0.87), which have almost identical excitation and emission spectra. In fluorescence lifetime-based FRET measurements, a donor, whose intrinsic lifetime has multiple components, can complicate the data analysis and should be avoided by choosing for example mCerulean or mCerulean3 over CFP. Other important factors for successful FRET imaging to be considered include photo-stability, brightness, and blinking issues.
FRET Microscopy and Spectroscopy
FRET depopulates the excited state of the donor, resulting in a decreased probability of photon emission from the donor and a shortening of the donor lifetime in the excited state; meanwhile, the probability of the photon emission from the acceptor increases (sensitization). Thus, FRET can be measured from changes in intensities (Figure 1B) or fluorescence lifetimes (Figure 1C) of the donor in the presence and absence of the acceptor, or by quantifying the sensitized emission of the acceptor. These FRET measurements typically require the donor and acceptor to be different fluorophores (called hetero-FRET), although the acceptor need not be fluorescent (e.g. dark quenchers) for measuring FRET based on the donor. FRET can also occur between identical fluorophores, called homo-FRET which can be measured by fluorescence anisotropy imaging (Figure 1D). Many FRET microscopy and spectroscopy techniques have been developed (63) - the basic concepts of several commonly used methods are summarized in Table 2 and explained in more details below.
Table 2.
FRET microscopy and spectroscopy imaging methods
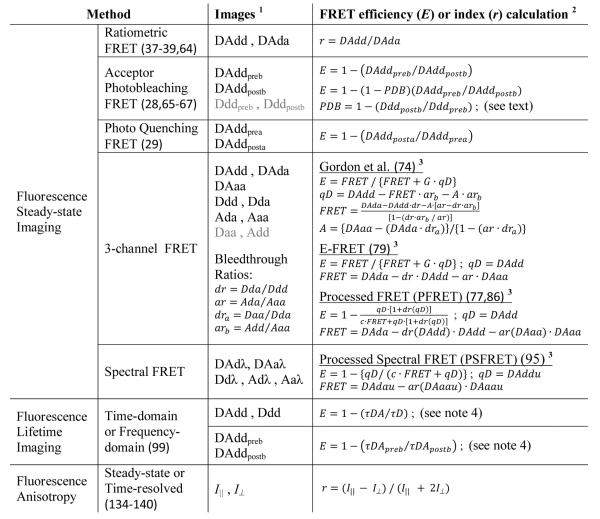
|
The capital letters denote the type of specimen: DA - the specimen containing both the donor and the acceptor; D - the donor-alone specimen; A - the acceptor-alone specimen. The first lower case letter represents the excitation for the donor (d) or the acceptor (a). The second lower case letter represents the detected emission range for the donor (d), the acceptor (a) or both as a λ-stack (λ). For subscripts, “preb” and “postb” refer to pre- and post-photobleaching of the acceptor, respectively; while “prea” and “posta” mean pre- and post-activation of the photo-activateable acceptor, respectively. In fluorescence lifetime imaging, measured data varies between acquisition methods (see text for details). In anisotropy imaging, the donor and acceptor are identical fluorophores, and images are acquired both, perpendicular (I⊥) and parallel (I∥) to the direction of the polarized excitation.
All E or r calculations can be performed on a pixel-by-pixel basis.
See text for details about each method. In spectral FRET imaging, `DAddu' and `DAdau' are the images obtained from the spectral linear unmixing of the `Dadλ' λ-stack; `DAaau' is the image obtained from unmixing the `DAaλ' λ-stack, with the donor (`Ddλ') and the acceptor (`Aaλ') reference spectra.
τDA (τDApreb − quenched donor lifetime) and τD (τDApostb − unquenched donor lifetime) are estimated from DAdd (DAddpreb) and Ddd (DAddpostb), respectively. See text for more details on each method.
2-channel Ratiometric FRET (rFRET) imaging
The rFRET approach is qualitative, only uses the donor excitation wavelength and measures the ratio of the acceptor emission and donor emission signals, representing a FRET index (Table 2) (37–39,64). This method is suitable for a fixed donor-acceptor stoichiometry (e.g. biosensor), in which case a higher `Acceptor:Donor' ratio indicates a larger E. This method cannot measure E directly, and also suffers from not being able to remove bleedthrough contaminations resulting from the donors and the acceptors of varied stoichiometry. On the plus-side, the `Acceptor:Donor' ratio readouts can be extremely fast by measuring both the donor and acceptor emission signals simultaneously, making the technique a major tool for FRET-based biosensor screening applications (37–39). To accurately track the dynamic FRET changes of a biosensor over time, one should carefully check if the ratio is affected by photobleaching perturbations, since the donor and acceptor molecules are likely to bleach at different rates at the donor excitation wavelength.
Acceptor photobleaching FRET (apFRET) imaging
In apFRET imaging, E can be quantified by measuring the donor intensities or fluorescence lifetimes before and after photobleaching the acceptor molecules (Table 2) (28,65–67). The specimen containing both the donor and the acceptor serves as its own control in apFRET imaging, making the technique straightforward.
However, the apFRET method is typically considered as an end-point assay, and thus is not suitable for intervention or time-lapse studies, although complete bleaching of the acceptor may not be required by some partial apFRET techniques (68,69). The intensive light illumination and the extensive time required for the photobleaching process may generate a few problems: (i) specimen movement and/or photo-damage to the specimen; (ii) bleaching of the donor; (iii) photo conversion of the donor or the acceptor (70). The percentage of donor bleached (PDB) factor given in Table 2 can be used to correct for unwanted photobleaching of donor molecules during acceptor photobleaching; this factor may be determined using the donor-only control specimen under the same photobleaching condition as the double-label specimen. Besides PDB, János et al. (67) also introduced factors to correct for incomplete photobleaching of the acceptor, the acceptor back bleedthrough to the donor channel and the post-bleached acceptor photo conversion to the donor channel; an ImageJ plugin called AccPbFRET developed by János et al. (67) is freely available at http://biophys.med.unideb.hu/accpbfret.
Photo-quenching FRET (pqFRET) imaging
The pqFRET approach uses photo-activateable fluorescence proteins as FRET acceptors, which turn from an original dark to a bright fluorescent state upon brief ultraviolet excitation (29). E can be quantified by measuring the donor intensities before and after activating the acceptor (Table 2). Other than measuring FRET, the pqFRET assay also allows monitoring the diffusion of the activated acceptor fluorophores over time, providing direct measurements of protein mobility, exchange and interactions in living cells (29).
3-channel FRET imaging
This approach measures FRET based on the acceptor sensitized emission - the FRET signal. The three imaging channels are defined as: the `Donor' channel uses the donor excitation and the donor emission filter; the `FRET' channel employs the donor excitation and the acceptor emission filter; the `Acceptor' channel applies the acceptor excitation and the acceptor emission filter (Table 2). The 3-channel FRET imaging is most widely used because it provides the capability for accurate and quantitative FRET measurements with any fluorescence microscopy and spectroscopy system. However, to correct the spectral bleedthrough (SBT) contaminations in the FRET signal, images of the donor-alone and the acceptor-alone control specimens in addition to the double-label specimens are acquired in the three imaging channels. Images are then processed by a computer algorithm to quantify FRET signals and efficiencies and correct potential SBT contaminations and autofluorescence, as described below.
Donor spectral bleedthrough (DSBT)
Cause: The donor fluorophore is excited by the donor excitation wavelength and emits in the acceptor emission range and is contained in the FRET signal in the FRET channel.
Check: Use the donor-alone control specimen and take images in the `FRET' channel.
Correction: Use the donor-alone control specimen to determine the DSBT ratio (dr), which will be used to estimate the amount of DSBT for the double-label in the `FRET' channel (Table 2).
Acceptor spectral bleedthrough (ASBT)
Cause: The acceptor fluorophore excited by the donor excitation wavelength yields its natural fluorescence, which is detected along with the FRET signal in the FRET channel.
Check: Excite the acceptor-alone control specimen with the donor wavelength and take images in the `FRET' channel.
Correction: Use the acceptor-alone control specimen to determine the ASBT ratio (ar), which will be used to estimate the amount of ASBT in the `FRET' channel (Table 2).
Donor spectral bleedthrough at the acceptor excitation wavelength (DSBTa)
Cause: The donor fluorophore is excited by the acceptor excitation wavelength and emits in the acceptor emission range, posing a problem for ASBT correction.
Check: Use the donor-alone control specimen and take images in the `Acceptor' channel.
Correction: Use the donor-alone control specimen to determine the DSBTa ratio (dra), which will be used to estimate the amount of DSBTa in the ASBT correction (Table 2). It is common to have DSBTa with multi-photon excitation. For single-photon excitation, however, it can usually be avoided by choosing an optimal FRET pair and a suitable combination of the donor and the acceptor excitation wavelengths.
Acceptor back spectral bleedthrough at the donor excitation wavelength (ASBTb)
Cause: The acceptor fluorophore is excited by the donor excitation wavelength and emits in the donor emission range, causing a problem for quantifying the quenched donor signal.
Check: Use the acceptor-alone control specimen and take images in the `Donor' channel.
Correction: Use the acceptor-alone control specimen to determine the ASBTb ratio (arb), which will be used to estimate the amount of ASBTb in the donor channel (Table 2). ASBTb can usually be avoided by choosing an optimal FRET pair.
Specimen autofluorescence
Cause: The specimen contains autofluorescent molecules, which are detected by acceptor or donor excitation wavelengths.
Check: Use the unlabeled specimen and take images at identical imaging conditions in all three imaging channels to verify if there are detectable autofluorescence signals in each channel.
Correction: For homogenous autofluorescence, one can determine its level using the unlabeled specimen for each imaging channel, and then subtract it from the corresponding images of the labeled specimen. Accurate removal of spatially varied autofluorescence can be challenging, but can be done by spectral imaging and linear unmixing (described below).
Comments
It should be stressed that specimen autofluorescence and some of the above-described SBT contaminations should be verified in other FRET imaging methods as well. For example, specimen autofluorescence and ASBTb should be verified in apFRET, pqFRET, rFRET imaging or fluorescence lifetime imaging (described below), where only the donor signals are measured in the `Donor' channel.
While the issues of SBT corrections may appear to be daunting, many 3-channel FRET imaging algorithms (71–87) were developed to ameliorate its effects. Commercial software solutions are widely available from companies including Olympus, Nikon, Zeiss, Leica, PerkinElmer, Molecular Devices (MetaMorph), Applied Precision Imaging, Bitplane, MediaCybernetics, and others. On the other hand, many researchers designed their own software based on published or their own algorithms, being fairly straightforward with basic programming skills (see Table 2). We developed the processed FRET (PFRET) algorithm (77,82,86), and created a proprietary ImageJ-based software package. The software can be obtained by contacting the University of Virginia Patent office (http://innovation.virginia.edu). Three free ImageJ-based software plugins are in the public domain: RiFRET (www.biophys.dote.hu/rifret), developed based on Ref. (85) can correct for SBT and autofluorescence contaminations and calculate FRET efficiencies pixel-by-pixel, and importantly, the source codes of this software are available for modification for other applications; PixFRET (www.unil.ch/cig/page16989.html), developed based on Ref. (80) also offers SBT corrections and calculates normalized FRET signals (by donor, acceptor, the product of both, or the square root of the product of both) as well as FRET efficiencies; the FRET and Colocalization Analyzer plugin (http://rsbweb.nih.gov/ij/plugins/fret-analyzer/fret-analyzer.htm), developed based on Ref. (83) has a simple interface to estimate SBT contaminations and produce a FRET index for each pixel, which is defined as the increase of intensity in the FRET channel over the donor and the acceptor SBTs; however this software can only be used for 8-bit images.
Three representative algorithms are compared in Table 2: (i) the Gordon et al. method (74) considers all possible SBT contaminations; (ii) the E-FRET approach (79) only considers DSBT and ASBT, which are the major SBT components for the majority of FRET experiments; (iii) different from these two methods, the PFRET algorithm used here does not assume a constant SBT ratio for the whole intensity dynamic range (e.g. 0 ~ 4095 for the 12-bit imaging), but rather estimates the SBT ratios based on varying signal levels (77,86). The PFRET software allows users define the number of the intensity ranges to estimate the corresponding SBT ratios and specify an intensity threshold value above which the pixels are only used for calculating SBT ratios. A proper threshold value should be chose upon the imaging depth to avoid poor signal-to-noise effects and this is especially critical for using a constant SBT ratio. We shall emphasize that it is important to collect a number of single-label control images (dim-to-bright), which cover the entire intensity dynamic range. A comparison of using constant vs. dynamic SBT ratios to produce the FRET results of a same data set is presented by Figure 2. A more thorough comparison between the PFRET and Gordon et al. methods was made to demonstrate the advantage of using dynamic rather than constant SBT ratios (88). In 3-channel FRET imaging, E is determined using the FRET signal and the quenched donor (qD) signal as the basis for the calculation (Table 2). The FRET signal is produced by the sensitized acceptor and measured in the `FRET' channel, while the qD signal is produced by the donor and measured in the `Donor' channel. Thus, a correction factor `G' is used in the Gordon et al. and E-FRET methods, and `c' is used in the PFRET method (Table 2) to compensate for the difference between the donor and acceptor quantum yields as well as different detection efficiencies (e.g. detector sensitivity) for the donor and the acceptor. Theoretical modeling of the `G' factor (74) and coefficient `c' (82,86) are described in the literature, and several methods were deployed to experimentally measure them (76,79,86,89). It should be noted that the PFRET method considers DSBT as part of qD for a more accurate E estimation (86) (Table 2).
Figure 2.

The comparison between the FRET results of a same data set obtained with the constant SBT ratios estimated in a single intensity range or the dynamic SBT ratios in 20 intensity ranges. A threshold value of 100 (>100) was used for calculating the dynamic SBT ratios, and two different threshold values of 100 (>100) and 500 (>500) to calculate the corresponding constant SBT ratios. (A) The mean values and the standard deviations of the dynamic donor (dot) and acceptor (square) SBT ratios at different intensity ranges are plotted along with the constant donor (solid line – 100 and dotted line - 500) and acceptor (short dashed line – 100 and long dashed line - 500) SBT ratios. (B) The table shows the intensity levels for each intensity range used to estimate the dynamic SBT ratios. (C) For a same field as an example, the E% images obtained using the dynamic SBT ratios vs. the constant SBT ratios with a threshold value of 100 are compared – the absolute difference image is made by taking the absolute value of the difference between the two E% images at the same pixel. (D) The pixel-by-pixel correlations between E% images obtained using the dynamic and constant SBT ratios are plotted. (The data set used here is the same as in Figure 4, where the experimental details are explained. It should be noted that - background is removed from raw images to obtain zero-background images prior to the FRET processing, i.e. SBT estimation, removal of SBT, FRET and E% calculations; saturated pixels are meaningless and thus ignored.)
Spectral FRET (sFRET) imaging
In vitro FRET experiments are commonly conducted in solution in cuvettes using a spectrofluorometer, applying donor excitation and measuring signals for the whole `donor-and-acceptor' emission range in 1-nm increments. FRET can be quantified by evaluating the double-label spectra data in comparison to the donor-alone and the acceptor-alone reference spectra (56,90). With specimens on a substrate (cells, tissue), spectral microscopy imaging produces a λ-stack consisting of spatial and spectral dimensions to measure emission signals in a series of spectral intervals equally sampled over a spectral range, at each pixel location. In sFRET imaging, the signals emitted from the donor, the acceptor or the autofluorescent fluorophores can be accurately separated by the spectral unmixing based on the corresponding reference spectra, which are obtained from the donor-alone, the acceptor-alone or the unlabeled specimens, respectively (91–97). Thus, DSBT, DSBTa, ASBTb and autofluorescence can be accurately removed by spectral unmixing, making the sFRET technique particularly suitable for multiphoton excitation FRET applications (93,97,98). However, the FRET signal may still contain ASBT after spectral unmixing. The processed spectral FRET (psFRET) method employs a similar strategy as PFRET for removing ASBT in sFRET microscopy imaging (95) (Table 2).
Fluorescence lifetime imaging (FLIM) FRET
Fluorescence lifetime is the average time a molecule spends in the excited state before returning to ground state. The fluorescence lifetime of a fluorophore carries information about events in its local microenvironment which affects its photophysical processes. FRET adds a non-radiative dissipation pathway for the excited state energy of the donor and thus shortens its fluorescence lifetime. Using FLIM, E can be quantified by measuring the reduction of the donor fluorescence lifetime, resulting from quenching in the presence of an acceptor (99) (Table 2). The FLIM-FRET approach has several advantages over intensity-based FRET imaging: (i) FLIM-FRET measurements typically do not require corrections for spectral bleedthrough, necessary for intensity-based measurements of acceptor sensitized emission; (ii) fluorescence lifetime measurements are insensitive to the change in fluorophore concentration, excitation intensity, or light scattering and to some extent of photobleaching – all these factors potentially induce artifacts in intensity-based imaging; and (iii) FLIM-FRET methods have the capability to estimate the percentage of `FRETing' and `non-FRETing' donor populations (100), which cannot be distinguished by most intensity-based FRET methods; the E measured in most intensity-based FRET methods are often called apparent E, which inter alia includes `non-FRETing' donors in the calculation.
FLIM-FRET measurements require advanced instrumentation as well as understanding of the basic physics for data analysis and interpretation. In the past 15 years, rapid developments in FLIM have greatly advanced and simplified the technique, and various FLIM methods have been developed for biological and clinical applications (32,101,102). More importantly, commercial stand-alone FLIM systems or integrated with existing multiphoton, confocal or wide-field microscopes have become available from Picoquant, Becker & Hickl, Lambert Instruments, ISS, Intelligent Imaging Innovations and others. FLIM therefore has become more of a routine tool for many laboratories for FRET studies (32,34,59,103–121). A recent development by Leray et al. (114) applied a 3-D polar plot analysis to spectrally resolved FLIM data, demonstrating that this multi-model fitting-free approach yields more accurate FRET measurements.
FLIM techniques are generally subdivided into the time-domain (TD) and the frequency-domain (FD), although the basic physics for both are essentially identical (32,99,102,122). TD FLIM uses a pulsed light source synchronized to high-speed detectors and electronics to directly measure the fluorescence decay profile to estimate the fluorescence lifetime. FD FLIM employs a modulated excitation light source, and measures the phase shift(s) and amplitude attenuation(s) of the emission relative to the excitation to estimate the fluorescence lifetime. The repetition rate of the excitation source in TD FLIM or the fundamental modulation frequency of the excitation source in FD FLIM are chosen according to the fluorescence lifetime to be measured, e.g. megahertz for measuring nanosecond lifetimes. Several TD and FD FLIM techniques are briefly described below.
TD FLIM by time correlated single photon counting (TCSPC)
TCPSC typically uses a pulsed laser, a photon-counting detector and TCSPC electronics (32,108,123–125). A spectrofluorometer synchronizes the detector to the excitation pulse and records the photon arrival time relative to the excitation pulse. By accumulating photons for a period of time, a `photon counts' histogram, called fluorescence decay, is generated to project the fluorescence lifetime. For laser scanning microscopes, the TCSPC device synchronizes both the detector and the scanning clock to the excitation pulse, and records the arrival time as well as spatial information for each detected photon; the fluorescence decay profile is shown for each pixel.
TD FLIM using a gated image-intensifier camera
Gating-camera FLIM is typically found on wide-field and spinning-disk confocal microscopes using a pulsed laser, an image-intensifier camera and gating-control electronics. The gating camera can be operated at superfast speeds to detect photons within a time (gating) window for hundred picoseconds to milliseconds relative to the excitation pulse (105,126). A number of images are acquired in sequential gating windows to estimate the fluorescence lifetime. Extracting the single-component lifetime requires collecting two gated images at a minimum – this may only take a couple of seconds or less (105,127), making the gating-camera FLIM technique suitable for high-content screening FRET applications (121,128).
TD FLIM using a streak-camera
A streak camera system, consisting of a streak scope and a fast CCD camera, can be operated to transform the temporal profile of a light pulse into a spatial profile on a detector by causing a time-varying deflection of the light across the width of the detector (129). In this FLIM method, a pulsed (one- or multi-photon) laser is synchronized with a streak camera system, and a stack of images is acquired to generate a 2-D fluorescence lifetime image: each line of an image consists of the decay profile to establish the fluorescence lifetime for each pixel along the x-dimension; each image contains the decay profiles for one line of pixels along the y-dimension (107,109,129).
Digital FD FLIM
In traditional heterodyne FD FLIM, both the light source and the detector are modulated, but at slightly different frequencies, e.g. a few hundred hertz. The digital FD FLIM employs a modulated pulsed excitation source, but does not require modulating the detector (130). In this technique, the detector is working in a photon-counting manner, and all operations including the generation of the light modulation frequency, the generation of the cross-correlation sampling frequency and the assignment of the time of arrival of a photon to a bin are digital, allowing multi-frequency measurements (for extracting multi-component lifetimes) to be done simultaneously, greatly improving photon efficiency and data acquisition speed (32,130).
FD FLIM using an image-intensifier camera
Camera-based FD FLIM is typically applied in widefield or spinning-disk confocal microscopes, using a LED or diode laser excitation light source and an image-intensifier camera, both modulated at slightly different frequencies (Heterodyne) (131) or at the same frequency (Homodyne) (132,133). Similar to the gating-camera TD FLIM, the camera-based FD FLIM technique also provides fast imaging speeds (54).
A few precautions need to be taken with FLIM-FRET measurements: (i) a FLIM system should always be carefully calibrated prior to a FRET study, using fluorescence lifetime standards (32); (ii) it is important to make sure that the donor fluorophores reside in the same microenvironment in both donor-alone control and double-label specimens, since the fluorescence lifetime of a fluorophore can be affected by its microenvironment (e.g. pH, temperature); and (iii) most of FLIM data analysis methods estimate the fluorescence lifetimes through least-square fittings of measured data based on a single- or multi-exponential decay model; it is important to have reproducibility of data for a particular data processing model (32).
Fluorescence anisotropy imaging FRET
For a randomly oriented population of fluorophores excited by linearly polarized light, those molecules with their absorption dipole oriented parallel to the polarization axis are preferentially excited. In fluorescence anisotropy (polarization) microscopy, the photons emitted from fluorophores excited by a linearly polarized light source are measured using two linear polarizer filters – one parallel to the direction of the excitation and the other perpendicular to that direction; the anisotropy value is determined using both the parallel and the perpendicular emission signals (Table 2), indicating the degree of the fluorophore orientation. The anisotropy value will decrease (called depolarization) if the fluorophore changes its orientation between excitation and emission, which can be caused by FRET, the basic concept for measuring FRET by fluorescence anisotropy (Figure 1D). Anisotropy has been employed to measure monomer-dimer transition of GFP-tagged proteins (134) and to quantify protein cluster sizes with sub-cellular resolution (135). There are advantages of using fluorescence anisotropy FRET measurements: (i) both the donor and the acceptor are tagged with the same fluorophore (homo-FRET), and thus there is no bleedthrough problem; and (ii) the parallel and perpendicular emission signals can be simultaneously measured by extremely fast anisotropy readouts, making the technique suitable for FRET screening applications (136,137). However, one should also keep in mind that depolarization can also be caused by an objective lens of a high numerical aperture (> 1.0). The fluorophore rotation can also change its orientation resulting in depolarization, although this effect can usually be ignored for large molecules like fluorescent proteins since energy transfer takes place more rapidly than their motions (134–140). It has been demonstrated that time-resolved anisotropy imaging can overcome many of the limitations of intensity-based anisotropy imaging for FRET measurements (134,135,138–140).
A FRET example for localizing protein-protein interactions in living cells
Prior to biological studies, a FRET imaging approach/system should be calibrated. A comparative method to determine the accuracy of FRET measurements was developed by the Vogel laboratory (NIH) (141). The approach uses “standards” in the form of genetic constructs encoding fusions between donor and acceptor fluorescent proteins separated by defined amino acid (aa) linker sequences. A series of FRET-standard constructs were generated through encoding Cerulean and Venus, directly coupled by either a 5, 17 or 32 aa linker - named as C5V, C17V and C32V correspondingly (141). In addition, a construct of a low FRET efficiency (CTV) was also made by separating Cerulean and Venus with a 229-aa TRAF domain (93). These plasmids are available at www.addgene.org/Steven_Vogel. Employing these FRET standards, we calibrated our PFRET, spectral FRET, time- and frequency-domain FLIM-FRET techniques, which roughly produced the same FRET efficiency of each construct: the C5V and CTV expressed in live cells gave 40~50% and 5–10% FRET efficiencies, respectively (12,32,86).
A biological FRET application used here concerns the basic region-leucine zipper (bZip) domain of the CCAAT/enhancer binding protein alpha (C/EBPα) transcription factor. The bZip family proteins form obligate dimers through their leucine-zipper domains, which positions the basic region residues for binding to specific DNA elements. Immunocytochemical staining of differentiated mouse adipocyte cells showed that endogenous C/EBPα is preferentially bound to satellite DNA-repeat sequences located in regions of centromeric heterochromatin (142,143). When the C/EBPα bZip domain is expressed as a fusion fluorescent protein in cells of mouse origin, it is localized to the well-defined regions of centromeric heterochromatin in the cell nucleus (Figures 3–5) (32). A FRET system for investigating this biological model in living cells was built by fusing the C/EBPα bZip domain to Cerulean (bZip-Cerulean, FRET donor) and Venus (bZip-Venus, FRET acceptor) fluorescence proteins separately.
Figure 3. Acceptor photobleaching spectral FRET microscopy.

Live cells co-expressing Cerulean (donor) and Venus (acceptor) tagged bZip were excited by a 458 nm laser line, and spectral images (λ-stack for 470 ~ 640 nm) were acquired at the same imaging conditions, before and after photobleaching the acceptor with the 514 nm laser line (bleaching time: ~120 seconds). Comparing the pre- and post-bleach spectra shows successful photobleaching of the acceptor and also an intensity increase (de-quenching) of the donor, indicating FRET between them. Using the donor and acceptor reference spectra obtained from cells expressing either bZip-Cerulean or bZip-Venus, spectral linear unmixing of a λ-stack produced two unmixed images: one only contains emission signals from the donor, the other only the acceptor emission signals. The same contrast adjustment was applied for the pre-bleach and post-bleach unmixed images of each channel (Donor: 0 ~ 1500, Acceptor: 0 ~ 4000, 12-bit). The FRET efficiency (E%) image was calculated using the pre- and post-bleach unmixed images of the donor channel (see Table 2). However, the E% calculation was limited to the centromeric regions by setting an intensity threshold of 500 for the pre-bleach acceptor image – clearly seen from the E% profile plotted as a line in the E% image. (Zeiss 510 Meta confocal microscope and 63X / 1.4NA oil immersion objective)
Figure 5. Two-photon excitation (TPE) time correlated single photon counting (TCSPC) fluorescence lifetime imaging (FLIM) FRET microscopy.

TCSPC FLIM data sets were acquired from cells co-expressing bZip-Cerulean and bZip-Venus and cells that only express bZip-Cerulean as the donor-alone control. The fluorescent lifetime decay kinetics for the bZip-Cerulean (FRET donor) in the absence and presence of bZip-Venus (acceptor) were determined by fitting the decay data into a single or double exponential decay model, respectively, with the estimated instrument response function (IRF). By applying a suitable intensity threshold, fitting was only applied to pixels in the centromeric heterochromatin regions of the cell nucleus. The comparison between the representative decay data points, fitting curves and fluorescence lifetime images and distributions of the two cases clearly shows bZip-Cerulean in the presence of bZip-Venus decays faster (i.e. has a shorter lifetime) than that in the donor-alone control cells, demonstrating bZip-Cerulean was quenched by bZip-Venus due to FRET. (Biorad Radiance 2100 Confocal / Multiphoton imaging system and Nikon 60X / 1.2NA water immersion objective; a Coherent Mira-900 ultrafast (repetition rate of ~78 MHz) pulsed (pulse width of ~150 femtoseconds) laser was tuned to 820 nm for the TPE wavelength; a Becker & Hickl (BH) SPC-150 board was used to synchronize the laser pulse to the scanning clock; photons were counted by using a BH PMH-100-0 photomultiplier tube detector; all decay data sets were analyzed using the BH SPCImage software Version 3.8.9)
Acceptor photobleaching spectral FRET
The acceptor photobleaching FRET approach combined with the spectral imaging microscopy provides a quick way to verify whether FRET is occurring. The donor de-quenching (if FRET occurred) can be directly observed from comparing the pre- and post-spectra of bleaching the acceptor, without data processing. For cells co-expressing bZip-Cerulean and bZip-Venus, the intensity increase (de-quenching) of the donor bZip-Cerulean was observed after bleaching the acceptor bZip-Venus, confirming a FRET event (Figure 3) and encouragement to proceed. However, the bleaching process takes time (typically one minute per cell in this experiment) during which the cell might move slightly or the molecules of interest moved, causing inaccuracies for quantitative data analysis. Thus, we chose the PFRET approach to collect data for extensive quantitative data analysis.
Confocal FRET using the PFRET method
Confocal FRET microscopy in combination with the PFRET method was used to simultaneously quantify the donor, acceptor, FRET signals and efficiencies (E%s) (Figure 4). For 230 regions of interest (ROIs) selected from 15 cells, E%s vary from ~10% to ~30% (Figure 4). The coefficient `c' (0.635) for the E% calculation (described above) was experimentally estimated using FRET-standard constructs (86). Plotting E% against the `acceptor-to-donor' ratios for all selected ROIs shows a positive correlation (R = 0.8) between the two (Figure 4). This range of E%s was expected, based on the fact that homo-dimerization can produce dimers with either two bZip-Cerulean or bZip-Venus monomers (no hetero-FRET), when only dimers with one donor (bZip-Cerulean) and one acceptor (bZip-Venus) may produce FRET signals. For example, ROIs containing a large proportion of dimers with two bZip-Cerulean monomers will result in a low `acceptor-to-donor' ratio, because of a high level of unquenched donor signals, affecting the E% calculation and thus producing low E%s. In contrast, when the `acceptor-to-donor' ratio is high, the likelihood of the E% being based on bZip-Cerulean monomers having formed dimers with bZip-Venus monomers and becoming quenched is much greater. The presence of bZip-Venus/bZip-Venus dimers may affect the `acceptor-to-donor' ratio but will not affect the E% calculation.
Figure 4. Confocal FRET microscopy using the PFRET method.

Images of live cells co-expressing Cerulean- (donor) and Venus- (acceptor) tagged bZip were acquired in the Donor, FRET and Acceptor imaging channels. Together with the images acquired from cells only expressing bZip-Cerulean or bZip-Venus (not shown), the images of the double-expressing cells were processed using the PFRET algorithm to remove spectral bleedthrough contaminations and calculate the pFRET and the FRET efficiency (E%) images (see text and Table 2). Due to different acceptor : donor (`A/D') ratios, a range of E%s emerged (see text for discussions). As an example, two sets of images of double-expressing cells having different `A/D' ratios are shown for a comparison: for each set, the raw and pFRET images are contrasted in the same range (top: 0~1500, bottom: 0~3000, 12-bit); the `A / D' ratio image was obtained by calculating pixel-by-pixel ratios between the two images acquired in the acceptor and the donor imaging channels, respectively; the ratio (0~4) or E% (0–40) images of both sets are color-contrasted in the same range, indicating larger `A / D' ratios yielding higher E%s. This is confirmed by plotting E% against the `acceptor -to- unquenched donor' ratios for 230 regions of interest (ROIs) selected from 15 cells. The unquenched donor is determined by adding the pFRET signal (multiplied by the coefficient `c', see text) to the quenched donor signal. The graph shows an increasing trend of E% with an increased `acceptor -to- unquenched donor' ratio, by either a 2nd order polynomial (solid) or linear (dashed) curve fitting. (Zeiss 510 Meta confocal microscope and 63X / 1.4NA oil immersion objective; the Donor channel: the 458 nm laser line and the 470–500 nm band-pass filter; the FRET channel: the 458 nm laser line and the 535–590 nm band-pass filter; the Acceptor channel: the 514 nm laser line and the 535–590 nm band-pass filter)
TCSPC FLIM-FRET
FLIM can provide a robust verification of intensity-based FRET measurements. The two-photon excitation TCSPC FLIM method was used to measure the quenched donor (Cerulean) fluorescence lifetimes due to the energy transfer from bZip-Cerulean to bZip-Venus (Figure 5). The average unquenched Cerulean lifetime obtained from 10 cells only expressing bZip-Cerulean was 2.75 ns, and the quenched bZip-Cerulean lifetimes measured from 10 cells co-expressing bZip-Cerulean and bZip-Venus ranged from 2.0 to 2.6 ns, producing an E% range of 5.5% to 27.3% (32). FLIM results clearly demonstrated FRET between bZip-Cerulean and bZip-Venus, confirming the ranges produced by intensity-based FRET.
Conclusion
Based on our literature analysis of the FRET-related publications, FRET applications have grown exponentially as shown by the number of publications in many diverse fields of the life sciences since the 1990's (details at www.kcci.virginia.edu/Literature). The exponential growth will continue, driven by the development of novel and advanced FRET techniques and the demonstration of their utilities to address a variety of biological questions, of which three-color FRET is one example (31). While we have explained in great detail the various SBT correction steps required for quantitative analysis, in reality, advanced algorithms in available software make these corrections a matter of routine. On the other hand, how to quantitatively interpret the FRET data in a meaningful biological way can be challenging: (i) positive or negative control experiments are always helpful if not actually required; (ii) a FRET technique is best calibrated using e.g. “FRET standards”, prior to being applied to a biological study; and (iii) optimization of both specimen- and imaging-related variables in a FRET experiment can be time consuming but play an important role in having a successful FRET assay.
Acknowledgements
The authors acknowledge funding from the University of Virginia and National Heart, Lung, and Blood Institute (NHLBI-NIH) PO1HL101871. The authors thank Ms. Kay Christopher for preparing the samples, Mr. Horst Wallrabe for providing suggestions and Dr. Richard Day (Indiana University School of Medicine) for providing the bZip constructs.
References
- 1.Förster T. Energy transport and fluorescence [in German] Naturwissenschafien. 1946;6:166–175. [Google Scholar]
- 2.Förster T. Zwischenmolekulare Energiewanderung und Fluoreszenz. Annalen Der Physik. 1948;437:55–75. [Google Scholar]
- 3.Förster T. Delocalized excitation and excitation transfer. In: Sinanoglu O, editor. InModern Quantum Chemistry. Academic Press Inc; 1965. pp. 93–137. [Google Scholar]
- 4.Clegg RM. Fluorescence resonance energy transfer. In: Wang XF, Herman B, editors. Fluorescence Imaging Spectroscopy and Microscopy. John Wiley & Sons Inc.; New York: 1996. pp. 179–251. [Google Scholar]
- 5.Szollosi J, Damjanovich S, Matyus L. Application of fluorescence resonance energy transfer in the clinical laboratory: routine and research. Cytometry. 1998;34:159–179. [PubMed] [Google Scholar]
- 6.Jares-Erijman EA, Jovin TM. FRET imaging. Nat Biotechnol. 2003;21:1387–1395. doi: 10.1038/nbt896. [DOI] [PubMed] [Google Scholar]
- 7.Sekar RB, Periasamy A. Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J Cell Biol. 2003;160:629–633. doi: 10.1083/jcb.200210140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagy P, Vereb G, Damjanovich S, Matyus L, Szollosi J. Measuring FRET in flow cytometry and microscopy. Curr Protoc Cytom. 2006;Chapter 12(Unit12.8) doi: 10.1002/0471142956.cy1208s38. [DOI] [PubMed] [Google Scholar]
- 9.Vogel SS, Thaler C, Koushik SV. Fanciful FRET. Sci STKE. 2006;2006:re2. doi: 10.1126/stke.3312006re2. [DOI] [PubMed] [Google Scholar]
- 10.Jares-Erijman EA, Jovin TM. Imaging molecular interactions in living cells by FRET microscopy. Curr Opin Chem Biol. 2006;10:409–416. doi: 10.1016/j.cbpa.2006.08.021. [DOI] [PubMed] [Google Scholar]
- 11.Piston DW, Kremers GJ. Fluorescent protein FRET: the good, the bad and the ugly. Trends Biochem Sci. 2007;32:407–414. doi: 10.1016/j.tibs.2007.08.003. [DOI] [PubMed] [Google Scholar]
- 12.Sun Y, Wallrabe H, Seo SA, Periasamy A. FRET Microscopy in 2010: The Legacy of Theodor Forster on the 100th Anniversary of his Birth. Chemphyschem. 2011;12:462–474. doi: 10.1002/cphc.201000664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pietraszewska-Bogiel A, Gadella TW. FRET microscopy: from principle to routine technology in cell biology. J Microsc. 2011;241:111–118. doi: 10.1111/j.1365-2818.2010.03437.x. [DOI] [PubMed] [Google Scholar]
- 14.Padilla-Parra S, Tramier M. FRET microscopy in the living cell: different approaches, strengths and weaknesses. Bioessays. 2012;34:369–376. doi: 10.1002/bies.201100086. [DOI] [PubMed] [Google Scholar]
- 15.Roy R, Hohng S, Ha T. A practical guide to single-molecule FRET. Nat Methods. 2008;5:507–516. doi: 10.1038/nmeth.1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mills JD, Stone JR, Rubin DG, Melon DE, Okonkwo DO, Periasamy A, Helm GA. Illuminating protein interactions in tissue using confocal and two-photon excitation fluorescent resonance energy transfer microscopy. J Biomed Opt. 2003;8:347–356. doi: 10.1117/1.1584443. [DOI] [PubMed] [Google Scholar]
- 17.McGinty J, Stuckey DW, Soloviev VY, Laine R, Wylezinska-Arridge M, Wells DJ, Arridge SR, French PM, Hajnal JV, Sardini A. In vivo fluorescence lifetime tomography of a FRET probe expressed in mouse. Biomed Opt Express. 2011;2:1907–1917. doi: 10.1364/BOE.2.001907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Banning C, Votteler J, Hoffmann D, Koppensteiner H, Warmer M, Reimer R, Kirchhoff F, Schubert U, Hauber J, Schindler M. A flow cytometry-based FRET assay to identify and analyse protein-protein interactions in living cells. PLoS One. 2010;5:e9344. doi: 10.1371/journal.pone.0009344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan FK. Monitoring molecular interactions in living cells using flow cytometric analysis of fluorescence resonance energy transfer. Methods Mol Biol. 2004;261:371–382. doi: 10.1385/1-59259-762-9:371. [DOI] [PubMed] [Google Scholar]
- 20.Dye BT, Schell K, Miller DJ, Ahlquist P. Detecting protein-protein interaction in live yeast by flow cytometry. Cytometry A. 2005;63A:77–86. doi: 10.1002/cyto.a.20112. [DOI] [PubMed] [Google Scholar]
- 21.Nagy P, Bene L, Balazs M, Hyun WC, Lockett SJ, Chiang NY, Waldman F, Feuerstein BG, Damjanovich S, Szollosi J. EGF-induced redistribution of erbB2 on breast tumor cells: flow and image cytometric energy transfer measurements. Cytometry. 1998;32:120–131. doi: 10.1002/(sici)1097-0320(19980601)32:2<120::aid-cyto7>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 22.Vereb G, Nagy P, Szollosi J. Flow cytometric FRET analysis of protein interaction. Methods Mol Biol. 2011;699:371–392. doi: 10.1007/978-1-61737-950-5_18. [DOI] [PubMed] [Google Scholar]
- 23.Miyawaki A, Tsien RY. Monitoring protein conformations and interactions by fluorescence resonance energy transfer between mutants of green fluorescent protein. Methods Enzymol. 2000;327:472–500. doi: 10.1016/s0076-6879(00)27297-2. [DOI] [PubMed] [Google Scholar]
- 24.Kraynov VS, Chamberlain C, Bokoch GM, Schwartz MA, Slabaugh S, Hahn KM. Localized Rac activation dynamics visualized in living cells. Science. 2000;290:333–337. doi: 10.1126/science.290.5490.333. [DOI] [PubMed] [Google Scholar]
- 25.Day RN, Periasamy A, Schaufele F. Fluorescence resonance energy transfer microscopy of localized protein interactions in the living cell nucleus. Methods. 2001;25:4–18. doi: 10.1006/meth.2001.1211. [DOI] [PubMed] [Google Scholar]
- 26.Day RN, Voss TC, Enwright JF, 3rd, Booker CF, Periasamy A, Schaufele F. Imaging the localized protein interactions between Pit-1 and the CCAAT/enhancer binding protein alpha in the living pituitary cell nucleus. Mol Endocrinol. 2003;17:333–345. doi: 10.1210/me.2002-0136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wallrabe H, Elangovan M, Burchard A, Periasamy A, Barroso M. Confocal FRET microscopy to measure clustering of ligand-receptor complexes in endocytic membranes. Biophys J. 2003;85:559–571. doi: 10.1016/S0006-3495(03)74500-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Demarco IA, Voss TC, Booker CF, Day RN. Dynamic interactions between Pit-1 and C/EBPalpha in the pituitary cell nucleus. Mol Cell Biol. 2006;26:8087–8098. doi: 10.1128/MCB.02410-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Demarco IA, Periasamy A, Booker CF, Day RN. Monitoring dynamic protein interactions with photoquenching FRET. Nat Methods. 2006;3:519–524. doi: 10.1038/nmeth889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cai D, Hoppe AD, Swanson JA, Verhey KJ. Kinesin-1 structural organization and conformational changes revealed by FRET stoichiometry in live cells. J Cell Biol. 2007;176:51–63. doi: 10.1083/jcb.200605097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun Y, Wallrabe H, Booker CF, Day RN, Periasamy A. Three-color spectral FRET microscopy localizes three interacting proteins in living cells. Biophys J. 2010;99:1274–1283. doi: 10.1016/j.bpj.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun Y, Day RN, Periasamy A. Investigating protein-protein interactions in living cells using fluorescence lifetime imaging microscopy. Nat Protoc. 2011;6:1324–1340. doi: 10.1038/nprot.2011.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wallrabe H, Bonamy G, Periasamy A, Barroso M. Receptor complexes cotransported via polarized endocytic pathways form clusters with distinct organizations. Mol Biol Cell. 2007;18:2226–2243. doi: 10.1091/mbc.E06-08-0700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berezovska O, Lleo A, Herl LD, Frosch MP, Stern EA, Bacskai BJ, Hyman BT. Familial Alzheimer's disease presenilin 1 mutations cause alterations in the conformation of presenilin and interactions with amyloid precursor protein. J Neurosci. 2005;25:3009–3017. doi: 10.1523/JNEUROSCI.0364-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou F, Xing D, Wu S, Chen WR. Intravital Imaging of Tumor Apoptosis with FRET Probes During Tumor Therapy. Mol Imaging Biol. 2009 doi: 10.1007/s11307-009-0235-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larionov S, Wielgat P, Wang Y, Thal DR, Neumann H. Spatially pathogenic forms of tau detected in Alzheimer's disease brain tissue by fluorescence lifetime-based Forster resonance energy transfer. J Neurosci Methods. 2010;192:127–137. doi: 10.1016/j.jneumeth.2010.07.021. [DOI] [PubMed] [Google Scholar]
- 37.Allen MD, DiPilato LM, Rahdar M, Ren YR, Chong C, Liu JO, Zhang J. Reading Dynamic Kinase Activity in Living Cells for High-Throughput Screening. ACS Chemical Biology. 2006;1:371–376. doi: 10.1021/cb600202f. [DOI] [PubMed] [Google Scholar]
- 38.Tian H, Ip L, Luo H, Chang DC, Luo KQ. A high throughput drug screen based on fluorescence resonance energy transfer (FRET) for anticancer activity of compounds from herbal medicine. Br J Pharmacol. 2007;150:321–334. doi: 10.1038/sj.bjp.0706988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peng J, Gong L, Si K, Bai X, Du G. Fluorescence resonance energy transfer assay for high-throughput screening of ADAMTS1 inhibitors. Molecules. 2011;16:10709–10721. doi: 10.3390/molecules161210709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.He L, Bradrick TD, Karpova TS, Wu X, Fox MH, Fischer R, McNally JG, Knutson JR, Grammer AC, Lipsky PE. Flow cytometric measurement of fluorescence (Forster) resonance energy transfer from cyan fluorescent protein to yellow fluorescent protein using single-laser excitation at 458 nm. Cytometry A. 2003;53A:39–54. doi: 10.1002/cyto.a.10037. [DOI] [PubMed] [Google Scholar]
- 41.Rizzo MA, Springer GH, Granada B, Piston DW. An improved cyan fluorescent protein variant useful for FRET. Nat Biotechnol. 2004;22:445–449. doi: 10.1038/nbt945. [DOI] [PubMed] [Google Scholar]
- 42.Kremers GJ, Goedhart J, van Munster EB, Gadella TW., Jr. Cyan and yellow super fluorescent proteins with improved brightness, protein folding, and FRET Forster radius. Biochemistry. 2006;45:6570–6580. doi: 10.1021/bi0516273. [DOI] [PubMed] [Google Scholar]
- 43.You X, Nguyen AW, Jabaiah A, Sheff MA, Thorn KS, Daugherty PS. Intracellular protein interaction mapping with FRET hybrids. Proc Natl Acad Sci U S A. 2006;103:18458–18463. doi: 10.1073/pnas.0605422103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ai HW, Henderson JN, Remington SJ, Campbell RE. Directed evolution of a monomeric, bright and photostable version of Clavularia cyan fluorescent protein: structural characterization and applications in fluorescence imaging. Biochem J. 2006;400:531–540. doi: 10.1042/BJ20060874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van der Krogt GN, Ogink J, Ponsioen B, Jalink K. A comparison of donor-acceptor pairs for genetically encoded FRET sensors: application to the Epac cAMP sensor as an example. PLoS One. 2008;3:e1916. doi: 10.1371/journal.pone.0001916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Day RN, Schaufele F. Fluorescent protein tools for studying protein dynamics in living cells: a review. J Biomed Opt. 2008;13:031202. doi: 10.1117/1.2939093. [DOI] [PubMed] [Google Scholar]
- 47.Day RN, Booker CF, Periasamy A. Characterization of an improved donor fluorescent protein for Forster resonance energy transfer microscopy. J Biomed Opt. 2008;13:031203. doi: 10.1117/1.2939094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Day RN, Davidson MW. The fluorescent protein palette: tools for cellular imaging. Chem Soc Rev. 2009;38:2887–2921. doi: 10.1039/b901966a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shcherbo D, Souslova EA, Goedhart J, Chepurnykh TV, Gaintzeva A, Shemiakina II, Gadella TW, Lukyanov S, Chudakov DM. Practical and reliable FRET/FLIM pair of fluorescent proteins. BMC Biotechnol. 2009;9:24. doi: 10.1186/1472-6750-9-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Visser AJ, Laptenok SP, Visser NV, van Hoek A, Birch DJ, Brochon JC, Borst JW. Time-resolved FRET fluorescence spectroscopy of visible fluorescent protein pairs. Eur Biophys J. 2009 doi: 10.1007/s00249-009-0528-8. [DOI] [PubMed] [Google Scholar]
- 51.Sun Y, Booker CF, Kumari S, Day RN, Davidson M, Periasamy A. Characterization of an orange acceptor fluorescent protein for sensitized spectral fluorescence resonance energy transfer microscopy using a white-light laser. J Biomed Opt. 2009;14:054009. doi: 10.1117/1.3227036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Padilla-Parra S, Auduge N, Lalucque H, Mevel JC, Coppey-Moisan M, Tramier M. Quantitative comparison of different fluorescent protein couples for fast FRET-FLIM acquisition. Biophys J. 2009;97:2368–2376. doi: 10.1016/j.bpj.2009.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Albertazzi L, Arosio D, Marchetti L, Ricci F, Beltram F. Quantitative FRET analysis with the EGFP-mCherry fluorescent protein pair. Photochem Photobiol. 2009;85:287–297. doi: 10.1111/j.1751-1097.2008.00435.x. [DOI] [PubMed] [Google Scholar]
- 54.Goedhart J, van Weeren L, Hink MA, Vischer NO, Jalink K, Gadella TW., Jr. Bright cyan fluorescent protein variants identified by fluorescence lifetime screening. Nat Methods. 2010;7:137–139. doi: 10.1038/nmeth.1415. [DOI] [PubMed] [Google Scholar]
- 55.Markwardt ML, Kremers GJ, Kraft CA, Ray K, Cranfill PJ, Wilson KA, Day RN, Wachter RM, Davidson MW, Rizzo MA. An improved cerulean fluorescent protein with enhanced brightness and reduced reversible photoswitching. PLoS One. 2011;6:e17896. doi: 10.1371/journal.pone.0017896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hiller DA, Fogg JM, Martin AM, Beechem JM, Reich NO, Perona JJ. Simultaneous DNA binding and bending by EcoRV endonuclease observed by real-time fluorescence. Biochemistry. 2003;42:14375–14385. doi: 10.1021/bi035520w. [DOI] [PubMed] [Google Scholar]
- 57.Panyi G, Bagdany M, Bodnar A, Vamosi G, Szentesi G, Jenei A, Matyus L, Varga S, Waldmann TA, Gaspar R, Damjanovich S. Colocalization and nonrandom distribution of Kv1.3 potassium channels and CD3 molecules in the plasma membrane of human T lymphocytes. Proc Natl Acad Sci U S A. 2003;100:2592–2597. doi: 10.1073/pnas.0438057100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hohng S, Joo C, Ha T. Single-molecule three-color FRET. Biophys J. 2004;87:1328–1337. doi: 10.1529/biophysj.104.043935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wallrabe H, Periasamy A. Imaging protein molecules using FRET and FLIM microscopy. Curr Opin Biotechnol. 2005;16:19–27. doi: 10.1016/j.copbio.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 60.Horvath G, Petras M, Szentesi G, Fabian A, Park JW, Vereb G, Szollosi J. Selecting the right fluorophores and flow cytometer for fluorescence resonance energy transfer measurements. Cytometry A. 2005;65A:148–157. doi: 10.1002/cyto.a.20142. [DOI] [PubMed] [Google Scholar]
- 61.Algar WR, Krull UJ. Quantum dots as donors in fluorescence resonance energy transfer for the bioanalysis of nucleic acids, proteins, and other biological molecules. Anal Bioanal Chem. 2008;391:1609–1618. doi: 10.1007/s00216-007-1703-3. [DOI] [PubMed] [Google Scholar]
- 62.Medintz IL, Mattoussi H. Quantum dot-based resonance energy transfer and its growing application in biology. Phys Chem Chem Phys. 2009;11:17–45. doi: 10.1039/b813919a. [DOI] [PubMed] [Google Scholar]
- 63.Periasamy A, Day RN, editors. Molecular Imaging: FRET Microscopy and Spectroscopy. Oxford University Press; New York: 2005. [Google Scholar]
- 64.Vinkenborg JL, Nicolson TJ, Bellomo EA, Koay MS, Rutter GA, Merkx M. Genetically encoded FRET sensors to monitor intracellular Zn2+ homeostasis. Nat Methods. 2009;6:737–740. doi: 10.1038/nmeth.1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gu Y, Di WL, Kelsell DP, Zicha D. Quantitative fluorescence resonance energy transfer (FRET) measurement with acceptor photobleaching and spectral unmixing. J Microsc. 2004;215:162–173. doi: 10.1111/j.0022-2720.2004.01365.x. [DOI] [PubMed] [Google Scholar]
- 66.Kenworthy AK. Imaging protein-protein interactions using fluorescence resonance energy transfer microscopy. Methods. 2001;24:289–296. doi: 10.1006/meth.2001.1189. [DOI] [PubMed] [Google Scholar]
- 67.Roszik J, Szollosi J, Vereb G. AccPbFRET: an ImageJ plugin for semi-automatic, fully corrected analysis of acceptor photobleaching FRET images. BMC Bioinformatics. 2008;9:346. doi: 10.1186/1471-2105-9-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Van Munster EB, Kremers GJ, Adjobo-Hermans MJ, Gadella TW., Jr. Fluorescence resonance energy transfer (FRET) measurement by gradual acceptor photobleaching. J Microsc. 2005;218:253–262. doi: 10.1111/j.1365-2818.2005.01483.x. [DOI] [PubMed] [Google Scholar]
- 69.Wang L, Chen T, Qu J, Wei X. Photobleaching-based quantitative analysis of fluorescence resonance energy transfer inside single living cell. J Fluoresc. 2010;20:27–35. doi: 10.1007/s10895-009-0518-3. [DOI] [PubMed] [Google Scholar]
- 70.Kremers GJ, Hazelwood KL, Murphy CS, Davidson MW, Piston DW. Photoconversion in orange and red fluorescent proteins. Nat Methods. 2009;6:355–358. doi: 10.1038/nmeth.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Tron L, Szollosi J, Damjanovich S, Helliwell SH, Arndt-Jovin DJ, Jovin TM. Flow cytometric measurement of fluorescence resonance energy transfer on cell surfaces. Quantitative evaluation of the transfer efficiency on a cell-by-cell basis. Biophys J. 1984;45:939–946. doi: 10.1016/S0006-3495(84)84240-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Matyus L. Fluorescence resonance energy transfer measurements on cell surfaces. A spectroscopic tool for determining protein interactions. J Photochem Photobiol B. 1992;12:323–337. doi: 10.1016/1011-1344(92)85039-w. [DOI] [PubMed] [Google Scholar]
- 73.Nagy P, Vamosi G, Bodnar A, Lockett SJ, Szollosi J. Intensity-based energy transfer measurements in digital imaging microscopy. Eur Biophys J. 1998;27:377–389. doi: 10.1007/s002490050145. [DOI] [PubMed] [Google Scholar]
- 74.Gordon GW, Berry G, Liang XH, Levine B, Herman B. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophys J. 1998;74:2702–2713. doi: 10.1016/S0006-3495(98)77976-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xia Z, Liu Y. Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys J. 2001;81:2395–2402. doi: 10.1016/S0006-3495(01)75886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hoppe A, Christensen K, Swanson JA. Fluorescence resonance energy transfer-based stoichiometry in living cells. Biophys J. 2002;83:3652–3664. doi: 10.1016/S0006-3495(02)75365-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Elangovan M, Wallrabe H, Chen Y, Day RN, Barroso M, Periasamy A. Characterization of one- and two-photon excitation fluorescence resonance energy transfer microscopy. Methods. 2003;29:58–73. doi: 10.1016/s1046-2023(02)00283-9. [DOI] [PubMed] [Google Scholar]
- 78.van Rheenen J, Langeslag M, Jalink K. Correcting confocal acquisition to optimize imaging of fluorescence resonance energy transfer by sensitized emission. Biophys J. 2004;86:2517–2529. doi: 10.1016/S0006-3495(04)74307-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zal T, Gascoigne NR. Photobleaching-corrected FRET efficiency imaging of live cells. Biophys J. 2004;86:3923–3939. doi: 10.1529/biophysj.103.022087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Feige JN, Sage D, Wahli W, Desvergne B, Gelman L. PixFRET, an ImageJ plug-in for FRET calculation that can accommodate variations in spectral bleed-throughs. Microsc Res Tech. 2005;68:51–58. doi: 10.1002/jemt.20215. [DOI] [PubMed] [Google Scholar]
- 81.Nagy P, Bene L, Hyun WC, Vereb G, Braun M, Antz C, Paysan J, Damjanovich S, Park JW, Szollsi J. Novel calibration method for flow cytometric fluorescence resonance energy transfer measurements between visible fluorescent proteins. Cytometry A. 2005;67A:86–96. doi: 10.1002/cyto.a.20164. [DOI] [PubMed] [Google Scholar]
- 82.Periasamy A, Chen Y, Elangovan M. FRET data analysis—the algorithm. In: Periasamy A, Day RN, editors. Molecular Imaging: FRET Microscopy and Spectroscopy. Oxford University Press; New York: 2005. pp. 126–145. [Google Scholar]
- 83.Hachet-Haas M, Converset N, Marchal O, Matthes H, Gioria S, Galzi JL, Lecat S. FRET and colocalization analyzer--a method to validate measurements of sensitized emission FRET acquired by confocal microscopy and available as an ImageJ Plug-in. Microsc Res Tech. 2006;69:941–956. doi: 10.1002/jemt.20376. [DOI] [PubMed] [Google Scholar]
- 84.Elder AD, Domin A, Kaminski Schierle GS, Lindon C, Pines J, Esposito A, Kaminski CF. A quantitative protocol for dynamic measurements of protein interactions by Förster resonance energy transfer-sensitized fluorescence emission. Journal of the Royal Society Interface. 2009;6:S59–S81. [Google Scholar]
- 85.Roszik J, Lisboa D, Szollosi J, Vereb G. Evaluation of intensity-based ratiometric FRET in image cytometry--approaches and a software solution. Cytometry A. 2009;75A:761–767. doi: 10.1002/cyto.a.20747. [DOI] [PubMed] [Google Scholar]
- 86.Sun Y, Periasamy A. Additional correction for energy transfer efficiency calculation in filter-based Forster resonance energy transfer microscopy for more accurate results. J Biomed Opt. 2010;15:020513. doi: 10.1117/1.3407655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Berney C, Danuser G. FRET or no FRET: a quantitative comparison. Biophys J. 2003;84:3992–4010. doi: 10.1016/S0006-3495(03)75126-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chen Y, Periasamy A. Intensity range based quantitative FRET data analysis to localize protein molecules in live cell nuclei. J Fluoresc. 2006;16:95–104. doi: 10.1007/s10895-005-0024-1. [DOI] [PubMed] [Google Scholar]
- 89.Chen H, Puhl HL, 3rd, Koushik SV, Vogel SS, Ikeda SR. Measurement of FRET efficiency and ratio of donor to acceptor concentration in living cells. Biophys J. 2006;91:L39–41. doi: 10.1529/biophysj.106.088773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gustiananda M, Liggins JR, Cummins PL, Gready JE. Conformation of prion protein repeat peptides probed by FRET measurements and molecular dynamics simulations. Biophys J. 2004;86:2467–2483. doi: 10.1016/S0006-3495(04)74303-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zimmermann T, Rietdorf J, Girod A, Georget V, Pepperkok R. Spectral imaging and linear un-mixing enables improved FRET efficiency with a novel GFP2–YFP FRET pair. FEBS Lett. 2002;531:245–249. doi: 10.1016/s0014-5793(02)03508-1. [DOI] [PubMed] [Google Scholar]
- 92.Neher RA, Neher E. Applying spectral fingerprinting to the analysis of FRET images. Microsc Res Tech. 2004;64:185–195. doi: 10.1002/jemt.20078. [DOI] [PubMed] [Google Scholar]
- 93.Thaler C, Koushik SV, Blank PS, Vogel SS. Quantitative multiphoton spectral imaging and its use for measuring resonance energy transfer. Biophys J. 2005;89:2736–2749. doi: 10.1529/biophysj.105.061853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Takanishi CL, Bykova EA, Cheng W, Zheng J. GFP-based FRET analysis in live cells. Brain Res. 2006;1091:132–139. doi: 10.1016/j.brainres.2006.01.119. [DOI] [PubMed] [Google Scholar]
- 95.Chen Y, Mauldin JP, Day RN, Periasamy A. Characterization of spectral FRET imaging microscopy for monitoring nuclear protein interactions. J Microsc. 2007;228:139–152. doi: 10.1111/j.1365-2818.2007.01838.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Megias D, Marrero R, Martinez Del Peso B, Garcia MA, Bravo-Cordero JJ, Garcia-Grande A, Santos A, Montoya MC. Novel lambda FRET spectral confocal microscopy imaging method. Microsc Res Tech. 2009;72:1–11. doi: 10.1002/jemt.20633. [DOI] [PubMed] [Google Scholar]
- 97.Raicu V, Stoneman MR, Fung R, Melnichuk M, Jansma DB, Pisterzi LF, Rath S, Fox M, Wells JW, Saldin DK. Determination of supramolecular structure and spatial distribution of protein complexes in living cells. Nat Photon. 2009;3:107–113. [Google Scholar]
- 98.Kim J, Li X, Kang MS, Im KB, Genovesio A, Grailhe R. Quantification of protein interaction in living cells by two-photon spectral imaging with fluorescent protein fluorescence resonance energy transfer pair devoid of acceptor bleed-through. Cytometry A. 2012;81A:112–119. doi: 10.1002/cyto.a.21150. [DOI] [PubMed] [Google Scholar]
- 99.Lakowicz JR. Principles of fluorescence spectroscopy. Springer; New York: 2006. p. 954. [Google Scholar]
- 100.Digman MA, Caiolfa VR, Zamai M, Gratton E. The phasor approach to fluorescence lifetime imaging analysis. Biophys J. 2008;94:L14–6. doi: 10.1529/biophysj.107.120154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Periasamy A, Clegg RM. FLIM Applications in the Biomedical Sciences. In: Periasamy A, Clegg RM, editors. FLIM microscopy in biology and medicine. CRC Press; London: 2009. pp. 385–400. [Google Scholar]
- 102.Gadella TWJ, editor. FRET and FLIM Techniques. Elsevier; Oxford, UK: 2009. p. 534. [Google Scholar]
- 103.Wouters FS, Bastiaens PI. Fluorescence lifetime imaging of receptor tyrosine kinase activity in cells. Curr Biol. 1999;9:1127–1130. doi: 10.1016/s0960-9822(99)80484-9. [DOI] [PubMed] [Google Scholar]
- 104.Verveer PJ, Wouters FS, Reynolds AR, Bastiaens PI. Quantitative imaging of lateral ErbB1 receptor signal propagation in the plasma membrane. Science. 2000;290:1567–1570. doi: 10.1126/science.290.5496.1567. [DOI] [PubMed] [Google Scholar]
- 105.Elangovan M, Day RN, Periasamy A. Nanosecond fluorescence resonance energy transfer-fluorescence lifetime imaging microscopy to localize the protein interactions in a single living cell. J Microsc. 2002;205:3–14. doi: 10.1046/j.0022-2720.2001.00984.x. [DOI] [PubMed] [Google Scholar]
- 106.Chen Y, Mills JD, Periasamy A. Protein localization in living cells and tissues using FRET and FLIM. Differentiation. 2003;71:528–541. doi: 10.1111/j.1432-0436.2003.07109007.x. [DOI] [PubMed] [Google Scholar]
- 107.Krishnan RV, Masuda A, Centonze VE, Herman B. Quantitative imaging of protein-protein interactions by multiphoton fluorescence lifetime imaging microscopy using a streak camera. J Biomed Opt. 2003;8:362–367. doi: 10.1117/1.1577574. [DOI] [PubMed] [Google Scholar]
- 108.Chen Y, Periasamy A. Characterization of two-photon excitation fluorescence lifetime imaging microscopy for protein localization. Microsc Res Tech. 2004;63:72–80. doi: 10.1002/jemt.10430. [DOI] [PubMed] [Google Scholar]
- 109.Biskup C, Zimmer T, Benndorf K. FRET between cardiac Na+ channel subunits measured with a confocal microscope and a streak camera. Nat Biotechnol. 2004;22:220–224. doi: 10.1038/nbt935. [DOI] [PubMed] [Google Scholar]
- 110.Biskup C, Zimmer T, Kelbauskas L, Hoffmann B, Klocker N, Becker W, Bergmann A, Benndorf K. Multi-dimensional fluorescence lifetime and FRET measurements. Microsc Res Tech. 2007;70:442–451. doi: 10.1002/jemt.20431. [DOI] [PubMed] [Google Scholar]
- 111.Li Q, Seeger S. Label-free detection of protein interactions using deep UV fluorescence lifetime microscopy. Anal Biochem. 2007;367:104–110. doi: 10.1016/j.ab.2007.04.050. [DOI] [PubMed] [Google Scholar]
- 112.Li H, Li HF, Felder RA, Periasamy A, Jose PA. Rab4 and Rab11 coordinately regulate the recycling of angiotensin II type I receptor as demonstrated by fluorescence resonance energy transfer microscopy. J Biomed Opt. 2008;13:031206. doi: 10.1117/1.2943286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Murakoshi H, Lee SJ, Yasuda R. Highly sensitive and quantitative FRET-FLIM imaging in single dendritic spines using improved non-radiative YFP. Brain Cell Biol. 2008;36:31–42. doi: 10.1007/s11068-008-9024-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Leray A, Spriet C, Trinel D, Heliot L. Three-dimensional polar representation for multispectral fluorescence lifetime imaging microscopy. Cytometry A. 2009;75A:1007–1014. doi: 10.1002/cyto.a.20802. [DOI] [PubMed] [Google Scholar]
- 115.Li H, Yu P, Sun Y, Felder RA, Periasamy A, Jose PA. Actin cytoskeleton-dependent Rab GTPase-regulated angiotensin type I receptor lysosomal degradation studied by fluorescence lifetime imaging microscopy. J Biomed Opt. 2010;15:056003. doi: 10.1117/1.3484751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Boutant E, Didier P, Niehl A, Mely Y, Ritzenthaler C, Heinlein M. Fluorescent protein recruitment assay for demonstration and analysis of in vivo protein interactions in plant cells and its application to Tobacco mosaic virus movement protein. Plant J. 2010;62:171–177. doi: 10.1111/j.1365-313X.2010.04126.x. [DOI] [PubMed] [Google Scholar]
- 117.Bu W, Lim KB, Yu YH, Chou AM, Sudhaharan T, Ahmed S. Cdc42 interaction with N-WASP and Toca-1 regulates membrane tubulation, vesicle formation and vesicle motility: implications for endocytosis. PLoS One. 2010;5:e12153. doi: 10.1371/journal.pone.0012153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Martin-Villar E, Fernandez-Munoz B, Parsons M, Yurrita MM, Megias D, Perez-Gomez E, Jones GE, Quintanilla M. Podoplanin associates with CD44 to promote directional cell migration. Mol Biol Cell. 2010;21:4387–4399. doi: 10.1091/mbc.E10-06-0489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jones PB, Adams KW, Rozkalne A, Spires-Jones TL, Hshieh TT, Hashimoto T, von Armin CA, Mielke M, Bacskai BJ, Hyman BT. Apolipoprotein E: isoform specific differences in tertiary structure and interaction with amyloid-beta in human Alzheimer brain. PLoS One. 2011;6:e14586. doi: 10.1371/journal.pone.0014586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Timpson P, McGhee EJ, Morton JP, von Kriegsheim A, Schwarz JP, Karim SA, Doyle B, Quinn JA, Carragher NO, Edward M, Olson MF, Frame MC, Brunton VG, Sansom OJ, Anderson KI. Spatial regulation of RhoA activity during pancreatic cancer cell invasion driven by mutant p53. Cancer Res. 2011;71:747–757. doi: 10.1158/0008-5472.CAN-10-2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kumar S, Alibhai D, Margineanu A, Laine R, Kennedy G, McGinty J, Warren S, Kelly D, Alexandrov Y, Munro I, Talbot C, Stuckey DW, Kimberly C, Viellerobe B, Lacombe F, Lam EW, Taylor H, Dallman MJ, Stamp G, Murray EJ, Stuhmeier F, Sardini A, Katan M, Elson DS, Neil MA, Dunsby C, French PM. FLIM FRET technology for drug discovery: automated multiwell-plate high-content analysis, multiplexed readouts and application in situ. Chemphyschem. 2011;12:609–626. doi: 10.1002/cphc.201000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Periasamy A, Clegg RM, editors. FLIM microscopy in biology and medicine. CRC Press; London: 2009. [Google Scholar]
- 123.Becker W. Advanced Time-Correlated Single Photon Counting Techniques. Springer; Berlin: 2005. p. 401. [Google Scholar]
- 124.Becker W, Bergmann A, Hink MA, Konig K, Benndorf K, Biskup C. Fluorescence lifetime imaging by time-correlated single-photon counting. Microsc Res Tech. 2004;63:58–66. doi: 10.1002/jemt.10421. [DOI] [PubMed] [Google Scholar]
- 125.Becker W, Bergmann A, Biskup C. Multispectral fluorescence lifetime imaging by TCSPC. Microsc Res Tech. 2007;70:403–409. doi: 10.1002/jemt.20432. [DOI] [PubMed] [Google Scholar]
- 126.Periasamy A, Wodnicki P, Wang XF, Kwon S, Gordon GW, Herman B. Time-resolved fluorescence lifetime imaging microscopy using a picosecond pulsed tunable dye laser system. Rev Sci Instrum. 1996;67:3722–3731. [Google Scholar]
- 127.Agronskaia AV, Tertoolen L, Gerritsen HC. High frame rate fluorescence lifetime imaging. J Phys D. 2003;36:1655–1662. [Google Scholar]
- 128.Talbot CB, McGinty J, Grant DM, McGhee EJ, Owen DM, Zhang W, Bunney TD, Munro I, Isherwood B, Eagle R, Hargreaves A, Dunsby C, Neil MA, French PM. High speed unsupervised fluorescence lifetime imaging confocal multiwell plate reader for high content analysis. J Biophotonics. 2008;1:514–521. doi: 10.1002/jbio.200810054. [DOI] [PubMed] [Google Scholar]
- 129.Krishnan RV, Saitoh H, Terada H, Centonze VE, Herman B. Development of a multiphoton fluorescence lifetime imaging microscopy system using a streak camera. Rev Sci Instrum. 2003;74:2714–2721. [Google Scholar]
- 130.Colyer RA, Lee C, Gratton E. A novel fluorescence lifetime imaging system that optimizes photon efficiency. Microsc Res Tech. 2008;71:201–213. doi: 10.1002/jemt.20540. [DOI] [PubMed] [Google Scholar]
- 131.Gadella TWJ, Jr., Jovin TM, Clegg RM. Fluorescence lifetime imaging microscopy (FLIM): Spatial resolution of microstructures on the nanosecond time scale. Biophys Chem. 1993;48:221–239. [Google Scholar]
- 132.Buranachai C, Kamiyama D, Chiba A, Williams BD, Clegg RM. Rapid frequency-domain FLIM spinning disk confocal microscope: lifetime resolution, image improvement and wavelet analysis. J Fluoresc. 2008;18:929–942. doi: 10.1007/s10895-008-0332-3. [DOI] [PubMed] [Google Scholar]
- 133.Chen YC, Clegg RM. Spectral resolution in conjunction with polar plots improves the accuracy and reliability of FLIM measurements and estimates of FRET efficiency. J Microsc. 2011;244:21–37. doi: 10.1111/j.1365-2818.2011.03488.x. [DOI] [PubMed] [Google Scholar]
- 134.Gautier I, Tramier M, Durieux C, Coppey J, Pansu RB, Nicolas JC, Kemnitz K, Coppey-Moisan M. Homo-FRET microscopy in living cells to measure monomer-dimer transition of GFP-tagged proteins. Biophys J. 2001;80:3000–3008. doi: 10.1016/S0006-3495(01)76265-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bader AN, Hofman EG, Voortman J, en Henegouwen PM, Gerritsen HC. Homo-FRET imaging enables quantification of protein cluster sizes with subcellular resolution. Biophys J. 2009;97:2613–2622. doi: 10.1016/j.bpj.2009.07.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rizzo MA, Piston DW. High-contrast imaging of fluorescent protein FRET by fluorescence polarization microscopy. Biophys J. 2005;88:L14–6. doi: 10.1529/biophysj.104.055442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Piston DW, Rizzo MA. FRET by fluorescence polarization microscopy. Methods Cell Biol. 2008;85:415–430. doi: 10.1016/S0091-679X(08)85018-2. [DOI] [PubMed] [Google Scholar]
- 138.Clayton AH, Hanley QS, Arndt-Jovin DJ, Subramaniam V, Jovin TM. Dynamic fluorescence anisotropy imaging microscopy in the frequency domain (rFLIM) Biophys J. 2002;83:1631–1649. doi: 10.1016/S0006-3495(02)73932-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lidke DS, Nagy P, Barisas BG, Heintzmann R, Post JN, Lidke KA, Clayton AH, Arndt-Jovin DJ, Jovin TM. Imaging molecular interactions in cells by dynamic and static fluorescence anisotropy (rFLIM and emFRET) Biochem Soc Trans. 2003;31:1020–1027. doi: 10.1042/bst0311020. [DOI] [PubMed] [Google Scholar]
- 140.Thaler C, Koushik SV, Puhl HL, 3rd, Blank PS, Vogel SS. Structural rearrangement of CaMKIIalpha catalytic domains encodes activation. Proc Natl Acad Sci U S A. 2009;106:6369–6374. doi: 10.1073/pnas.0901913106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Koushik SV, Chen H, Thaler C, Puhl HL, 3rd, Vogel SS. Cerulean, Venus, and VenusY67C FRET reference standards. Biophys J. 2006;91:L99–L101. doi: 10.1529/biophysj.106.096206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Tang QQ, Lane MD. Role of C/EBP homologous protein (CHOP-10) in the programmed activation of CCAAT/enhancer-binding protein-beta during adipogenesis. Proc Natl Acad Sci U S A. 2000;97:12446–12450. doi: 10.1073/pnas.220425597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Tang QQ, Lane MD. Activation and centromeric localization of CCAAT/enhancer-binding proteins during the mitotic clonal expansion of adipocyte differentiation. Genes Dev. 1999;13:2231–2241. doi: 10.1101/gad.13.17.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]