

Abstract
In this study, we attempted to develop functional liposomes loaded with camptothecin and attached to α-melanocyte-stimulating hormone (α-MSH) to target melanoma cells. The liposomes were mainly composed of phosphatidylcholine, cholesterol, and stearylamine, and were characterized by the vesicle size, zeta potential, camptothecin encapsulation efficiency, and release behavior. Results revealed that α-MSH liposomes possessed an average size of approximately 250 nm with a surface charge of 60 mV. Camptothecin was successfully entrapped by the targeted liposomes with an encapsulation percentage of nearly 95%. The liposomes provided sustained and controlled camptothecin release. Non-targeted liposomes with the drug exerted superior cytotoxicity against melanomas compared to the free control. Cell viability was reduced from 48% to 32% compared to conventional liposomes. Peptide ligand conjugation further promoted cytotoxicity to 18% viability, which was a 2.7-fold decrease versus the free control. According to the images of fluorescence microscopy, α-MSH liposomes exhibited greater cell endocytosis than did non-targeted liposomes and the free control. α-MSH liposomes were predominantly internalized in the cytoplasm. These findings demonstrate that α-MSH liposomes could enhance the anti-melanoma activity of camptothecin owing to their targeting ability and controlled drug delivery.
Keywords: Camptothecin, Controlled release, Liposomes, α-melanocyte-stimulating hormone, Melanoma
INTRODUCTION
Melanomas are one of the most deadly cancers in the world. The rapid increase in their incidence is a serious public health problem.[1] Melanomas contribute to >50% of deaths involving skin cancers, although they only account for 5% of all skin cancers.[2] Because of their aggressive property, melanomas present high therapeutic resistance to classical radiotherapy and chemotherapy.[3] Liposomes, as carriers for drug delivery, can prolong circulation times and accelerate tumor uptake, thus improving therapeutic resistance. Liposomes are nano-sized vesicles consisting of membrane-like phospholipid bilayers in an aqueous solution. Their biocompatibility, low toxicity, and protection of drugs provide an effective strategy of drug delivery to action sites.[4,5]
Currently approved liposomes for anticancer drugs still possess some problems such as cardiotoxicity, myelosuppression, and alopecia because of insufficient targeting to tumors.[6] A promising method to resolve those shortcomings is to decorate the liposomal surface with specific proteins or antibodies that can bind to receptors on cell membranes.[7] The melanocortin-1 receptor (MC1R) belongs to the superfamily of G protein-coupled receptors, which is overexpressed in melanomas. Over 80% of metastatic melanomas were identified as displaying the MC1R.[8] α-Melanocyte-stimulating hormone (α-MSH) is a tricapeptide that selectively activates the MC1R. It is well known for its role in regulating skin pigmentation.[9] It is anticipated that carriers can efficiently bind to melanoma cells by conjugating peptide ligands. A previous study[10] developed gold nanoparticles conjugated with α-MSH for successful melanoma targeting. Nevertheless, toxicity is a concern for nanoparticles made of heavy metals. Yagi, et al.,[11] prepared liposomes with α-MSH via C-terminal conjugation with lipids, which successfully bound to melanomas. However, no drug was loaded in the liposomes for examining cytotoxicity to melanomas in that study. We tried to develop a drug-loaded liposomal system embedding α-MSH in the present work. Raposinho, et al.,[12] demonstrated that α-MSH derivatization via disulfide bonds displayed increased binding affinity and proteolysis resistance. We linked sulfosuccinimidyl 6-[3′-(2-pyridyldithio)-propionamide] hexanoate (sulfo-LC-SPDP) to the N-terminus of α-MSH via disulfide bonds to prepare α-MSH–containing liposomes.
Camptothecin is a natural product used as the model anticancer drug in this report due to its ability to kill melanoma cells.[13] The insolubility of camptothecin in most biocompatible solvents makes it difficult to deliver it to the body by an intravenous injection. Many camptothecin analogs were synthesized for clinical use. However, camptothecin always shows more potent activity than its semi-synthetic analogs.[14] Liposomes may act as an ideal vehicle for camptothecin to expand its clinical applications. We attempted to synthesize α-MSH–containing liposomes and evaluate their physicochemical properties, including size, zeta potential, vesicle morphology, drug encapsulation, and release profiles. To elucidate their targeting ability, the cytotoxicity and cellular uptake were examined in the B16F10 melanoma cell line. Free camptothecin in dimethyl sulfoxide (DMSO) and liposomes without peptide ligands were also used for comparison.
MATERIALS AND METHODS
Materials
Camptothecin, cholesterol, stearylamine, α-MSH, dithiothreitol (DTT), rhodamine 123, and Sephadex G25 were purchased from Sigma-Aldrich (St. Louis, MO, USA). Hydrogenated soybean phosphatidylcholine (SPC, Phospholipon® 80H) was supplied by American Lecithin (Oxford, CT, USA). Sulfo-LC-SPDP was provided by Thermo Scientific (Rockford, IL, USA). Cellulose membrane (CelluSep® T1, with a molecular weight cutoff of 3500) was purchased from Membrane Filtration Products (Seguin, TX, USA).
Preparation of liposomes
Liposomes were prepared by a thin-film hydration method. SPC (1.6%, w/v), cholesterol (0.4%), stearylamine (0.2%), and camptothecin (0.04%) were dissolved in chloroform: Methanol (2:1) solution for complete solubilization. The solvent was then evaporated in a rotary evaporator at 50°C for 30 min. The residual solvent was removed under a vacuum for 6 h. The film was hydrated with double-distilled water using a high-shear homogenizer (Pro 250, Pro Scientific, Oxford, CT, USA) for 10 min. The resulting systems were then subjected to a probe-type sonicator (VCX 600, Sonics and Materials, Danbury, CT, USA) at 35 W for 30 min. The total volume of the final products was 10 ml.
Preparation of α-MSH liposomes
α-MSH (0.5 ml) in a phosphate-buffered saline (PBS) solution (2 mg/ml) was first modified at its N-terminus by sulfo-LC-SPDP. α-MSH was mixed with 0.5 ml sulfo-LC-SPDP in DMSO (2 mg/ml) for 5 h at 37°C. Subsequently, 0.1 M DTT was added to the mixture at 37°C for 2 h to link the sulfhydryl moiety to the N-terminus of α-MSH (sulfhydryl-α-MSH). Liposomes (2 ml) were reacted with sulfhydryl-α-MSH (20-fold molar excess of sulfhydryl-α-MSH: Liposomes) at 4°C for 12 h. The -NH2 group in stearylamine provided the reaction site for the carboxylate moiety of sulfhydryl-α-MSH. Then the α-MSH liposomes were purified through a Sephadex G25 column and validated by matrix-assisted laser desorption/ionization-time of flight/time of flight (MALDI-TOF/TOF) mass spectrometry (Ultraflex, Bruker-Daltonik, Bellerica, MA, USA). Figure 1 shows the complete reaction scheme for preparing α-MSH liposomes.
Figure 1.
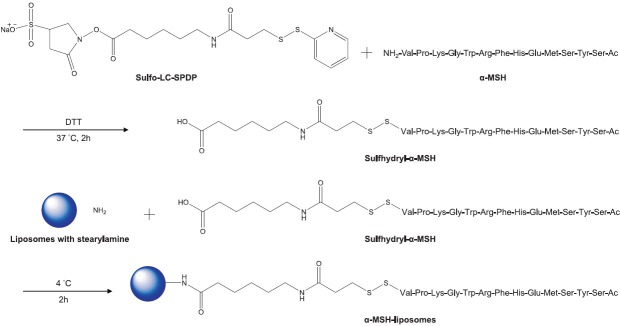
Scheme of the chemical reaction for synthesizing sulfhydryl-α-melanocyte-stimulating hormone (sulfhydryl-α-MSH) and preparing α-MSH liposomes
Vesicle size and zeta potential of liposomes
The average size and zeta potential were determined by photon correlation spectrometry (Nano ZS90, Malvern, Worcestershire, UK) using a helium–neon laser at a wavelength of 633 nm. A 1:100 dilution with double-distilled water was performed before the measurement. The determination was repeated three times per sample for three different batches.
Transmission electron microscopy
The morphology of liposomes was observed by TEM. A drop of liposomal dispersion was pipetted onto a carbon film-covered copper grid to form a thin-film specimen. Phosphotungstic acid at 1% was used to stain the samples. The prepared samples were examined and photographed with transmission electron microscopy (TEM; H-7500, Hitachi, Tokyo, Japan).
Encapsulation efficiency of camptothecin in liposomes
The encapsulation percentage of camptothecin in liposomes was measured by an ultracentrifugation method. The liposomal dispersion was centrifuged at 48,000 × g at 4°C for 8 min in a Beckman Optima MAX® (Beckman Coulter, Fullerton, CA, USA) to separate the encapsulated camptothecin from the free form. After centrifugation, both the supernatant and precipitate were analyzed by high-performance liquid chromatography (HPLC) to determine the encapsulation percentage (%) of the total camptothecin load in the vesicles. The analytical method for camptothecin was described in our previous study.[13]
Camptothecin release from liposomes
Camptothecin release was determined using a Franz diffusion assembly. A cellulose membrane was mounted between the donor and receptor compartments. The donor medium consisted of either 0.5 ml camptothecin (0.04%) in 30% ethanol/double-distilled water (free control), conventional liposomes, or α-MSH liposomes. The receptor medium was 5.5 ml of 30% ethanol in pH 7.4 phosphate-citrate buffer. The available area for release between the compartments was 0.785 cm2. The stirring rate and temperature of the receptor were kept at 600 rpm and 37°C, respectively. At appropriate intervals, 300-μl aliquots of the receptor medium were withdrawn and immediately replaced with an equal volume of fresh buffer. The amount of camptothecin release was quantified by HPLC.
Cell viability assay
The mouse melanoma B16F10 cell line was purchased from American Type Culture Collection (Rockville, MD, USA). Cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) heat-inactivated fetal bovine serum and 1% antibiotics (100 U/ml penicillin and 100 U/ml streptomycin) at 37°C in a humidified atmosphere with 5% CO2. Cells (104) were sealed into 96-well plates and cultured for 24 h. The free control or liposomes with or without α-MSH were used to treat cells. For the cell viability determination, intracellular ATP was detected by a bioluminescence assay based on an ATP-dependent luciferase reaction using a commercial kit as described previously.[15] After a 24-h culture with camptothecin vehicles, 100 μl of the CellTiter-Glo® reagent was added to each well. The mixture was shaken for 2 min on an orbital shaker to induce cell lysis. The plate was allowed to incubate at room temperature for 10 min to stabilize the luminescent signal. Luminescence was measured with a luminometer (Chameleon V, Hidex, Turku, Finland).
Cellular uptake assay
To study the cellular uptake by melanoma cells, 0.03% rhodamine 123 instead of camptothecin was loaded in liposomes by the same preparation process as described in the section “Preparation of Liposomes.” Rhodamine 123 is a fluorescent dye that shows excitation at 511 nm and emission at 534 nm. Melanoma cells (105) were seeded in 24-well plates (1 ml) and cultured for 24 h. Then, either the free control, liposomes, or α-MSH liposomes with rhodamine 123 were added to the plate and incubated for 2 h at 37°C. The medium was removed, and cells were washed twice with PBS. B16F10 cell uptake of rhodamine 123 was imaged under fluorescent microscopy (DP-70, Olympus, Tokyo, Japan). The excitation wavelength of the microscopy was set to 488 nm, and the green emission of rhodamine 123 was monitored.
Statistical analysis
Unpaired Student's t-test was utilized to examine the statistical difference between samples. A P value of < 0.05 was considered a significant difference. Data are presented as the mean and standard deviation (SD). All experiments were independently repeated at least three times.
RESULTS
Physicochemical properties
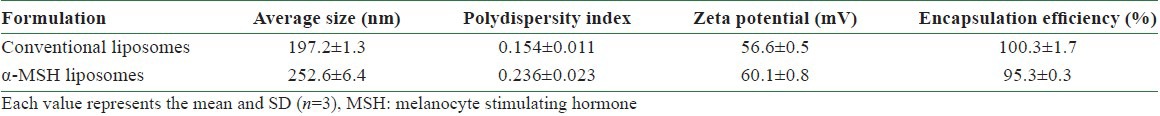
The vesicle size, polydispersity, and zeta potential of liposomes with or without α-MSH were detected by the Zetasizer. Results are shown in Table 1. The average size of non-targeted liposomes was estimated to be about 200 nm. The polydispersity index could be controlled to a narrow distribution of 0.15, suggesting a homogeneous population of vesicles. Compared to conventional liposomes, the size of α-MSH liposomes was significantly larger (253 nm) and the size distribution was wider (0.24). Stearylamine produced a positive charge on the liposomal bilayers [Table 1]. The zeta potential of conventional liposomes was about 57 mV. α-MSH slightly but significantly (P < 0.05) increased the zeta potential to a value of 60 mV.
Table 1.
The characterization of the liposomes and α-MSH liposomes by size, zeta potential, and encapsulation efficiency
In order to obtain more information about the size and morphology, TEM imaging was used to analyze both types of liposomes. As depicted in Figure 2a, the prepared conventional liposomes were spherical and compact. The outer morphology was smooth. On the other hand, the entire structure of α-MSH liposomes was heterogeneous in shape as shown in Figure 2b. The liposomal size estimated by TEM was well correlated with that measured with the Zetasizer. An important concern with respect to liposomes as drug nanocarriers is the drug loading capacity. The camptothecin encapsulation percentages of both formulations exceeded 95% of the total drug added to the system [Table 1]. Nearly complete entrapment of camptothecin was found for conventional liposomes. The encapsulation percentage dropped from 100% to 95% after conjugation with α-MSH [Figure 2].
Figure 2.

Transmission electron microscopic (TEM) photographs of (a) conventional liposomes and (b) α-melanocyte-stimulating hormone (α-MSH) liposomes at 120,000×. The scale bar is 100 nm
Camptothecin release from liposomes
A key issue of liposomes as carriers is the feasibility of delivering a drug. The camptothecin release percentage was determined as a function of time as shown in Figure 3. DMSO was used as the free control for camptothecin. The control group exhibited the greatest release. A burst release was shown for the free control. Camptothecin release from 30% ethanol was almost complete in 24 h. About 59% of the camptothecin was released during 48 h. From Figure 3 it can be seen that the release rate significantly decreased following liposomal application. Camptothecin was released in a sustained manner when loaded in liposomes. The release percentage–time curve of liposomes was fitted by a zero-order release function. Incorporating α-MSH in the systems slightly promoted camptothecin release; nevertheless, this enhancement was not significant (P > 0.05). Non-targeted and targeted liposomes respectively showed 32% and 34% drug release after 48 h of application [Figure 3].
Figure 3.
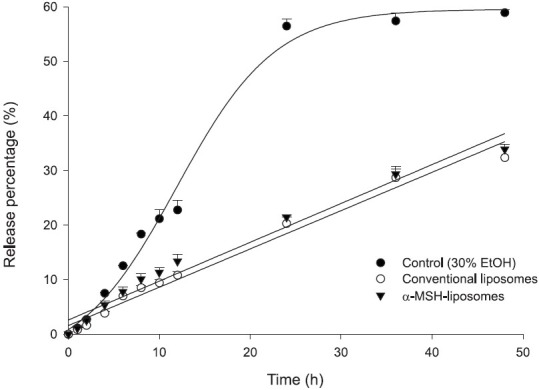
In vitro release percentage (%)–time profiles of camptothecin (at a 0.04% dose) across cellulose membranes from the free control (30% ethanol in double-distilled water), conventional liposomes, and α-melanocyte-stimulating hormone (α-MSH) liposomes. Each value represents the mean and standard deviation (n= 4)
Cell viability assay
The cytotoxicity of camptothecin liposomes against B16F10 melanomas was examined in vitro. An ATP assay was applied for the cell viability test, which follows the cellular ATP level as an indicator of cell viability. At first, the cell viability was determined by free camptothecin in DMSO at different concentrations. Free camptothecin induced a dose-dependent growth inhibition of the melanoma cell line with an IC50 of about 50 μM as shown in Figure 4. Actually, a further increment of the dose to 100 μM did not significantly (P > 0.05) increase the cytotoxicity compared to the level at 50 μM. Melanoma cells were exposed to 50 μM camptothecin to examine the cytotoxicity of liposomes. As demonstrated in Figure 5, both liposomal dispersions displayed significantly lower (P < 0.05) cell viability compared to the free control. The results indicate that α-MSH liposomes bearing camptothecin were more active against melanomas than the conventional systems. After 24 h of incubation, cell viabilities after treatment with the free control, non-targeted liposomes, and targeted liposomes were 48%, 32%, and 18%, respectively. Blank liposomes without the drug were also tested for cytotoxicity. There was no obvious effect of the blank liposomes on growth inhibition of the melanoma cells (data not shown). This suggests that the cytotoxicity toward the melanoma was predominantly a result of camptothecin itself [Figures 4 and 5].
Figure 4.
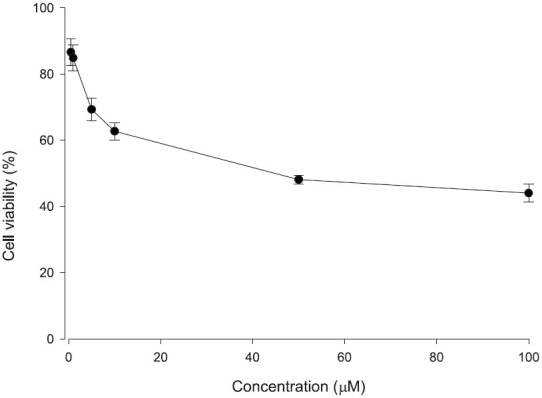
Viability percentage (%) of B16F10 melanoma cells treated with free camptothecin in DMSO at various concentrations. Each value represents the mean and standard deviation (n = 3)
Figure 5.
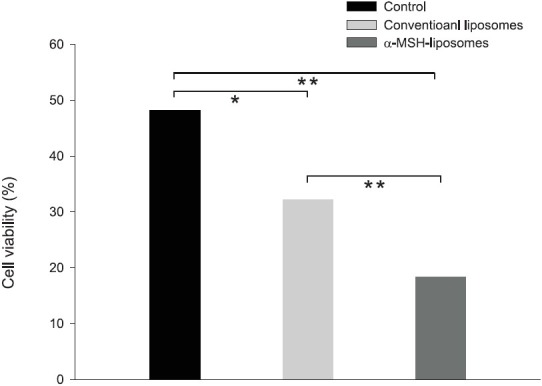
Viability percentage (%) of B16F10 melanoma cells treated with camptothecin in the free control, conventional liposomes, and α-melanocyte-stimulating hormone (α-MSH) liposomes at a concentration of 50 μM. Each value represents the mean and standard deviation (n = 3). *P < 0.01; **P < 0.001
Cellular uptake assay
To track the cellular internalization of liposomes, rhodamine 123 was included in the liposomes for imaging. Figure 6 summarizes the imaging profiles of cellular uptake by free and liposome-encapsulated rhodamine 123 under fluorescent microscopy. As can be observed in Figure 6A, there was nearly no fluorescence signal in melanoma cells after free rhodamine 123 treatment. The fluorescence intensity was significantly enhanced by the addition of liposomes to melanoma cells [Figure 6b and c]. The fluorescence was mainly derived from the cytoplasm rather than the cell nuclei or membranes. This indicates that rhodamine 123 included in liposomes had been introduced into B16F10 cells. More intense fluorescence from α-MSH liposomes in the cytoplasm was seen compared to non-targeted liposomes, suggesting that targeted liposomes efficiently bound to melanoma cells via the α-MSH ligands. Cells appeared evenly fluorescent, demonstrating widespread distribution of the vesicles [Figure 6].
Figure 6.
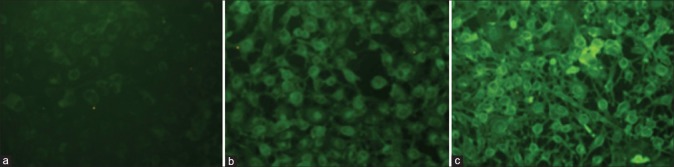
Images of fluorescent microscopy of rhodamine 123 uptake by the (a) free control, (b) conventional liposomes, and (c) α-melanocyte-stimulating hormone (α-MSH) liposomes by B16F10 melanoma cells
DISCUSSION
The success of cancer therapy is greatly dependent upon carriers developed to deliver the drugs. Liposomes and related nanomaterials are increasingly being employed as nanocarriers for drugs due to their ability to lessen the adverse effects and maximize their therapeutic activity. Efficient tumor targeting is promising by conjugating the liposomal surface with ligands capable of binding to cell receptors. Based on the capability of α-MSH to selectively bind to melanomas but not normal cells, we attempted to develop α-MSH–conjugated liposomes incorporating camptothecin, which is not a regular anticancer drug candidate because of the lack of feasible carrier systems. The experimental results showed a targeting efficiency of α-MSH liposomes to melanomas according to the cell-viability and cellular-uptake studies, whereas conventional liposomes exhibited less-efficient activity. Controlled and sustained camptothecin release was detected with the liposomal systems.
Sulfo-LC-SPDP was used to form amine-to-sulfhydryl linkages for α-MSH conjugation to stearylamine-containing liposomes. The liposomes were prepared by thin-film hydration and subsequent high-pressure homogenization and sonication. α-MSH may reside on the liposomal bilayers, resulting in an increment in the vesicle size from 200 to 250 nm. α-MSH on the liposomal surface also contributed to the heterogeneous shape of the vesicles shown in the TEM image [Figure 2]. The polydispersity values indicated a narrow and unimodal vesicle size distribution (<0.25). Stearylamine produced a positive charge on the surface of the liposomes. No remarkable difference in the zeta potential was observed after α-MSH conjugation, although the value slightly increased from 57 to 60 mV. This could have been due to the presence of some amine groups located in α-MSH's structure. The zeta potential can act as a predictor of physical stability. Liposomal aggregation and fusion are less likely to occur with a zeta potential of >│30│ mV because of electrical repulsion.[3,16] Our liposomes fulfilled this criterion. Another benefit of cationic liposomes is that they encourage interactions with negatively charged cellular surfaces.
The liposomes showed an entrapment efficiency of >95% for drug loading. Since camptothecin is a lipophilic molecule with a log P (partition coefficient) of 2.42,[17] it can readily be intercalated in the phospholipid bilayers of liposomes with high affinity. Encapsulation was found to be slightly reduced when coupling to vesicles with α-MSH. The rigidity of liposomal SPC bilayers might be disrupted by mixing with α-MSH, leading to the greater permeability of the α-MSH–containing membrane.
Drug release was evaluated by determining the release percentage across cellulose membranes. The mean pore size of the membrane was <10 nm,[18] and so only very small vesicles could diffuse through it. Almost only free camptothecin molecules were able to permeate through the membrane. Camptothecin release from the free control did not reach 100% by the end of the experiment. A ~60% release percentage was detected over 48 h. This may have been due to utilization of the Franz diffusion assembly. Since the definite receptor volume (5.5 ml) and diffusion area (0.785 cm2) are limited, drug loading in the receptor is restricted. There is no longer a concentration gradient between the donor and receptor compartments. Yet, this release platform is still valuable for distinguishing release profiles from different formulations. Phospholipid bilayers are a diffusion barrier for drug delivery. Cholesterol interacting with SPC also plays an essential role in increasing membrane rigidity and reducing the permeability of the bilayers.[19,20] The sustained and controlled release of camptothecin from liposomes may be attributed to high encapsulation, strong intercalation of camptothecin within the bilayers, and the barrier function of the bilayers. The slightly higher drug that leached from α-MSH liposomes compared to conventional liposomes was due to lower encapsulation in this targeted carrier. However, the difference was not large and could be neglected. In a previous investigation,[21] α-MSH with an amphiphilic property had a tendency to increase leakage of liposomal membranes. This phenomenon was not observed in our case. The release profiles demonstrated that α-MSH liposomes are a promising carrier for controlled release to administer camptothecin. Sustained release is a feature often correlated with improved drug efficacy and pharmacokinetics.[22] It is especially important for camptothecin since serum albumin in the blood can open the camptothecin lactone ring and shorten the circulation half-life.[23] Liposomal incorporation of camptothecin can help retain the active lactone form. Liposomes also reduce camptothecin toxicity,[24] since liposomes prevent a burst effect of drug exposure in the body.
Camptothecin can induce apoptosis of melanoma cells, which is essentially dependent upon p53 and mitochondrial pathways. Caspase-2 and p73 are also involved in apoptotic pathways with camptothecin.[25] An in vitro cell viability assay of camptothecin showed that the cytotoxicity of α-MSH liposomes was the greatest among all formulations tested, followed by non-targeted liposomes and the free control. The cytotoxicity of drug-loaded liposomes may be dominated by extracellular drug release from liposomes and the intracellular uptake of intact liposomes.[26] The direct penetration of liposomes may be a predominant route for cytotoxicity because of the slow camptothecin release and limited growth-inhibition activity of free camptothecin.
According to the results of the cellular uptake assay, α-MSH ligands promoted cell internalization via the MC1R. The uptake performance was greater than that of non-targeted liposomes which appeared to enter cells by less-efficient pathways. The therapeutic efficiency of anticancer drugs is largely dependent on the activity of cellular uptake.[27] Cellular uptake by various formulations was well correlated to the cytotoxic activity of camptothecin. This indicates that inhibition of cell proliferation was predominated by the actual intracellular drug amount. The C- or N-terminal derivatization of α-MSH is an important character in the specific binding efficiency to the MC1R. Schiöth et al.,[28] demonstrated that C-terminal modification weakened α-MSH's affinity to the receptor, whereas an N-terminal addition did not influence the binding affinity. In this study, we developed liposomes conjugated with α-MSH at the N-terminus. The success in eliciting effective cellular internalization was most likely due to liposomal endocytosis. It is necessary for the liposomes to enter cells and diffuse through the viscous cytosol to access particular targets where the action sites are located. Endocytosis is involved in the fusion of liposomal surfaces with cell membranes. Receptor-mediated endocytosis basically results in the entry of internalized ligands by the lysosomal route, leading to their destruction.[29]
The failure of cellular uptake by liposomes is usually attributed to anionic vesicles that are repelled by negatively charged cell membranes.[30] Cationic liposomes are thought to easily interact with cell plasma membranes, subsequently enhancing their uptake into cells.[31,32] Another potential of cationic liposomes is their preferential uptake by interstitial tumor tissues and leaky tumor vasculature.[2,33] When administered by an intravenous route, the cationic form can obviate plasma protein adsorption and the complement system,[5,34] thus prolonging the half-life of the carriers. Helper lipids such as cholesterol also assist the cellular internalization of liposomes.[2,35] It was suggested that even in the absence of specific ligands attached to the liposomal surface (conventional liposomes), vesicles could localize into cells due to the positive charge and cholesterol on the liposomes. In addition to the above-mentioned mechanisms, liposomes can bypass efflux transporters, thus increasing the anticancer activity of the drugs.[36] Further study is necessary to elucidate this mechanism.
When entering the cancer cells, drug distribution between the cytoplasm and nuclei can largely influence the anticancer activity.[37] Camptothecin is a topoisomerase inhibitor that interferes with topoisomerase function of DNA replication in the nuclei. According to fluorescence imaging of cellular uptake, the green signal was mainly located in the cytoplasm, not in the nuclei. The proposed process for the anticancer effect of α-MSH liposomes is that after liposomal internalization to the cytoplasm, degradative enzymes in the cytosol break down the liposomal membranes and release the drug. Or drug release from liposomes in the cytoplasm may become faster in the presence of endolysosomes.[37] Subsequently, the drug diffuses to the nuclei to trigger apoptotic procedures. p53 and its transactivated targets may induce camptothecin-related apoptosis of melanoma cells by translocating into mitochondria,[25,38] where they form complexes with proteins and enhance the loss of the mitochondrial membrane potential and its transition. This indicates that camptothecin might not necessarily have to target nuclei, but could reside in the cytoplasm to elicit its cytotoxicity against melanomas. A great accumulation of liposomes in the cytoplasm would be helpful to achieve this.
CONCLUSIONS
The experimental data presented in this study indicate that α-MSH can be utilized to efficiently ferry liposomes for melanoma targeting. Potent cytotoxicity against melanoma cells was observed after loading camptothecin as the model drug. Fluorescent microscopic imaging showed that most liposomes were located in the cytoplasm after cell internalization. Sustained and controlled camptothecin release was achieved after being loaded into liposomes, which was due to the high encapsulation efficiency and strong interaction with the phospholipid bilayers. It is possible that the adverse effect of camptothecin can be reduced by α-MSH liposomes because peptide ligands can selectively and specifically target melanoma cells, while minimizing the distribution to normal cells and tissues. The high efficiency of cellular uptake and cytotoxicity of α-MSH liposomes are particularly important for melanomas, since these malignant tumors are highly resistant to conventional chemotherapy and lack a satisfactory therapy with sustained responses. Although our findings mainly refer to in vitro cell line experiments, it is evident that the liposomal application with α-MSH is a promising approach to overcome the drug resistance of melanomas. Further in vivo studies are needed and are in progress to explore the efficacy of α-MSH liposomes for future applicability.
REFERENCES
- 1.Thompson JF, Scolyer RA, Kefford RF. Cutaneous melanoma. Lancet. 2005;365:687–701. doi: 10.1016/S0140-6736(05)17951-3. [DOI] [PubMed] [Google Scholar]
- 2.Tran MA, Watts RJ, Robertson GP. Use of liposomes as drug delivery vesicles for treatment of melanoma. Pigment Cell Melanoma Res. 2009;22:388–99. doi: 10.1111/j.1755-148X.2009.00581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barbugli PA, Siqueira-Moura MP, Espreafico EM, Tedesco AC. In vitro phototoxicity of liposomes and nanocapsules containing chloroaluminum phthalocyanine on human melanoma cell line. J Nanosci Nanotechnol. 2010;10:569–73. doi: 10.1166/jnn.2010.1741. [DOI] [PubMed] [Google Scholar]
- 4.Fenske DB, Cullis PR. Liposomal nanomedicines. Exp Opin Drug Deliv. 2008;5:25–44. doi: 10.1517/17425247.5.1.25. [DOI] [PubMed] [Google Scholar]
- 5.Hsu SH, Al-Suwayeh SA, Chen CC, Chi CH, Fang JY. PEGylated liposomes incorporated with nonionic surfactants as an apomorphine delivery system targeting the brain: In vitro release and in vivo real-time imaging. Curr Nanosci. 2011;7:91–9. [Google Scholar]
- 6.Gupta Y, Ganesh N, Kohli DV, Jain SK. Development and characterization of doxorubicin bearing vitamin B12 coupled sterically stabilized liposomes for tumor targeting. Curr Nanosci. 2011;7:427–35. [Google Scholar]
- 7.Elbayoumi TA, Torchilin VP. Tumor-targeted nanomedicines: enhanced antitumor efficacy in vivo of doxorubicin-loaded, long-circulating liposomes modified with cancer-specific monoclonal antibody. Clin Cancer Res. 2009;15:1973–80. doi: 10.1158/1078-0432.CCR-08-2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miao Y, Quinn TP. Peptide-targeted radionuclide therapy for melanoma. Crit Rev Oncol/Hematol. 2008;67:213–28. doi: 10.1016/j.critrevonc.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schiöth HB, Mutulis F, Muceniece R, Prusis P, Wikberg JES. Discovery of novel melanocortin4 receptor selective MSH analogues. Br J Pharmacol. 1998;124:75–82. doi: 10.1038/sj.bjp.0701804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu W, Xiong C, Zhang G, Huang Q, Zhang R, Zhang JZ, et al. Targeted photothermal ablation of murine melanomas with melanocyte-stimulating hormone analog-conjugated hollow gold nanospheres. Clin Cancer Res. 2009;15:876–85. doi: 10.1158/1078-0432.CCR-08-1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yagi N, Ogawa Y, Kodaka M, Okada T, Tomohiro T, Konakahara T, et al. Preparation of functional liposomes with peptide ligands and their binding to cell membranes. Lipids. 2000;35:673–9. doi: 10.1007/s11745-000-0572-4. [DOI] [PubMed] [Google Scholar]
- 12.Raposinho PD, Correia JD, Oliveira MC, Santos I. Melanocortin-1 receptor-targeting with radiolabeled cyclic α-melanocyte-stimulating hormone analogs for melanoma imaging. Biopolymers. 2010;94:820–9. doi: 10.1002/bip.21490. [DOI] [PubMed] [Google Scholar]
- 13.Huang ZR, Hua SC, Yang YL, Fang JY. Development and evaluation of lipid nanoparticles for camptothecin delivery: A comparison of solid lipid nanoparticles, nanostructured lipid carriers, and lipid emulsion. Acta Pharmacol Sin. 2008;29:1094–102. doi: 10.1111/j.1745-7254.2008.00829.x. [DOI] [PubMed] [Google Scholar]
- 14.Thomas CJ, Rahier NJ, Hecht SM. Camptothecin: Current perspectives. Bioorg Med Chem. 2004;12:1585–604. doi: 10.1016/j.bmc.2003.11.036. [DOI] [PubMed] [Google Scholar]
- 15.Hidalgo E, Dominguez C. Study of cytotoxicity mechanisms of silver nitrate in human dermal fibroblasts. Toxicol Lett. 1998;98:169–79. doi: 10.1016/s0378-4274(98)00114-3. [DOI] [PubMed] [Google Scholar]
- 16.Hwang TL, Lee WR, Hua SC, Fang JY. Cisplatin encapsulated in phosphatidylethanolamine liposomes enhances the in vitro cytotoxicity and in vivo intratumor drug accumulation against melanomas. J Dermatol Sci. 2007;46:11–20. doi: 10.1016/j.jdermsci.2006.12.011. [DOI] [PubMed] [Google Scholar]
- 17.Carrigan SW, Fox PC, Wall ME, Wani MC, Bowen JP. Comparative molecular field analysis and molecular modeling studies of 20-(s)-camptothecin analogs as inhibitors of DNA topoisomerase I and anticancer/antitumor agents. J Comput-Aided Mol Des. 1997;11:71–8. doi: 10.1023/a:1008027528218. [DOI] [PubMed] [Google Scholar]
- 18.Fang JY, Hung CF, Hwang TL, Huang YL. Physicochemical characteristics and in vivo deposition of liposome-encapsulated tea catechins by topical and intratumor administrations. J Drug Target. 2005;13:19–27. doi: 10.1080/10611860400015977. [DOI] [PubMed] [Google Scholar]
- 19.Li H, Song JH, Park JS, Han K. Polyethylene glycol-coated liposomes for oral delivery of recombinant human epidermal growth factor. Int J Pharm. 2003;258:11–9. doi: 10.1016/s0378-5173(03)00158-3. [DOI] [PubMed] [Google Scholar]
- 20.Huang YB, Tsai MJ, Wu PC, Tsai YH, Wu YH, Fang JY. Elastic liposomes as carriers for oral delivery and the brain distribution of (+)-catechin. J Drug Target. 2011;19:709–18. doi: 10.3109/1061186X.2010.551402. [DOI] [PubMed] [Google Scholar]
- 21.Ogawa Y, Kawahara H, Yagi N, Kodaka M, Tomohiro T, Okada T, et al. Synthesis of a novel lipopeptide withα-melanocyte-stimulating hormone peptide ligand and its effect on liposome stability. Lipids. 1999;34:387–94. doi: 10.1007/s11745-999-0377-5. [DOI] [PubMed] [Google Scholar]
- 22.Marcato PD, Durán N. New aspects of nanopharmaceutical delivery systems. J Nanosci Nanotechnol. 2008;8:2216–29. doi: 10.1166/jnn.2008.274. [DOI] [PubMed] [Google Scholar]
- 23.Fang JY, Hung CF, Hua SC, Hwang TL. Acoustically active perfluorocarbon nanoemulsions as drug delivery carriers for camptothecin: Drug release and cytotoxicity against cancer cells. Ultrasonics. 2009;49:39–46. doi: 10.1016/j.ultras.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 24.Watanabe M, Kawano K, Toma K, Hattori Y, Maitani Y. In vivo antitumor activity of camptothecin incorporated in liposomes formulated with an artificial lipid and human serum albumin. J Control Release. 2008;127:231–8. doi: 10.1016/j.jconrel.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 25.Rudolf E, Rudolf K, Cervinka M. Camptothecin induces p53-dependent and -independent apoptogenic signaling in melanoma cells. Apoptosis. 2011;16:1165–76. doi: 10.1007/s10495-011-0635-8. [DOI] [PubMed] [Google Scholar]
- 26.Niu R, Zhao P, Wang H, Yu M, Cao S, Zhang F, et al. Preparation, characterization, and antitumor activity of paclitaxel-loaded folic acid modified and TAT peptide conjugated PEGylated polymeric liposomes. J Drug Target. 2011;19:373–81. doi: 10.3109/1061186X.2010.504266. [DOI] [PubMed] [Google Scholar]
- 27.Zhao P, Wang H, Yu M, Cao S, Zhang F, Chang J, et al. Paclitaxel-loaded, folic-acid-targeted and TAT-peptide-conjugated polymeric liposomes: In vitro and in vivo evaluation. Pharm Res. 2010;27:1914–26. doi: 10.1007/s11095-010-0196-5. [DOI] [PubMed] [Google Scholar]
- 28.Schiöth HB, Mutulis F, Muceniece R, Prusis P, Wikberg JE. Selective properties of C- and N-terminals and core residues of the melanocyte-stimulating hormone on binding to the human melanocortin receptor subtypes. Eur J Pharmacol. 1998;349:359–66. doi: 10.1016/s0014-2999(98)00212-x. [DOI] [PubMed] [Google Scholar]
- 29.Glover DJ, Ng SM, Mechler A, Martin LL, Jans DA. Multifunctional protein nanocarriers for targeted nuclear gene delivery in nondividing cells. FASEB J. 2009;23:2996–3006. doi: 10.1096/fj.09-131425. [DOI] [PubMed] [Google Scholar]
- 30.Wang H, Zhao P, Liang X, Gong X, Song T, Niu R, et al. Folate-PEG coated cationic modified chitosan-cholesterol liposomes for tumor-targeted drug delivery. Biomaterials. 2010;31:4129–38. doi: 10.1016/j.biomaterials.2010.01.089. [DOI] [PubMed] [Google Scholar]
- 31.Santel A, Aleku M, Keil O, Endruschat J, Esche V, Burieux B, et al. RNA interference in the mouse vascular endothelium by systemic administration of siRNA-lipoplexes for cancer therapy. Gene Ther. 2006;13:1360–70. doi: 10.1038/sj.gt.3302778. [DOI] [PubMed] [Google Scholar]
- 32.Ito A, Fujioka M, Yoshida T, Wakamatsu K, Ito S, Yamashita T, et al. 4-S-Cysteaminylphenol-loaded magnetite cationic liposomes for combination therapy of hyperthermia with chemotherapy against malignant melanoma. Cancer Sci. 2007;98:424–30. doi: 10.1111/j.1349-7006.2006.00382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hsu SH, Wen CJ, Al-Suwayeh SA, Chang HW, Yen TC, Fang JY. Physicochemical characterization and in vivo bioluminescence imaging of nanostructured lipid carriers (NLCs) for targeting the brain: Apomorphine as a model drug. Nanotechnology. 2010;21 doi: 10.1088/0957-4484/21/40/405101. 405101. [DOI] [PubMed] [Google Scholar]
- 34.Kaur IP, Bhandari R, Bhandari S, Kakkar V. Potential of solid lipid nanoparticles in brain targeting. J Control Release. 2008;127:97–109. doi: 10.1016/j.jconrel.2007.12.018. [DOI] [PubMed] [Google Scholar]
- 35.Xu L, Wempe MF, Anchordoquy TJ. The effect of cholesterol domains on PEGylated liposomal gene delivery in vitro. Ther Deliv. 2011;2:451–60. doi: 10.4155/tde.11.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2:751–60. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 37.Tippayamontri T, Kotb R, Paquette B, Sanche L. Cellular uptake and cytoplasm/DNA distribution of cisplatin and oxaliplatin and their liposomal formulation in human colorectal cancer cell HCT116. Invest New Drugs. 2011;29:1321–7. doi: 10.1007/s10637-010-9494-3. [DOI] [PubMed] [Google Scholar]
- 38.Ferrin G, Linares CI, Muntane J. Mitochondrial drug targets in cell death and cancer. Curr Pharm Des. 2011;17:2002–16. doi: 10.2174/138161211796904803. [DOI] [PubMed] [Google Scholar]