

Abstract
Background and Purpose
Crotalphine is an antinociceptive peptide that, despite its opioid-like activity, does not induce some of the characteristic side effects of opioids, and its amino acid sequence has no homology to any known opioid peptide. Here, we evaluated the involvement of the peripheral cannabinoid system in the crotalphine effect and its interaction with the opioid system.
Experimental Approach
Hyperalgesia was evaluated using the rat paw pressure test. Involvement of the cannabinoid system was determined using a selective cannabinoid receptor antagonist. Cannabinoid and opioid receptor activation were evaluated in paw slices by immunofluorescence assays using conformation state-sensitive antibodies. The release of endogenous opioid peptides from skin tissue was measured using a commercial enzyme immunoassay (EIA).
Key Results
Both p.o. (0.008–1.0 μg·kg−1) and intraplantar (0.0006 μg per paw) administration of crotalphine induced antinociception in PGE2-induced hyperalgesia. Antinociception by p.o. crotalphine (1 μg·kg−1) was blocked by AM630 (50 μg per paw), a CB2 receptor antagonist, and by antiserum anti-dynorphin A (1 μg per paw). Immunoassay studies confirmed that crotalphine increased the activation of both κ-opioid (51.7%) and CB2 (28.5%) receptors in paw tissue. The local release of dynorphin A from paw skin was confirmed by in vitro EIA and blocked by AM630.
Conclusions and Implications
Crotalphine-induced antinociception involves peripheral CB2 cannabinoid receptors and local release of dynorphin A, which is dependent on CB2 receptor activation. These results enhance our understanding of the mechanisms involved in the peripheral effect of crotalphine, as well as the interaction between the opioid and cannabinoid systems.
Keywords: crotalphine, antinociception, endogenous opioid release, dynorphin A, CB2 cannabinoid receptors, cannabinoid–opioid interaction
Introduction
Crotalphine is a 14 amino acid synthetic peptide that has recently been described as a new compound with an antinociceptive effect (Gutierrez et al., 2008; Konno et al., 2008). Its sequence is based on the structure of the natural analgesic factor isolated from the venom of the South American rattlesnake Crotalus durissus terrificus (Konno et al., 2008). Previous work from our group demonstrated that crotalphine, when administered in low doses by the p.o., i.v. or intraplantar (i.pl.) routes, induced potent antinociceptive effects that lasted for 5 and 3 days in acute (Konno et al., 2008) and chronic (Gutierrez et al., 2008) pain models respectively. Additionally, this antinociceptive effect was blocked by i.pl. pretreatment with selective antagonists of either the κ (acute pain models) (Konno et al., 2008) or κ and δ (chronic constriction injury of rat sciatic nerve) (Gutierrez et al., 2012) opioid receptors, suggesting the involvement of peripheral opioid receptors in crotalphine's effect. Despite its opioid-like activity, crotalphine does not manifest some of the side effects that are characteristics of opioid drugs such as tolerance (Gutierrez et al., 2008) and constipation (G.A. Rae, unpubl. data), even after prolonged treatment. Also, its amino acid sequence displays no homology to any known opioid peptide (Konno et al., 2008), suggesting that opioid receptors might not be the target of crotalphine.
A significant interaction between endogenous opioid and cannabinoid systems in pain management (Cichewicz, 2004; Anand et al., 2009) has been reported in the literature. Although opioids and cannabinoids bind to distinct receptors, it appears that one system may potentiate the other, which suggests that these two systems may operate synergistically (Cichewicz, 2004). Moreover, behavioural and molecular studies have demonstrated that cannabinoids induce the release of endogenous opioids just as opioids may induce the release of endocannabinoids (Ibrahim et al., 2005; Welch, 2009). Furthermore, these studies have suggested the existence of direct receptor–receptor interactions as well as interactions in their intracellular signalling pathways when cannabinoid and opioid receptors are co-expressed in the same cells (Desroches and Beaulieu, 2010; Parolaro et al., 2011).
The aim of the present study was to characterize the mechanisms involved in the antinociceptive effect of crotalphine by investigating the possible involvement of CB2 cannabinoid receptors as well as the possible interaction of these receptors with the opioid system. Our results indicate that peripheral CB2 cannabinoid receptors are involved in the antinociceptive effect of crotalphine in the PGE2-induced hyperalgesia model in rats. Moreover, this effect was mediated by the release of peripheral dynorphin A, an endogenous agonist of κ-opioid receptors, and this release was dependent on CB2 receptor activation.
Methods
Peptide synthesis
Crotalphine (<E-F-S-P-E-N-C-Q-G-E-S-Q-P-C, where <E is pyroglutamic acid and 7C-14C forms a disulphide bond; MW 1534.6 Da) was synthesized by the American Peptide Co. (Sunnyvale, CA, USA) as described by Konno et al. (2008). The synthesized crotalphine was stored at −20°C until used. The peptide was diluted in sterile saline (0.85% NaCl) and administered either p.o. (gastric cannula, 2 mL) or i.pl., with sterile saline used as a control. The doses of crotalphine were based on previously published results (Konno et al., 2008).
Animals
Male Wistar rats (170–190 g) from the Butantan Institute (São Paulo, Brazil) were used throughout this study. Animals were housed four to five per cage (length, 45 cm; width, 30 cm; height, 20 cm) in a temperature-controlled (21 ± 2°C) and light-controlled (12/12 h light/dark cycle) room. Animals were adapted to these conditions for at least 4 days before the beginning of the experiments. All behavioural tests were performed between 09:00 and 16:00 h. Standard food and water were available ad libitum until 2 h prior to p.o. crotalphine administration. After this period, only water was available for a period of no longer than 5 h. All procedures were performed in accordance with the guidelines for the ethical use of conscious animals in pain research published by the International Association for the Study of Pain (Zimmermann, 1983) and were approved by the Institutional Animal Care Committee of the Butantan Institute (CEUAIB, protocol number 622/2009). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Induction of hyperalgesia by PGE2
Hyperalgesia was induced by i.pl. administration of PGE2 (100 ng per paw in 0.1 mL of sterile saline) into either one or, when indicated, both hind paws (Picolo et al., 2000; Picolo and Cury, 2004). The nociceptive threshold was measured before and 3 h after injection of the hyperalgesic agent.
Mechanical hyperalgesia evaluation
To assess the nociceptive threshold, the rat paw pressure test was used (Randall and Selitto, 1957) (Ugo Basile, VA, Italy). The test was applied before and 3 h after the i.pl. injection of PGE2 (100 ng per paw). Testing was blind in regard to group designation. In this test, an increasing force (measured in g) was applied to the hind paw of the rat and interrupted when the animal withdrew its paw. The force necessary to induce this reaction was recorded as the nociceptive threshold. A maximum pressure of 250 g (i.e. cut-off) was established to minimize damage to the paw. To reduce stress, rats were habituated to the testing procedure a day before experimentation.
Experimental design
To evaluate the antinociceptive effect, crotalphine was administered 1 h after PGE2 (100 ng per paw, 50 μL), by either p.o. (0.008–1.0 μg·kg−1, 2 mL) or i.pl. (0.0006 μg per paw, 50 μL) routes (Konno et al., 2008). Behavioural tests were conducted 3 h after PGE2 injection, which corresponds to the peak of the hypernociceptive response induced by PGE2. To evaluate the involvement of CB2 receptors in crotalphine-induced antinociception, AM630 (50 μg per paw, 50 μL), a CB2 receptor antagonist, was injected i.pl. 30 min before crotalphine administration. To determine the involvement of endogenous opioid peptides in this antinociceptive effect, anti-β-endorphin (0.05–5.0 μg per paw), anti-dynorphin A (0.01–1.0 μg per paw) or anti-met-enkephalin (0.1–50.0 μg per paw) antibodies were given i.pl. 15 min before crotalphine administration. The animals were allocated randomly to each group.
Evaluation of cannabinoid and opioid receptor activation using conformation-sensitive receptor antibodies
To determine which receptor type is activated by crotalphine, conformation state-sensitive anti-CB1, anti-CB2, anti-μ, anti-δ and anti-κ antibodies were used. These antibodies are sensitive to activity-mediated conformational changes in the receptors being able to recognize the activated state of the receptor (Gupta et al., 2007; Heimann et al., 2007; Supporting Information Figs S1–S5). For this study, crotalphine was injected by the i.pl. route (0.0006 μg per paw, right paw) 2 h after PGE2 administration (100 ng per paw, in 50 μL, right paw) in either the presence or absence of norbinaltorphimine (nor-BNI) and AM630, antagonists of the κ and CB2 receptors respectively. The antagonists were injected i.pl. 30 min before crotalphine administration. One hour after crotalphine treatment, the rats were anaesthetized with an overdose of ketamine (10%) and xylazine (2%) (1:1 v v−1) i.p., and transcardially perfused with buffered saline followed by 4% paraformaldehyde in 0.1 M PBS (pH 7.4) at 4°C. The paw tissue was removed, and the samples were postfixed at 4°C in the perfusion fixative solution for 4 h, cryoprotected in 40% sucrose in PBS for at least 48 h and sectioned at 14 μm on a cryostat in a plane perpendicular to the skin surface and parallel to the long axis of the paw. The sections were mounted on gelatine-treated slides, blocked with 1% BSA + 5% sucrose and processed for immunostaining. The material was incubated overnight with anti-CB1, anti-CB2, anti-μ, anti-κ and anti-δ primary antibodies that were selective for the active conformation state of the receptors (Proteimax Biotechnology, Cotia, São Paulo, Brazil), which were labelled with fluorescent Alexa Fluor dyes (682 or 800 nm; Dyomics GmbH, Jena, Germany) and diluted 1:4000. The fluorescence analysis and quantification was performed using the Odyssey system (Li-Cor, Lincoln, NE, USA). The increase in receptor activation was evaluated by accounting for the total number of each receptor expressed in the same tissue using non-conformation-specific antibodies to the κ-opioid receptor-1 (N-19), μ-opioid receptor (N-terminus), δ-opioid receptor (N-terminus), CB1 receptor (N-terminus) and CB2 receptor (C-terminus), all of which were diluted 1:100 (Santa Cruz Biotechnology, Santa Cruz, CA, USA).
Measurement of the release of endogenous opioid peptides from rat skin tissue
Measurement of endogenous opioid peptide release was performed as previously described by Ibrahim et al. (2005). Crotalphine was dissolved in HBSS (1.26 mM CaCl2, 5.33 mM KCl, 0.44 mM KH2PO4, 0.5 mM MgCl2, 0.41 mM MgSO4, 138 mM NaCl, 4 mM NaHCO3, 0.3 mM Na2HPO4, 5.6 mM glucose, pH 7.4) containing 1% BSA. AM630 was dissolved in HBSS/BSA and 0.4% DMSO. Subsequent dilutions were made in HBSS/BSA to obtain the desired final concentration. After the animals were killed using 4% isoflurane inhalation, skin tissue from the plantar surface of the hind paw of both naïve rats and rats pretreated with PGE2 (100 ng per paw) for 2 h was quickly removed using a 4 mm punch and equilibrated for 30 min at 37°C in HBSS/BSA. Each skin sample was transferred to a 1.5 mL polypropylene tube containing 240 μL HBSS/BSA either with or without AM630 (10 μM). Five minutes later, 60 μL of crotalphine solution was added to achieve the desired final concentration. The following groups were incubated at a final volume of 300 μL at 37°C for 30 min with periodic gentle agitation to improve oxygenation: (i) HBSS/BSA alone; (ii) HBSS/BSA containing crotalphine (2.6 nM); (iii) HBSS/BSA containing crotalphine (1 μM); (iv) HBSS/BSA containing crotalphine (2.6 nM) + AM630 (10 μM); (v) HBSS/BSA containing crotalphine (1 μM) + AM630 (10 μM); and (vi) HBSS/BSA containing AM630 (10 μM). The supernatant was collected and kept on ice. β-Endorphin, dynorphin A and met-enkephalin levels in the supernatant were measured immediately using a commercially available enzyme immunoassay (EIA; Bachem, San Carlos, CA, USA).
Materials
AM1241, a CB2 receptor agonist, was purchased from Cayman Chemical (Ann Arbor, MI, USA). AM630, a CB2 receptor antagonist, was supplied by Tocris Bioscience (Ellisville, MO, USA). Anti-dynorphin A, anti-β-endorphin and anti-met-enkephalin antisera, as well as the β-endorphin EIA kit, met-enkephalin EIA kit and dynorphin A EIA kit, were obtained from Bachem. PGE2 was purchased from the Sigma Chemical Co. (St. Louis, MO, USA). Morphine sulphate was kindly provided by União Química (Embu Guaçu, São Paulo, Brazil). AM1241 and AM630 were dissolved in DMSO and then diluted in sterile saline and sterile distilled water, respectively, for injection into the rat paw. The percentage of DMSO in both final solutions was 6%, which was low enough to have no detectable effect in the assay. The anti-dynorphin A, anti-β-endorphin and anti-met-enkephalin antisera were diluted in sterile saline (0.85% NaCl solution). A stock solution of PGE2 was prepared by dissolving 500 μg PGE2 in 1 mL of 100% ethanol and then diluting it in sterile saline before injection into the rat paw. The percentage of ethanol in the solution injected in the hind paw was 0.2%.
Nomenclature
The drug and molecular target nomenclature conforms to the British Journal of Pharmacology's Concise Guide to PHARMACOLOGY (Alexander et al., 2013).
Data analysis
The results are expressed as mean ± SEM. Comparisons between pretreatment and post-treatment as well as between different treatment groups were carried out using the anova (Gad and Weil, 1989) followed by Tukey's post hoc test. The results were considered to be statistically significant when P < 0.05.
Results
Antinociceptive effect of crotalphine on PGE2-induced hyperalgesia
Our results demonstrated that p.o. (0.008–1.0 μg·kg−1; Figure 1A) and i.pl. (0.0006 μg per paw; Figure 1B) administration of crotalphine caused antinociception in PGE2-induced mechanical hyperalgesia (Figure 1A and B). The intensity of the increase in the nociceptive threshold of the animals treated with crotalphine was comparable with the increase induced by morphine (5 mg·kg−1, s.c.) (Figure 1A). After injection of PGE2 into both hind paws and crotalphine (0.0006 μg) into one hind paw, the antihyperalgesic effect of the peptide was detected only in the ipsilateral paw but not in the contralateral one (Figure 1B), indicating that at this dose, the peptide exerts a localized effect.
Figure 1.
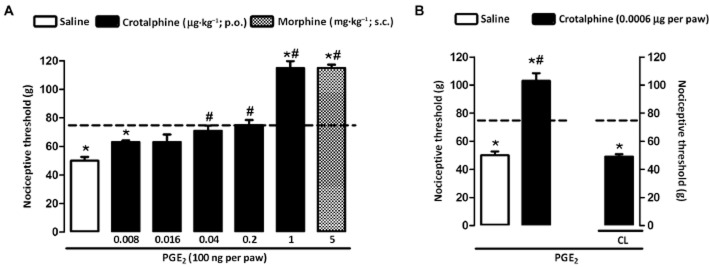
Antinociceptive effect of crotalphine on PGE2-induced hyperalgesia. Nociceptive threshold was obtained in the rat paw pressure test before (baseline – dotted line) and 3 h after PGE2 (100 ng per paw) injection. Crotalphine and saline were administered 1 h prior the threshold assessment. (A) Crotalphine (0.008, 0.016, 0.04, 0.2, 1.0 μg·kg−1) administered by p.o. route. Morphine (5 mg·kg−1), administered by s.c. route, was used as a control. (B) Crotalphine (0.0006 μg per paw) administered by i.pl. route. CL, contralateral paw. The results are expressed as means ± SEM (n = 5 animals for control group and 6 animals for experimental group). *P < 0.05 significantly different from values of the baseline measurement (baseline – dotted line); #P < 0.05 significantly different from values of the control group (saline).
CB2 cannabinoid receptors are involved in the antinociceptive effect of crotalphine
The antinociceptive effect of crotalphine administered by either p.o. (1 μg·kg−1; Figure 2A) or i.pl. routes (0.0006 μg; Figure 2B) was blocked by i.pl. injection of AM630, a selective CB2 cannabinoid receptor antagonist. The peripheral (local) effect of the antagonist was confirmed in experiments in which the CB2 cannabinoid receptor agonist AM1241 (5 μg per paw), used as positive control, was injected into both hind paws of the rats and the antagonist AM630 into one hind paw. The results demonstrated that AM630 abolishes the antinociceptive effect of the agonist in the ipsilateral paw but not in the contralateral one (Figure 2C).
Figure 2.

Participation of CB2 cannabinoid receptors in the antinociceptive effect of crotalphine. Nociceptive threshold was obtained in the rat paw pressure test before (baseline – dotted line) and 3 h after PGE2 (100 ng per paw) injection. (A) Crotalphine (1 μg·kg−1) was administered by p.o. route 1 h before the threshold assessment. (B) Crotalphine (0.0006 μg per paw) was administered by i.pl. route 1 h before the threshold assessment. (C) AM1241 (10 μg per paw) was administered by i.pl. route 1 h before the threshold assessment. AM630 (CB2 antagonist, 50 μg per paw, i.pl.) was administered 30 min before crotalphine (A and B) or AM1241 (C). CL, contralateral paw. The results are expressed as means ± SEM (n = 5 animals for control group and 6 animals for experimental group). *P < 0.05 compared with the values of the baseline measurement (dashed line); #P < 0.05 compared with the control group (saline).
Crotalphine induces peripheral activation of opioid and cannabinoid receptors
Because our results suggest the involvement of CB2 receptors in crotalphine-induced antinociceptive effect and previous data from our group showed that peripheral κ-opioid receptors are involved in the effect of the peptide in PGE2-induced hyperalgesia (Konno et al., 2008), we sought to determine whether crotalphine interferes with the activation state of either cannabinoid or opioid receptors. Paw tissue from animals treated with the peptide (0.0006 μg per paw, i.pl.) and PGE2 (100 ng per paw) was incubated with anti-CB1, anti-CB2, anti-μ, anti-κ and anti-δ receptor antibodies that recognize the activated state of the receptor. These antibodies are able to distinguish conformational changes in the N-terminus of CB1, CB2, μ-, κ-and δ-receptors following activation and are thus sensitive to the activity-mediated conformational states of their respective receptors (Gupta et al., 2007; Heimann et al., 2007; Supporting Information Figs S1–S5). The results showed that i.pl. injection of PGE2 did not alter the activation of CB1, CB2, κ-and δ-receptors but decreased the activation of μ-opioid receptors (Figure 3A and B). Intraplantar. injection of crotalphine in the same paw increased the activated state of both CB2 cannabinoid and κ-opioid receptors (Figure 3A and B). Crotalphine-induced activation of the κ-opioid receptors, but not CB2 receptors, was blocked by the κ-antagonist nor-BNI. Furthermore, both κ-opioid and CB2 receptor activation induced by crotalphine was blocked by the CB2 antagonist AM630 (Figure 3C and D), and crotalphine did not affect the decrease in the activation of μ-opioid receptors induced by PGE2 (Figure 3B). The fluorescence of the tissue obtained from naïve rats was taken as 100%, and the fluorescence of saline and crotalphine groups, acquired using the Odyssey system (Li-Cor), was represented as the percentage of activation in relation to the control group (Figure 3B). The results here presented were normalized to total level of opioid receptors.
Figure 3.

Crotalphine induces an increase in the activation of κ-opioid and CB2 cannabinoid receptors in skin tissue. PGE2 (100 ng per paw, i.pl.) and crotalphine (0.0006 μg per paw, i.pl.) were administered in the rat paw 3 and 1 h before the perfusion respectively. (A,B) The immunostaining technique was performed in slices of tissue from the rat paw using conformation state-sensitive anti-CB1, anti-CB2, anti-μ, anti-δ and anti-κ antibodies. (C,D) The immunostaining technique was performed in slice of tissue from the rat paw using conformation state-sensitive anti-CB2 and anti-κ antibodies, in the presence of nor-BNI or AM630, antagonists of κ and CB2 receptors, respectively, administered by i.pl. route 30 min before crotalphine. (A,C) Images of the sections. Analysis and quantification of fluorescence was performed using the Odyssey system (Li-Cor). (B,D) The fluorescence of the naïve rats was taken as 100% (dashed line), and the fluorescence of the saline and crotalphine groups was represented as percentage of activation compared with the control group. The control group (negative control) represents slices treated only with secondary antibody labelled with fluorescent Alexa Fluor dyes (682 and 800 nm). The results are expressed as means ± SEM (four tissue sections from each rat in each treatment group; n = 6 animals for control group and 7 animals for experimental group).*P < 0.05 compared with the control group measurement (dashed line).
Dynorphin A release is responsible for the antinociceptive effect of crotalphine
Our results demonstrated that both CB2 cannabinoid and κ-opioid receptors are activated after crotalphine injection. Based on data from the literature demonstrating that CB2 cannabinoid receptor activation produces antinociception by stimulating peripheral endogenous opioid release (Ibrahim et al., 2005), we sought to determine whether endogenous opioids could mediate the antinociceptive effect of crotalphine. Our results suggested that i.pl. administration of the anti-dynorphin A antibody prevented the effect of p.o. administered crotalphine (Figure 4C), whereas anti-β-endorphin and anti-met-enkephalin antibodies did not interfere with this effect (Figure 4A and B). The dose of the anti-dynorphin A antibody induced a localized effect because its injection abolished the antinociceptive effect of the peptide in the injected paw but not in the contralateral one (Figure 4C).
Figure 4.
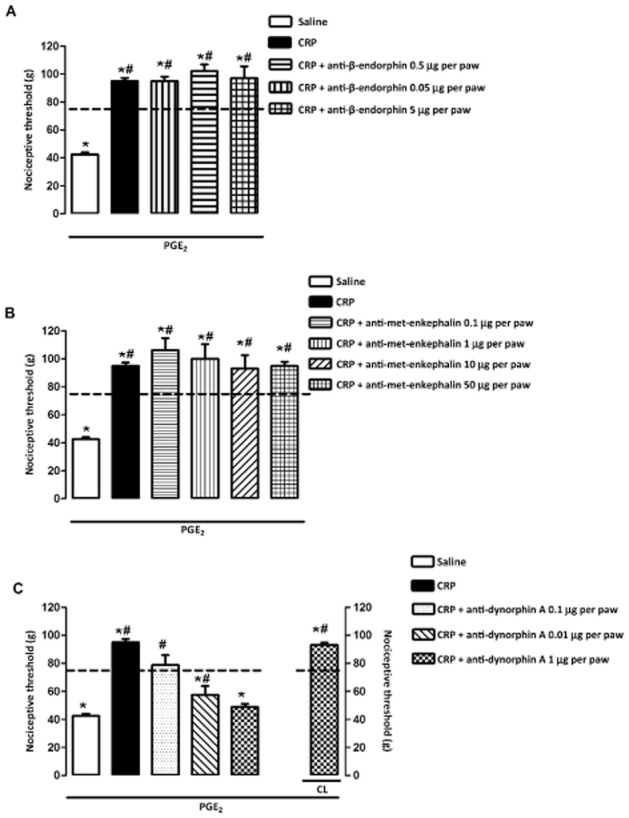
Participation of endogenous opioid in the antinociceptive effect of crotalphine on PGE2-induced hyperalgesia. Nociceptive threshold was obtained in the rat paw pressure test before (baseline – dotted line) and 3 h after PGE2 (100 ng per paw) injection. Crotalphine (CRP – 1 μg·kg−1, p.o.) was administered 1 h before the threshold assessment. Antibodies anti-β-endorphin (0.05, 0.5, 5.0 μg per paw, i.pl.; A), anti-met-enkephalin (0.1, 1, 10, 50 μg per paw, i.pl.; B) and anti-dynorphin A (0.01, 0.1, 1.0 μg per paw, i.pl.; C) were administered 15 min before crotalphine. The results are expressed as means ± SEM (n = 5 animals for control group and 6 animals for experimental group).*P < 0.05 compared with the values of the baseline measurement (dashed line); #P < 0.05 compared with the control group (saline).
Release of dynorphin A induced by crotalphine depends on CB2 receptor activation
Because crotalphine induces a peripheral (local) effect, we sought to determine whether the endogenous opioids involved in this effect could be released from the rat paw skin tissue. Tissues obtained from the paws of both naïve animals and rats pretreated with PGE2 were incubated with crotalphine, and the amounts of dynorphin A, β-endorphin and met-enkephalin released into the incubation medium were measured. Incubation of the tissue with two concentrations of crotalphine – 2.6 nM, which corresponds to a dose of 0.0006 μg per paw, and 1 μM, a concentration that activates the intracellular signalling cascade of mediators related to MAPKs in vitro (V.O. Zambelli, unpubl. data) – caused the release of dynorphin A from tissues obtained from naïve (Figure 5A) and PGE2 pretreated (Figure 5B) rats. The amount of dynorphin A released in the presence of PGE2 (Figure 5B) was significantly higher compared with the amount released by tissues obtained from naïve rats (Figure 5A). This release was prevented by treatment with AM630 (Figure 5).
Figure 5.
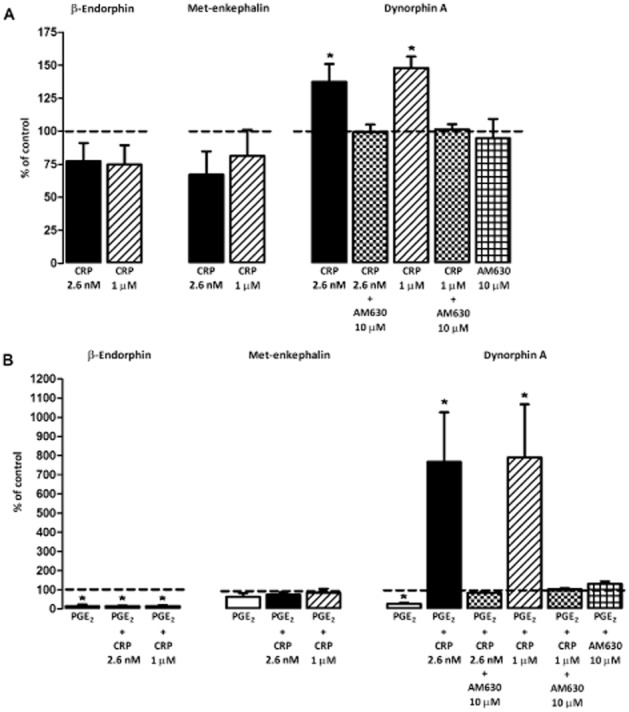
Release of endogenous opioid dynorphin A from rat paw skin tissue induced by crotalphine. For this in vitro assay, skin from the plantar surface of the hind paw of naïve (A) or PGE2 pretreated (100 μg per paw, 2 h before; B) rats were collected using a punch (4 mm in diameter to prepare skin samples of equivalent surface area) and placed in HBSS containing crotalphine (2.6 and 1.0 μM) or crotalphine (2.6 and 1.0 μM) plus AM630 (10 μM). β-Endorphin, met-enkephalin and dynorphin A content in the supernatant was measured using a commercially available EIA (Peninsula Laboratories). The endogenous opioid release from the control group (saline rats) was represented as 100% (baseline – dotted line), and the release of crotalphine and crotalphine + AM630 groups was represented as percentage of release in relation to control group. The results are expressed as the means ± SEM (n = 6 animals for control group and 7 animals for experimental group). *P < 0.05 compared with the control group (dashed line).
Discussion
The results presented herein demonstrate for the first time the involvement of the cannabinoid system in the antinociceptive effect of crotalphine. Our results clearly showed that (i) the antinociceptive effect of crotalphine is mediated by dynorphin A, and (ii) the peptide is able to release dynorphin A from rat paw skin tissue via a CB2 receptor-dependent mechanism. Taken together, these results suggest that CB2 receptor activation induced by crotalphine stimulates the release of endogenous dynorphin A from paw tissue, which then may act on κ-opioid receptors of primary afferent neurons to inhibit nociception.
Cannabinoids exert their analgesic effects acting in the brain via descending modulation, by a direct spinal action and/or by an action on the peripheral nerve (Zogopoulos et al., 2013). The cannabinoid GPCRs CB1 and CB2 are the best-studied molecular targets of cannabinoids, which bind to and activate them with different affinity (Basu and Dittel, 2011; Graham et al., 2009). CB1 is the most abundant GPCR found in the brain, being highly expressed in regions associated with emotion, cognition, memory, motor and executive function (such as cortex, limbic system, hippocampus, cerebellum, brainstem and several nuclei in the basal ganglia) (Ameri, 1999; Piomelli, 2003; Mackie, 2008; Svizenska et al., 2008) and in brain areas involved in nociceptive transmission and processing (including the periaqueductal grey, anterior cingulate cortex and thalamus, in addition to the dorsal horn of the spinal cord and dorsal root ganglion) (Herkenham et al., 1991; Farquhar-Smith et al., 2000; Zogopoulos et al., 2013). CB1 is also found in peripheral tissues, co-expressed with CB2, including immune cells, bone cells, adipose tissue, liver, renal tissue and skeletal muscle (Galiègue et al., 1995; Farquhar-Smith et al., 2000; Turu and Hunyady, 2010). CB2 is predominantly expressed in immune cells throughout the whole body (i.e. B lymphocytes, macrophages, NK cells), and they are also expressed in the myocardium, the human coronary endothelial cells, the smooth muscle cells, keratinocytes, adipocytes and the liver (Galiègue et al., 1995; Klein et al., 2003; Ashton et al., 2006; Zogopoulos et al., 2013). CB2 is also present at low levels in some areas of the brain (like perivascular microglial cells, astrocytes, cerebromicrovascular endothelial cells and in brainstem), where it is markedly activated upon insults (Carrier et al., 2004; Gong et al., 2006; Onaivi et al., 2006; Zogopoulos et al., 2013). Considering the predominant expression of CB2 receptors in the periphery and the previous demonstration of the endogenous opioid release induced by peripheral CB2 activation (Ibrahim et al., 2005; Su et al., 2011; Katsuyama et al., 2013), we focused this study on the involvement of CB2 receptors in the antinociceptive activity of crotalphine. Despite the data showing that the analgesic effect of crotalphine is completely reversed by the CB2 receptor antagonist, we cannot rule out the involvement of CB1 receptors in this mechanism.
Anyway, the results obtained in the assays using antibodies that are sensitive to active GPCR conformations demonstrated that the CB2, but not CB1 receptors, exert increased activity after administration of crotalphine in the PGE2-induced hyperalgesia. In addition to the activation of CB2 receptors, these assays also demonstrated an increased activation of κ-opioid receptors induced by crotalphine. These results are in agreement with previous data from our group where it was observed that κ-, but not μ-and δ-, opioid receptors are involved in the crotalphine antinociceptive effect in both PGE2-and carrageenan-induced hyperalgesia (Konno et al., 2008). It is important to note that PGE2, per se, did not alter the activation of both receptors but inhibited the activity of μ-opioid receptors. Several lines of evidence have indicated that during an inflammatory response, distinct peripheral nociceptive stimuli could activate and/or inhibit the transcription and/or expression of specific opioid receptors in nociceptive fibres (Stein, 1993; Antonijevic et al., 1995; Obara et al., 2009; Stein and Lang, 2009). The decrease in the activation of μ-opioid receptors induced by PGE2 was not altered by crotalphine.
The investigation of the possible interaction between opioid and cannabinoid systems clearly indicated that crotalphine-induced activation of κ-opioid receptors depends on CB2 receptor activation, as nor-BNI blocked crotalphine-induced κ-opioid receptor activation but not CB2 receptor activation, whereas the CB2 antagonist blocked crotalphine-induced activation of both the κ-opioid receptors and the CB2 receptors. These results suggest that crotalphine-induced κ-opioid receptor activation is subsequent to the CB2 cannabinoid receptor activation.
Several biochemical, molecular and pharmacological studies have demonstrated reciprocal interactions between cannabinoid and opioid systems, suggesting a common underlying mechanism (Cichewicz, 2004; Desroches and Beaulieu, 2010). In fact, the endogenous opioid system could be involved in cannabinoid antinociception, and the endogenous cannabinoid system could play a role in opioid antinociception (Smith et al., 1994; Pugh et al., 1996; Ibrahim et al., 2005; da Fonseca Pacheco et al., 2008; Welch, 2009; Desroches and Beaulieu, 2010). As CB2 and κ-opioid receptors are activated after crotalphine administration and because CB2 activation may induce the release of endogenous opioids (Ibrahim et al., 2005), we hypothesized that the antinociceptive effect of crotalphine could involve the release of endogenous opioids. This hypothesis was confirmed by the results indicating that dynorphin A is involved in the antinociceptive effect of crotalphine in the PGE2-induced hyperalgesia. We also demonstrated through ex vivo experiments that the action of crotalphine involves the release of dynorphin A from rat paw tissue, which is blocked by a CB2 receptor antagonist. These data may explain the involvement of the κ-opioid receptors in crotalphine-induced antinociceptive effect because dynorphin A is an endogenous agonist of these opioid receptors (Chavkin et al., 1982).
The release of endogenous opioids by cannabinoid receptor activation has been shown previously by Ibrahim et al. (2005). These authors demonstrated that CB2 receptor activation produces antinociception by stimulating β-endorphin release from keratinocytes, which, in turn, produces antinociception by acting on μ-opioid receptors. Our data suggest, for the first time, that activation of the CB2 receptors, which are present in skin tissue, may induce the local release of dynorphin A, and this release may occur in non-injured tissue. The results showing that the amount of β-endorphin and met-enkephalin released from the paw tissue was not altered by crotalphine support our previous data demonstrating that μ-and δ-opioid receptors are not involved in the antinociceptive action of crotalphine in PGE2-induced hyperalgesia (Konno et al., 2008).
The cell type involved in crotalphine-induced dynorphin A release has not been characterized. Both resident and migrated cells may release endogenous opioids after different types of stimuli (Cabot et al., 1997; 2001,; Cabot, 2001; Ibrahim et al., 2005; Sehgal et al., 2011). Aside from keratinocytes (Ibrahim et al., 2005), other components of skin such as resident immune and inflammatory cells may release opioid peptides. Immune cells, including T and B lymphocytes, granulocytes, monocytes and macrophages, are the major source of endogenous opioid ligands in the skin due to their abundance compared with resident keratinocytes and peripheral sensory neurons (Stein et al., 1990; Hassan et al., 1992; Schauer et al., 1994; Hadley and Haskell-Luevano, 1999; Cabot, 2001; Cabot et al., 2001). Further investigation is needed to determine the cells responsible for the release of dynorphin A by crotalphine.
In conclusion, our results demonstrated that the antinociceptive effect of crotalphine involves activation of peripheral CB2 receptors, which stimulates dynorphin A release from peripheral skin tissues. Dynorphin A, in turn, induces antinociception by acting on κ-opioid receptors expressed on primary afferent neurons. These results contribute to the understanding of the mechanisms involved in crotalphine-induced antinociception and corroborate the existence of a close connection between the opioid and cannabinoid systems in the control of pain.
Acknowledgments
The authors are grateful to Dr Lakshmi A. Devi (Mount Sinai School of Medicine, New York) for critical suggestions on these studies. We also express our gratitude to Dr Luiz Roberto G. de Britto for allowing us to perform the rat paw slices in his laboratory, and to Adilson da S. Alves and Miriam Aline Geigner for their technical assistance. This work was supported by funds from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, Brazil, Grant Nos. 2009/14203-5 and 2010/12917-8), Instituto Nacional de Ciencia e Tecnologia em Toxinologia (INCTTOX PROGRAM) of Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq)/FAPESP (Grant No. 2008/57898-0), and Fundação do Desenvolvimento Administrativo (FUNDAP).
Glossary
- AM1241
(3-iodo-5-nitrophenyl)-[1-[(1-methylpiperidin-2-yl)methyl]indol-3-yl]-methanone
- AM630
[6-iodo-2-methyl-1-[2-(4-morpholinyl)ethyl]-1H-indol-3-yl](4-methoxyphenyl)-methanone (also known as iodopravadoline)
- EIA
enzyme immunoassay; i.pl., intraplantar
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site: http://dx.doi.org/10.1111/bph.12488
Figure S1 SKNSH cells (human neuroblastoma) treated with (±)-U-50488 hydrochloride (a κ agonist). SKNSH cells (106 cells per well) endogenously expressing κ-receptors were treated with the indicated concentrations of (±)-U-50488 hydrochloride. Cells were incubated overnight at 4°C with 1 μg per well of antireceptor antibodies, then washed three times with PBS and incubated with a secondary antibody (dilution 1:500) of alkaline phosphatase-conjugated antirabbit antibodies for 60 min at room temperature. After washing three times with PBS, the amount of binding was detected using phosphatase substrate. The extent of receptor recognition by the antibodies was assayed by ELISA. Data from the vehicle-treated cells were taken as 100%. All of the results of the experiments are represented as the percentage of the control. The results are expressed as mean ± SEM (n = 3).
Figure S2 Rat spleen membranes treated with Δ9-THC. Rat spleen membranes (5 μg per well) were treated with 1 μM concentration of either Δ9-THC (CB2 agonist) or WIN 55212-3 (CB2 antagonist). Cells were incubated overnight at 4°C with 1 μg per well of antireceptor antibodies, washed three times with PBS and incubated with a secondary antibody (dilution 1:500) of alkaline phosphatase-conjugated anti-rabbit antibodies for 60 min at room temperature. After washing three times with PBS, the amount of binding was detected using phosphatase substrate. The extent of receptor recognition by the antibodies was assayed by ELISA. Data from the vehicletreated cells were taken as 100%. All of the results of the experiments are represented as the percentage of the control. The results are expressed as means ± SEM (n = 3).
Figure S3 In vivo assay for the κ-opioid receptor. Mice were injected with either saline or 10 mg·kg−1 of dynorphin A (i.p.) and killed 30 min after the injection. Brains were immediately collected and stored at −80°C until used. The immunostaining technique was performed in tissue slices from the mouse brain using conformation state-sensitive anti-κ antibodies. Primary antibodies were labelled with fluorescent dyes, and fluorescence was measured using Odyssey equipment from Li-Cor. (A) Images from brain sections representing striatum from the control and treated mice were probed with conformation-specific anti-κ-opioid receptor antibodies. (B) Data quantification from striatum-treated and control mice. Vehicle-treated mice were taken as 100%. The results are expressed as means ± SEM (n = 3).
Figure S4 HEK293 cells expressing the CB2 receptor treated with Δ9-THC. HEK293 cells expressing the CB2 receptor were treated with 1 μM concentration of agonist (Δ9-THC). The cells were incubated overnight at 4°C with 1 μg per well of anti-receptor antibodies, washed three times with PBS and incubated with a secondary antibody (dilution 1:500) of alkaline phosphatase-conjugated anti-rabbit antibodies for 60 min at room temperature. After washing three times with PBS, the amount of binding was detected using phosphatase substrate. The extent of receptor recognition by the antibodies was assayed by ELISA. Data from the vehicle-treated cells were taken as 100%. All of the results of the experiments are represented as a percentage of the control. The results are expressed as means ± SEM (n = 3).
Figure S5 Specificity of the conformation-sensitive receptor antibodies. Paw tissues from animals treated with either ACEA (CB1 agonist) or AM1241 (CB2 agonist) 2 h after PGE2 (100 ng per paw) injection were incubated with anti-CB1 and anti-CB2 conformation-sensitive receptor antibodies (Figure 5A). Paw tissues from animals treated with DAMGO (μ-agonist), U50.488 (κ-agonist) or DPDPE (Δ agonist) 2 h after PGE2 (100 ng per paw) injection were incubated with anti-μ, anti-κ and anti-Δ conformation-sensitive receptor antibodies (Figure 5B). The analysis and quantification of the fluorescence was performed using the Odyssey system (Li-Cor). The fluorescence of the naïve rats was taken as 100% (dashed line), and the fluorescence of the treated groups was represented as percentage of activation compared with the control group. This control group (negative control) represents slices treated only with secondary antibody labelled with fluorescent Alexa Fluor dyes (682 and 800 nm). The results are expressed as means ± SEM (n = 5 animals for control group and 6 animals for experimental group). *P < 0.05 compared with the control group measurement (dashed line).
Table S1 Cross-reactivity assay. SKNSH cells (human neuroblastoma) were treated with various agonists (1 μM). Cells were incubated overnight at 4°C with 1 μg per well of anti-receptor antibodies, washed three times with PBS and incubated with a secondary antibody (dilution 1:500) of alkaline phosphatase-conjugated anti-rabbit antibodies for 60 min at room temperature. After being washed three times with PBS, the amount of binding by the cells was detected by a phosphatase substrate. The extent of receptor recognition by the antibodies was assayed by ELISA. Data from the vehicle-treated cells were taken as 100%. The results are represented as the percentage of the control (buffer treatment). Data from the vehicle-treated cells were taken as 100%. The results are expressed as means ± SD (n = 3).
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, Spedding M, Peters JA, Harmar AJ, Collaborators CGTP. The Concise Guide to PHARMACOLOGY 2013/14: Overview. Br J Pharmacol. 2013;170:1449–1867. [Google Scholar]
- Ameri A. The effects of cannabinoids on the brain. Prog Neurobiol. 1999;58:315–348. doi: 10.1016/s0301-0082(98)00087-2. [DOI] [PubMed] [Google Scholar]
- Anand P, Whiteside G, Fowler CJ, Hohmann AG. Targeting CB2 receptors and the endocannabinoid system for the treatment of pain. Brain Res Rev. 2009;60:255–266. doi: 10.1016/j.brainresrev.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonijevic I, Mousa SA, Schafer M, Stein C. Perineurial defect and peripheral opioid analgesia in inflammation. J Neurosci. 1995;15:165–172. doi: 10.1523/JNEUROSCI.15-01-00165.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton JC, Friberg D, Darlington CL, Smith PF. Expression of the cannabinoid CB2 receptor in the rat cerebellum: an immunohistochemical study. Neurosci Lett. 2006;396:113–116. doi: 10.1016/j.neulet.2005.11.038. [DOI] [PubMed] [Google Scholar]
- Basu S, Dittel BN. Unraveling the complexities of cannabinoid receptor 2 (CB2) immune regulation in health and disease. Immunol Res. 2011;51:26–38. doi: 10.1007/s12026-011-8210-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabot PJ. Immune-derived opioids and peripheral antinociception. Clin Exp Pharmacol Physiol. 2001;28:230–232. doi: 10.1046/j.1440-1681.2001.03425.x. [DOI] [PubMed] [Google Scholar]
- Cabot PJ, Carter L, Gaiddon C, Zhang Q, Schafer M, Loeffler JP, et al. Immune cell-derived beta-endorphin. Production, release, and control of inflammatory pain in rats. J Clin Invest. 1997;100:142–148. doi: 10.1172/JCI119506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabot PJ, Carter L, Schafer M, Stein C. Methionine-enkephalin-and Dynorphin A-release from immune cells and control of inflammatory pain. Pain. 2001;93:207–212. doi: 10.1016/S0304-3959(01)00322-0. [DOI] [PubMed] [Google Scholar]
- Carrier EJ, Kearn CS, Barkmeier AJ, Breese NM, Yang W, Nithipatikom K, et al. Cultured rat microglial cells synthesize the endocannabinoid 2-arachidonylglycerol, which increases proliferation via a CB2 receptor-dependent mechanism. Mol Pharmacol. 2004;65:999–1007. doi: 10.1124/mol.65.4.999. [DOI] [PubMed] [Google Scholar]
- Chavkin C, James IF, Goldstein A. Dynorphin is a specific endogenous ligand of the kappa opioid receptor. Science. 1982;215:413–415. doi: 10.1126/science.6120570. [DOI] [PubMed] [Google Scholar]
- Cichewicz DL. Synergistic interactions between cannabinoid and opioid analgesics. Life Sci. 2004;74:1317–1324. doi: 10.1016/j.lfs.2003.09.038. [DOI] [PubMed] [Google Scholar]
- Desroches J, Beaulieu P. Opioids and cannabinoids interactions: involvement in pain management. Curr Drug Targets. 2010;11:462–473. doi: 10.2174/138945010790980303. [DOI] [PubMed] [Google Scholar]
- Farquhar-Smith WP, Egertova M, Bradbury EJ, McMahon SB, Rice AS, Elphick MR. Cannabinoid CB(1) receptor expression in rat spinal cord. Mol Cell Neurosci. 2000;15:510–521. doi: 10.1006/mcne.2000.0844. [DOI] [PubMed] [Google Scholar]
- da Fonseca Pacheco D, Klein A, de Castro Perez A, da Fonseca Pacheco CM, de Francischi JN, Duarte ID. The mu-opioid receptor agonist morphine, but not agonists at delta-or kappa-opioid receptors, induces peripheral antinociception mediated by cannabinoid receptors. Br J Pharmacol. 2008;154:1143–1149. doi: 10.1038/bjp.2008.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gad S, Weil C. Statistics for toxinologists. In: Hayer A, editor. Principles and Methods of Toxicology. New York: Raven Press Ltd; 1989. pp. 435–483. [Google Scholar]
- Galiègue S, Mary S, Marchand J, Dussossoy D, Carrière D, Carayon P, et al. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem. 1995;232:54–61. doi: 10.1111/j.1432-1033.1995.tb20780.x. [DOI] [PubMed] [Google Scholar]
- Gong JP, Onaivi ES, Ishiguro H, Liu QR, Tagliaferro PA, Brusco A. Cannabinoid CB2 receptors: immunohistochemical localization in rat brain. Brain Res. 2006;1071:10–23. doi: 10.1016/j.brainres.2005.11.035. [DOI] [PubMed] [Google Scholar]
- Graham ES, Ashton JC, Glass M. Cannabinoid receptors: a brief history and ‘what's hot’. Front Biosci. 2009;14:944–957. doi: 10.2741/3288. [DOI] [PubMed] [Google Scholar]
- Gupta A, Decaillot FM, Gomes I, Tkalych O, Heimann AS, Ferro ES, et al. Conformation state-sensitive antibodies to G-protein-coupled receptors. J Biol Chem. 2007;282:5116–5124. doi: 10.1074/jbc.M609254200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez VP, Konno K, Chacur M, Sampaio SC, Picolo G, Brigatte P, et al. Crotalphine induces potent antinociception in neuropathic pain by acting at peripheral opioid receptors. Eur J Pharmacol. 2008;594:84–92. doi: 10.1016/j.ejphar.2008.07.053. [DOI] [PubMed] [Google Scholar]
- Gutierrez VP, Zambelli VO, Picolo G, Chacur M, Sampaio SC, Brigatte P, et al. The peripheral L-arginine-nitric oxide-cyclic GMP pathway and ATP-sensitive K+ channels are involved in the antinociceptive effect of crotalphine on neuropathic pain in rats. Behav Pharmacol. 2012;23:14–24. doi: 10.1097/FBP.0b013e32834eafbc. [DOI] [PubMed] [Google Scholar]
- Hadley ME, Haskell-Luevano C. The proopiomelanocortin system. Ann N Y Acad Sci. 1999;885:1–21. doi: 10.1111/j.1749-6632.1999.tb08662.x. [DOI] [PubMed] [Google Scholar]
- Hassan AH, Pzewlocki R, Herz A, Stein C. Dynorphin, a preferential ligand for kappa-opioid receptors, is present in nerve fibers and immune cells within inflamed tissue of the rat. Neurosci Lett. 1992;140:85–88. doi: 10.1016/0304-3940(92)90688-4. [DOI] [PubMed] [Google Scholar]
- Heimann AS, Gomes I, Dale CS, Pagano RL, Gupta A, de Souza LL, et al. Hemopressin is an inverse agonist of CB1 cannabinoid receptors. Proc Natl Acad Sci U S A. 2007;104:20588–20593. doi: 10.1073/pnas.0706980105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC. Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci. 1991;11:563–583. doi: 10.1523/JNEUROSCI.11-02-00563.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim MM, Porreca F, Lai J, Albrecht PJ, Rice FL, Khodorova A, et al. CB2 cannabinoid receptor activation produces antinociception by stimulating peripheral release of endogenous opioids. Proc Natl Acad Sci U S A. 2005;102:3093–3098. doi: 10.1073/pnas.0409888102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuyama S, Mizoguchi H, Kuwahata H, Komatsu T, Nagaoka K, Nakamura H, et al. Involvement of peripheral cannabinoid and opioid receptors in β-caryophyllene-induced antinociception. Eur J Pain. 2013;17:664–675. doi: 10.1002/j.1532-2149.2012.00242.x. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein TW, Newton C, Larsen K, Lu L, Perkins I, Nong L, et al. The cannabinoid system and immune modulation. J Leukoc Biol. 2003;74:486–496. doi: 10.1189/jlb.0303101. [DOI] [PubMed] [Google Scholar]
- Konno K, Picolo G, Gutierrez VP, Brigatte P, Zambelli VO, Camargo AC, et al. Crotalphine, a novel potent analgesic peptide from the venom of the South American rattlesnake Crotalus durissus terrificus. Peptides. 2008;29:1293–1304. doi: 10.1016/j.peptides.2008.04.003. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie K. Signaling via CNS cannabinoid receptors. Mol Cell Endocrinol. 2008;286:S60–S65. doi: 10.1016/j.mce.2008.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obara I, Parkitna JR, Korostynski M, Makuch W, Kaminska D, Przewlocka B, et al. Local peripheral opioid effects and expression of opioid genes in the spinal cord and dorsal root ganglia in neuropathic and inflammatory pain. Pain. 2009;141:283–291. doi: 10.1016/j.pain.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Onaivi ES, Ishiguro H, Gong JP, Patel S, Perchuk A, Meozzi PA, et al. Discovery of the presence and functional expression of cannabinoid CB2 receptors in brain. Ann N Y Acad Sci. 2006;1074:514–536. doi: 10.1196/annals.1369.052. [DOI] [PubMed] [Google Scholar]
- Parolaro D, Rubino T, Vigano D, Massi P, Guidali C, Realini N. Cellular mechanisms underlying the interaction between cannabinoid and opioid system. Curr Drug Targets. 2011;11:393–405. doi: 10.2174/138945010790980367. [DOI] [PubMed] [Google Scholar]
- Picolo G, Cury Y. Peripheral neuronal nitric oxide synthase activity mediates the antinociceptive effect of Crotalus durissus terrificus snake venom, a delta-and kappa-opioid receptor agonist. Life Sci. 2004;75:559–573. doi: 10.1016/j.lfs.2003.12.024. [DOI] [PubMed] [Google Scholar]
- Picolo G, Giorgi R, Cury Y. delta-opioid receptors and nitric oxide mediate the analgesic effect of Crotalus durissus terrificus snake venom. Eur J Pharmacol. 2000;391:55–62. doi: 10.1016/s0014-2999(99)00934-6. [DOI] [PubMed] [Google Scholar]
- Piomelli D. The molecular logic of endocannabinoid signalling. Nat Rev Neurosci. 2003;4:873–884. doi: 10.1038/nrn1247. [DOI] [PubMed] [Google Scholar]
- Pugh G, Jr, Smith PB, Dombrowski DS, Welch SP. The role of endogenous opioids in enhancing the antinociception produced by the combination of delta 9-tetrahydrocannabinol and morphine in the spinal cord. J Pharmacol Exp Ther. 1996;279:608–616. [PubMed] [Google Scholar]
- Randall LO, Selitto JJ. A method for measurement of analgesia activity on inflamed tissue. Arch Inst Pharmacodyn. 1957;111:209–219. [PubMed] [Google Scholar]
- Schauer E, Trautinger F, Kock A, Schwarz A, Bhardwaj R, Simon M, et al. Proopiomelanocortin-derived peptides are synthesized and released by human keratinocytes. J Clin Invest. 1994;93:2258–2262. doi: 10.1172/JCI117224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehgal N, Smith H, Manchikanti L. Peripherally acting opioids and clinical implications for pain control. Pain Physician. 2011;14:249–258. [PubMed] [Google Scholar]
- Smith PB, Welch SP, Martin BR. Interactions between delta 9-tetrahydrocannabinol and kappa opioids in mice. J Pharmacol Exp Ther. 1994;268:1381–1387. [PubMed] [Google Scholar]
- Stein C. Peripheral mechanisms of opioid analgesia. Anesth Analg. 1993;76:182–191. doi: 10.1213/00000539-199301000-00031. [DOI] [PubMed] [Google Scholar]
- Stein C, Lang LJ. Peripheral mechanisms of opioid analgesia. Curr Opin Pharmacol. 2009;9:3–8. doi: 10.1016/j.coph.2008.12.009. [DOI] [PubMed] [Google Scholar]
- Stein C, Hassan AH, Przewlocki R, Gramsch C, Peter K, Herz A. Opioids from immunocytes interact with receptors on sensory nerves to inhibit nociception in inflammation. Proc Natl Acad Sci U S A. 1990;87:5935–5939. doi: 10.1073/pnas.87.15.5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su TF, Zhang LH, Peng M, Wu CH, Pan W, Tian B, et al. Cannabinoid CB2 receptors contribute to upregulation of β-endorphin in inflamed skin tissues by electroacupuncture. Mol Pain. 2011;7:98. doi: 10.1186/1744-8069-7-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svizenska I, Dubovy P, Sulcova A. Cannabinoid receptors 1 and 2 (CB1 and CB2), their distribution, ligands and functional involvement in nervous system structures – a short review. Pharmacol Biochem Behav. 2008;90:501–511. doi: 10.1016/j.pbb.2008.05.010. [DOI] [PubMed] [Google Scholar]
- Turu G, Hunyady L. Signal transduction of the CB1 cannabinoid receptor. J Mol Endocrinol. 2010;44:75–85. doi: 10.1677/JME-08-0190. [DOI] [PubMed] [Google Scholar]
- Welch SP. Interaction of the cannabinoid and opioid systems in the modulation of nociception. Int Rev Psychiatry. 2009;21:143–151. doi: 10.1080/09540260902782794. [DOI] [PubMed] [Google Scholar]
- Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]
- Zogopoulos P, Vasileiou I, Patsouris E, Theocharis SE. The role of endocannabinoids in pain modulation. Fundam Clin Pharmacol. 2013;27:64–80. doi: 10.1111/fcp.12008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 SKNSH cells (human neuroblastoma) treated with (±)-U-50488 hydrochloride (a κ agonist). SKNSH cells (106 cells per well) endogenously expressing κ-receptors were treated with the indicated concentrations of (±)-U-50488 hydrochloride. Cells were incubated overnight at 4°C with 1 μg per well of antireceptor antibodies, then washed three times with PBS and incubated with a secondary antibody (dilution 1:500) of alkaline phosphatase-conjugated antirabbit antibodies for 60 min at room temperature. After washing three times with PBS, the amount of binding was detected using phosphatase substrate. The extent of receptor recognition by the antibodies was assayed by ELISA. Data from the vehicle-treated cells were taken as 100%. All of the results of the experiments are represented as the percentage of the control. The results are expressed as mean ± SEM (n = 3).
Figure S2 Rat spleen membranes treated with Δ9-THC. Rat spleen membranes (5 μg per well) were treated with 1 μM concentration of either Δ9-THC (CB2 agonist) or WIN 55212-3 (CB2 antagonist). Cells were incubated overnight at 4°C with 1 μg per well of antireceptor antibodies, washed three times with PBS and incubated with a secondary antibody (dilution 1:500) of alkaline phosphatase-conjugated anti-rabbit antibodies for 60 min at room temperature. After washing three times with PBS, the amount of binding was detected using phosphatase substrate. The extent of receptor recognition by the antibodies was assayed by ELISA. Data from the vehicletreated cells were taken as 100%. All of the results of the experiments are represented as the percentage of the control. The results are expressed as means ± SEM (n = 3).
Figure S3 In vivo assay for the κ-opioid receptor. Mice were injected with either saline or 10 mg·kg−1 of dynorphin A (i.p.) and killed 30 min after the injection. Brains were immediately collected and stored at −80°C until used. The immunostaining technique was performed in tissue slices from the mouse brain using conformation state-sensitive anti-κ antibodies. Primary antibodies were labelled with fluorescent dyes, and fluorescence was measured using Odyssey equipment from Li-Cor. (A) Images from brain sections representing striatum from the control and treated mice were probed with conformation-specific anti-κ-opioid receptor antibodies. (B) Data quantification from striatum-treated and control mice. Vehicle-treated mice were taken as 100%. The results are expressed as means ± SEM (n = 3).
Figure S4 HEK293 cells expressing the CB2 receptor treated with Δ9-THC. HEK293 cells expressing the CB2 receptor were treated with 1 μM concentration of agonist (Δ9-THC). The cells were incubated overnight at 4°C with 1 μg per well of anti-receptor antibodies, washed three times with PBS and incubated with a secondary antibody (dilution 1:500) of alkaline phosphatase-conjugated anti-rabbit antibodies for 60 min at room temperature. After washing three times with PBS, the amount of binding was detected using phosphatase substrate. The extent of receptor recognition by the antibodies was assayed by ELISA. Data from the vehicle-treated cells were taken as 100%. All of the results of the experiments are represented as a percentage of the control. The results are expressed as means ± SEM (n = 3).
Figure S5 Specificity of the conformation-sensitive receptor antibodies. Paw tissues from animals treated with either ACEA (CB1 agonist) or AM1241 (CB2 agonist) 2 h after PGE2 (100 ng per paw) injection were incubated with anti-CB1 and anti-CB2 conformation-sensitive receptor antibodies (Figure 5A). Paw tissues from animals treated with DAMGO (μ-agonist), U50.488 (κ-agonist) or DPDPE (Δ agonist) 2 h after PGE2 (100 ng per paw) injection were incubated with anti-μ, anti-κ and anti-Δ conformation-sensitive receptor antibodies (Figure 5B). The analysis and quantification of the fluorescence was performed using the Odyssey system (Li-Cor). The fluorescence of the naïve rats was taken as 100% (dashed line), and the fluorescence of the treated groups was represented as percentage of activation compared with the control group. This control group (negative control) represents slices treated only with secondary antibody labelled with fluorescent Alexa Fluor dyes (682 and 800 nm). The results are expressed as means ± SEM (n = 5 animals for control group and 6 animals for experimental group). *P < 0.05 compared with the control group measurement (dashed line).
Table S1 Cross-reactivity assay. SKNSH cells (human neuroblastoma) were treated with various agonists (1 μM). Cells were incubated overnight at 4°C with 1 μg per well of anti-receptor antibodies, washed three times with PBS and incubated with a secondary antibody (dilution 1:500) of alkaline phosphatase-conjugated anti-rabbit antibodies for 60 min at room temperature. After being washed three times with PBS, the amount of binding by the cells was detected by a phosphatase substrate. The extent of receptor recognition by the antibodies was assayed by ELISA. Data from the vehicle-treated cells were taken as 100%. The results are represented as the percentage of the control (buffer treatment). Data from the vehicle-treated cells were taken as 100%. The results are expressed as means ± SD (n = 3).