

It has been observed that there is an imperative to develop predictive human systems that more effectively study and/or prevent the onset of liver disease and decompensated organ function. A serum-free, scalable, and shippable cell-based model that faithfully predicts the potential for human liver injury has been developed. Such a resource has direct application in human modeling, and, in the future, could play an important role in developing renewable cell-based therapies.
Keywords: Human embryonic stem cells, iPSC, Cytochrome P450, Drug metabolism, Drug toxicity, Liver
Abstract
Despite major progress in the knowledge and management of human liver injury, there are millions of people suffering from chronic liver disease. Currently, the only cure for end-stage liver disease is orthotopic liver transplantation; however, this approach is severely limited by organ donation. Alternative approaches to restoring liver function have therefore been pursued, including the use of somatic and stem cell populations. Although such approaches are essential in developing scalable treatments, there is also an imperative to develop predictive human systems that more effectively study and/or prevent the onset of liver disease and decompensated organ function. We used a renewable human stem cell resource, from defined genetic backgrounds, and drove them through developmental intermediates to yield highly active, drug-inducible, and predictive human hepatocyte populations. Most importantly, stem cell-derived hepatocytes displayed equivalence to primary adult hepatocytes, following incubation with known hepatotoxins. In summary, we have developed a serum-free, scalable, and shippable cell-based model that faithfully predicts the potential for human liver injury. Such a resource has direct application in human modeling and, in the future, could play an important role in developing renewable cell-based therapies.
Introduction
There are millions of people suffering from chronic liver disease worldwide. This is highlighted in the United Kingdom, where liver disease is the fifth most common cause of death and on the rise [1]. Currently, the only cure for end-stage liver disease is orthotopic liver transplantation. Although successful, liver transplantation is severely limited by the number of donors [2, 3]. This has led scientists, physicians, and surgeons to search for alternative therapies. Recently, liver specialists at King’s College Hospital successfully pioneered a technique to encapsulate human hepatocytes in alginate before infusion into the peritoneal cavity [4]. In addition, recent studies have shown that macrophages and neutrophils play an essential role in tissue remodeling during recovery from liver injury and represent tractable targets [5–7].
There are significant morbidity, mortality, and economic burden associated with human liver disease. Therefore, developing new systems that improve the study and prediction of liver disease is likely to lead to more effective intervention in the future. One such example is the development of simple cell-based models that closely reflect human physiology. In the future, such models will undoubtedly offer new insight into liver diseases, idiosyncrasies, and the influence of patient genetics (reviewed in [8]). Although current models will still have an important role to play, the development of human liver cells, from renewable and genetically defined origins, is likely to be a game-changing addition to the field.
For new models to have significant impact in the field, they must be derived from renewable cell populations and delivered at scale. For this reason, we have used pluripotent stem cells (PSCs) to model human liver biology. Human embryonic stem cells (hESCs) and induced pluripotent stem cells (iPSCs) are examples of renewable cell populations [9, 10]. Both populations have been shown to efficiently differentiate to hepatocytes by either spontaneous or directed differentiation and, therefore, offer significant promise in the quest to deliver a new wave of predictive human models [11–19]. In this vein, we have developed highly efficient, scalable, and serum-free differentiation procedures that are compatible with pluripotent stem cells. Importantly, the cell populations derived from these procedures are shippable and display high level of accuracy in predicting human drug-induced liver injury. We believe that such a resource has direct application in modeling human biology “in a dish” and, in the future, could aid the delivery of renewable cell-based therapies.
Materials and Methods
Cell Culture and Differentiation
hESCs (H9) and iPSCs (33D6) were cultured as described [16, 18–20] and maintained in a humidified 37°C, 5% CO2 incubator. At day 9, differentiating cells from hESCs and iPSCs were cultured in hepatocyte maturation medium HepatoZYME (Life Technologies, Carlsbad, CA, http://www.lifetech.com) containing 1% Glutamax (Life Technologies), supplemented with 10 μM hydrocortisone 21-hemisuccinate (Sigma-Aldrich, St. Louis, MO, http://www.sigmaaldrich.com), 10 ng/ml hepatocyte growth factor (Peprotech, Rocky Hill, NJ, http://www.peprotech.com), and 20 ng/ml oncostatin M (Peprotech). Cryoplateable human hepatocytes were used as the gold standard comparison and purchased from Celsis/In Vitro Technologies (catalog no. F00995, lot 124). Primary hepatocytes were plated per vendor’s instructions in InVitroGRO HI medium (Celsis, Baltimore, MD, http://www.celsis.com) plus 10% fetal bovine serum (Gibco, Grand Island, NY, http://www.invitrogen.com) on 96-well collagen I-coated plates (BD Biosciences, San Diego, CA, http://www.bdbiosciences.com) at a concentration of 20,000 cells per well 24 hours before compound addition and maintained in a humidified 37°C, 5% CO2 incubator.
Western Blot
A total of 4 × 105 cells from each time point during differentiation was lysed in 150 µl of SUMO buffer (2% SDS, 50 mM Tris-HCl, pH 8, 1 mM ethylenediaminetetraacetic acid [EDTA], and 10 mM iodoacetamide [all from Sigma-Aldrich]). Protein concentrations were estimated using the standard bicinchoninic acid (BCA) assay (Pierce, Rockford, IL, http://www.piercenet.com). A quantity amounting to 10 µg of protein was then used for Western blotting studies. The lysates were separated in 4%–12% Bis-Tris gradient gel. The XCell SureLock Mini-Cell system from Novex (Life Technologies) was used, as per the manufacturer’s instructions. The membranes were blocked for 1 hour at room temperature with phosphate-buffered saline (PBS)/0.1% Tween 20/10% nonfat milk. Following blocking, the membranes were probed with primary antibody (diluted in PBS/0.1%Tween/10% milk) at 4°C overnight under constant rotation. Following primary antibody incubation, membranes were washed three times in PBS/0.1% Tween. The secondary antibody was then incubated for 1 hour at room temperature and washed three times in PBS/0.1% Tween. Enhanced chemiluminescence (Pierce) was used to detect immobilized complexes. The primary and secondary antibodies are listed in supplemental online Table 1.
RNA Isolation and Quantitative Polymerase Chain Reaction
Total RNA was isolated from cells using TRIzol reagent (Life Technologies). RNA quality was assessed by NanoDrop (Thermo Scientific, Wilmington, DE, http://www.nanodrop.com). Total RNA was reverse transcribed from each time point using Superscript III reverse-transcription kit, as per manufacturer’s instructions (Life Technologies). Quantitative polymerase chain reaction (qPCR) was carried out using TaqMan Fast Advance Master Mix and appropriate primers (Applied Biosystems, Foster City, CA, http://www.appliedbiosystems.com). The primers are listed in supplemental online Table 2. The samples were analyzed using Roche LightCycler 480 Real-Time PCR System. Results were normalized to β-2 microglobulin and expressed as relative expression over the control sample (hESC or iPSC). The qPCRs were run in triplicate. Data analysis was performed using Roche LightCycler 480 software (version 1.5). Levels of significance were measured by Student’s t test.
Flow Cytometry
Single-cell populations were prepared from days 0 to 6 of the differentiation process and resuspended in PBS supplemented with 0.1% bovine serum albumin (BSA) and 0.1% sodium azide. The cells were counted and diluted to a concentration of 1 × 107 cells per milliliter. A total of 1 × 106 cells was incubated for 30 minutes at 4°C with conjugated antibodies to TRA-1-60 (eBioscience, San Diego, CA, http://www.ebioscience.com; 1:50 dilution) and CXCR4 (eBioscience; 1:20 dilution). Cells were washed extensively to remove any unbound antibody, before analysis. Data for 100,000 live events were acquired for each sample using a FACSCalibur cytometer.
Immunofluorescence
Cell cultures at different points during cellular differentiation were fixed in 100% ice-cold methanol at −20°C for 30 minutes. Postfixation, cell monolayers were washed twice with PBS at room temperature. After blocking with 0.1% PBS-Tween containing 10% BSA for 1 hour, the cells were incubated with primary antibodies diluted in PBS-0.1% Tween/1% BSA at 4°C overnight. The following day, the primary antibody was removed, and the fixed monolayers were washed three times with PBS-0.1% Tween/1% BSA. Following this, the cells were incubated with the appropriate secondary antibody diluted in PBS-0.1% Tween/1% BSA for 1 hour at room temperature and washed three times, as before. Cultures were subsequently mounted with Mowiol 4-88 (Calbiochem, San Diego, CA, http://www.emdbiosciences.com) containing glycerol (Fisher Chemicals, Leicestershire, U.K.), 0.2 M [pH 8] Tris (Sigma-Aldrich), and 4′,6-diamidino-2-phenylindole (1:1,000) (Sigma-Aldrich) overnight at 4°C. The cells were analyzed by Olympus TH4-200 microscope and Volocity 4 software. The percentage of positive cells and standard deviation were estimated from at least four random fields of view. The primary and secondary antibodies are listed in supplemental online Table 1.
Cytochrome p450 (CYP3A and CYP1A2) Functional Assay
CYP3A and CYP1A2 activity were measured by pGlo kit from Promega (Madison, WI, http://www.promega.com) and carried out as per the manufacturer's instructions for nonlytic CYP450 activity estimation (http://http://www.promega.com/tbs/tb325/tb325.pdf). CYP activities are expressed as relative light units (RLU) per milligram of protein (BCA assay). Cultures were incubated in the presence and absence of the CYP3A inducer, phenobarbital (PB), at 1 mM concentration for 48 hours before measurement of CYP3A and CYP1A2 activity. Levels of significance were measured by Student’s t test, where p < .01 is denoted as **, n = 3.
Compound Incubation
Compounds were dissolved in Hybri-Max dimethyl sulfoxide (DMSO) from Sigma-Aldrich. The 20 mM stock solutions were made and further diluted in DMSO before being added to HepatoZYME culture medium (hESC-derived hepatocytes) or InVitroGRO HI medium (cryoplateable hepatocytes), so that a consistent final concentration of 1% DMSO was maintained. Medium was aspirated from 96-well plates and replaced with 100 µl of medium containing the appropriate concentration of compound (vehicle control, 0.1, 1, 10, 25, 50, 100, or 200 µM) in triplicate. Plates were placed in a humidified 37°C, 5% CO2 incubator for 6 hours or 24 hours for the Apo-ONE Homogeneous Caspase-3/7 Assay, and 24 hours, 4 days, or 7 days for the CellTiter-Glo Luminescent Cell Viability Assay. Old medium was aspirated, and fresh medium-containing compound was added to the remaining plates on day 4.
Caspase-3/7 Assay
Caspase 3/7 levels were measured using the Apo-ONE Homogeneous Caspase-3/7 Assay kit from Promega and carried out as per the manufacturer's instructions for cells in culture. Fluorescence was detected using an EnVision plate reader (PerkinElmer, Waltham, MA, http://www.perkinelmer.com) and expressed as a percentage of vehicle control.
Cell Viability Assay
The CellTiter-Glo Luminescent Cell Viability Assay from Promega was used to measure ATP levels and carried out as per the manufacturer’s instructions for cells in culture. Luminescence was detected using an EnVision plate reader (PerkinElmer) and expressed as a percentage of vehicle control.
Analysis of Results
Concentration-response curves were plotted using the mean of the three replicates for each of the cell lines, and the concentration corresponding to a 50% viability on the four-parameter logistic regression line (50% inhibition/inhibitory concentration [IC50]) was determined using XLfit model 205.
Results
Driving Hepatocellular Differentiation Through Important Developmental Intermediates Using a Serum-Free Process
To deliver cell-based models at scale, we developed a serum-free approach to hepatocellular differentiation. During differentiation, hESCs underwent a series of profound morphological changes (Fig. 1). hESC morphology (Fig. 1Aa) changed as cells exited pluripotency and transited through definitive endoderm (Fig. 1Ab) and upon hepatic specification (Fig. 1Ac, 1Ad). In line with changes in cell morphology, we observed changes in gene expression throughout cellular differentiation. At 3 days postdifferentiation, hESCs had committed to a population of cells reminiscent of the definitive endoderm. In support of this, we detected significant decreases in pluripotent gene transcription (octamer 4 [Oct4], p < .0001 and Nanog, p < .01) and increases in definitive endoderm marker gene expression (SRY-box containing gene 17 [Sox17], p < .001 and Nodal, p < .001; CXCR4, p < .001) (Fig. 1B). This was highly efficient with ∼93% and ∼84% of cells expressing Sox17 and Forkhead box protein 1 (Foxa1) (Fig. 2).
Figure 1.
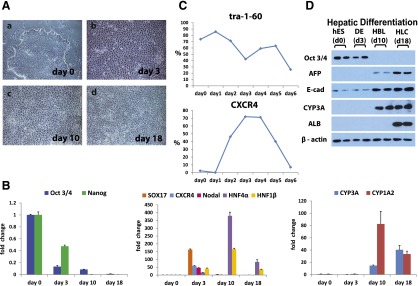
Driving efficient hepatocellular differentiation using a serum-free system. Human embryonic stem cells were differentiated in a stagewise fashion toward the hepatocyte lineage. (A): Phase-contrast imaging demonstrated the cells were undergoing sequential morphological changes during transit from stem cell (day 0) (Aa) to definitive endoderm (day 3) (Ab) to hepatoblast (day 10) (Ac) to hepatocyte (day 18) (Ad). (B): The quantitative polymerase chain reaction (qPCR) gene expression analysis demonstrated that changes in morphology were paralleled by changes in pluripotent (Oct4, Nanog), definitive endoderm (Sox17, CXCR4, Nodal), and hepatic (HNF4α, HNF1β, CYP3A, and 1A2) transcripts in line with human development. (C): Cell surface marker expression was also significantly altered during cellular differentiation, as assessed by flow cytometry. The downregulation of TRA 1-60, marking onset of cellular differentiation at day 6, and the increase in CXCR4 staining confirmed definitive endoderm specification at days 3 and 4. (D): Western blotting was used to assess protein expression of a number of differentiation markers. As the cells differentiated toward the hepatocyte lineage, we detected key changes in Oct4, AFP, albumin, and E-cadherin gene expression. Images were taken at ×10 magnification. The qPCRs were run in triplicates. Error bars represent the standard deviation. Abbreviations: d, day; DE, definitive endoderm; HBC, hepatocyte; HBL, hepatoblast; hES, human embryonic stem cells.
Figure 2.

Stagewise human embryonic stem cell (hESC) differentiation to the hepatocyte lineage. hESCs were differentiated in a stagewise fashion toward the hepatocyte lineage. Immunocytochemistry showing downregulation of pluripotent marker Oct4, as the cells expressed endodermal transcripts (Sox17, Foxa1). Upon hepatic specification, HNF4α expression increased. Immunoglobulin G controls demonstrated the specificity of immunostaining. The percentage of positive cells is provided in the top right of each panel. This was calculated from four random fields of view and is quoted as ±standard error. The images were taken at ×20 magnification. Abbreviations: Foxa1, Forkhead box protein 1; HNF4α, hepatocyte nuclear factor 4α; Oct4, octamer 4; Sox 17, SRY-box containing gene 17.
In addition to snapshot analysis, we performed flow cytometry over a 6-day time course to measure marker expression indicative of either pluripotency or differentiation. In line with previous reports, TRA 1-60 was elevated in hESC populations and decreased as the cells differentiated toward definitive endoderm. This was in contrast to CXCR4, which increased as the cells transited from pluripotency to definitive endoderm, peaking at ∼72% positive by day 3 (Fig. 1C). Postspecification, definitive endoderm was directly differentiated to hepatoblast and hepatocytic populations. In support of this, we observed increases in hepatic gene expression for hepatocyte nuclear factor 4α (HNF4α) (p < .001), HNF1β (p < .001), CYP3A4 (p < .01), and CYP1A2 (p < .01) (Fig. 1B), and, upon hepatic specification, ∼94% of cells expressed HNF4α (Fig. 2). Further support of these data was provided by Western blotting. As differentiation proceeded, we observed a decrease in Oct4 and an increase in epithelial (E-cadherin) and functional gene expression (a-fetoprotein [AFP], albumin, CYP3A) (Fig. 1D).
Translating Serum-Free Differentiation to Induced Pluripotent Stem Cells
We were keen to assess whether our serum-free approach was applicable to other stem cell lines, in particular iPSCs. For this purpose, we used a well-established iPSC line [19, 20]. To direct efficient hepatic differentiation, it was necessary to make simple changes to the iPSC differentiation process. As with hESCs, we detected significant changes in cell morphology as differentiation progressed (Fig. 3A). Similar to hESC populations, iPSCs demonstrated downregulation of pluripotent transcripts (Oct4, p < .001 and Nanog, p < .001) as the cells differentiated toward definitive endoderm (day 5), determined by Sox17 (p < .001), Foxa2 (p < .0001), and CXCR4 (p < .001). Following hepatocyte specification (day 9), we observed increases in hepatocyte markers, HNF4α (p < .0001), albumin (p < .001), and CYP1A2 (p < .05) (Fig. 3B) with CYP3A4 (p < .05) gene expression apparent at the day 14 time point. iPSC differentiation to hepatic endoderm was highly efficient. In support of this, we observed a downregulation of Oct4-positive cells as differentiation ensued. Upon hepatic specification, ∼98% cells expressed HNF4α, AFP, and E-cadherin (Fig. 4). Further support of these data was provided by Western blotting. As differentiation progressed, we observed a decrease in Oct4 and an increase in α1-antitrypsin expression (Fig. 3C).
Figure 3.
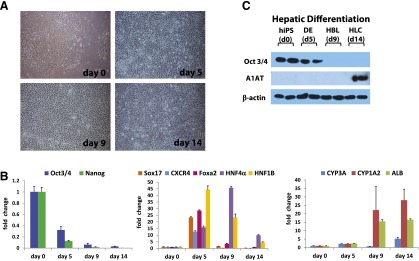
Translating new model to human induced pluripotent stem cells (iPSCs). iPSCs were differentiated in a stagewise fashion toward the hepatocyte lineage. (A): Imaging showing sequential morphological changes during iPSC differentiation. The light microscopy demonstrated transition from iPSC (day 0), to definitive endoderm (day 5), to hepatoblast (day 9), and to hepatocyte (day 14). (B): The quantitative polymerase chain reaction (qPCR) gene expression analysis demonstrated that changes in morphology were paralleled by changes in pluripotent (Oct4, Nanog), definitive endoderm (Sox17, CXCR4, Foxa 2), and hepatic (HNF4α, HNF1β, CYP3A, CYP1A2, ALB) transcripts, which is in line with human development. (C): Western blotting was used to assess protein expression of a number of differentiation markers. As the cells differentiated toward hepatic lineage, Oct4 protein expression was reduced and α1 antitrypsin gene expression increased. Images were taken at ×10 magnification. The qPCRs were run in triplicates. Error bars represent the standard deviation. Abbreviations: d, day; DE, definitive endoderm; HBC, hepatocyte; HBL, hepatoblast; hIPS, human induced pluripotent cells.
Figure 4.

Stagewise induced pluripotent stem cell (iPSC) differentiation to the hepatocyte lineage. iPSCs were differentiated in a stagewise fashion toward the hepatocyte lineage. Immunocytochemistry shows downregulation of pluripotent marker Oct4 during cellular differentiation. Upon hepatic specification, HNF4α, AFP, and E-cad expression increased. Immunoglobulin G controls demonstrated the specificity of immunostaining. The percentage of positive cells is provided in the top right of each panel. This was calculated from four random fields of view and is quoted as ±standard error. The images were taken at ×20 magnification. Abbreviations: AFP, a-fetoprotein; Ecad, E cadherin; HNF4α, hepatocyte nuclear factor 4α; Oct4, octamer 4.
Selecting Hepatocyte Populations That Demonstrate Mature Hepatic Function
The liver plays a central role in many biochemical processes. Our interest was specifically in drug metabolism and how stem cell-derived models could be used to study compound pharmacology and hepatotoxicity. The first step was to assess which, if any, cell-based assay was suitable for large-scale screening within the pharmaceutical industry. The criteria we applied were stem cell-derived hepatocytes exhibit inducible cytochrome P450 activity to progress to large-scale manufacture and industrial screening. Using our serum-free process, hESC and iPSC hepatocytes demonstrated significantly improved metabolic capacity and were more stable in character, when compared with previous approaches using serum-containing medium [17, 20]. Interestingly, hESC-derived hepatocytes displayed better metabolic capacity than their iPSC counterparts (Fig. 5).
Figure 5.
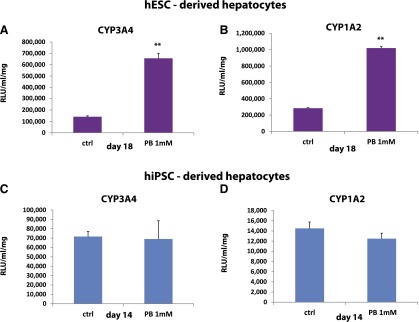
Identification of mature hepatocyte populations for scale-up and drug toxicity testing. Pluripotent stem cells were differentiated to hepatocytes as previously described. (A, B): hESC hepatocytes were induced with 1 mM phenobarbital drug for 48 hours prior to analysis of CYP3A and CYP1A2 function. (C, D): iPSC hepatocytes were induced with 1 mM phenobarbital drug for 48 hours prior to analysis of CYP3A and CYP1A2 function. The activity of CYP3A and CYP1A2 was analyzed using Promega pGlo technology, and activity was measured on a luminometer (POLARstar Optima). Units of activity are expressed as relative light units (RLU)/milliliter (ml)/milligram (mg) of protein (RLU/ml/mg). Levels of significance were measured by Student's t test. A p value <.01 is denoted as ∗∗, n = 3. Abbreviations: ctrl, control; hESC, human embryonic stem cell; hiPSC, human induced pluripotent stem cell; PB, phenobarbital.
Whereas basal activities of the cytochrome P450 enzymes provided important background information, we were keen to assess whether our PSC-derived hepatocytes were drug inducible, a hallmark of a more mature hepatocyte. To assess this, we used a pleiotropic P450 inducer, phenobarbital. PSC-derived hepatocytes were induced with 1 mM PB for 48 hours before the assessment of P450 activity. It was clear that hESC-derived hepatocyte populations exhibited inducible CYP3A (∼fivefold) and CYP1A2 (∼fourfold) activity (Fig. 5A, 5B), whereas the iPSC-derived hepatocytes did not (Fig. 5C, 5D). Therefore, we elected to use hESC-derived hepatocytes in future experiments as they fulfilled the criteria for large-scale manufacture and industrial application.
hESC Hepatocytes Predict Potential Liver Injury in a Manner Equivalent to Current Gold Standards
hESC-derived hepatocytes, chosen on the basis of metabolic competence, were compared to a well-characterized batch of cryoplateable human hepatocytes (hereafter referred to as primary hepatocytes). hESC were scaled up and differentiated in 96-well format. Post-hepatocyte commitment (day 14), hESC-derived hepatocytes were shipped to Bristol-Myers Squibb (Princeton, NJ, http://www.bms.com). Upon arrival (day 16), hESC-derived hepatocytes were reconstituted and left to recover for an additional 72 hours. At the 72-hour time point, no contamination or major cell death was evident and the cells were released for screening. Twenty known hepatotoxins were screened using primary and stem cell-derived hepatocytes. Each cell type was incubated with the specific compound for 1, 4, or 7 days. In general, toxicity increased with time of exposure (Fig. 6). All compounds eventually identified as toxic (IC50 < 200 µM) in primary hepatocyte cultures were also toxic in hESC hepatocytes. Initially (day 1), fewer compounds were toxic to hESC hepatocytes (45%) as compared with primary hepatocytes (60%), but by day 4 hESC hepatocytes displayed increased sensitivity (65%) over primary hepatocytes (60%) (Fig. 6). One compound, benzbromarone, was substantially more toxic in hESC hepatocyte cultures than in primary hepatocytes, but most compounds (8 of 20) were more toxic in primary hepatocyte cultures. By day 7, both assays identified 75% of the hepatotoxins as toxic in vitro.
Figure 6.

IC50 for 20 human hepatotoxicants cultured with hESC-derived and cryopreserved human hepatocytes for 1, 4, or 7 days. A comparison of hESC-derived and cryoplateable human hepatocytes exposed to 20 known human hepatotoxicants for 1, 4, or 7 days. Cellular ATP levels were measured and used to calculate IC50 (the concentration of the compound resulting in 50% toxicity). Wells were plated in triplicate for each concentration and each compound tested. If a compound failed to generate an IC50 with the maximum concentration tested (200 µM), it was expressed as >200. In general, toxicity increased with time of exposure. All compounds eventually identified as toxic (i.e., an IC50 < 200 µM) in primary hepatocyte cultures were also toxic in hESC hepatocytes. Abbreviation: hESC, human embryonic stem cell.
Nine of the 15 toxic compounds (amiodarone, benzbromarone, hycanthone, ketoconazole, nefazodone, perhexiline, puromycin, simvastatin, and troglitazone) induced increases in caspase 3/7 activity over the same range of concentrations that reduced ATP (supplemental online Fig. 1). The remaining six toxic compounds (bicalutamide, danazol, dantrolene, flutamide, nitrofurantoin, and tolcapone) did not induce caspase activity (supplemental online Fig. 1). Of the five compounds that were not toxic (dacarbazine, diclofenac, felbamate, isoniazid, and valproic acid), concentration-related increases in hESC hepatocyte ATP were observed for dacarbazine, felbamate, and valproic acid, indicative of hormesis (supplemental online Fig. 1). Of note, troglitazone also induced a substantial increase in hESC hepatocyte ATP on day 7, at lower concentrations, before sustaining a complete loss of cell ATP between 10 and 100 µM (supplemental online Fig. 1).
Discussion
The liver plays an important role in human physiology. It has been estimated to perform more than 500 functions in vivo [21]. Liver disease leads to decompensated organ function and results in the derangement of normal bodily functions. Therefore, it is important that early diagnosis and intervention take place. In this vein, we have developed defined human hepatocyte models from hESCs and iPSCs. Importantly, our novel serum-free approach drives efficient hepatic differentiation (Figs. 1–4) in a format suitable for high-throughput screening. Interestingly, hESC hepatocytes exhibited greater metabolic and drug-inducible activity when compared with iPSC hepatocytes (Fig. 5). The reason for this is unknown, and we do not claim that this is a generic iPSC trait, but merely that it is evident in our best-performing retrovirally derived iPSC line. The failure to respond to phenobarbital may be attributable to a number of factors, including stresses inherent with cellular reprogramming, genetic background, epigenetic modifications, and/or retroviral DNA insertion, and those will be the focus of future investigations.
On the basis of metabolic competence, hESC hepatocytes were selected for scale-up and evaluated as a tool for hepatotoxicity studies. hESC hepatocyte populations were robust, survived transit, and accurately predicted human compound toxicity, which was comparable to cryopreserved human hepatocytes (Fig. 6; supplemental online Fig. 1). Within the group of 20 compounds tested, we determined 15 as toxic. Two mechanisms of cytotoxicity appeared to be involved, caspase-dependent apoptosis (9 compounds) and caspase-independent necrosis (6 compounds) (supplemental online Fig. 1). Of note, ATP synthesis was induced at lower compound concentrations in response to some compounds. This is an example of hormesis, which is associated with cellular stress [22]. Of note, there was clear evidence of hormesis in hESC-derived hepatocytes postexposure to valproic acid, felbamate, and dacarbazine. Interestingly, these three compounds were not detected as cytotoxic, and further investigation of stress-related endpoints, for example, reactive oxygen species, heat shock proteins, and unfolded protein-response transcription factors, will most likely be useful in defining the mechanism of toxicity associated with these compounds. The two remaining compounds, isoniazid and diclofenac, did not induce toxicity in either primary or hESC hepatocytes. Isoniazid hepatotoxicity appears to be immune mediated [23], whereas diclofenac toxicity in human patients appears over weeks to months, making it possible that models of short duration or lacking immune components do not reveal the cumulative effects of these compounds. These data support that hESC hepatocytes performed on a par with gold standard assays, promising in the future that they may serve as a partial or full replacement for primary cells in safety screening.
Conclusion
We demonstrate that it is possible to model the potential for human drug-induced liver injury using stem cell-derived hepatocytes. This is an important step to using stem cell-derived somatic cells in tailoring human medicines for different genetic backgrounds and improving the efficiency of drug development. In the future, it may also be possible to use such a resource for developing cell-based therapies [24]. Whereas current models are equivalent to primary hepatocytes, there is a requirement to improve their sensitivity and prediction rates of drug toxicity. Therefore, in the future it will be important to sophisticate cell-based models and endpoint measurements to deliver better success in predicting human liver injury.
Supplementary Material
Footnotes
Contributed equally as first authors.
Acknowledgments
We thank Dr. Ian Wilmut, Dr. George Daley, Dr. In Hyun Park, and Dr. Gareth Sullivan for providing the iPSC line used in these studies. We thank Dr. Claire Medine for providing training on iPSC culture and differentiation. D.C.H. was supported by a Research Councils UK (RCUK) fellowship. D.S. and B.L.-V. were supported by Medical Research Council studentships. S.F. was supported by a Biotechnology and Biological Sciences Research Council BioSkape award. W.Z. was supported by a grant from the Chinese government. J.P.I. was supported by a Medical Research Council Programme grant.
Author Contributions
D.S.: collection and/or assembly of data, data analysis and interpretation, manuscript writing; S.F.: collection and/or assembly of data, data analysis and interpretation, manuscript writing; B.L.-V.: collection and/or assembly of data, data analysis and interpretation; C.S.: collection and/or assembly of data, data analysis and interpretation; W.Z.: collection and/or assembly of data, data analysis and interpretation; J.P.I.: conception and design, manuscript writing; O.F.: conception and design, data analysis and interpretation, manuscript writing; D.C.H.: conception and design, data analysis and interpretation, manuscript writing, financial support.
Disclosure of Potential Conflicts of Interest
D.C.H. and J.P.I. are founders and shareholders in FibromEd Products Ltd. C.S. and O.F. have compensated employment from Bristol-Myers Squibb.
References
- 1.British Association for the Study of the Liver. A Time to Act: Improving Liver Health and Outcomes in Liver Disease. Available at http://www.bsg.org.uk/attachments/1004_National%20Liver%20Plan%202009.pdf. Accessed August 1, 2014.
- 2.Adam R, Cailliez V, Majno P, et al. Normalised intrinsic mortality risk in liver transplantation: European Liver Transplant Registry study. Lancet. 2000;356:621–627. doi: 10.1016/s0140-6736(00)02603-9. [DOI] [PubMed] [Google Scholar]
- 3.Faber W, Seehofer D, Puhl G, et al. Donor age does not influence 12-month outcome after orthotopic liver transplantation. Transplant Proc. 2011;43:3789–3795. doi: 10.1016/j.transproceed.2011.10.048. [DOI] [PubMed] [Google Scholar]
- 4.Dhawan A, Strom SC, Sokal E, et al. Human hepatocyte transplantation. Methods Mol Biol. 2010;640:525–534. doi: 10.1007/978-1-60761-688-7_29. [DOI] [PubMed] [Google Scholar]
- 5.Boulter L, Govaere O, Bird TG, et al. Macrophage-derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat Med. 2012;18:572–579. doi: 10.1038/nm.2667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thomas JA, Pope C, Wojtacha D, et al. Macrophage therapy for murine liver fibrosis recruits host effector cells improving fibrosis, regeneration, and function. Hepatology. 2011;53:2003–2015. doi: 10.1002/hep.24315. [DOI] [PubMed] [Google Scholar]
- 7.Bird TG, Lu WY, Boulter L, et al. Bone marrow injection stimulates hepatic ductular reactions in the absence of injury via macrophage-mediated TWEAK signaling. Proc Natl Acad Sci USA. 2013;110:6542–6547. doi: 10.1073/pnas.1302168110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Szkolnicka D, Zhou W, Lucendo-Villarin B, et al. Pluripotent stem-cell derived hepatocytes: Potential and challenges in pharmacology. Annu Rev Pharmacol Toxicol. 2013;53:147–159. doi: 10.1146/annurev-pharmtox-011112-140306. [DOI] [PubMed] [Google Scholar]
- 9.Reubinoff BE, Pera MF, Fong CY, et al. Embryonic stem cell lines from human blastocysts: Somatic differentiation in vitro. Nat Biotechnol. 2000;18:399–404. doi: 10.1038/74447. [DOI] [PubMed] [Google Scholar]
- 10.Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 11.Asahina K, Fujimori H, Shimizu-Saito K, et al. Expression of the liver-specific gene Cyp7a1 reveals hepatic differentiation in embryoid bodies derived from mouse embryonic stem cells. Genes Cells. 2004;9:1297–1308. doi: 10.1111/j.1365-2443.2004.00809.x. [DOI] [PubMed] [Google Scholar]
- 12.Lavon N, Yanuka O, Benvenisty N. Differentiation and isolation of hepatic-like cells from human embryonic stem cells. Differentiation. 2004;72:230–238. doi: 10.1111/j.1432-0436.2004.07205002.x. [DOI] [PubMed] [Google Scholar]
- 13.Basma H, Soto-Gutiérrez A, Yannam GR, et al. Differentiation and transplantation of human embryonic stem cell-derived hepatocytes. Gastroenterology. 2009;136:990–999. doi: 10.1053/j.gastro.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cai J, Zhao Y, Liu Y, et al. Directed differentiation of human embryonic stem cells into functional hepatic cells. Hepatology. 2007;45:1229–1239. doi: 10.1002/hep.21582. [DOI] [PubMed] [Google Scholar]
- 15.Hay DC, Zhao D, Ross A, et al. Direct differentiation of human embryonic stem cells to hepatocyte-like cells exhibiting functional activities. Cloning Stem Cells. 2007;9:51–62. doi: 10.1089/clo.2006.0045. [DOI] [PubMed] [Google Scholar]
- 16.Hay DC, Fletcher J, Payne C, et al. Highly efficient differentiation of hESCs to functional hepatic endoderm requires ActivinA and Wnt3a signaling. Proc Natl Acad Sci USA. 2008a;105:12301–12306. doi: 10.1073/pnas.0806522105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Greenhough S, Bradburn H, Gardner J, et al. Development of an embryoid body-based screening strategy for assessing the hepatocyte differentiation potential of human embryonic stem cells following single-cell dissociation. Cell Reprogram. 2013;15:9–14. doi: 10.1089/cell.2012.0049. [DOI] [PubMed] [Google Scholar]
- 18.Hay DC, Pernagallo S, Diaz-Mochon JJ, et al. Unbiased screening of polymer libraries to define novel substrates for functional hepatocytes with inducible drug metabolism. Stem Cell Res. 2011;6:92–102. doi: 10.1016/j.scr.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 19.Medine CN, Lucendo-Villarin B, Storck C, et al. Developing high-fidelity hepatotoxicity models from pluripotent stem cells. Stem Cells Translational Medicine. 2013;2:505–509. doi: 10.5966/sctm.2012-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sullivan GJ, Hay DC, Park IH, et al. Generation of functional human hepatic endoderm from human induced pluripotent stem cells. Hepatology. 2010;51:329–335. doi: 10.1002/hep.23335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maton A, Hopkins J, McLaughhlin CW, et al. Human Biology and Health. Englewood Cliffs, NJ: Prentice Hall; 1997. [Google Scholar]
- 22.Damelin LH, Vokes S, Whitcutt JM, et al. Hormesis: A stress response in cells exposed to low levels of heavy metals. Hum Exp Toxicol. 2000;19:420–430. doi: 10.1191/096032700678816133. [DOI] [PubMed] [Google Scholar]
- 23.Metushi IG, Cai P, Zhu X, et al. A fresh look at the mechanism of isoniazid-induced hepatotoxicity. Clin Pharmacol Ther. 2011;89:911–914. doi: 10.1038/clpt.2010.355. [DOI] [PubMed] [Google Scholar]
- 24.Hay DC. Rapid and scalable human stem cell differentiation: Now in 3D. Stem Cells Dev. 2013;22:2691–2692. doi: 10.1089/scd.2013.1500. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.