

Abstract
Five members of the KMT2 family of lysine methyltransferases, originally named the mixed lineage leukemia (MLL1-5) proteins, regulate gene expression during embryogenesis and development. Each KMT2A-E contains a catalytic SET domain that methylates lysine 4 of histone H3, and one or several PHD fingers. Over the past few years a growing number of studies have uncovered diverse biological roles of the KMT2A-E PHD fingers, implicating them in binding to methylated histones and other nuclear proteins, and in mediating the E3 ligase activity and dimerization. Mutations in the PHD fingers or deletion of these modules are linked to human diseases including cancer and Kabuki syndrome. In this work, we summarize recently identified biological functions of the KMT2A-E PHD fingers, discuss mechanisms of their action, and examine preference of these domains for histone and non-histone ligands.
Keywords: PHD finger, KMT2, MLL, methyltransferase, histone, chromatin, structure
1. Introduction
The lysine methyltransferases KMT2A-E are evolutionarily conserved transcriptional regulators. They play a key role in embryogenesis and maintenance of the expression levels of HOX and other developmental genes and are essential in the cell cycle control, hormone signaling and reproduction [1–5]. Chromosomal translocations, tandem duplications, deletions and mutations in the KMT2A-E genes have been directly linked to leukemogenesis and other human diseases [2, 6–8]. The KMT2A-E subfamily consists of five members, including KMT2A (or MLL1), KMT2B (MLL2), KMT2C (MLL3), KMT2D (MLL4) and KMT2E (MLL5), which together with KMT2F (SET1A) and KMT2G (SET1B) constitute a larger family of KMT2 methyltransferases. The KMT2 enzymes are highly specific and catalyze the transfer of methyl groups from S-adenosyl-L-methionine (SAM) to the ε-amino group of lysine 4 of histone H3, generating mono-, di- or tri-methylated H3K4me1/2/3 marks.
To say that KMT2A-E are complex macromolecules is understatement. These very large, ~300–600 kDa multimodular proteins interact with numerous ligands, at times recruiting co-factors with apparently opposing functions. KMT2A alone has over a dozen of binding partners and is cleaved into two pieces, a larger N-terminal fragment, involved in gene repression, and a smaller C-terminal fragment, which is a transcriptional activator [9, 10]. The cleavage, followed by the association of the two fragments, is necessary for KMT2A to be fully active. Like many other methyltransferases, the KMT2 family members exist in multisubunit nuclear complexes (human COMPASS), where other subunits also mediate the enzymatic activity [11, 12]. Furthermore, the KMT2 function can be fine-tuned by posttranslational modifications (PTMs). For example, KMT2E has no intrinsic histone lysine methyltransferase activity [13], however, once the SET domain is glycosylated, it is capable of generating mono- and dimethylated H3K4 marks [14].
KMT2A-E can be distinguished through the catalytic Su(var)3–9, Enhancer of Zeste, Trithorax (SET) domain, however the number of PHD fingers found in these proteins differs considerably. Four PHD fingers are present in KMT2A and KMT2B, whereas KMT2C, KMT2D and KMT2E have eight, seven, and one, respectively (Fig. 1a). Overall, the KMT2A-E subfamily contains 24 PHD fingers, each of which is characterized by the canonical C4HC2C/H sequence that coordinates two zinc ions (Fig. 1b). A number of studies in the last few years revealed diverse roles of the KMT2A-E PHD fingers. Here, we compare known biological activities of the PHD fingers and discuss the molecular mechanisms underlying these functions. We also explore the conservation of the PHD-ligand interactions within the KMT2A-E subfamily.
Figure 1. The KMT2A-E subfamily of lysine methyltransferases.

(a) Schematic representation of the KMT2A-E subfamily. The 24 PHD fingers and single zinc-fingers are depicted as orange ovals and small brown circles, respectively. (b) Alignment of the KMT2A-E PHD finger sequences: absolutely, moderately and weakly conserved residues are colored orange, yellow and blue, respectively. Zinc-coordinating cysteine/histidine residues are labeled.
2. PHD fingers of KMT2A and KMT2B
KMT2A and KMT2B have similar domain architecture and both contain three consecutive PHD fingers (PHD1-PHD3), followed by a bromodomain (BD) and the fourth PHD4 finger. The first and second modules in the triple PHD1-3 finger cassette are closely linked, whereas the third module is separated from PHD1-PHD2 by an additional single C4-type zinc finger. The PHD finger region plays a regulatory role in KMT2A function and suppresses KMT2A-mediated leukaemogenesis. Inclusion of the PHD2-PHD3 fingers in the chimeric KMT2A-AF9 inhibits transformation of mouse bone marrow and leads to hematopoietic cell differentiation and downregulation of Hoxa9 [15]. Incorporation of PHD3 into the KMT2A-ENL chimera suppresses KMT2A-ENL-induced immortalization of murine bone marrow progenitor cells [16].
Although the precise function of the PHD1 finger remains poorly understood, it cooperates with PHD4 in mediating intramolecular interactions between the N-terminal and C-terminal fragments of KMT2A [17]. The PHD2 finger of KMT2A and KMT2B shows the E3 ubiquitin ligase activity in the presence of the E2-conjugating enzyme CDC34 [18]. It ubiquitinates histones H3 and H4 in vitro and plays a role in ubiquitination of KMT2A itself, which may provide a mechanism for the regulation of KMT2A degradation during cell cycle [18]. In agreement, disruption of the PHD2 finger fold or deletion of this domain enhances recruitment of KMT2A to target genes and augments its transactivation ability, most likely due to the increased protein stability. Additionally, the PHD2 finger of KMT2A is involved in homo-dimerization [19].
The third PHD3 finger of KMT2A is most structurally and functionally characterized. This unique zinc finger has two binding partners: it recognizes histone H3K4me3, the product of the KMT2A enzymatic activity, and interacts with the RNA recognition motif (RRM) of the nuclear cyclophilin Cyp33, an KMT2A-associated co-repressor [16, 19–24]. Several recent studies shed light on the molecular basis of these interactions [20–22, 24] and Wang et. al. have uncovered a remarkably complex mechanism for the association of PHD3 with H3K4me3 and Cyp33 [20].
The crystal structure of the PHD3-BD region of KMT2A reveals that in the apo-state the two domains are in close contact, mediated by the bended α1-helix of PHD3 and the extended αZ helix and the C-terminal loop of BD [20]. A proline residue in the short linker connecting PHD3 and BD adopts a cis conformation, thus allowing for the formation of a pair of inter-domain salt bridges and leading to the overall compact PHD3-BD assembly. Unlike other acetyllysine-recognizing BDs, the bromodomain of KMT2A does not bind acetylated lysine residues, however it plays an essential role in modulating functions of the PHD3 finger, influencing its interaction with both H3K4me3 and Cyp33. In the presence of BD, binding affinity of the PHD3 finger for H3K4me3 is enhanced ~20 fold, whereas association with an isolated Cyp33 RRM domain is abolished because BD blocks the RRM-binding site. The latter interaction can be restored if full-length Cyp33 is used. Cyp33 is a peptidyl-prolyl isomerase (PPIase) that catalyzes cis-trans isomerization of proline residues. Here, it converts a Pro-His peptide bond in the PHD-BD linker from the cis to trans conformation, consequently disrupting the PHD3-BD contacts and releasing the previously occluded RRM-binding site of PHD3 [20].
As many other epigenetic readers [25], the KMT2A PHD3 finger exhibits a low μM binding affinity for H3K4me3 (Kd=4 μM, 19 μM and 30 μM, as measured by ITC and tryptophan fluorescence [20–22]) and binds weaker to di- or mono-methylated H3K4. The 1.9 Å-resolution crystal structure of PHD3-BD in complex with histone H3K4me3 peptide demonstrates that the KMT2A PHD3 finger recognizes this PTM through a conserved mechanism [20] (Fig. 2a, b). H3K4me3 adopts an extended conformation and pairs with the β-sheet of PHD3, forming characteristic backbone-backbone intermolecular hydrogen bonds. The fully extended side chain of trimethylated K4 occupies an aromatic/hydrophobic cage, consisting of two tyrosine residues (Y1576 and Y1581), a methionine (M1585), and a tryptophan (W1594) that make cation-π, hydrophobic and van der Waals contacts with the trimethylammonium group of K4. The bulky side chain of W1594 separates the K4me3-binding cage from the adjacent R2-binding groove, where N1587 of the protein is hydrogen bonded to the ε guanidinium proton of R2. Yet, the H3K4me3-binding mechanism of KMT2A PHD3 has several unique features: the bound peptide makes a β turn at the K4me3-Q5 step, and this interaction induces a conformational change in the Y1576-M1585 loop whereby Y1581 flips toward the aromatic cage, almost fully enclosing bound K4me3. Substitution of the aromatic cage residues disrupts the interaction with H3K4me3.
Figure 2. The structural basis of the interactions of KMT2A PHD3 with H3K4me3 and Cyp33.

(a) The 1.9 Å-resolution crystal structure of the PHD3-BD region of KMT2A in complex with histone H3K4me3 peptide (PDB: 3LQJ). (b) The H3K4me3-binding site of the KMT2A PHD3 finger. (c) The solution structure of the Cyp33 RRM domain fused with the α1-helix of the KMT2A PHD3 finger (PDB: 2KU7).
Binding of KMT2A PHD3 to RRM involves α1-helix of the PHD3 finger, the same helix that associates with BD. This helix lays across the central β-sheet of RRM, as seen in the solution structure of the Cyp33 RRM domain fused with the α1-helix of the PHD3 finger [20] and from chemical shift perturbation analyses [20, 22, 24] (Fig. 2c). This interaction is mediated primarily through hydrophobic contacts involving the side chains of M1606, I1609 and L1610 of PHD3. The RRM-binding site is adjacent to and does not visibly overlap with the H3K4me3-binding site, however the PHD-BD region binds to H3K4me3 ~3-fold weaker when full-length Cyp33 is present [20]. Likewise binding of an isolated PHD3 finger is reduced ~6-fold in the presence of RRM, and the interaction of PHD3 with Cyp33 is ~4-fold weaker in the presence of H3K4me3 [22]. It will be interesting to explore the interplay between these two interactions in detail and establish whether the PHD3 finger is able to concomitantly associate with both binding partners in vivo.
The KMT2A PHD3 finger is a striking example of a PTM reader whose function depends on the epigenetic environment and can be switched from transcriptional activation to repression through binding to H3K4me3 or Cyp33. Surprisingly, the Cyp33 binding activity is not conserved in the PHD3 finger of KMT2B, despite high sequence similarity of these domains (see below and [20]). The loss of binding can be attributed to the replacement of a single residue M1606 in the α1-helix of KMT2A PHD3 by an aspartate in KMT2B PHD3. Swapping mutations (M1606D in KMT2A and D1375M in KMT2B, respectively) confirmed that the methionine is required for the interaction with Cyp33 [20].
3. PHD fingers of KMT2C and KMT2D
KMT2C and KMT2D contain eight and seven PHD fingers, respectively (Fig. 1a). Although the biological role of the KMT2C PHD fingers is unknown, the atomic-resolution NMR structure of the tandem PHD2-PHD3 fingers of KMT2C (PDB: 2YSM, unpublished, RIKEN) implies that these domains may function in concert. In the structure, both domains adopt a typical zinc-finger fold [26] but have a large interface that holds the PHD fingers together in a bean-shaped scaffold (Fig. 3). Notably, PHD2, and the concave surface of the tandem domains in general, is highly negatively charged, whereas the opposite side (PHD3 particularly) is highly positively charged (Fig. 3b). Such charge distribution may indicate a role of electrostatic contacts in functioning of the KMT2C PHD2-PHD3 fingers.
Figure 3. Solution structure of the tandem PHD2-PHD3 fingers of KMT2C.

(a) A ribbon diagram of the structure (PDB: 2YSM, unpublished, RIKEN). (b) Electrostatic potential map of PHD2-PHD3. Red and blue color represents negative charge and positive charge, respectively.
A triple PHD finger cassette (PHD4-6) of KMT2D associates with histone H4 either unmodified or asymmetrically dimethylated at R3 (H4R3me2a) [27]. This interaction is required for methyltransferase activity of KMT2D and for KMT2D-mediated differentiation, and is inhibited by symmetric dimethylation of R3. Kabuki syndrome mutations C1430R and C1471Y [8], which most likely disrupt the PHD5 finger fold, reduce histone binding and catalytic activity of KMT2D [27].
4. PHD finger of KMT2E
A single PHD finger of KMT2E recognizes histone H3K4me3 with high specificity and affinity [28, 29]. The 1.48 Å resolution crystal structure of the PHD-H3K4me3 complex shows that the histone peptide lays antiparallel to and pairs with the double-stranded β sheet of the protein [28]. The side chain of trimethylated K4 is bound in an elongated groove formed by the D128, M132 and W141 residues of the PHD finger (Fig. 4a). The N-terminal amino group of A1 is hydrogen bonded to two backbone carbonyl groups of the protein, whereas the methyl group of A1 is buried in a small hydrophobic cavity. Additional intermolecular hydrogen bonds restrain Q5 and T6 of the peptide.
Figure 4. The molecular mechanism of H3K4me3 recognition by the KMT2E PHD finger.
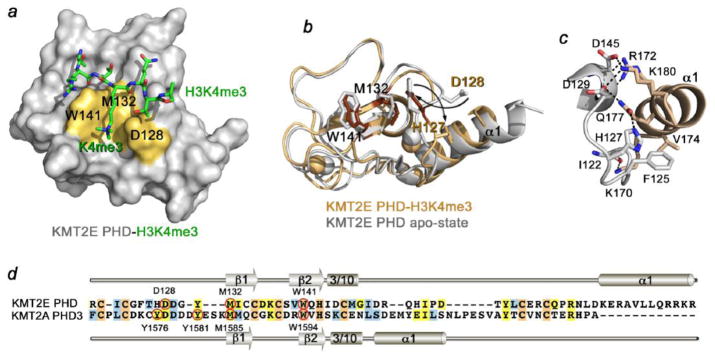
(a) The 1.48 Å resolution crystal structure of the KMT2E PHD finger in complex with H3K4me3 peptide (PDB: 4L58). The K4me3-binding groove residues are colored yellow and labeled. (b) An overlay of the structure of the H3K4me3-bound KMT2E PHD finger (dark yellow) with the structure of KMT2E PHD in the apo-state (grey) (PDB: 2LV9). The H3K4me3 peptide is omitted for clarity. (c) A zoom-in view of the KMT2E PHD structure showing close contacts between the N-terminal loop and the C-terminal α1-helix. Dashed lines represent hydrogen bonds. (d) Alignment of sequences of the KMT2E and KMT2A PHD fingers. The K4me3-binding groove residues are indicated by red circles and labeled. Secondary structure elements of KMT2E PHD and KMT2A PHD3 are shown.
The three unique aspects of the H3K4me3-binding mode of the KMT2E PHD finger are evident [28] (Fig. 4). In contrast to other PHD fingers, the K4me3-binding groove of KMT2E PHD contains only one aromatic residue, W141, with the acidic residue D128 forming an opposite (to W141) wall of the groove (Fig. 4d). The presence of an acidic residue in the aromatic cage of epigenetic readers is known to drive their preference for di- or monomethylated over trimethylated lysine substrates (reviewed in [25]). The carboxyl group of an aspartate can form additional favorable hydrogen bonding contacts with di- or mono- but not with tri-methylammonium group of K4. In spite of this, the KMT2E PHD finger selects for H3K4me3.
An overlay of the crystal structure of the KMT2E PHD finger in complex with H3K4me3 peptide and the solution structure of the apo-state reveals that binding is accompanied by large conformational changes in the protein N-terminal loop [28, 29] (Fig. 4b). In the ligand-free state, D128 points toward the C-terminal α1-helix, whereas the preceding residue H127 points toward the K4me3-binding groove. Upon binding to H3K4me3, H127 and D128 swap their positions. The side chains of H127 and D128 swing in opposite directions, each by almost 180 degrees. H127 moves away from the groove and interacts with the α1-helix. Concomitantly, D128 dissociates from the α1-helix and flips inward to complete the K4me3-binding pocket. The H127-D128 swapping rearrangement causes conformational changes in other residues of the G124-Y131 loop.
The involvement of the α1-helix of KMT2E PHD underlines another distinctive feature of this binding mechanism. The unusually long C-terminal α1-helix of KMT2E PHD is in direct contact with and stabilizes the core of the domain. It packs against the N-terminal loop and the 310 helix, creating an intricate set of hydrogen bonding, electrostatic and hydrophobic interactions at the interface (Fig. 4c). This helix is located further downstream in sequence as compared to a typical α1-helix of KMT2A PHD3, which is involved in the interaction with Cyp33 and BD (Fig. 4d).
5. Conservation of histone and RRM binding activities within the KMT2A-E subfamily
Analysis of amino acid sequences of the H3K4me3-specific PHD fingers indicates a critical role of a tryptophan residue at the position −2 in respect to zinc-coordinating histidine [26, 30]. Because six of the 24 PHD fingers of the KMT2A-E subfamily contain the tryptophan residue (Fig. 5a), we sought to examine whether they are capable of histone binding. We expressed and purified KMT2A PHD3, KMT2B PHD3, KMT2C PHD7, KMT2D PHD6 and KMT2E PHD fingers as 15N-labeled proteins and tested their interactions with H3K4me3 peptide using 1H,15N HSQC experiments (we were unable to obtain a cDNA of KMT2C PHD4) (Fig. 5). As expected, H3K4me3 induced chemical shift changes in the KMT2A PHD3 and KMT2E PHD fingers, corroborating previous findings that the two PHD fingers recognize H3K4me3 [20–22, 28, 29] (Fig. 5b, first row, first panel, and second row, second panel). Likewise, KMT2B PHD3 exhibited substantial resonance perturbations and thus associated with H3K4me3 (Fig. 5b, first row, second panel). Surprisingly we found that the KMT2C PHD7 and KMT2D PHD6 fingers do not bind H3K4me3 as their 1H,15N HSQC spectra remain unperturbed upon addition of the peptide (Fig. 5b, first row, third panel, and second row, first panel). These results suggest that not only the signature tryptophan but also other residues are necessary for this interaction. The obvious candidates for further studies are the isoleucine and leucine residues, present in the KMT2C PHD7 and KMT2D PHD6 sequences instead of a methionine in the KMT2A PHD3 and KMT2E PHD sequences. We note however, that this methionine is not strictly conserved and is replaced by a tyrosine in BPTF PHD or by a tryptophan in KDM5A PHD3, and both these domains are well known to bind H3K4me3 [31–33] (Fig. 5a).
Figure 5. Conservation of histone and RRM binding activities within the KMT2A-E subfamily.

(a) Alignment of the PHD finger sequences: absolutely, moderately and weakly conserved residues are colored orange, yellow and blue, respectively. The K4me3-binding site residues are indicated by red circles. The M1606 residue of KMT2A PHD3, critical for the interaction with Cyp33 is marked with a purple square. (b, c) Superimposed 1H,15N HSQC spectra of the indicated PHD fingers collected upon titration with H3K4me3 peptide (b) or the Cyp33 RRM domain (c). Spectra are color coded according to the protein:ligand molar ratio. The following constructs: KMT2A PHD1 (1431–1481), KMT2A PHD3 (1565–1627), KMT2B PHD3 (1334–1397), KMT2C PHD7 (1083–1143), KMT2D PHD6 (1486–1572), KMT2E PHD (117–181) and Cyp33 RRM (1–83) were expressed and purified as described in [21, 24]. The NMR titration experiments were performed at 298K on Varian INOVA 500 MHz and 600 MHz spectrometers. The binding was characterized by monitoring chemical shift changes in 1H,15N HSQC spectra of 0.1 – 0.2 mM uniformly 15N-labelled PHD fingers of KTM2A-E as the unlabeled histone H3K4me3 peptide or the Cyp33 RRM domain was gradually added. (d) Summary of binding activities of the tested KMT2A-E PHD fingers. (e) Control: the KMT2A PHD1 finger is folded and stable but has no detectable histone binding activity. Superimposed 1H,15N HSQC spectra of the KMT2A PHD1 finger recorded upon titration of H3K9me2 peptide. No binding was observed to unmodified H3 as well (data not shown).
On the other hand and also unexpectedly, KMT2C PHD7 was able to associate with the RRM domain of Cyp33, despite it lacks the methionine residue in a putative α1-helix, which is required for binding of KMT2A PHD3 to Cyp33 (Fig. 5c). Indeed, substitution of this methionine with an aspartic acid in KMT2B PHD3 finger completely disrupted the interaction with Cyp33 (Fig. 5c, second panel).
The binding properties of the five tested PHD fingers are summarized in the table (Fig. 5d). However to fully understand the biological activities of KMT2A-E and various cellular processes they facilitate, it is imperative to identify and characterize functions of all 24 PHD fingers present in this subfamily. As binding partners for the majority of these domains are challenging to predict, high-throughput assays and combinatorial approaches can be of substantial help in the identification of both physiological ligands and synthetic drugs for the PHD fingers.
Highlights.
Biological activities of the PHD fingers of the MLL/KMT2 subfamily are compared.
The mechanisms underlying functions of the KMT2A-E PHD fingers are discussed.
The conservation of the KMT2A-E PHD-histone interactions is explored.
Acknowledgments
This work is supported by grants from the NIH, GM096863 and GM101664 (T.G.K.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, Hess JL. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002;10:1107–1117. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- 2.Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nature reviews. 2007;7:823–833. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- 3.Ansari KI, Hussain I, Shrestha B, Kasiri S, Mandal SS. HOXC6 Is transcriptionally regulated via coordination of MLL histone methylase and estrogen receptor in an estrogen environment. J Mol Biol. 2011;411:334–349. doi: 10.1016/j.jmb.2011.05.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Andreu-Vieyra CV, Chen R, Agno JE, Glaser S, Anastassiadis K, Stewart AF, Matzuk MM. MLL2 is required in oocytes for bulk histone 3 lysine 4 trimethylation and transcriptional silencing. PLoS biology. 2010;8 doi: 10.1371/journal.pbio.1000453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herz HM, Garruss A, Shilatifard A. SET for life: biochemical activities and biological functions of SET domain-containing proteins. Trends Biochem Sci. 2013 doi: 10.1016/j.tibs.2013.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Y, Bogershausen N, Alanay Y, Simsek Kiper PO, Plume N, Keupp K, Pohl E, Pawlik B, Rachwalski M, Milz E, Thoenes M, Albrecht B, Prott EC, Lehmkuhler M, Demuth S, Utine GE, Boduroglu K, Frankenbusch K, Borck G, Gillessen-Kaesbach G, Yigit G, Wieczorek D, Wollnik B. A mutation screen in patients with Kabuki syndrome. Human genetics. 2011;130:715–724. doi: 10.1007/s00439-011-1004-y. [DOI] [PubMed] [Google Scholar]
- 7.Hess JL. MLL: a histone methyltransferase disrupted in leukemia. Trends in molecular medicine. 2004;10:500–507. doi: 10.1016/j.molmed.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 8.Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, Beck AE, Tabor HK, Cooper GM, Mefford HC, Lee C, Turner EH, Smith JD, Rieder MJ, Yoshiura K, Matsumoto N, Ohta T, Niikawa N, Nickerson DA, Bamshad MJ, Shendure J. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42:790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yokoyama A, Kitabayashi I, Ayton PM, Cleary ML, Ohki M. Leukemia proto-oncoprotein MLL is proteolytically processed into 2 fragments with opposite transcriptional properties. Blood. 2002;100:3710–3718. doi: 10.1182/blood-2002-04-1015. [DOI] [PubMed] [Google Scholar]
- 10.Yokoyama A, Wang Z, Wysocka J, Sanyal M, Aufiero DJ, Kitabayashi I, Herr W, Cleary ML. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol. 2004;24:5639–5649. doi: 10.1128/MCB.24.13.5639-5649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takahashi YH, Westfield GH, Oleskie AN, Trievel RC, Shilatifard A, Skiniotis G. Structural analysis of the core COMPASS family of histone H3K4 methylases from yeast to human. Proc Natl Acad Sci U S A. 2011;108:20526–20531. doi: 10.1073/pnas.1109360108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohan M, Herz HM, Shilatifard A. SnapShot: Histone lysine methylase complexes. Cell. 2012;149:498–498. e491. doi: 10.1016/j.cell.2012.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sebastian S, Sreenivas P, Sambasivan R, Cheedipudi S, Kandalla P, Pavlath GK, Dhawan J. MLL5, a trithorax homolog, indirectly regulates H3K4 methylation, represses cyclin A2 expression, and promotes myogenic differentiation. Proc Natl Acad Sci U S A. 2009;106:4719–4724. doi: 10.1073/pnas.0807136106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fujiki R, Chikanishi T, Hashiba W, Ito H, Takada I, Roeder RG, Kitagawa H, Kato S. GlcNAcylation of a histone methyltransferase in retinoic-acid-induced granulopoiesis. Nature. 2009;459:455–459. doi: 10.1038/nature07954. [DOI] [PubMed] [Google Scholar]
- 15.Muntean AG, Giannola D, Udager AM, Hess JL. The PHD fingers of MLL block MLL fusion protein-mediated transformation. Blood. 2008;112:4690–4693. doi: 10.1182/blood-2008-01-134056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen J, Santillan DA, Koonce M, Wei W, Luo R, Thirman MJ, Zeleznik-Le NJ, Diaz MO. Loss of MLL PHD finger 3 is necessary for MLL-ENL-induced hematopoietic stem cell immortalization. Cancer research. 2008;68:6199–6207. doi: 10.1158/0008-5472.CAN-07-6514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yokoyama A, Ficara F, Murphy MJ, Meisel C, Naresh A, Kitabayashi I, Cleary ML. Proteolytically cleaved MLL subunits are susceptible to distinct degradation pathways. Journal of cell science. 2011;124:2208–2219. doi: 10.1242/jcs.080523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J, Muntean AG, Wu L, Hess JL. A subset of mixed lineage leukemia proteins has plant homeodomain (PHD)-mediated E3 ligase activity. J Biol Chem. 2012;287:43410–43416. doi: 10.1074/jbc.M112.423855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fair K, Anderson M, Bulanova E, Mi H, Tropschug M, Diaz MO. Protein interactions of the MLL PHD fingers modulate MLL target gene regulation in human cells. Mol Cell Biol. 2001;21:3589–3597. doi: 10.1128/MCB.21.10.3589-3597.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Z, Song J, Milne TA, Wang GG, Li H, Allis CD, Patel DJ. Pro Isomerization in MLL1 PHD3-Bromo Cassette Connects H3K4me Readout to CyP33 and HDAC-Mediated Repression. Cell. 2010;141:1183–1194. doi: 10.1016/j.cell.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang PY, Hom RA, Musselman CA, Zhu L, Kuo A, Gozani O, Kutateladze TG, Cleary ML. Binding of the MLL PHD3 finger to histone H3K4me3 is required for MLL-dependent gene transcription. J Mol Biol. 2010;400:137–144. doi: 10.1016/j.jmb.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park S, Osmers U, Raman G, Schwantes RH, Diaz MO, Bushweller JH. The PHD3 domain of MLL acts as a CYP33-regulated switch between MLL-mediated activation and repression. Biochemistry. 2010;49:6576–6586. doi: 10.1021/bi1009387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xia ZB, Anderson M, Diaz MO, Zeleznik-Le NJ. MLL repression domain interacts with histone deacetylases, the polycomb group proteins HPC2 and BMI-1, and the corepressor C-terminal-binding protein. Proc Natl Acad Sci U S A. 2003;100:8342–8347. doi: 10.1073/pnas.1436338100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hom RA, Chang PY, Roy S, Musselman CA, Glass KC, Selezneva AI, Gozani O, Ismagilov RF, Cleary ML, Kutateladze TG. Molecular mechanism of MLL PHD3 and RNA recognition by the Cyp33 RRM domain. J Mol Biol. 2010;400:145–154. doi: 10.1016/j.jmb.2010.04.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Musselman CA, Lalonde ME, Cote J, Kutateladze TG. Perceiving the epigenetic landscape through histone readers. Nat Struct Mol Biol. 2012;19:1218–1227. doi: 10.1038/nsmb.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Musselman CA, Kutateladze TG. Handpicking epigenetic marks with PHD fingers. Nucleic Acids Res. 2011;39:9061–9071. doi: 10.1093/nar/gkr613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dhar SS, Lee SH, Kan PY, Voigt P, Ma L, Shi X, Reinberg D, Lee MG. Trans-tail regulation of MLL4-catalyzed H3K4 methylation by H4R3 symmetric dimethylation is mediated by a tandem PHD of MLL4. Genes Dev. 2012;26:2749–2762. doi: 10.1101/gad.203356.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ali M, Rincon-Arano H, Zhao W, Rothbart SB, Tong Q, Parkhurst SM, Strahl BD, Deng LW, Groudine M, Kutateladze TG. Molecular basis for chromatin binding and regulation of MLL5. Proc Natl Acad Sci U S A. 2013;110:11296–11301. doi: 10.1073/pnas.1310156110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lemak A, Yee A, Wu H, Yap D, Zeng H, Dombrovski L, Houliston S, Aparicio S, Arrowsmith CH. Solution NMR Structure and Histone Binding of the PHD Domain of Human MLL5. PLoS One. 2013;8:e77020. doi: 10.1371/journal.pone.0077020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Musselman CA, Kutateladze TG. PHD fingers: epigenetic effectors and potential drug targets. Mol Interv. 2009;9:314–323. doi: 10.1124/mi.9.6.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li H, Ilin S, Wang W, Duncan EM, Wysocka J, Allis CD, Patel DJ. Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature. 2006;442:91–95. doi: 10.1038/nature04802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wysocka J, Swigut T, Xiao H, Milne TA, Kwon SY, Landry J, Kauer M, Tackett AJ, Chait BT, Badenhorst P, Wu C, Allis CD. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006;442:86–90. doi: 10.1038/nature04815. [DOI] [PubMed] [Google Scholar]
- 33.Wang GG, Song J, Wang Z, Dormann HL, Casadio F, Li H, Luo JL, Patel DJ, Allis CD. Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature. 2009;459:847–851. doi: 10.1038/nature08036. [DOI] [PMC free article] [PubMed] [Google Scholar]