

Abstract
RRP1B (ribosomal RNA processing 1 homolog B) was first identified as a metastasis susceptibility gene in breast cancer through its ability to modulate gene expression in a manner that can be used to accurately predict prognosis in breast cancer. However, the mechanism(s) by which RRP1B modulates gene expression is currently unclear. Many RRP1B binding candidates are involved in alternative splicing, a mechanism of gene expression regulation that is increasingly recognized to be involved in cancer progression and metastasis. One such target is SRSF1 (SF2/ASF), an essential splicing regulator that also functions as an oncoprotein. Earlier studies demonstrated that splicing and transcription occur concurrently and are coupled processes. Given that RRP1B regulates transcriptional activity, we hypothesized that RRP1B also regulates the expression of alternative mRNA isoforms through its interaction with SRSF1. Interaction between RRP1B and SRSF1 was verified by co-immunoprecipitation and co-immunofluorescence. Treatment of cells with transcriptional inhibitors significantly increased this interaction, demonstrating that the association of these two proteins is transcriptionally regulated. To assess the role of RRP1B in the regulation of alternative isoform expression, RNA-seq data were generated from control and Rrp1b-knockdown cells. Knockdown of Rrp1b induced a significant change in isoform expression in over 600 genes compared to control cell lines. This was verified by qRT-PCR using isoform-specific primers. Pathway enrichment analyses identified cell cycle and checkpoint regulation to be those most affected by Rrp1b knockdown. These data suggest that RRP1B suppresses metastatic progression by altering the transcriptome through its interaction with splicing regulators such as SRSF1.
Keywords: Metastasis, RRP1B, germline modifiers, RNA-seq, alternative splicing, breast cancer
Introduction
RRP1B (ribosomal RNA processing 1 homolog B) was first identified as a metastasis susceptibility gene through expression quantitative trait locus analysis of polyoma middle-T transgene-induced mammary tumors. Studies of these tumors revealed that the expression of Rrp1b highly correlated with the expression of extracellular matrix genes (1). Abnormal regulation of extracellular matrix genes is commonly observed in human and mouse tumors, and serves a predictive signature of metastases (2, 3). Microarray analyses demonstrated that ectopic expression of Rrp1b in mouse mammary tumor cell lines has profound effects on global gene expression. In addition, an expression signature generated from these microarray data accurately predicted overall survival in multiple breast cancer patient datasets (1). Finally, the importance of this gene as a germline suppressor of tumor progression and metastasis in human breast cancer was further underscored by the observation that a non-synonymous coding polymorphism in RRP1B (1421C>T; P436L; rs9306160) is reproducibly associated with metastasis-free survival in multiple human breast cancer cohorts (1, 4).
Human RRP1B is composed of 758 amino acids, and contains an amino-terminal NOP52 domain, and three nuclear localization signals located near the carboxy-terminus. Mouse RRP1B is 724 amino acids long and shares 62% sequence similarity with human RRP1B. RRP1B interacts with chromatin as well as rRNA transcripts, and its localization is almost entirely nuclear, with the majority of RRP1B being found in the nucleolus (5, 6). The mechanism underlying the functions of RRP1B largely remains to be identified. Characterization of potential binding candidates of RRP1B through tandem affinity purification and mass spectrometry provided some insight on this matter (5). A large number of these candidates were involved in alternative mRNA splicing, two prominent examples of which are SRSF1 (SF2/ASF) and CROP. SRSF1 is an essential splicing regulator that assists in the formation of the spliceosome by binding with U1 snRNP (7), and influences the selection of alternative splice sites (8, 9). CROP is the human homolog of yeast Luc7p, which is an essential component of yeast U1 snRNP (10), and it has been shown to interact with SRSF1 through yeast two-hybrid assays (11).
Alternative mRNA splicing is an essential method of regulating eukaryotic gene expression in which a single pre-mRNA is subsequently spliced to generate various transcripts yielding different protein products. Cells or tissues control alternative splicing in response to different physiological states. Dysregulation of mRNA splicing in most cases leads to severe diseases, including cancer (12). In cancerous tissues, it was found that the level of mRNA splicing and the number of transcript variants are comparable to those of normal tissues (13). Yet, neoplastic tissues do express an altered transcriptome characterized by tumor-specific isoforms (14–16). It is unclear whether this is a direct cause or a secondary effect of tumor progression (17).
The aim of this study is to examine whether RRP1B plays a role in regulating mRNA splicing. RNA-sequencing (RNA-seq) was performed to measure changes in isoform expression in response to Rrp1b knockdown, and biochemical approaches were used to assess the interaction of RRP1B and additional proteins involved in splicing. RRP1B was confirmed to interact with the splicing regulator SRSF1 and spliceosomal protein CROP. Knockdown of Rrp1b increased the metastatic activity of mouse mammary tumor cells and caused a significant change in isoform expression in 658 different genes. Based on these findings, we conclude that RRP1B functions as a metastasis suppressor by regulating gene expression, most likely at the mRNA level.
Results
RRP1B interacts with the splicing regulator SRSF1
The splicing regulator and oncoprotein SRSF1 (18) was identified as a potential binding factor of RRP1B (5, 6). Co-immunoprecipitation of endogenous RRP1B and SRSF1 was confirmed in MDA-MB-231 cells (Figure 1A). Co-localization of the two proteins was observed in speckles in the nucleus of cells overexpressing wild-type RRP1B (Figure 1B). To identify the region of RRP1B that interacts with SRSF1, 293T cells were transfected with a series of HA-tagged RRP1B deletion constructs; ΔN (Δ9–220), in which the highly conserved NOP52 domain was deleted, ΔMID (Δ221–435), which does not have the middle region containing the first nuclear localization signal, and ΔC (Δ452–749), in which the two nuclear localization signals in the C-terminus were deleted (5). Interaction with SRSF1 that was observed with wild-type RRP1B was lost upon deletion of the C-terminus of RRP1B, implying that this portion of RRP1B mediates the interaction between these two proteins (Figure 1C). However, deletion of the C-terminus of RRP1B has been shown to disrupt its localization and cause partial redistribution into the cytoplasm (5). Therefore, although a significant amount remains in the nucleus, the loss of interaction between RRP1B and SRSF1 observed with the ΔC construct might be due, in part, to the redistribution of RRP1B to the cytoplasm.
Figure 1. Co-immunoprecipitation and co-localization of RRP1B with SRSF1 and CROP.

A. Western blot analysis of co-immunoprecipitation of endogenous RRP1B and SRSF1. B. Co-immunofluorescence of full-length HA-tagged RRP1B and endogenous SRSF1. Scale bar measures 10 μM. C. Co-immunoprecipitation of HA-tagged RRP1B constructs with endogenous SRSF1 and CROP. Lysates from 293T cells transfected with HA-tagged RRP1B were incubated with anti-HA for immunoprecipitation and blotted with anti-SRSF1 or anti-CROP. D. Co-immunofluorescence of full-length HA-tagged RRP1B and endogenous CROP. Scale bar measures 10 μM.
In addition to SRSF1, RRP1B was found to interact with another factor involved in splicing, the spliceosomal protein CROP (Figure 1C, D). Co-immunoprecipitation patterns of CROP with RRP1B were similar to that of SRSF1, where interaction was lost with the deletion of the C-terminus, but not with the deletion of the N-terminus (Figure 1C).
The interaction between RRP1B and SRSF1 is transcriptionally regulated
Many studies have shown that transcription and splicing occur concurrently and that their mutual progression is highly intertwined (19–23). With this in mind, and given the fact that RRP1B is a critical regulator of metastasis-related transcriptional programs (1), we hypothesized that the interaction between RRP1B and SRSF1 is transcriptionally dependent. To test this hypothesis, transcription in MDA-MB-231 cells was inhibited using either 5,6-Dichloro-1-β-D-ribofuranosylbenzimidazole (DRB) or Actinomycin D (Act D). DRB inhibits transcription by binding near the ATP binding site of CDK9 (24), an essential kinase subunit of Positive Transcription Elongation Factor. Act D is a DNA intercalator that inhibits transcription by interfering with the progression of RNA polymerase (25).
In cells treated with 25 μg/mL DRB or 2.5 μg/mL and 5 μg/mL Act D, interaction between endogenous RRP1B and SRSF1 increased significantly compared to control (Figure 2A, lane 1–4). These concentrations inhibit transcription mediated by RNA polymerase I and II when added to the medium (26). Longer DRB treatment increased the degree of interaction between the two proteins (Figure 2A, lane 5, 6). This increase in interaction was confirmed through immunofluorescence as well, where co-localization of endogenous RRP1B and SRSF1 occurred in larger aggregates within the nuclei with 2 h DRB treatment (Figure 2B). We also observed redistribution of RRP1B from the nucleoli to the nucleoplasm with DRB treatment, which causes disruption of the nucleoli, as previously described (6). As the two inhibitors, DRB and Act D, function through distinct pathways to inhibit transcription, it is most likely that the interaction between RRP1B and SRSF1 is affected downstream of the transcriptional process, rather than through a common upstream pathway.
Figure 2. Inhibition of transcription enhances the interaction between RRP1B and SRSF1.

A. Co-immunoprecipitation of RRP1B and SRSF1 with DRB or Act D treatment. 293T cells treated with DMSO, 25 μg/mL DRB, or 2.5 μg/mL and 5 μg/mL Act D as indicated were used for immunoprecipitation with anti-RRP1B. Normal rabbit IgG was used as a negative control. B. Co-immunofluorescence of RRP1B and SRSF1 after DMSO or 25 μg/mL DRB treatment. 293T cells were treated with 2 h DMSO or DRB and collected for immunofluorescence. Co-localization was confirmed using confocal fluorescence microscopy. Scale bar measures 10 μM.
Knockdown of Rrp1b increases cell invasiveness in vitro and metastasis in vivo
The effects of stable shRNA-mediated knockdown of Rrp1b were examined in the highly metastatic mouse mammary tumor cell lines Mvt-1 (27) and 4T1 (28). For the Mvt-1 cell line, clonal isolates were selected by serial dilution. RRP1B knockdown in both Mvt-1 clones and 4T1 cells was confirmed by qRT-PCR and Western blotting (Figure 3A, B). Knockdown of RRP1B caused an increase in cell proliferation in both cell lines in culture (Figure 3C, D). When injected into the mammary fat pads of syngenic FVB/NJ mice, Rrp1b knockdown in the Mvt-1 cell line significantly reduced primary tumor burden and produced an approximately six-fold increase in pulmonary surface metastasis compared to controls (Figure 3E). This increase in metastasis with Rrp1b knockdown was reproduced with the orthotopic injection of the 4T1 cell cultures into the mammary fat pads of syngenic BALB/c mice (Figure 3F). To examine in vitro cell invasiveness, Mvt-1 clonal isolates were plated in control or Matrigel chambers. With overnight incubation, Rrp1b-knockdown cell lines displayed an average two-fold increase in invasion compared to control (Figure 3G). In addition, Rrp1b-knockdown cell lines formed more colonies in soft agar compared to control, demonstrating an increase in anchorage-independent cell growth (Figure 3H, I). Increase in cell proliferation and cell invasiveness was observed with the human breast carcinoma MDA-MB-231 cell line as well (Figure S1). Such changes in cell behavior are likely either primary events or secondary to the in vitro increase in cell proliferation rate following Rrp1b knockdown.
Figure 3. Knockdown of Rrp1b increases cell proliferation and invasiveness in vitro, and metastasis in vivo.

A, B. RRP1B expression in stable knockdown cell lines. A shRNA targeting mouse Rrp1b was transfected to stably knockdown RRP1B expression in the highly metastatic Mvt-1 and 4T1 cell line. Expression of mRNA was confirmed by qRT-PCR and Western blot. In the Western blot for Mvt-1, numbers indicate different clones for each stable cell line. C, D. Stable knockdown of RRP1B increased cell growth rate. Cells were plated at 2.5 × 104 cells per 12-well in duplicate and counted each day as indicated. * and ** indicate p = 0.03 and p = 0.02, respectively. E, F. Knockdown of RRP1B increased metastasis in vivo. After 4 weeks of injection, lungs were collected for examination of surface metastasis sites (ctrl, n = 20; Rrp1b kd, n = 20 for each cell line). G. Matrigel invasion assay with Mvt-1 stable cell lines. Each cell line was plated in triplicate. H, I. Soft agar assay.
RNA-seq analysis demonstrates that knockdown of Rrp1b induces differential gene and isoform expression
Based on the metastatic activity observed in vivo, duplicate control and Rrp1b-knockdown Mvt-1 clonal isolate cell lines were selected for RNA-seq. Gene expression analysis of the RNA-seq data demonstrated that 615 genes displayed significant changes in transcript levels following Rrp1b knockdown, with 362 up-regulated and 253 down-regulated genes at a q ≤ 0.05 genome-wide significance level (Figure 4A, Table S1). Pathway enrichment analysis using Ingenuity Pathway Analysis (IPA, www.ingenuity.com) indicated that cell cycle regulation was a major function of these genes affected by Rrp1b expression (Table 1).
Figure 4. Rrp1b knockdown causes a significant change in global gene and isoform expression.

A. A heatmap of differentially expressed genes from RNA-seq data in control and Rrp1b-knockdown stable cell lines. B. Differential isoform expression in control and Rrp1b-knockdown stable cell lines. A heat map was generated to display the ratio of isoforms that were up-regulated in the control and/or up-regulated in the knockdown cell lines for each gene. Note the middle section, shown in a light blue color across both sets, has a ratio of 0.5 for the control and the knockdown cell lines, indicating a complete switch in isoform expression. C. A total of 497 genes overlapped between the differential gene expression analysis and differential isoform expression analysis.
Table 1.
Pathway enrichment analysis of differentially expressed genes with Rrp1b knockdown.
| Ingenuity Canonical Pathways | −log(B-H p- value) | Molecules |
|---|---|---|
| Cell Cycle Control of Chromosomal Replication | 4.96 | MCM5, MCM3, MCM2, CDC6 (includes EG:23834), RPA1, CHEK2, MCM4, ORC1 (includes EG:18392), RPA2 |
| Cell Cycle: G2/M DNA Damage Checkpoint Regulation | 4.34 | CDC25C, MDM4, GADD45A, TOP2A, CCNB 2, PLK1, BRCA1, CHEK2, CHEK1, EP300 |
| Role of BRCA1 in DNA Damage Response | 4.34 | GADD45A, E2F1, BARD1, BRCA2, PLK1, RP A1, RBL1, BRCA1, CHEK2, E2F2, CHEK1 |
| p53 Signaling | 3.73 | TOPBP1, PERP, CCND1, CHEK1, SERPINE2, EP300, CCNG1, PCNA, MDM4, GADD45A, E2F1, BRCA1, CHEK2 |
| Role of CHK Proteins in Cell Cycle Checkpoint Control | 3.71 | PCNA, CDC25C, E2F1, RPA1, BRCA1, CHEK 2, E2F2, CHEK1 |
| EIF2 Signaling | 3.54 | RPL11, RPL27A, RPL27, RPS8, RPL31, RPL23A, RPL14, RPL37A, RPL37, RPL6, RPS27, RP S10, RPS26, RPL39 (includes EG:25347), RPL19, RPS3, RPLP1, RPL13 |
| Hereditary Breast Cancer Signaling | 3.54 | CDC25C, TUBG1, BARD1, CREBBP, RPA1, CCND1, CHEK1, EP300, GADD45A, HDAC11, E2F1, BRCA2, BRCA1, CHEK2 |
| Mitotic Roles of Polo-Like Kinase | 3.43 | CDC25C, ESPL1, CDC20, TGFB1 (includes EG:21803), CCNB2, PLK1, ANAPC13, Ccnb1/Gm5593, CHEK2, KIF11 |
| Molecular Mechanisms of Cancer | 2.77 | ADCY9, CDC25C, HAT1, CREBBP, CDKN2 D, SMAD6, AURKA, RBL1, CCND1, MAPK11, CHEK1, EP300, MYC, FOXO1 (includes EG:2308), RABIF, TGFB1 (includes EG:21803), E2F1, FZD6, BMP7, BRCA1, FZD 2, CHEK2, PRKD1, E2F2 |
| ATM Signaling | 2.48 | CDC25C, MDM4, GADD45A, CCNB2, BRCA 1, MAPK11, CHEK2, CHEK1 |
| DNA Double-Strand Break Repair by Homologous Recombination | 1.83 | GEN1, BRCA2, RPA1, BRCA1 |
| TGF-β Signaling | 1.81 | TGFB1 (includes EG:21803), SKI, CREBBP, SMAD6, BMP7, V DR, MAPK11, INHBB, EP300 |
| Mismatch Repair in Eukaryotes | 1.66 | PCNA, FEN1, RPA1, EXO1 (includes EG:26909) |
| Cell Cycle: G1/S Checkpoint Regulation | 1.64 | MYC, TGFB1 (includes EG:21803), HDAC11, E2F1, RBL1, CCND1, E 2F2 |
| VDR/RXR Activation | 1.47 | CYP24A1, FOXO1 (includes EG:2308), GADD45A, NCOA1, CEBPB (includes EG:1051), VDR, PRKD1, EP300 |
To examine the role of RRP1B in the regulation of alternative isoforms, we analyzed differential isoform expression between control and knockdown cell lines. Similar to the results seen in gene expression, a substantial difference was observed in isoform expression; 699 isoforms, representing 658 different genes, were identified as having a significant change in expression with Rrp1b knockdown (Figure 4B, Table S2). Four hundred and ninety seven of the 699 isoforms displayed differential gene expression, with the remaining 202 isoforms not being identified in the aforementioned gene expression analysis (Figure 4C). The locations of isoform-specific exons were profiled by comparing them to a transcript constructed in silico that included all exons from annotated isoforms. These data demonstrated that the majority of differential exons regulated by RRP1B are located internally between the first and last exon. Changes in the first exon may be due to differential promoter utilization, whereas changes in internal exons indicate true differences in mRNA splicing. For all 699 isoforms profiled, 59% of differential exons were located internally within the transcript (e.g., Tapbp-001 exon 3; Figure 5), 27.2% were in the 5′ region or the first exon (e.g., Arl8a-001 exon 1; Figure 5), and 13.8% were in the 3′ region or the last exon.
Figure 5. Validation of isoform-specific regulation by RRP1B via qRT-PCR.

RNA from control and Rrp1b-knockdown stable Mvt-1 cell lines was used for qRT-PCR. Isoform-specific regions were selected for target genes identified through RNA-seq analysis. For each gene, the top isoform was identified by RNA-seq to have a significant fold-change in expression with Rrp1b knockdown. Primers were designed so that at least one primer extended across an exon junction. Exons are depicted in relation to their genomic location.
Pathway enrichment analysis was performed to gain more insight on the isoforms affected by Rrp1b knockdown. Depending on the number of molecules identified to support each pathway, there was some variation in the significance (determined by Benjamini-Hochberg correction of p-values) of each pathway between the gene and isoform expression. For example, p53 signaling ranked as the 4th most significant pathway in the gene analysis with a −log(B-H p-value) of 3.73, whereas it ranked 9th in the isoform analysis with a −log(B-H p-value) of 3.59. Despite these differences in p-values, the majority of the significant pathways, which was determined by a −log(B-H p-value) of 1.30 or higher (p-value ≤ 0.05), overlapped between the two analyses. The top pathways for differential gene and isoform expression analyses are shown in Table 1 and 2, respectively. As was the case with the gene expression analyses, the most highly represented molecular and cellular functions and pathways were those involved in cell-cycle and checkpoint regulation (Table 2). Surprisingly, the differentially expressed isoforms were not of genes involved in pathways involved in alternative mRNA splicing. Among the 54 splicing-related factors screened (29), which included hnRNPs and SR proteins, only two genes, Hnrnph2 (30, 31) and Srsf5 (Srp40) (32, 33) were identified to have differential isoform expression with the knockdown of Rrp1b. Thus, the large-scale change in isoform expression observed with Rrp1b knockdown is likely a direct effect of RRP1B itself, rather than a secondary effect caused by the factors regulated by RRP1B.
Table 2.
Pathway enrichment analysis of differentially expressed isoforms with Rrp1b knockdown.
| Ingenuity Canonical Pathways | −log(B-H p- value) | Molecules |
|---|---|---|
| Cell Cycle Control of Chromosomal Replication | 5.81 | MCM5, MCM3, MCM6, MCM2, CDC6 (includes EG:23834), RPA1, MCM4, DBF4 (includes EG:10926), ORC1 (includes EG:18392), RPA2 |
| EIF2 Signaling | 5.52 | RPL11, RPL22, EIF3H, RPL27, RPS19, RPS8, RPL14, RPL37A, RPL37, RPS27, RPL6, RPS10, RPS5, RPS26, RPL39 (includes EG:25347), RPS9, MRAS, RPL19, RPS3, RPL P1, RPL13A, RPL13, RPS14 |
| Cell Cycle: G2/M DNA Damage Checkpoint Regulation | 5.04 | CDC25C, GADD45A, CDKN1A, TOP2A, CC NB2, MDM2, PLK1, BRCA1, CDK1, CHEK1, EP300 |
| VDR/RXR Activation | 4.12 | SPP1 (includes EG:20750), CYP24A1, CEBPB (includes EG:1051), EP300, FOXO1 (includes EG:2308), NCOA2, GADD45A, PRKCD, CD KN1A, NCOA1, VDR, PRKD1, PRKCA |
| Mitotic Roles of Polo-Like Kinase | 3.66 | CDC25C, ESPL1, CDC20, HSP90AB1, TGFB 1 (includes EG:21803), CCNB2, HSP90AA1, PLK1, ANA PC13, CDK1, KIF11 |
| Hereditary Breast Cancer Signaling | 3.64 | CDC25C, TUBG1, CREBBP, RPA1, CCND1, CDK1, CHEK1, EP300, FANCB, GADD45A, HDAC11, CDKN1A, MRAS, BRCA2, BRCA1 |
| HER-2 Signaling in Breast Cancer | 3.63 | ITGB2, CCNE1, FOXO1 (includes EG:2308), PRKCD, CDKN1A, MRAS, MDM2, CCND1, ITGB5, PRKD1, AREG/AREGB, PR KCA |
| Aryl Hydrocarbon Receptor Signaling | 3.59 | MDM2, CCND1, RARG, CHEK1, EP300, CCN A2, CCNE1, CCND3, HSP90AB1, NCOA2, T GFB1 (includes EG:21803), RARA, CDKN1A, HSP90AA1, D HFR, ALDH5A1 |
| p53 Signaling | 3.59 | TOPBP1, PERP, MDM2, CCND1, BIRC5, CH EK1, SERPINE2, EP300, CCNG1, PCNA, GA DD45A, CDKN1A, BRCA1 |
| Molecular Mechanisms of Cancer | 3.41 | RBL1, CCND1, EP300, CHEK1, NLK, TGFB1 (includes EG:21803), MRAS RCA1, PRKD1, PRKCA, CDC25C, HAT1, CDKN2D, CREBBP, SMAD 6, MDM2, AURKA, CCNE1, CCND3, RHOQ, RABIF, FOXO1 (includes EG:2308), PRKCD, CDKN1A, FZD6, BMP7, ADCY7 |
| mTOR Signaling | 3.23 | EIF3H, RPS19, RPS8, VEGFC, PDGFC, RPS27, RPS10, RHOQ, RPS5, RPS26, PRKCD, RPS9, MRAS, RPS3, PRKD1, RPS14, PRKCA, EIF4 B |
| Role of BRCA1 in DNA Damage Response | 2.81 | FANCB, GADD45A, CDKN1A, BRCA2, PLK 1, RPA1, RBL1, BRCA1, CHEK1 |
| Role of CHK Proteins in Cell Cycle Checkpoint Control | 2.81 | PCNA, CDC25C, CDKN1A, RPA1, BRCA1, C DK1, CHEK1 |
| Mismatch Repair in Eukaryotes | 2.64 | PCNA, FEN1, RPA1, POLD1, EXO1 (includes EG:26909) |
| Cyclins and Cell Cycle Regulation | 2.40 | CCNA2, CCNE1, CCND3, TGFB1 (includes EG:21803), HDAC11, CDKN1A, CDKN2D, C CNB2 CCND1 CDK1 CNB2, CCND1, CDK1 |
| ATM Signaling | 2.38 | CDC25C, GADD45A, CDKN1A, CCNB2, MD M2, BRCA1, CDK1, CHEK1 |
To confirm the differential regulation of alternative isoforms by RRP1B, qRT-PCR expression analysis was performed with Mvt-1 control and Rrp1b-knockdown samples. Isoform-specific primers were designed to either extend across a unique exon junction or lie within an isoform-specific exon (Figure 5, arrows). For instance, for Tubb5-001 and Tubb5-003, the junction region spanning between exon 1 and 2 is unique for each isoform as the sequence for exon 2 differs between the two isoforms. For expression analysis, two isoforms were selected for each gene with one isoform being identified by RNA-seq as being dysregulated by Rrp1b knockdown (Figure 5, top isoform) and the other having no significant change (Figure 5, bottom isoform). These analyses confirmed that Rrp1b regulates gene expression in an isoform-specific manner.
Splicing patterns vary greatly across different species and tissues (34), and the level of conservation for orthologous genes ranges from 11 to 20% (35–37). To see if the change in splicing observed with Rrp1b knockdown was cell-line specific, the expression of the isoforms in Figure 5 were also examined in clonal isolates of stable 4T1 Rrp1b-knockdown cells and compared to that of the Mvt-1 cell lines (Figure S2). Many of the isoforms changed expression in the same direction with similar Log(fold change) values, while a few of them, such as Arl8a-003 and Rpa2-001, demonstrated divergent expression patterns. This variation is not unexpected given the different genetic background of the two cell lines. Further, it is unlikely that these minor differences in splicing have a significant impact upon metastasis since knockdown of Rrp1b in both Mvt-1 and 4T1 cell lines resulted in very similar in vitro and in vivo behaviors.
Discussion
With the discovery of the diverse functions of RNA transcripts, increasing interest has been placed upon the role of alternative splicing in tumor progression. Although neoplastic tissues have been reported to display a perturbation in mRNA isoform expression compared to matched normal tissues (14–16, 38), the precise role of this altered transcriptome and splicing pattern in tumorigenesis remains uncertain. Previously, it was shown that the metastasis suppressor Nm23-H1 regulates changes in the expression of RNA post-transcriptional modification proteins, including Gemin5, a component of the spliceosome (39). Yet, a direct role for metastasis modifiers in splicing has not yet been reported, although it seems that many factors involved in splicing have a variety of cellular functions (40). Here, using RNA-seq in combination with other biochemical methods, the metastasis modifier RRP1B has been demonstrated to directly regulate alternative isoform expression. This suggests that alternative splicing, and the diverse isoforms generated from that process, may play a role in the regulation of various factors implicated in metastasis.
Alternative mRNA splicing is a fundamental cellular mechanism involved in the regulation of gene expression, and it has been reported that the majority of human genes is alternatively spliced to express different isoforms (41). It is a precisely regulated process that consists of specific splice recognition sites, splicing machinery, and other regulatory factors. Defects in splicing can occur at any stage of the process, and have been shown to be features of a wide-range of diseases, including cancer. Since alternative splicing is a tightly-regulated process, mutations in most, but not all, splicing factors result in a change in splicing patterns (42). In addition, many studies demonstrate that relatively few genes have altered splicing patterns in primary tumor tissue compared to matched normal tissue (43, 44).
Considering these facts, the fundamental question posed by our research is whether the metastasis modifier RRP1B is modulating cellular splicing patterns. Our data does indeed indicate that this is the case since our analysis of the transcriptome of the highly metastatic Mvt-1 cell line clearly demonstrates that Rrp1b knockdown induces widespread changes in isoform expression. Specifically, the majority of the isoform-specific exons induced by knockdown of Rrp1b were found to be located internally within the mature mRNA. Given that splicing is used to generate different mature mRNA isoforms from the same pre-mRNA, we argue that these changes in internal exon composition upon Rrp1b knockdown can only arise due to changes in splicing regulation. In addition, interaction between RRP1B and the splicing regulator SRSF1, as well as with spliceosomal protein CROP (Figure 1), supports the direct involvement of RRP1B in splicing, rather than indirect regulation of splicing, such as that of Nm23-H1 (39). We would, however, like to stress that the mechanism of action of RRP1B is likely not entirely dependent on its role in splicing regulation. In particular, we acknowledge that a sizable proportion of the isoform-specific exons expressed as a consequence of Rrp1b knockdown are located at the 5′ region of the transcript. Expression of these isoforms has likely been induced through the utilization of alternative promoters. Our previous work has demonstrated that RRP1B interacts with a variety of chromatin-associated factors. Therefore, expression of this type of isoform with alternative 5′ exons is likely a consequence of RRP1B interacting with these chromatin-associated binding partners rather than splicing regulators.
An extensive study on the nuclear protein interactome of RRP1B by Chamousset et al. found that it was enriched with ribosomal subunit proteins, particularly those of the large subunit (6). This suggests that RRP1B plays an important role in cell growth and metabolism, as the ribosome is regulated in response to signals controlling these pathways. A large number of genes encoding components of both the 60S large and 40S small ribosomal subunits displayed differentially expressed isoforms with the knockdown of Rrp1b. Interestingly, all of the dysregulated components of the small ribosomal subunit except for Rps19, as well as 7 of the 12 dysregulated large ribosomal subunit genes were those encoding the previously identified components of the RRP1B protein interactome (highlighted in Table S2). This demonstrates that RRP1B not only interacts with ribosomal protein subunits in the nucleus, but also controls their expression. Not surprisingly, the cellular pathways most affected by the knockdown of Rrp1b were those involved in cell cycle regulation. Collectively, these findings suggest a greater role for RRP1B as a regulator of cell growth than previously reported, which explains the significant change in in vitro cell proliferation observed here.
Many studies demonstrate that splicing and transcription occur concurrently, rather than sequentially, and that splicing is affected by the rate and progression of transcription, which is affected by events at the chromatin, such as histone modification (19–23). An earlier study that identified potential binding factors of RRP1B through tandem affinity purification found that these proteins can be largely classified as either chromatin-associated or splicing-related factors, and confirmed RRP1B interaction with chromatin-associated factors (5). In addition, we observed interaction between RNA polymerase II and RRP1B through co-immunoprecipitation (Figure S3). These studies combined with our findings here suggest a role for RRP1B as a mediator between the transcription complex and splicing factors (Figure 6), and that differential levels of RRP1B impacts the transcriptome, and ultimately metastasis. To our knowledge, this study is the first to directly link a metastasis modifier to regulation of alternative splicing machinery and subsequent alterations in cellular mRNA splicing patterns.
Figure 6. RRP1B acts as a mediator between chromatin-associated transcription factors and splicing regulators.
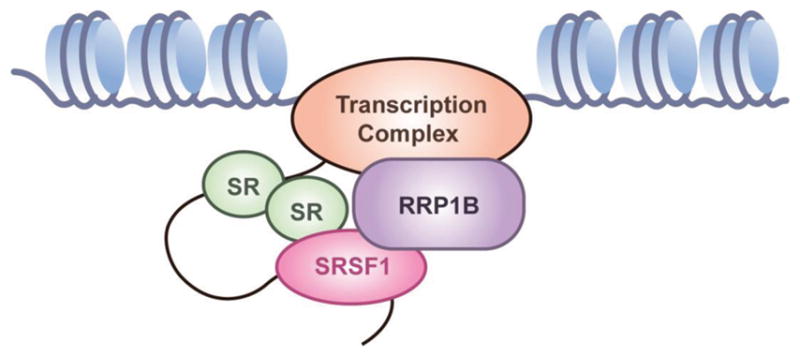
Previous studies and our results shown here collectively suggest a role for RRP1B at the chromatin as a mediator between transcription and splicing factors to regulate mRNA expression.
Materials and Methods
Cell culture and transient transfection
Cell lines were a gift from Dr. Lalage Wakefield, NCI/NIH. All cell lines were maintained in DMEM supplemented with 10% FBS and 1% Penicillin-Streptomycin (Gibco, Grand Island, NY). For transient transfections, cells were plated at 1 × 105 cells in 12-well plates for RNA isolation or at 2 × 106 cells in 100 mm plates for protein isolation. The next day, transfection was performed using SuperFect Transfection Reagent (QIAGEN, Valencia, CA) following manufacturer’s protocol. Cells were collected 48 h after transfection for analyses.
Generation of stable RRP1B-knockdown cell lines
For Rrp1b knockdown in the Mvt-1 and 4T1 cell lines, a shRNA targeting mouse Rrp1b inserted into the pSuper.retro.puro vector (OligoEngine, Seattle, WA) was transfected along with retroviral packaging vectors. Mvt-1 and 4T1 cells expressing the vector were selected in 10 μg/mL or 5 μg/mL of puromycin, respectively. Stable RRP1B-knockdown cells were also established in MDA-MB-231 cells using shRNA targeting human RRP1B. Knockdown at the protein level was confirmed using polyclonal antibodies against RRP1B (6). Oligo sequences for shRNAs are provided in Table S3.
RNA isolation and qRT-PCR
Total RNA was isolated from cells using the RNeasy mini kit (QIAGEN, Valencia, CA) following manufacturer’s protocol with a 15 min DNase treatment before elution. RNA concentration was measured using NanoDrop (Wilmington, DE), and 1 μg of total RNA was used per reverse transcription reaction in a final volume of 20 μL. Reverse transcription was performed using cDNA Synthesis Kit (Bio-Rad, Hercules, CA). The obtained cDNA was then diluted 10-fold, and 1 μL was used for each 5 μL real-time PCR reaction. Primers used for qRT-PCR are provided in Table S3.
Co-immunoprecipitation and Western blotting
For co-immunoprecipitation, cells in 100 mm plates were collected with 250 μL 1X lysis buffer (600 mM NaCl, 0.2 mM EGTA, 40 mM Tris pH 7.4, 4 mM MgCl2, 2% Triton-X). After 30 min incubation on ice, samples were centrifuged at 14,000 g at 4 °C for 5 min. From each sample, 5 μL of the supernatant was removed as Input, and the balance was diluted to a final volume of 800 μL and incubated with rotation at 4 °C with one of the following antibodies: anti-HA (Roche, Indianapolis, IN) for HA-tagged RRP1B constructs, anti-RRP1B (Sigma, St. Louis, MO) for endogenous RRP1B, or IgG (Millipore, Billerica, MA) as a negative control. After 2 h, 20 μL of DynaBeads (Invitrogen, Grand Island, NY) washed and resuspended in 1X lysis buffer were added to each immunoprecipitation and incubated with rotation overnight at 4 °C. The following day, samples were washed with 1X lysis buffer and resuspended in 2X sample buffer. Samples were separated on a 4–12% Bis-Tris gel (Invitrogen).
Immunofluorescence and microscopy
Cells were plated at 1 × 105 cells/mL in 2-well glass chamber slides. Following transfection with HA-tagged RRP1B or treatment with DMSO, DRB, or Act D, cells were washed three times with 1X PBS and fixed with Histochoice Tissue Fixative (AMRESCO, Solon, OH) for 15 min. Cells were washed three additional times with 1X PBS and permeated with Triton 100-X for 15 min. After blocking with 0.5% BSA in 0.1% Tween PBS for 30 min, cells were incubated with the appropriate antibodies (anti-HA, Roche 11 867 423 001; anti-RRP1B, Sigma HPA017893; anti-SF2, Invitrogen 32–4600; anti-CROP, Sigma HPA018475) for co-immunofluorescence in 0.1% BSA in 0.1% Tween PBS for 1 h at room temperature with rotation. After washing three times with 0.1% Tween PBS, cells were incubated with the appropriate secondary fluorescent antibodies (Alexa Fluor 488 or Alexa Fluor 594, Invitrogen) for 1 h at room temperature with rotation. Cells were then mounted with VECTASHIELD with DAPI (Vector Laboratories, Burlingame, CA) and visualized with LSM 510 NLO (Carl Zeiss, Thornwood, New York).
Orthotopic mammary fat pad injection
Female FVB/NJ and BALB/c mice were purchased from Jackson Laboratory (Bar Harbor, ME). Injections and metastasis assays were performed as previously described (1). Briefly, 1 × 105 cells in 100 μL saline were orthotopically implanted into the mammary fat pads of 6 to 8-week-old female mice. Lungs and tumors were collected 4 weeks later. All animal experiments were performed in compliance with the National Human Genome Research Institute Animal Care and Use Committee’s guidelines.
Soft agar and Matrigel invasion assays
For soft agar assays, Mvt-1 and 4T1 cells were plated at 2 × 103 cells per 24-well in 0.33% agar, and incubated for 7 days. MDA-MB-231 cells, 4 × 103 cells were plated in 0.33% agar in 24-well plates and allowed to grow for 15 days until significant colony formation was observed. Cells were then stained with 0.005% crystal violet for colony counting. For Matrigel invasion assays, control and Rrp1b-knockdown stable Mvt-1 cell lines were plated at 7.5 × 103 cells per 24-well control Transwell or Matrigel invasion chambers (BD Biosciences, San Jose, California) in DMEM 0% FBS and chambers were placed in DMEM 10% FBS. Chambers were collected and stained with crystal violet the next day for counting.
RNA-seq
For mRNA library construction, mRNA from 10 μg of total RNA was poly-A selected using Dynal Oligo (dT) beads (Invitrogen) and sheared to ~400 b using Covaris S2. The sample was concentrated to 25 μl using an Amicon Ultra 20K filter. Reverse transcription was performed using random hexamers and SuperScript® Double-Stranded cDNA Synthesis Kit (Invitrogen). A library was constructed from the resulting cDNA according to the Illumina protocol in the Paired-End DNA Sample Prep Oligo Only Kit. All additional enzymes were purchased from New England Biolabs (Ipswich, MA) with the exception of Platinum Pfx (Invitrogen). After paired-end adapter ligation, the sample was size selected on a 2% agarose gel. Test amplification was performed by removing aliquots of a PCR reaction every 2 cycles from 4–16 cycles to insure that the library was not over amplified. These aliquots were evaluated on a 2% agarose gel and an optimal cycle number was selected for subsequent large scale amplification. Amplification reactions were cleaned up using two rounds of Agencourt AMPure Beads (Beckman Coulter, Indianapolis, IN). Libraries were quantitated by qPCR using Power Sybr Green PCR Master Mix (Applied Biosystems, Grand Island, NY). Libraries were sequenced on the Illumina GAIIx platform using ver. 4 chemistry to obtain 76-base-paired reads. Data was processed using RTA1.8.70.0 and CASAVA 1.7.0. Those reads that passed the Illumina platform quality check were kept for downstream analyses.
RNA-seq transcript assembly and identification of differentially expressed genes and isoforms
A reference genome-guided strategy was used to assemble transcripts from the RNA-seq data. Sequence reads were aligned to the mouse genome (mm9; downloaded from ENSEMBL) using TopHat (v. 1.2.0) (45) with the following parameters: -p 3; -m 2; -r 348; -G (GTF of protein-coding transcripts downloaded from ENSEMBL (e64)). TopHat alignments were supplied to Cufflinks (v. 1.1.0) (46) for transcript assembly using the following parameters: -p 6; -N (upper quartile normalization); -b [mm9 genome FASTA sequence for bias correction]; -M (GTF from ENSEMBL (e61) of rRNA, tRNA, and mtRNA genomic locations for masking); GTF of protein-coding transcripts from ENSEMBL (e64). Differentially expressed genes and transcripts were identified using Cuffdiff (v. 1.1.0) with the following parameters: -p 6; -N (upper quartile normalization); -b (mm9 genome fasta sequence for bias correction (47)); -M (GTF of rRNA, tRNA, and mtRNA genomic locations for masking); GTF of protein-coding transcripts from ENSEMBL (e64). Genes and isoforms having a q-value ≤ 0.05 were considered differentially expressed, as reported in the gene_exp.diff and isoform_exp.diff output files from Cuffdiff. Pathway enrichment was analyzed using IPA (Ingenuity Systems, Redwood City, CA) using a Benjamini-Hochberg correction on p-values generated from a Fisher’s Exact test.
To determine the frequency at which the first exon, internal exons, or the last exon of a gene is alternatively expressed, a primary transcript was constructed in silico that included all exons from the annotated isoforms (ENSEMBL e64) for a given protein-coding gene. The differentially expressed isoforms were compared to these primary transcripts for differences in the ENSEMBL-annotated genomic positions of exon start and stop locations. A record was kept if differences in exon usage and/or exon junction positions occurred in the initial 5′ exon (first exon), final 3′ exon (last exon), or an exon that is not the initial 5′ or final 3′ exon (internal).
Supplementary Material
Knockdown of RRP1B in MDA-MB-231 cells increases cell proliferation and cell invasiveness. A. RRP1B expression in stable knockdown cell lines was confirmed by qRT-PCR and Western blot. B. Cell proliferation assay. Cells were plated at 2.5 × 104 cells per 12-well in duplicate and counted each day as indicated. C. Soft agar assay.
Comparison of isoform expression with the knockdown of Rrp1b in Mvt-1 and 4T1 cells.
RRP1B interacts with RNA polymerase II. Interaction between RRP1B and RNA polymerase II was determined by co-immunoprecipitation.
List of differentially expressed genes. 1FPKM (fragments per kilobase pair of exon per fragments mapped).
List of differentially expressed isoforms. Rpl and Rps genes that express proteins reported to interact with RRP1B are highlighted in pink and yellow, respectively.
Oligo and primer sequences.
Acknowledgments
This work was supported by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health, USA. We thank Stephen Wincovitch and Sujata Bupp for technical assistance, and Drs. Kent Hunter, Tom Misteli, and Larry Brody for critical review of the manuscript.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
References
- 1.Crawford NP, Qian X, Ziogas A, Papageorge AG, Boersma BJ, Walker RC, et al. Rrp1b, a new candidate susceptibility gene for breast cancer progression and metastasis. PLoS Genet. 2007 Nov;3(11):e214. doi: 10.1371/journal.pgen.0030214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003 Jan;33(1):49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- 3.van de Vijver MJ, He YD, van’t Veer LJ, Dai H, Hart AA, Voskuil DW, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002 Dec 19;347(25):1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- 4.Hsieh SM, Look MP, Sieuwerts AM, Foekens JA, Hunter KW. Distinct inherited metastasis susceptibility exists for different breast cancer subtypes: a prognosis study. Breast Cancer Res. 2009;11(5):R75. doi: 10.1186/bcr2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Crawford NP, Yang H, Mattaini KR, Hunter KW. The metastasis efficiency modifier ribosomal RNA processing 1 homolog B (RRP1B) is a chromatin-associated factor. J Biol Chem. 2009 Oct 16;284(42):28660–73. doi: 10.1074/jbc.M109.023457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chamousset D, De Wever V, Moorhead GB, Chen Y, Boisvert FM, Lamond AI, et al. RRP1B targets PP1 to mammalian cell nucleoli and is associated with Pre-60S ribosomal subunits. Mol Biol Cell. 2010 Dec;21(23):4212–26. doi: 10.1091/mbc.E10-04-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu JY, Maniatis T. Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell. 1993 Dec 17;75(6):1061–70. doi: 10.1016/0092-8674(93)90316-i. [DOI] [PubMed] [Google Scholar]
- 8.Krainer AR, Conway GC, Kozak D. The essential pre-mRNA splicing factor SF2 influences 5′ splice site selection by activating proximal sites. Cell. 1990 Jul 13;62(1):35–42. doi: 10.1016/0092-8674(90)90237-9. [DOI] [PubMed] [Google Scholar]
- 9.Zuo P, Manley JL. Functional domains of the human splicing factor ASF/SF2. EMBO J. 1993 Dec;12(12):4727–37. doi: 10.1002/j.1460-2075.1993.tb06161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Puig O, Bragado-Nilsson E, Koski T, Seraphin B. The U1 snRNP-associated factor Luc7p affects 5′ splice site selection in yeast and human. Nucleic Acids Res. 2007;35(17):5874–85. doi: 10.1093/nar/gkm505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Umehara H, Nishii Y, Morishima M, Kakehi Y, Kioka N, Amachi T, et al. Effect of cisplatin treatment on speckled distribution of a serine/arginine-rich nuclear protein CROP/Luc7A. Biochem Biophys Res Commun. 2003 Feb 7;301(2):324–9. doi: 10.1016/s0006-291x(02)03017-6. [DOI] [PubMed] [Google Scholar]
- 12.Venables JP. Aberrant and alternative splicing in cancer. Cancer Res. 2004 Nov 1;64(21):7647–54. doi: 10.1158/0008-5472.CAN-04-1910. [DOI] [PubMed] [Google Scholar]
- 13.Kim E, Goren A, Ast G. Insights into the connection between cancer and alternative splicing. Trends Genet. 2008 Jan;24(1):7–10. doi: 10.1016/j.tig.2007.10.001. Research Support, Non-U.S. Gov’t. [DOI] [PubMed] [Google Scholar]
- 14.Xu Q, Lee C. Discovery of novel splice forms and functional analysis of cancer-specific alternative splicing in human expressed sequences. Nucleic Acids Res. 2003 Oct 1;31(19):5635–43. doi: 10.1093/nar/gkg786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Z, Lo HS, Yang H, Gere S, Hu Y, Buetow KH, et al. Computational analysis and experimental validation of tumor-associated alternative RNA splicing in human cancer. Cancer Res. 2003 Feb 1;63(3):655–7. [PubMed] [Google Scholar]
- 16.Hui L, Zhang X, Wu X, Lin Z, Wang Q, Li Y, et al. Identification of alternatively spliced mRNA variants related to cancers by genome-wide ESTs alignment. Oncogene. 2004 Apr 15;23(17):3013–23. doi: 10.1038/sj.onc.1207362. [DOI] [PubMed] [Google Scholar]
- 17.Wang GS, Cooper TA. Splicing in disease: disruption of the splicing code and the decoding machinery. Nat Rev Genet. 2007 Oct;8(10):749–61. doi: 10.1038/nrg2164. [DOI] [PubMed] [Google Scholar]
- 18.Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol. 2007 Mar;14(3):185–93. doi: 10.1038/nsmb1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carrillo Oesterreich F, Preibisch S, Neugebauer KM. Global analysis of nascent RNA reveals transcriptional pausing in terminal exons. Mol Cell. 2010 Nov 24;40(4):571–81. doi: 10.1016/j.molcel.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 20.Alexander RD, Innocente SA, Barrass JD, Beggs JD. Splicing-dependent RNA polymerase pausing in yeast. Mol Cell. 2010 Nov 24;40(4):582–93. doi: 10.1016/j.molcel.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ip JY, Schmidt D, Pan Q, Ramani AK, Fraser AG, Odom DT, et al. Global impact of RNA polymerase II elongation inhibition on alternative splicing regulation. Genome Res. 2011 Mar;21(3):390–401. doi: 10.1101/gr.111070.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T. Regulation of alternative splicing by histone modifications. Science. 2010 Feb 19;327(5968):996–1000. doi: 10.1126/science.1184208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vargas DY, Shah K, Batish M, Levandoski M, Sinha S, Marras SA, et al. Single-molecule imaging of transcriptionally coupled and uncoupled splicing. Cell. 2011 Nov 23;147(5):1054–65. doi: 10.1016/j.cell.2011.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baumli S, Endicott JA, Johnson LN. Halogen bonds form the basis for selective P-TEFb inhibition by DRB. Chem Biol. 2010 Sep 24;17(9):931–6. doi: 10.1016/j.chembiol.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 25.Trask DK, Muller MT. Stabilization of type I topoisomerase-DNA covalent complexes by actinomycin D. Proc Natl Acad Sci U S A. 1988 Mar;85(5):1417–21. doi: 10.1073/pnas.85.5.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bensaude O. Inhibiting eukaryotic transcription: Which compound to choose? How to evaluate its activity? Transcription. 2011 May;2(3):103–8. doi: 10.4161/trns.2.3.16172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pei XF, Noble MS, Davoli MA, Rosfjord E, Tilli MT, Furth PA, et al. Explant-cell culture of primary mammary tumors from MMTV-c-Myc transgenic mice. In Vitro Cell Dev Biol Anim. 2004 Jan-Feb;40(1–2):14–21. doi: 10.1290/1543-706X(2004)40<14:ECOPMT>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 28.Aslakson CJ, Miller FR. Selective events in the metastatic process defined by analysis of the sequential dissemination of subpopulations of a mouse mammary tumor. Cancer Res. 1992 Mar 15;52(6):1399–405. [PubMed] [Google Scholar]
- 29.Piva F, Giulietti M, Nocchi L, Principato G. SpliceAid: a database of experimental RNA target motifs bound by splicing proteins in humans. Bioinformatics. 2009 May 1;25(9):1211–3. doi: 10.1093/bioinformatics/btp124. [DOI] [PubMed] [Google Scholar]
- 30.Bagga PS, Ford LP, Chen F, Wilusz J. The G-rich auxiliary downstream element has distinct sequence and position requirements and mediates efficient 3′ end pre-mRNA processing through a transacting factor. Nucleic Acids Res. 1995 May 11;23(9):1625–31. doi: 10.1093/nar/23.9.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arhin GK, Boots M, Bagga PS, Milcarek C, Wilusz J. Downstream sequence elements with different affinities for the hnRNP H/H’ protein influence the processing efficiency of mammalian polyadenylation signals. Nucleic Acids Res. 2002 Apr 15;30(8):1842–50. doi: 10.1093/nar/30.8.1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Du K, Leu JI, Peng Y, Taub R. Transcriptional up-regulation of the delayed early gene HRS/SRp40 during liver regeneration. Interactions among YY1, GA-binding proteins, and mitogenic signals. J Biol Chem. 1998 Dec 25;273(52):35208–15. doi: 10.1074/jbc.273.52.35208. [DOI] [PubMed] [Google Scholar]
- 33.Zahler AM, Neugebauer KM, Lane WS, Roth MB. Distinct functions of SR proteins in alternative pre-mRNA splicing. Science. 1993 Apr 9;260(5105):219–22. doi: 10.1126/science.8385799. [DOI] [PubMed] [Google Scholar]
- 34.Yeo G, Holste D, Kreiman G, Burge CB. Variation in alternative splicing across human tissues. Genome Biol. 2004;5(10):R74. doi: 10.1186/gb-2004-5-10-r74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yeo GW, Van Nostrand E, Holste D, Poggio T, Burge CB. Identification and analysis of alternative splicing events conserved in human and mouse. Proc Natl Acad Sci U S A. 2005 Feb 22;102(8):2850–5. doi: 10.1073/pnas.0409742102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lemischka IR, Pritsker M. Alternative splicing increases complexity of stem cell transcriptome. Cell Cycle. 2006 Feb;5(4):347–51. doi: 10.4161/cc.5.4.2424. [DOI] [PubMed] [Google Scholar]
- 37.Nurtdinov RN, Artamonova, Mironov AA, Gelfand MS. Low conservation of alternative splicing patterns in the human and mouse genomes. Hum Mol Genet. 2003 Jun 1;12(11):1313–20. doi: 10.1093/hmg/ddg137. [DOI] [PubMed] [Google Scholar]
- 38.Venables JP. Unbalanced alternative splicing and its significance in cancer. Bioessays. 2006 Apr;28(4):378–86. doi: 10.1002/bies.20390. [DOI] [PubMed] [Google Scholar]
- 39.Lee JH, Horak CE, Khanna C, Meng Z, Yu LR, Veenstra TD, et al. Alterations in Gemin5 expression contribute to alternative mRNA splicing patterns and tumor cell motility. Cancer Res. 2008 Feb 1;68(3):639–44. doi: 10.1158/0008-5472.CAN-07-2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grosso AR, Martins S, Carmo-Fonseca M. The emerging role of splicing factors in cancer. EMBO Rep. 2008 Nov;9(11):1087–93. doi: 10.1038/embor.2008.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008 Nov 27;456(7221):470–6. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Albulescu LO, Sabet N, Gudipati M, Stepankiw N, Bergman ZJ, Huffaker TC, et al. A Quantitative, High-Throughput Reverse Genetic Screen Reveals Novel Connections between Pre-mRNA Splicing and 5′ and 3′ End Transcript Determinants. PLoS Genet. 2012 Mar;8(3):e1002530. doi: 10.1371/journal.pgen.1002530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thorsen K, Sorensen KD, Brems-Eskildsen AS, Modin C, Gaustadnes M, Hein AM, et al. Alternative splicing in colon, bladder, and prostate cancer identified by exon array analysis. Mol Cell Proteomics. 2008 Jul;7(7):1214–24. doi: 10.1074/mcp.M700590-MCP200. [DOI] [PubMed] [Google Scholar]
- 44.Venables JP, Klinck R, Koh C, Gervais-Bird J, Bramard A, Inkel L, et al. Cancer-associated regulation of alternative splicing. Nat Struct Mol Biol. 2009 Jun;16(6):670–6. doi: 10.1038/nsmb.1608. [DOI] [PubMed] [Google Scholar]
- 45.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009 May 1;25(9):1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010 May;28(5):511–5. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roberts A, Trapnell C, Donaghey J, Rinn JL, Pachter L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011;12(3):R22. doi: 10.1186/gb-2011-12-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Knockdown of RRP1B in MDA-MB-231 cells increases cell proliferation and cell invasiveness. A. RRP1B expression in stable knockdown cell lines was confirmed by qRT-PCR and Western blot. B. Cell proliferation assay. Cells were plated at 2.5 × 104 cells per 12-well in duplicate and counted each day as indicated. C. Soft agar assay.
Comparison of isoform expression with the knockdown of Rrp1b in Mvt-1 and 4T1 cells.
RRP1B interacts with RNA polymerase II. Interaction between RRP1B and RNA polymerase II was determined by co-immunoprecipitation.
List of differentially expressed genes. 1FPKM (fragments per kilobase pair of exon per fragments mapped).
List of differentially expressed isoforms. Rpl and Rps genes that express proteins reported to interact with RRP1B are highlighted in pink and yellow, respectively.
Oligo and primer sequences.