

Abstract
The cosignaling network mediated by the herpesvirus entry mediator (HVEM; TNFRSF14) functions as a dual directional system that involves proinflammatory ligand, lymphotoxin that exhibits inducible expression and competes with HSV glycoprotein D for HVEM, a receptor expressed by T lymphocytes (LIGHT; TNFSF14), and the inhibitory Ig family member B and T lymphocyte attenuator (BTLA). To dissect the differential contributions of HVEM/BTLA and HVEM/LIGHT interactions, topographically-specific, competitive, and nonblocking anti-HVEM Abs that inhibit BTLA binding, but not LIGHT, were developed. We demonstrate that a BTLA-specific competitor attenuated the course of acute graft-versus-host reaction in a murine F1 transfer semiallogeneic model. Selective HVEM/BTLA blockade did not inhibit donor T cell infiltration into graft-versus-host reaction target organs, but decreased the functional activity of the alloreactive T cells. These results highlight the critical role of HVEM/BTLA pathway in the control of the allogeneic immune response and identify a new therapeutic target for transplantation and autoimmune diseases.
Introduction
Attenuation of the immune response is an obligated clinical intervention in the treatment of acute graft rejection and for the maintenance of long-term allograft survival as well as for the treatment of acute relapsing episodes of autoimmunity and in chronic autoimmune diseases (1, 2). Temporal and coordinate expression of membrane-bound receptors and soluble factors modulates the course of the immune response during an inflammatory process. Costimulation blockade of receptor/ligand interactions that participate in the exchange of information between APCs and T cells leads to the attenuation of the immune response due to the impaired communication between these two cell types. This approach represents a rational and promising therapeutic intervention to mitigate the deleterious consequences of the immune response in transplanted patients, including those undergoing graft-versus-host disease (GvHD) after bone marrow transplantation, and those suffering from autoimmune diseases (3).
Two major families of molecules are involved in the control of T cell activation, differentiation, and survival of terminally differentiated T cells, Ig superfamily (Ig SF) and the TNF/TNFR superfamily (4–7). In the early phase of T cell activation, interactions between molecules of the Ig SF predominate, whereas in the late phase of T cell activation, interactions between members of the TNF/TNFR superfamily molecules become responsible for the maintenance of the T cell response (5, 8). Herpesvirus entry mediator (HVEM; TNFRSF14) is widely expressed on hematopoietic and nonhematopoietic cells (9, 10), whereas B and T lymphocyte attenuator (BTLA) expression is more restricted to the hematopoietic cellular compartment (11–14). HVEM is a type I transmembrane molecule containing an extracellular domain composed of four cysteine-rich domains (CRD) (9, 15, 16) with distinct binding sites for its ligands. BTLA and CD160 bind to CRD1 domain of HVEM and compete with HSV gD for binding to this receptor (17, 18), whereas the binding site for lymphotoxin that exhibits inducible expression and competes with HSV glycoprotein D for HVEM, a receptor expressed by T lymphocytes (LIGHT; TNFSF14) is located at CRD2 and CRD3 domains of HVEM. Topographically, BTLA/CD160 and LIGHT interact with HVEM on opposite faces of its extracellular domain (19). CRD1 is an essential domain for the inhibitory function of soluble HVEM-Ig, because its deletion results in costimulation instead (17). HVEM represents a molecular switch depending on whether HVEM is functioning as a ligand of BTLA/CD160 (coinhibition) or LIGHT (costimulation) or a receptor of these molecules during the course of an immune response (costimulation). BTLA and CD160 engagement by HVEM expressed on the same cell (cis interaction) transmits inhibitory signals to resting lymphocytes and provides an intrinsic regulatory mechanism for T cell inhibition by impeding HVEM from receiving signals from the surrounding microenvironment (17, 18, 20), whereas engagement of HVEM by LIGHT in trans delivers T cell costimulatory signals (21, 22). An extra level of complexity to this cosignaling pathway was found in the ability of BTLA and CD160 to function as activating ligands of HVEM in trans-promoting T cell survival (22, 23). These authors demonstrated that engagement of HVEM by BTLA and CD160 agonist ligands induces IκBα degradation and activation of NF-κB RelA (p65) in epithelial and T cell subsets promoting their survival (22). Likewise, engagement of LIGHT on activated T cells by HVEM expressed in other T cell types costimulates their T cell proliferation (22, 23).
The dual specificity of the soluble receptors, LTβR-Fc or HVEM-Fc, leaves the interpretation of the most significant ligand-receptor pathway ambiguous. To overcome these difficulties and dissect better the role of each interaction separately and thus determine to which extent each interaction pathway is contributing to disease outcome, anti-HVEM mAbs that disrupt individual ligand-receptor interaction were developed. We demonstrate in this study that mAbs topographically specific for HVEM/BTLA interaction are capable of ameliorating graft-versus-host reaction (GvHR) by mitigating donor-alloreactive T cell effector function.
Materials and Methods
Mice and rats
Twelve- to 16-wk-old female Lewis rats (Harland) and 8- to 12-wk-old female C57BL/6 (B6), BALB/c (Charles River), and CB6F1 mice (offspring of BALB/c × B6, H-2d/b) were bred at the animal facility of the University of Leon. All experiments with rodents were handled and cared for in accordance with the Ethical Committee for Animal Research of the School of Veterinary Medicine (University of Leon) and the European Guidelines for Animal Care and Use of Laboratory Animals.
Cloning and expression of membrane-bound murine HVEM and their soluble ligands
Total RNA was extracted from B6 splenocytes using the RNeasy mini kit (Qiagen). Reverse transcription from mRNA to cDNA was performed with GeneAmp RNA PCR kit (Applied Biosystems). The cloning of soluble mouse B and T lymphocyte attenuator bound to mouse IgG2a Fc fragment (sBTLA.Ig) recombinant protein has been previously reported (24).
Full-length murine HVEM gene was PCR amplified with a proofreading Taq PFU polymerase and cloned into a modified pcDNA3.1+ vector (Invitrogen), upstream of the gene encoding for monster GFP (Clontech) inserted with EcoRV and XbaI restriction flanking sites. Flag-tagged soluble human LIGHT (hereafter, Flag-shLIGHT) and Flag-Foldon–tagged soluble murine LIGHT (from now on, Flag-Foldon-smLIGHT) (provided by C.F. Ware, La Jolla, CA, and Y. Shintani, Osaka, Japan, respectively) were used for the binding experiments (25).
Cell transfection
Chinese hamster ovary (CHO) cells were seeded on 6-well plates at 2.5 × 105 cells/well in complete RPMI 1640 medium containing 10% FCS, 2 mM L-glutamine, 1 mM sodium pyruvate, 10 mM HEPES, 50 μg/ml gentamicin, and 5 × 10−5 M 2-ME, and allowed to grow until they reached 60–70% confluence. The pSecTag2 Hygro b vector (Invitrogen) containing the extracellular domain of murine BTLA, Flag-shLIGHT and Flag-Foldon-smLIGHT, and HVEM-monster GFP constructs was purified using endotoxin-free Maxi-prep kit (Qiagen) and then transfected into CHO cells with 2 μg DNA/well of each construct/liposome complex (lipofectamine; Invitrogen) for 6–16 h (26).
Generation, characterization, and purification of anti-murine HVEM mAbs for in vivo use
Female Lewis rats were immunized i.p. with 0.5 ml of a 1:1.2 mixture of 5–10 × 106 HVEM stably transfected CHO cells expressing the membrane-bound murine HVEM-GFP fusion protein in IFA (Sigma-Aldrich). Six weeks after the first immunization, the animals were inoculated i.v. with 10 × 106 HVEM-transfected CHO cells and the hybridoma fusion protocol was carried out 3 d later, as previously described (24, 27). Twelve days after the fusion, culture supernatants from growing hybridomas were collected from 96-well plates and tested by flow cytometry against murine HVEM-GFP–transfected and control GFP-transfected CHO cells for 2 h at 37°C, washed, and subsequently incubated with an optimal dilution of Cy5-labeled polyclonal mouse anti-rat IgG (H+L; Jackson ImmunoResearch Laboratories).
Hybridomas secreting anti-HVEM mAbs and isotype control rat IgG2a (anti-plant cytokinin, clone AFRC-MAC-157, ECACC 93090997) were purified through a protein G-Sepharose affinity chromatography, quantified, filtered through 0.45 μm, and stored frozen at 1 mg/ml.
Flow cytometry-based competition-binding assays
A flow cytometry competition assay was established to define the epitopes recognized by anti-HVEM Abs. We first compared each possible pair of anti-HVEM mAbs, in which one of the Abs, the competitor, was unlabeled, whereas the other member of the pair, the developer, was biotinylated. Thus, by comparing each possible pair combination of anti-HVEM mAbs, we could determine whether the Abs competed with each other for binding to either identical or overlapping epitopes, or alternatively were recognizing distinct epitopes located at the extracellular domain of HVEM. Each unlabeled anti-HVEM mAb was confronted to the rest of biotinylated anti-HVEM mAbs of the panel. Thus, a saturating amount (10 μg) of each unlabeled anti-HVEM mAb (competitor Ab) or rat IgG2a isotype control was first incubated with 2.5 × 105 HVEM-GFP–transfected CHO cells, and each of the biotinylated anti-HVEM members of the panel was added later (developer Ab) to the immunological reaction.
To assess to what extent anti-HVEM mAbs generated in the present studies could interfere with the binding of murine sBTLA-mouse IgG2a.Fc (from now on sBTLA-Ig) (24), Flag-Foldon-smLIGHT or Flag-shLIGHT fusion proteins to HVEM-GFP–transfected CHO cells, each unlabeled anti-HVEM mAb, were first incubated with HVEM-transfected cells at saturating concentrations (10 μg Ab per 2.5 × 105 cells) for 30 min at room temperature. Then, in the presence of each anti-HVEM Ab as competitor, 10 μg of each soluble recombinant fusion protein was added and incubated for 2 h at 37°C, the supernatant was washed out, and the immunostaining was developed with either a biotinylated rat anti-mouse IgG2a isotype-specific mAb (R19-15; BD Biosciences) or biotinylated anti-Flag (clone M2; BioLegend), followed by streptavidin (SA)-PE.
GM-CSF–mediated bone marrow-derived dendritic cell differentiation
Syngeneic C57BL/6 (B6) and allogeneic BALB/c bone marrow cells were harvested from tibiae and differentiated with 30 ng/ml murine GM-CSF (Peprotech), as previously described (28). Bone marrow-derived dendritic cells (BM-DC) were finally matured upon overnight exposure to 1 μg/ml LPS O111:B4 (Sigma-Aldrich).
Parental into nonirradiated F1 acute GvHR murine model of alloreactivity
Donor B6 splenocytes were harvested, red cells were lysed, and cell suspensions were washed and resuspended in Dulbecco’s PBS. Then, 70 × 106 cells were adoptively transferred i.v. into CB6F1 recipient mice. Control mice were injected i.v. with 70 × 106 syngeneic F1 splenocytes. All donor cell suspensions were injected on the same day using cells processed simultaneously under the same conditions. F1 recipient mice received a single dose of 1 mg anti-HVEM mAb or isotype-matched rat IgG2a control administered i.p. Nineteen days after the adoptive transfer of donor B6 splenocytes into F1 recipients, mice were euthanized and the absolute number of hematopoietic cells was counted in each lymphoid compartment. Parental cell engraftment was assessed in distinct lymphoid compartments by flow cytometry. Single-cell suspensions were first incubated with Fc blocker (FcγRIII, clone 2.4G2) to prevent nonspecific staining, and then cells were stained with FITC-conjugated anti–H-2d (SF1-1.1) and Alexa 647-conjugated anti-H-2b (AF6-88.5). To further identify the different cell subsets, the following list of lineage-restricted Abs was used: anti-CD3 (145-2C11), anti-CD4 (L3T4), anti-CD8α (53-6.7), anti-CD19 (1D3), anti-Ly6G (1A8), and anti-Ly6C (Monts-1; provided by E. Butcher, Stanford University School of Medicine). All of these mAbs were purchased from BioLegend, except 2.4G2 and anti-Ly6C, which were purified and labeled in our laboratory.
In all flow cytometry experiments, dead cells were excluded by propidium iodide or DAPI staining. Flow cytometry acquisition was carried out on a Cyan 9 cytometer (Beckman Coulter). Data analysis was performed using the WinList 3D Version 7 (Verity Software House, Topsham, ME).
CD107a degranulation assay and frequency of IFN-γ–secreting T cells
Sixteen days after GvHR induction, 2 × 105 splenocytes were isolated from isotype control, 1H7-, and 6C9-treated F1 mice, and were restimulated in vitro with 1 × 105 either syngeneic (B6) or allogeneic BALB/c mature BM-DC. PE-conjugated anti-CD107a mAb (clone 1D4B; BioLegend) was added to the MLC and incubated for 1 h at 37°C in 5% CO2. Cells were incubated for an additional period of 4 h in the presence of 2 μM monensin and analyzed following CD107a degranulation assay gating on donor CD8+ T cells (29).
To determine the frequency of CD8+ T cells secreting IFN-γ, 2 × 105 splenocytes were restimulated in vitro with 1 × 105 either syngeneic (B6) or allogeneic BALB/c mature BM-DC in the presence of monensin. Cell cultures were then harvested, and donor CD8+ T cells were stained with FITC-labeled anti-CD8α and PE-labeled anti-Kd. Cells were then fixed for 20 min at room temperature (fixation buffer; BioLegend), centrifuged, and washed twice in permeabilization buffer (BioLegend). Finally, cells were stained with allophycocyanin-labeled anti-mouse IFN-γ (clone XMG1.2; BioLegend), washed, and collected for analysis. To confirm the specificity of the staining, cells were preincubated with unlabeled anti-mouse IFN-γ (clone XMG1.2; BioLegend) before allophycocyanin-labeled anti-mouse IFN-γ mAb was added (30).
In vivo cytotoxicity assay
Spleens from B6, BALB/c, and F1 mice were collected, and single-cell suspensions were prepared in RPMI 1640 complete medium. Target cells were washed twice in Dulbecco’s PBS and labeled with CFSE (Molecular Probes) at 10 μM (F1), 2 μM (BALB/c), or 0.4 μM (B6) for 10 min at 37°C. The reaction was stopped by adding 2 vol of cold RPMI 1640 containing 10% FCS, followed by two washes in Dulbecco’s PBS. A total of 30 × 106 of each target cell (B6, BALB/c, and F1 cells) was mixed at ratio 1:1:1 and was subsequently i.v. injected into F1 recipients at day 16 post-GvHR induction. Three days later (day 19 post-GvHR induction), recipient F1 mice were euthanized and target cells were analyzed in spleen and peripheral lymph nodes. The percentage of specific target lysis was calculated by comparing the survival of each target population with the survival of syngeneic population according to the following equation: percentage of specific killing of target cells = 100 − ([absolute number of target population in experiment/absolute number of syngeneic population in experiment]/[absolute number of target population in F1 recipient/absolute number of syngeneic population in F1 recipient]) × 100 (31, 32).
Quantification of Th1/Th2 cytokine by cytometric bead array
A total of 70 × 106 parental B6 splenocytes was injected i.v. into F1 recipients, which were treated at day 0 with a single saturating dose of 1 mg either isotype-matched control (rat IgG2a) or anti-HVEM mAbs. Sixteen days after treatment, 2 × 105 splenocytes from each experimental group were harvested and cocultured with 1 × 105 syngeneic B6 or allogeneic BALB/c BM-DC. Supernatants were collected and analyzed at 48 h after in vitro restimulation, and the amount of IL-2, IL-4, IL-5, IFN-γ, and TNF-α was quantified using a cytometric bead array following the manufacturer’s instructions (BD Biosciences).
Statistical analysis
Data are expressed as mean ± SD. Statistical significance was assessed using the parametric Student t test and nonparametric tests. A p value <0.05 was considered statistically significant.
Specificity and in vivo nondepleting activity of anti-HVEM mAbs recognizing the extracellular domain of murine HVEM receptor
A panel of four anti-HVEM mAbs (clones 1H7, 5B7, 6C9, and 10F3, all of them rat IgG2a, κ L chain) was obtained and characterized in the initial part of this study. The specificity of anti-HVEM mAbs was demonstrated via flow cytometry by their capacity to recognize CHO-HVEM-GFP–transfected cells, but not GFP-transfected CHO cells used as negative control (Fig. 1A).
FIGURE 1.
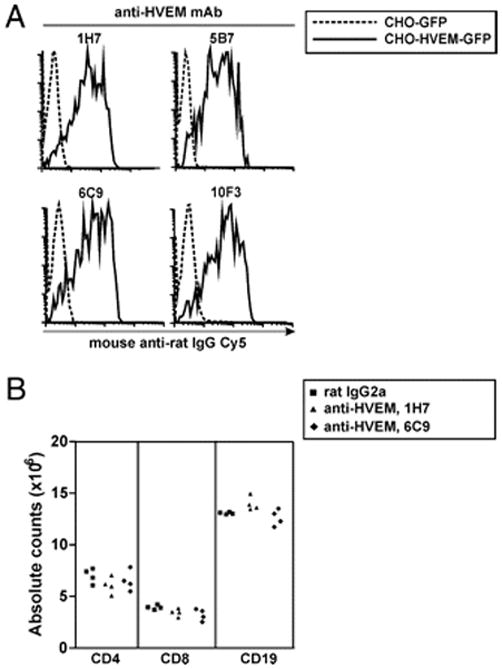
Anti-HVEM mAbs specifically recognize HVEM receptor on transfected cells. (A) The complete HVEM-encoding gene fused in frame to monster GFP was cloned into the mammalian pcDNA3.1 expression vector. pcDNA3.1-HVEM-GFP plasmid and empty vector pcDNA3.1-GFP were transiently transfected into CHO cell line and stained with rat anti-mouse HVEM mAbs, followed by Cy5-labeled mouse anti-rat IgG polyclonal conjugate. Cells were gated on propidium iodide-GFP+ and analyzed by flow cytometry. Dotted lines indicate control CHO-GFP cell line incubated with each anti-HVEM mAb. Solid lines show the reactivity pattern of anti-HVEM hybridoma supernatants (1H7 and 5B7, upper panel; 6C9 and 10F3, lower panel) against HVEM-transfected CHO cells. (B) In vivo treatment with anti-HVEM mAbs (clones 1H7 and 6C9) that were selected for the in vivo experiments depleted neither T cells nor B cells. F1 mice were i.p. injected with a single dose of 1 mg rat IgG2a isotype control (black squares), 1H7 mAb (black triangles), or 6C9 mAb (black diamonds) anti-HVEM mAbs. Total cell number of CD4, CD8, and CD19 cells was analyzed 5 d later in spleen.
To gain further insight into the potential in vivo functional activity of the anti-HVEM mAbs and to rule out any possible depleting activity, anti-HVEM Abs used in the in vivo studies were injected i.p. into F1 mice. This in vivo assay allowed us to determine simultaneously any depleting activity mediated by either complement- or Ab-dependent cellular cytotoxicity on T or B lymphocytes. No significant detectable decay in lymphocyte numbers 5 d after the i.p. injection of 1 mg anti-HVEM mAbs was observed, which was in agreement with the lack of depleting activity reported for the majority of rat Abs of this particular isotype (33) (Fig. 1B).
Anti-HVEM mAbs within group II exhibit competitive inhibition of sBTLA-Ig binding to HVEM-transfected cells, without preventing HVEM/LIGHT interaction
The extracellular domain of HVEM exhibits two opposite regions in its spatial conformation for the interaction with its ligands BTLA/CD160 or LIGHT (19, 34). A competitive binding assay was designed to dissect whether the panel of anti-HVEM mAb raised in our study was recognizing the same, overlapping, or different epitopes on the extracellular domain of HVEM. As shown in Fig. 2, anti-HVEM mAbs were classified into two distinct epitopes based on their competitive behavior, as follows: group I included clones 1H7 and 5B7 (Fig. 2A) and group II, 6C9, or 10F3 (Fig. 2B).
FIGURE 2.
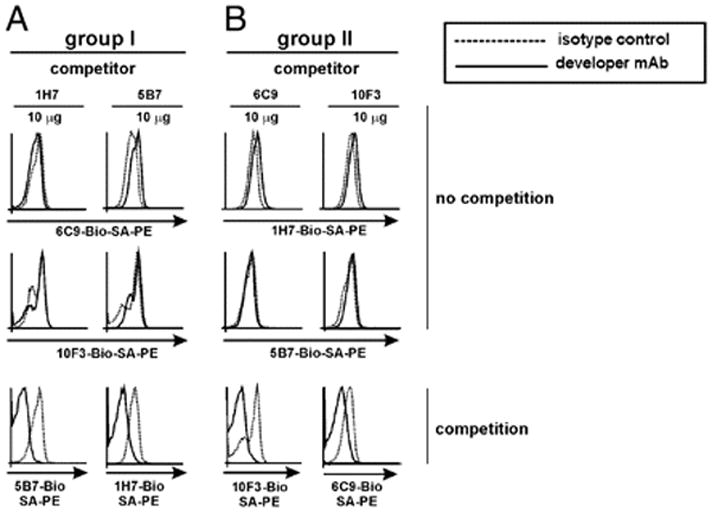
The panel of anti-HVEM mAbs defines the existence of at least two distinct epitopes on the extracellular domain of HVEM. With the purpose of mapping epitopes on HVEM receptor, HVEM-transfected cells (2 × 105 cells/well) were incubated with a saturating amount (10 μg/well) of each unlabeled anti-HVEM mAb or isotype-matched control. Anti-HVEM mAbs were classified within group I (clones 1H7 and 5B7) (A) or group II (clones 6C9 and 10F3) (B) based on their competition profile. Stably transfected HVEM-GFP CHO cells were incubated for 1 h at 4°C with a saturating amount of 10 μg/well of each unlabeled anti-murine HVEM mAb (competitor Ab). To detect competition among Abs recognizing the same molecule, each biotinylated anti-HVEM Ab (developer Ab, solid lines) was added to HVEM-transfected CHO cells, and the immunological reactions were revealed using an optimal dilution of SA-PE. Dashed lines depict HVEM-transfected cells incubated with an irrelevant competitor isotype-matched rat IgG2a control. The two first rows of dot plots display no competition profiles, whereas the third row exhibits those anti-HVEM Abs that competed with each other. One representative experiment of three with identical results is shown.
This panel of anti-HVEM mAbs showed differential competition with HVEM ligands. sBTLA-Fc construct specifically bound to HVEM-transfected cells, but not to control GFP-transfected cells (Fig. 3A) (17–19, 24, 35). Anti-HVEM Abs within group I (clones 1H7 and 5B7) did not compete with sBTLA-Fc (nonblocking Abs) (Fig. 3B, upper panel), although group II, clones 6C9 and 10F3, completely abrogated sBTLA-Ig binding to HVEM on transfected cells (blocking Abs) (Fig. 3B, lower panel).
FIGURE 3.
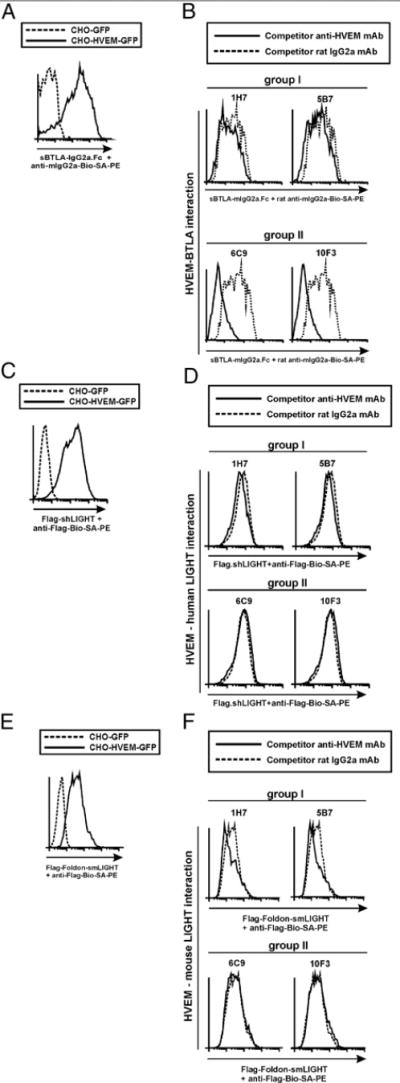
Group II anti-HVEM mAbs exhibit antagonist activity and abrogate HVEM/BTLA interaction without affecting HVEM/LIGHT binding. (A) The specificity and binding affinity of recombinant sBTLA-Ig fusion protein to membrane-bound HVEM-GFP stably transfected CHO cells or control CHO-GFP cells are shown on gated GFP-positive cells. Dashed line corresponds to binding of sBTLA-Ig to CHO-GFP control-transfected cells, and solid line depicts the binding of sBTLA-Ig to HVEM-GFP–transfected cells. (B) A total of 2.5 × 105 stably HVEM-GFP–transfected CHO cells was incubated with 10 μg/ml either rat IgG2a competitor control (dashed lines) or anti-HVEM mAbs (solid lines) following group I (upper panel) or group II (lower panel) for 30 min at room temperature. Then, 10 μg/ml sBTLA-Ig was added to the cells and incubated at 37°C for 2 h. The reaction was washed and further incubated with biotinylated rat anti-mouse IgG2a mAb, and the staining was finally developed with SA-PE. (C) The specificity of Flag-shLIGHT binding to HVEM-GFP–transfected cells (solid line) is shown. Dashed line displays the background binding of Flag-shLIGHT to control GFP-transfected cells. (D) Stably transfected HVEM-GFP cells were incubated with 10 μg/ml either rat IgG2a competitor Ab control (dashed lines) or anti-HVEM mAbs (solid lines) following group I (D, upper panel) or group II (D, lower panel) for 30 min at room temperature. Then, 10 μg/ml Flag-shLIGHT was added to the reaction and incubated at 37°C for 2 h. Biotinylated anti-Flag mAb followed by SA-PE was used to develop the immunological reactions. (E) The specificity of Flag-Foldon-smLIGHT binding to HVEM-GFP–transfected cells (solid line) is depicted. Dotted line represents the background binding of Flag-Foldon-smLIGHT to control GFP-transfected cells. (F) Stably transfected HVEM-GFP cells were incubated with 10 μg/ml either rat IgG2a competitor Ab control (dashed lines) or anti-HVEM mAbs (solid lines) following group I (F, upper panel) or group II (F, lower panel) for 30 min at room temperature. Then, 10 μg/ml Flag-Foldon smLIGHT was added to the reaction and incubated at 37°C for 2 h. Biotinylated anti-Flag mAb followed by SA-PE was used to develop the immunological reactions. A cartoon of the competition assay setup is shown. One representative experiment of three with identical results is shown.
Flag-soluble human LIGHT recombinant fusion proteins bound specifically to HVEM-transfected cells in flow cytometry (Fig. 3C). Neither group I nor II anti-HVEM Abs (Fig. 3D) interfered with Flag-shLIGHT binding to HVEM. Similar results were obtained with mouse version Flag-Foldon soluble LIGHT recombinant fusion proteins (Fig. 3E, 3F).
Altogether, the data indicate that the set of anti-HVEM Abs mapped to two topographically different epitopes on the extracellular domain of HVEM. The anti-HVEM mAbs in group II that blocked HVEM-BTLA interaction may be the most suitable therapeutic tool for the in vivo evaluation of the biological consequences derived from selective Ab-mediated blockade of HVEM/BTLA interaction.
Donor antihost alloreactive T cells downmodulate HVEM receptor expression following alloantigen recognition
To monitor the impact of the alloreactive T cell response on the modulation of HVEM receptor expression, B6 splenocytes were adoptively transferred to F1 recipients (B6-F1), whereas baseline expression of HVEM was monitored in syngeneic F1 recipients adoptively transferred with F1 splenocytes (F1-F1). Under homeostatic conditions, naive resting B cells expressed HVEM to a lower extent than T cells (Fig. 4A), which is in agreement with previous reports (14, 36, 37). The expression of HVEM was quickly downregulated on alloreactive CD4 and CD8 T cells as soon as day 3 after the adoptive transfer, and the reduced expression was sustained when compared with the amount of HVEM expressed on host T cells of syngeneic recipients at the same time points (Fig. 4A, 4B). Interestingly, expression of HVEM on host T cells of F1 recipients adoptively transferred with allogeneic B6 splenocytes did not undergo any apparent change in HVEM expression (Fig. 4A, 4B). Indeed, the amount of HVEM expressed on host T cells of F1 recipients was similar regardless of whether they were adoptively transferred with either syngeneic F1 or allogeneic B6 splenocytes. In contrast, BTLA was reciprocally upregulated early after T cell activation, and its expression then declined gradually and more rapidly on CD8 T cells than on CD4 T cells (Fig. 4C).
FIGURE 4.
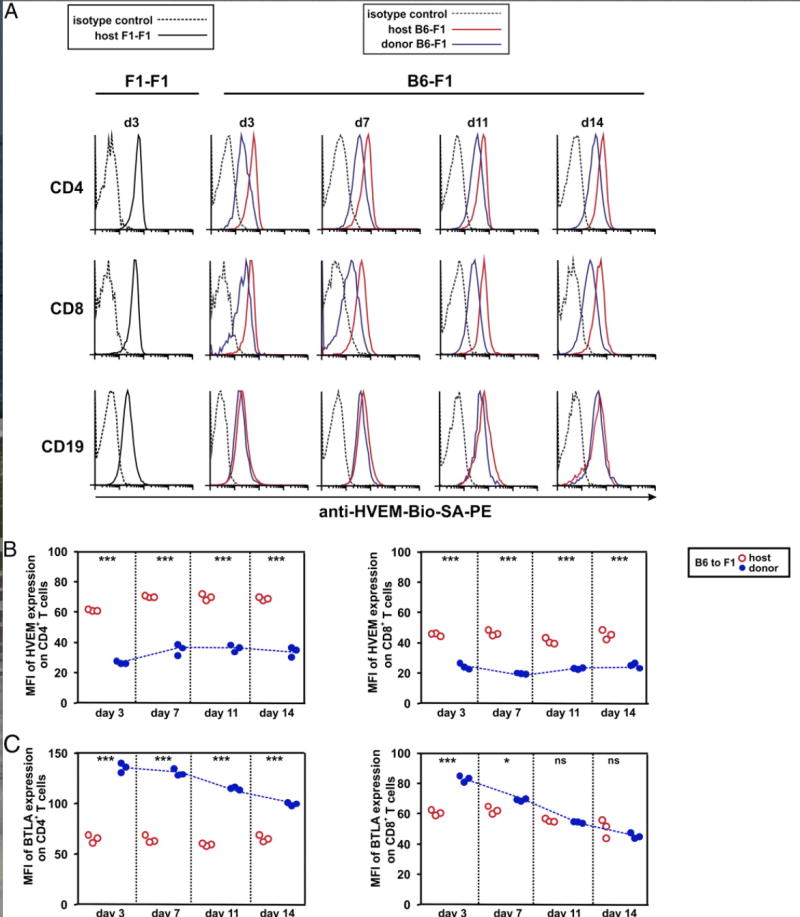
Reciprocal regulation of HVEM and BTLA expression is dependent on alloantigen recognition by donor T cells during the course of GvHR. (A) A total of 70 × 106 of B6 splenocytes was adoptively transferred into F1 recipients (B6-F1), and the time course of HVEM expression was monitored at days 3, 7, 11, and 14 after GvHR induction on host and donor CD4+ T cells, CD8+ T cells, and CD19+ cells of spleen. Syngeneic adoptive transfer of F1 splenocytes to F1 recipients (F1-F1) served as control group to determine the basal level of expression of these molecules at resting state under noninflammatory conditions. Black solid lines represent basal expression of HVEM on host cells of F1 mice receiving syngeneic F1 splenocytes; black dashed lines display isotype-matched control. Allogeneic adoptive transfer of B6 splenocytes to F1 recipients allowed the monitoring of surface expression of HVEM receptor on host (red solid lines) and donor (blue solid lines) lymphocytes during the course of GvHR. Black dashed lines indicate isotype-matched control. Biotinylated anti-HVEM mAb (10F3) followed by SA-PE was used to develop the reaction. The mean fluorescence intensity (MFI) of HVEM (B) and BTLA (C) expression on CD4 and CD8 T cells was calculated at different time points after the adoptive transfer of allogeneic splenocytes to F1 recipient mice. Red open circles and blue closed circles depict values of MFI of host and donor T cells, respectively. At each time point, MFI of HVEM and BTLA expression on donor and host CD4 and CD8 T cells was compared, and the statistical significant differences are indicated inside the plots (*p < 0.05, **p < 0.005, ***p < 0.0005; ns, nonsignificant). Blue dotted line highlights the expression trend of HVEM and BTLA on donor CD4 and CD8 T cells.
These observations indicate that bystander stimulation by the inflammatory milieu created by donor alloreactive T cells reacting against host tissues did not influence HVEM expression on host T cells or B cells. More importantly, modulation of HVEM and BTLA following donor T cell activation was dependent on TCR recognition of host alloantigens. These data support the idea that HVEM/BTLA cis prevents nonspecific activation in an inflammatory milieu.
Blockade of HVEM/BTLA ameliorates the course of rejection of host hematopoietic cells during the acute phase of the GvHR
The adoptive transfer of donor B6 splenocytes into nonirradiated F1 recipients (B6 × BALB/c) induces a progressive alloreactive response against host BALB/c H-2d histoincompatible Ags (38). Flow cytometry was used to measure the loss of host hematopoietic cells in primary hematopoietic (bone marrow and thymus) and secondary (spleen) organs using a combination of Alexa 647-labeled anti-murine Kb and FITC-labeled anti-murine Kd.
We examined the panel of mAbs to mouse HVEM to address the question of whether epitope-selective HVEM blockade will attenuate the GvHR. An increase in host H-2d–positive leukocytes indicated that treatment of mice with blocking anti-HVEM mAb (6C9) efficiently protected the host bone marrow compartment from donor T cell-mediated rejection in the acute phase of GvHR. In contrast, a dramatic loss of H-2d cells in the bone marrow was observed in F1 mice receiving semiallogeneic splenocytes treated with either nonblocking anti-HVEM mAb (clone 1H7) or rat IgG2a isotype control (Fig. 5A). This result was a clear indication that competitive blockade of HVEM/BTLA interaction was sufficient to attenuate the course of graft rejection in host bone marrow cells. The global prevention of host bone marrow rejection after 6C9 Ab-mediated blockade was also reflected in different host leukocyte subsets present in this compartment, when compared with isotype- or 1H7-treated F1 recipient mice receiving B6 donor allogeneic splenocytes. Thus, host granulocytes (Fig. 5B) and monocytes (Fig. 5C) were significantly more protected in 6C9-treated F1 mice than in 1H7- or isotype-treated allogeneic F1 recipients. Treatment with blocking anti-HVEM mAb (clone 6C9) conferred substantial, but incomplete protection against rejection of B cells when compared with B cells in the syngeneic F1-F1 recipients (Fig. 5D). Host thymocytes and double-positive thymocytes were fully protected from rejection by the blocking anti-HVEM mAb (6C9) (Fig. 5E, 5F), as were lymphocytes in the spleen (Fig. 5G).
FIGURE 5.
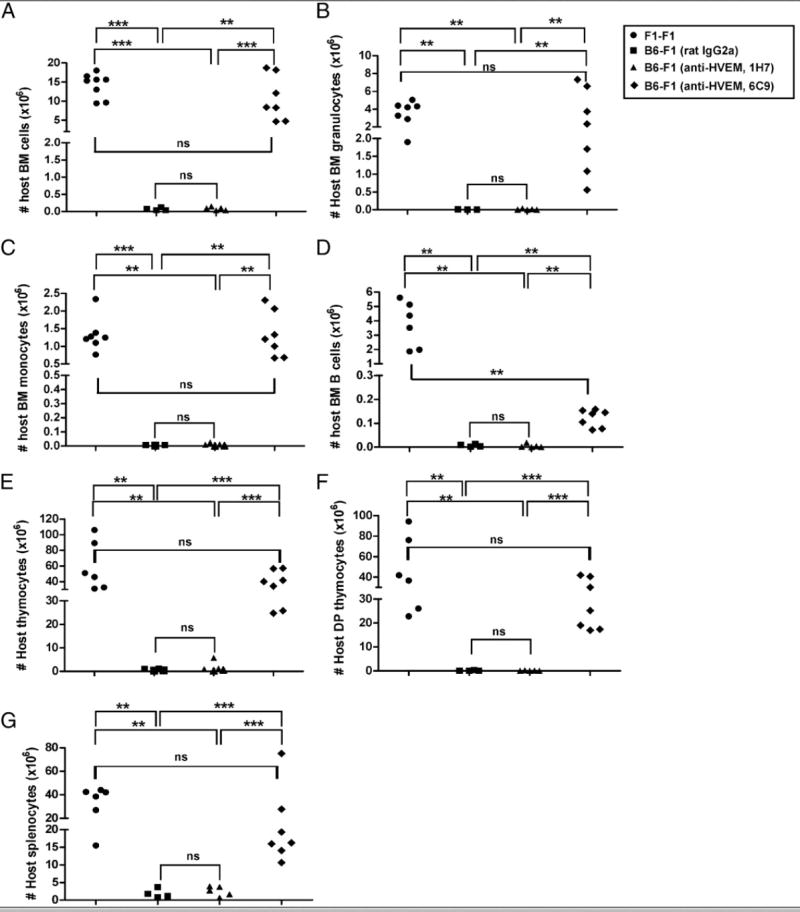
Blockade of HVEM/BTLA interaction attenuates the rejection of host target tissues. A total of 70 × 106 of allogeneic donor B6 splenocytes was adoptively transferred into F1 recipients, which were treated with a single dose of 1 mg either rat IgG2a isotype control (black squares), nonblocking anti-HVEM mAb (1H7, black triangles), or blocking anti-HVEM mAb (6C9, black diamond) at day 0. A syngeneic control group, in which 70 × 106 F1 splenocytes were adoptively transferred to F1 recipients (black circles), was included in the experimental setup. The absolute number of host bone marrow cells (A), granulocytes (B), monocytes (C), B cells (D), as well as the total number of host F1 thymocytes (E), host F1 double-positive thymocytes (F), and host splenocytes (G), are depicted at 19 d after the adoptive transfer of syngeneic or allogeneic B6 splenocytes. Statistical significance and the p value were calculated using unpaired Student t test and Mann-Whitney U test. The following criterion of significance was used, as follows: *p < 0.05, **p < 0.005, ***p < 0.0005. The experiment was repeated twice with similar results.
These data indicate that blockade of HVEM/BTLA interaction mitigates the acute phase of GvHR by attenuating the rejection of host hematopoietic cells.
Blockade of HVEM/BTLA interaction reduces the frequency of donor CD8+ T cells expressing CD107a and secreting IFN-γ
To dissect the contribution of HVEM/BTLA interaction to the course of GvH development, donor T cell infiltration on distinct hematopoietic target tissues was assessed. We found that CD4+ and CD8+ T cell infiltration in 6C9-treated F1 recipient mice was similar to that of isotype- or 1H7-treated F1 mice in distinct host hematopoietic compartments. Thus, in the bone marrow, thymus, and spleen, the absolute number of donor CD4+ or CD8+ T cells infiltrating this tissue was similar for all experimental groups (Fig. 6). Thus, the blockade of HVEM/BTLA interaction by anti-HVEM 6C9 did not impact in the absolute number of T cells infiltrating host hematopoietic GvHR major target organs, suggesting perhaps that the effector function was blocked. To test this idea, we measured CD107a degranulation in CD8 T cells and expression of intracellular IFN-γ as well as soluble cytokines characteristic of differentiated Th1 and Th2 cells. Splenocytes from anti-HVEM– or rat IgG2a-treated F1 recipients (at day 16 posttransplantation) were restimulated in vitro with allogeneic BALB/c mature BM-DC to monitor donor antihost CD8+ T cell-mediated allosensitization in response to alloantigen following the CD107a degranulation assay. Donor-alloreactive and host CD8+ T cells were gated separately, and CD107a expression was analyzed in isotype control- and in anti-HVEM–treated F1 mice.
FIGURE 6.
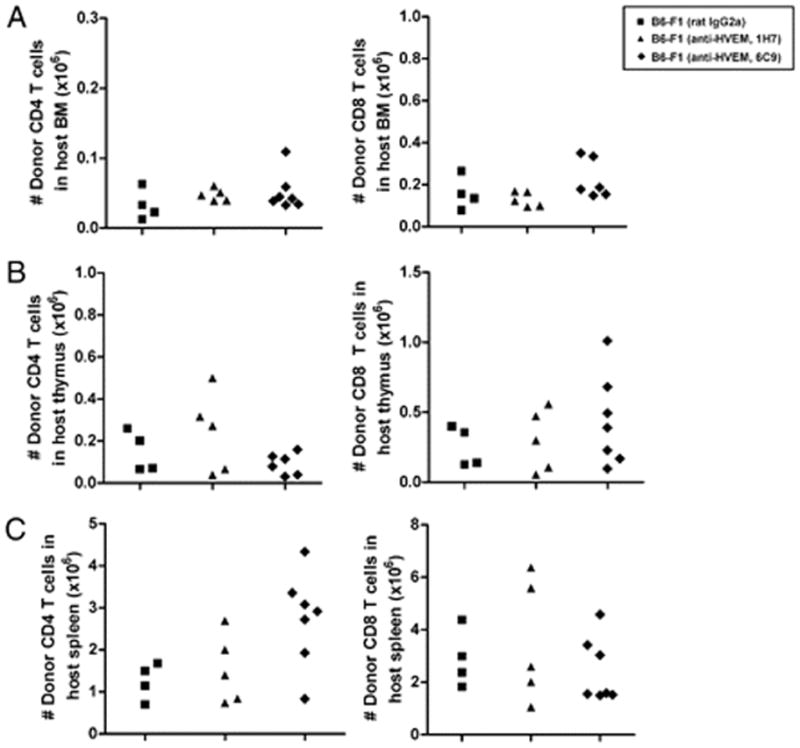
Ab targeting of BTLA does not reduce donor T cell number infiltrating host hematopoietic tissues during the acute phase of GvHR. Donor B6 splenocytes were adoptively transferred into allogeneic F1 recipient mice treated with irrelevant isotype rat IgG2a control (black square), nonblocking anti-HVEM mAb (clone 1H7, black triangle), or blocking anti-HVEM mAb (clone 6C9, black diamond). The absolute number of donor CD4+ cells and CD8+ cells infiltrating host bone marrow (A), thymus (B), and spleen (C) at day 19 of the acute phase of GvHR is depicted. No statistically significant differences were found between isotype- and anti-HVEM–treated F1 recipients. The experiment was repeated twice with a similar number of mice, and similar findings were recorded.
The frequency of CD8+ T cells expressing CD107a was suppressed in donor cells from mice treated with anti-HVEM 6C9 compared with mice receiving rat IgG2a control or the anti-HVEM 1H7 mAb, which were significantly elevated compared with the syngeneic F1/F1 control graft (Fig. 7A). Similarly, the frequency and absolute number of IFN-γ–secreting cells decreased in anti-HVEM 6C9-treated mice when compared with controls (Fig. 7B).
FIGURE 7.
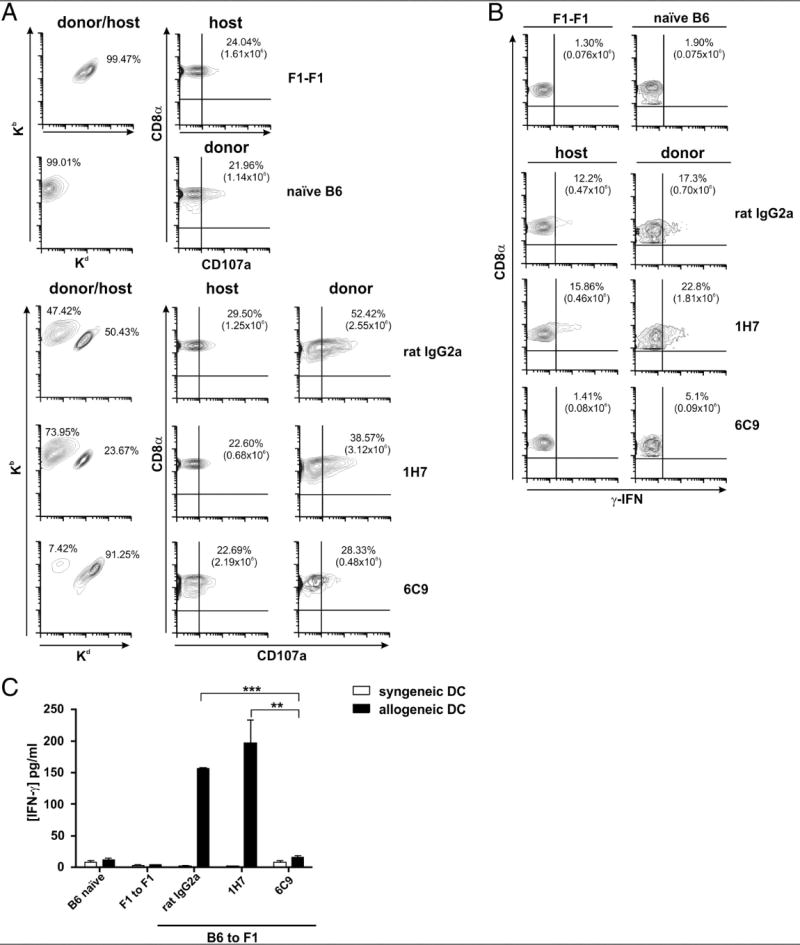
Reduced frequency and absolute number of donor alloantigen-specific CD8+ T cells expressing CD107a and secreting IFN-γ, and diminished Th1 cytokine production after blockade of HVEM/BTLA interaction. (A) Sixteen days after GvHR induction, 2 × 105 splenocytes isolated from either isotype control or anti-HVEM–treated F1 mice were restimulated in vitro with 1 × 105 allogeneic BALB/c mature BM-DC per well for 1 h in the presence of PE-conjugated anti-CD107a mAb or PE-conjugated isotype control and additional 4 h in the presence of monensin. A five-color flow cytometry panel was used to simultaneously analyze surface markers. Cells were stained with Alexa 647-labeled anti-mouse Kb and FITC-labeled anti-mouse Kd to distinguish donor and host T cells, and the percentage of each population was calculated. After gating on donor and host CD8+ T cells, the percentage and absolute number of CD8+ T cells expressing CD107a were assessed in isotype- and anti-HVEM–treated F1 recipients. PE-labeled isotype-matched nonbinding control Ig was used to set the quadrant lines. This figure is a representative experiment of two performed with similar results. (B) Splenocytes were restimulated in vitro with allogeneic APCs, as described in (A), and then stained with FITC-labeled anti-CD8α and PE-labeled anti-Kd. After surface staining, splenocytes were washed, fixed, and permeabilized, and intracellular IFN-γ staining was performed. Quadrant lines represent appropriate negative control to confirm the specificity of the anticytokine staining that was set preincubating splenocytes with unlabeled anti-mouse IFN-γ (blocker), followed by allophycocyanin-labeled anti-mouse IFN-γ. The percentage and absolute number of donor CD8+ IFN+-γ T cells in each experimental group were calculated in the absence of blocker, followed by allophycocyanin-labeled anti-mouse IFN-γ. One representative experiment of two performed is displayed in this figure. (C) A total of 2 × 105 splenocytes was collected at day 16 from isotype- or anti-HVEM–treated F1 mice undergoing GvHR and was cocultured with 1 × 105 syngeneic B6 (white bars) or allogeneic BALB/c (black bars) BM-DC for 48 h. Cell culture supernatants were then harvested, and Th1/Th2 cytokines were monitored by cytometric bead array. Mean fluorescence intensity values were obtained for each given cytokine, and cytokine concentrations (pg/ml) were calculated relative to the appropriate calibration curves with standard dilutions. A significant increase of IFN-γ was found in isotype- or 1H7-treated F1 mice compared with F1 mice receiving blocking 6C9 mAb. No significant differences were seen between mice treated with nonblocking and blocking Abs against HVEM for the rest of cytokines tested in the same assay (IL-2, IL-4, IL-5, and TNF-α). Bars indicate mean ± SEM, and t test was used to compare differences between groups. The degree of significance was indicated as follows: **p < 0.005, ***p < 0.0005.
IL-2, IL-4, IL-5, IFN-γ, and TNF-α secreted by CD4+ Th1 and Th2 were also measured in supernatants of splenocytes of F1 recipients adoptively transferred with B6 splenocytes, which were restimulated in vitro with syngeneic and allogeneic mature BM-DC for 48 h. Cytokines of Th1 cells, particularly IFN-γ, were significantly augmented in F1 mice treated with rat IgG2a isotype or 1H7 mAbs compared with recipients treated with 6C9 mAb, but no significant differences were seen when IL-2, IL-4, IL-5, or TNF-α was analyzed in the same in vitro restimulation assay (Fig. 7C).
These data indicate that blockade of HVEM/BTLA interaction mitigates the course of GvHR by inhibiting the frequency and cytotoxic function of donor-alloreactive CD8 and CD4 T cells.
In vivo donor antihost CTL activity is significantly reduced after HVEM/BTLA blockade during the acute phase of GvHR
Donor CTL-mediated allogeneic responses play a critical role in the rejection of host hematopoietic tissues that occurs after the adoptive transfer of allogeneic parental splenocytes into F1 recipients (38, 39). To investigate the consequences of in vivo donor antihost CTL response after selective HVEM/BTLA blockade, F1 recipient mice were injected with an identical number of B6, BALB/c, and F1 target cells that were differentially labeled with different amounts of CFSE, as described in Materials and Methods. A quantitative analysis of the percentage of killing of target BALB/c and F1 cells in host spleen and peripheral lymph nodes was evaluated. A significant reduction of killing of F1 and BALB/c target cells was observed in F1 mice after selective blockade of HVEM/BTLA with 6C9 mAb compared with either isotype- or 1H7-treated groups in spleen (Fig. 8A) and peripheral lymph nodes (Fig. 8B) (p < 0.0005).
FIGURE 8.
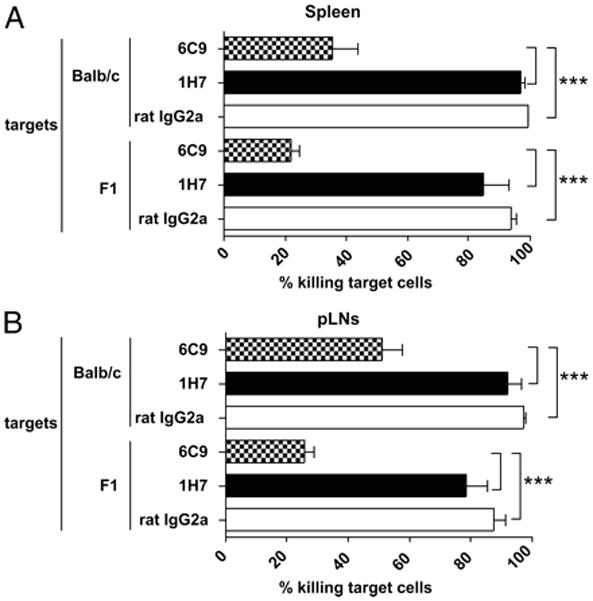
Significant reduction of in vivo donor antihost cytotoxic response after HVEM/BTLA blockade. Recipient F1 mice were adoptively transferred with 70 × 106 of B6 splenocytes and treated with either isotype control (rat IgG2a, 1 mg), anti-HVEM (nonblocking, clone 1H7, 1 mg), or anti-HVEM (blocking, clone 6C9, 1 mg) mAbs. Sixteen days later, recipient mice received 30 × 106 splenocytes of each CFSE-labeled target cell, as follows: B6 (0.4 μM), BALB/c (2 μM), and F1 (10 μM). The percentage of specific lysis in spleen (A) and peripheral lymph node (inguinals plus axilars) (B) was calculated at 72 h, according to the equation described in Materials and Methods. Data are representative of two independent experiments with three mice per group. Bars indicate mean ± SEM, and t test was used to compare differences between groups. Statistical significance was indicated, as follows: ***p < 0.0005.
Discussion
Hematopoietic stem cell transplantation and global and long-lasting immunosuppression are associated with delayed recovery of host immunocompetence, frequent opportunistic infections, and GvHD. More specific pharmacological interventions are necessary for the treatment of hematological disorders after bone marrow transplantation and for the treatment of the GvHD-related side effects (40). Molecules of the Ig SF and molecules of TNF/TNFR superfamily play a nonredundant and complementary role in T cell activation, differentiation, and the acquisition of effector function (41, 42).
In this study, we define a panel of topographically distinct Abs to mouse HVEM that segregate into two groups, as follows: HVEM-BTLA competitive group and noncompetitive group; neither group competes with the binding of LIGHT. We demonstrate that the competitive blocking HVEM Ab 6C9 suppresses the immune rejection in an allogeneic GvHR murine model. Our results indicate the effector mechanisms of CD4 and CD8 T cells require HVEM/BTLA signaling.
The importance and scientific relevance of the role of HVEM/BTLA/CD160 and HVEM/LIGHT interaction in transplantation and autoimmune disease have been evaluated in vitro and in vivo using several experimental approaches. The costimulatory function of the HVEM receptor has been revealed in transplantation experiments, in which graft rejection was attenuated, such as in allogeneic donor infusion of HVEM or LIGHT knockout (KO) T cells to lethally irradiated histoincompatible hosts, rescued with a syngeneic or allogeneic bone marrow transplant (43–45) or transplantation of MHC-mismatched tissues into HVEM or LIGHT KO recipients (46). In line with the costimulatory function of HVEM, Xu et al. (43) have provided evidence that Ab-mediated blockade of both HVEM/BTLA and HVEM/LIGHT interactions with an antagonist hamster anti-HVEM mAb (clone LBH1) in lethally irradiated mice that were rescued with allogeneic T cell-depleted bone marrow cells plus allogeneic splenocytes effectively protected host hematopoiesis from rejection in various bone marrow transplantation settings across distinct histocompatibility barriers. However, these results did not definitely elucidate the dilemma of whether HVEM/BTLA or HVEM/LIGHT blockade was the most crucial interaction for the prevention of disease and to which extent each pathway acting separately contributes to the overall protective effect of GvHD (43). In their studies, LBH1 Ab-mediated blockade of both HVEM/BTLA and HVEM/LIGHT pathways would prevent all possible signals through HVEM receptor (19, 22). In a parallel setting, these authors also adoptively transferred allogeneic HVEM KO or LIGHT KO splenocytes to semiallogeneic nonirradiated or irradiated F1 recipients (43), but HVEM deficiency precludes LIGHT and BTLA costimulatory signaling in trans through HVEM, and also prevents HVEM from delivering negative signals upon engaging BTLA. The same applies to donor LIGHT KO T cells that cannot receive signals from HVEM or LTβR or costimulate other T cells through HVEM (43, 47–50).
The use of decoy receptors and receptor-specific–deficient mice to address the role of complex pathways of interactions, in which multiple ligand/receptor cross-interactions are involved, provides limited mechanistic information. An interpretative dilemma often emerges, for instance, with the use of soluble HVEM-Ig that would interfere with both HVEM/BTLA/CD160 coinhibitory/costimulatory interactions and also HVEM/LIGHT costimulatory axis. The same occurs with the use of donor HVEM KO or LIGHT KO T cells and HVEM KO or LIGHT KO mice as recipients, because all potential bidirectional interactions of HVEM expressed on hematopoietic and nonhematopoietic cells with their ligands are completely abolished. Therefore, the in vivo consequences derived from these experimental settings should be taken with caution before drawing definitive conclusions.
To overcome these difficulties and gain insight into this complex network of HVEM interactions, we propose the utilization of highly specific nondepleting mAbs against epitopes located at the extracellular domain of HVEM permitting the targeting of only one potential interaction, without perturbing other possible interactions between HVEM and its ligands. Following these premises, and taking into account that BTLA and LIGHT bind to nonoverlapping and opposite sites on HVEM molecule (LIGHT binds to CRD2/3 domains of HVEM, whereas BTLA binds to CRD1 domain of HVEM) (18, 19), it was postulated that the specific blockade of BTLA/HVEM could be accomplished specifically with an Ab-based strategy, without perturbing HVEM/LIGHT interaction. With that goal in mind, we have characterized a set of anti-HVEM Abs that followed two different patterns of HVEM recognition. Thus, anti-HVEM Abs within group II are likely recognizing an epitope located on the binding site of interaction between domain CRD1 of HVEM and BTLA because they exhibited blocking activity, whereas anti-HVEM Abs of group I would be recognizing other epitopes than those located on the HVEM binding site of BTLA or LIGHT. The identification of blocking and nonblocking epitopes on the extracellular domain of HVEM provided us with an extraordinary investigative tool to determine the functional relevance of blocking specifically HVEM/BTLA without altering HVEM/LIGHT binding site and determine the biological consequences in preventing GvHR development.
It should not be overlooked that HVEM/BTLA Ab blockade is probably interrupting a bidirectional pathway of signal transduction between HVEM/BTLA. That is, both costimulatory signals transduced in trans after the engagement of HVEM on T cells by BTLA expressed on other immune cell types as well as coinhibitory signals transduced in cis upon interaction between HVEM and BTLA on the same cell type might be blocked (51). Because the outcome of HVEM/BTLA blockade is disease prevention in our experimental setting, instead of promotion of disease development, the data support that the costimulatory function of HVEM dominates over the coinhibitory activity. Moreover, in the context of an allogeneic response, T cell activation upregulates BTLA expression and downregulates HVEM expression on the same cell. This would decompensate the stoichiometry of cis inhibitory interactions on the same cell and would promote BTLA trans costimulatory interactions with other surrounding cells expressing HVEM, and Ab-mediated blockade of HVEM/BTLA would prevent them (18).
The adoptive transfer of donor allogeneic BTLA KO, HVEM KO, or LIGHT KO splenocytes to F1 recipients has been frequently associated with the attenuation of rejection of host hematopoietic target tissues due to poor survival and increased apoptosis of the donor T cells, despite donor T cell proliferation proceeding normally (43–45, 52). The use of 6C9 Ab-mediated blockade of HVEM/BTLA in a similar murine model of GvHR led to the same outcome, that is, protection against rejection of host hematopoietic target tissues. This protection was not, however, accompanied by decreased donor T cell infiltration in host hematopoietic target tissues in 6C9-treated mice compared with those mice receiving nonblocking anti-HVEM mAb or isotype-treated control. To reconcile the results of poor survival of HVEM-deficient donor T cells adoptively transferred to allogeneic F1 recipients (43) and the good survival of donor T cells after Ab-mediated blockade of HVEM/BTLA interaction, one possible explanation is that HVEM-deficient donor T cells would not receive costimulatory signals from both BTLA and LIGHT, whereas 6C9 Ab treatment would only interfere with BTLA/HVEM interaction, but would allow the transmission of LIGHT-mediated costimulatory survival signals upon engagement with HVEM. Moreover, blockade of HVEM/BTLA interaction did not affect the absolute number of T cells infiltrating the graft-versus-host target tissues, but diminished the frequency and absolute number of donor alloantigen-specific CD8 T cells secreting IFN-γ or expressing CD107a compared with those receiving nonblocking Abs or isotype-matched control. Besides, the amount of IFN-γ released by donor alloantigen-specific T cells was also decreased after HVEM/BTLA blockade. These observations were in accordance with in vivo decreased cytolytic function of donor T cells after competitive blockade of HVEM/BTLA interaction. Altogether, our data may well account for the attenuation of the cytotoxic donor antihost response after HVEM/BTLA blockade during the course of parental to F1 GvHR by either affecting the differentiation of naive CD8 T cells to effector CD8 T cells due to a decreased of T cell help or compromising the survival and maintenance of donor alloantigen-specific effector CD8 T cells.
Parent into nonirradiated or lethally irradiated F1 recipients have been extensively used as preclinical models for the study of alloreactivity and bone marrow rejection. The limitations of the nonirradiated model are that splenocytes adoptively transferred to allogeneic or semiallogeneic recipients undergo acute GvHD and immunoincompetence is only transient, mimicking the situation in humans under nonmyeloablative conditioning regimens (53). In contrast, the irradiated model resembles more the myeloablative conditioning regimens, in which allogeneic T cells adoptively transferred along with T cell-depleted bone marrow cells lead to development of different patterns of acute GvHD depending on the differences in class I and class II MHC Ags between donor and recipient, and involve the effector functional activity of CD4+ and CD8+ T cells (54). The space created after irradiation is fully permissive to alloreactive T cells to freely expand very quickly in response to two type of stimuli; one is alloantigen, and the other is homeostatic proliferation in response to an emptied hematopoietic compartment, which induces a conversion of the phenotype of the alloreactive and nonalloreactive T cell repertoire into memory-like T cells with less costimulatory requirements (55, 56). Because of those influences, T cells become more refractory to therapeutic manipulation, as it has been demonstrated for CD28/B7 blockade with CTLA-4.Ig (57). Another limitation of the nonirradiated model is that skin pathology and weight loss are reduced compared with this pathology in the irradiated model. Acute GvHD in nonirradiated model is largely manifested as immune deficiency and mixed chimerism, with less mortality than in irradiated model (39, 58–60). Despite the above mentioned limitations of the nonirradiated model, this was chosen because it does not require manipulation of the recipient and avoids the many variables introduced by a lymphopenic environment that affect the course of the alloreactive T cell response. Thus, we could interrogate the experimental system to assess the influence of HVEM/BTLA blockade on the initiation and progression of the cytotoxic response in a peripheral environment, in which alloreactive T cells are not homeostatically proliferating. Disease pathogenesis at day 19 after the adoptive transfer of allogeneic splenocytes is largely dependent on alloreactive effector CD8 T cells and not on cytotoxic autoreactive Abs, which appear later along with the chronic manifestations of the disease (38).
Therefore, our data favor the notion that attenuation of donor antihost cytotoxicity accounts for the reduced donor antihost rejection in a parent to nonirradiated F1 murine model of GvHR under blockade of only HVEM/BTLA interaction. This refined Ab-based strategy presented in this work, compared with previous studies, allowed us to demonstrate the contribution of HVEM/BTLA pathway to the attenuation of graft rejection in a GvHR murine model.
Acknowledgments
We are particularly grateful to Leonides Alaiz for outstanding animal husbandry. We also thank Dr. Fermín Sánchez-Guijo (Department of Hematology, Salamanca University Hospital, Salamanca, Spain) for suggestions and critical reading of the manuscript.
This work has been supported by Grants FIS PI#10/01039 (Fondo de Investigaciones Sanitarias, Ministry of Health, Spanish Government) and LE007A10-2 (Department of Education of the Regional Government, Junta de Castilla y Leon) to J.-I.R.-B.
Abbreviations used in this article
- BM-DC
bone marrow-derived dendritic cell
- BTLA
B and T lymphocyte attenuator
- CHO
Chinese hamster ovary
- CRD
cysteine-rich domain
- GvHD
graft-versus-host disease
- GvHR
graft-versus-host reaction
- HVEM
herpesvirus entry mediator
- Ig SF
Ig superfamily
- KO
knockout
- LIGHT
lymphotoxin that exhibits inducible expression and competes with
- HSV
glycoprotein D for HVEM, a receptor expressed by T lymphocytes
- SA
streptavidin
- sBTLA.Ig
soluble mouse B and T lymphocyte attenuator bound to mouse IgG2a Fc fragment
- shLIGHT
soluble human LIGHT
- smLIGHT
soluble murine LIGHT
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Getts DR, Shankar S, Chastain EM, Martin A, Getts MT, Wood K, Miller SD. Current landscape for T-cell targeting in autoimmunity and transplantation. Immunotherapy. 2011;3:853–870. doi: 10.2217/imt.11.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Starzl TE, Todo S, Fung J, Demetris AJ, Venkataramman R, Jain A. FK 506 for liver, kidney, and pancreas transplantation. Lancet. 1989;2:1000–1004. doi: 10.1016/s0140-6736(89)91014-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goldstein DR. T cell costimulation blockade and organ transplantation: a change of philosophy for transplant immunologists? J Immunol. 2011;186:2691–2692. doi: 10.4049/jimmunol.1190002. [DOI] [PubMed] [Google Scholar]
- 4.Adams AB, Larsen CP, Pearson TC, Newell KA. The role of TNF receptor and TNF superfamily molecules in organ transplantation. Am J Transplant. 2002;2:12–18. doi: 10.1034/j.1600-6143.2002.020104.x. [DOI] [PubMed] [Google Scholar]
- 5.Watts TH. TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol. 2005;23:23–68. doi: 10.1146/annurev.immunol.23.021704.115839. [DOI] [PubMed] [Google Scholar]
- 6.del Rio ML, Buhler L, Gibbons C, Tian J, Rodriguez-Barbosa JI. PD-1/PD-L1, PD-1/PD-L2, and other co-inhibitory signaling pathways in transplantation. Transpl Int. 2008;21:1015–1028. doi: 10.1111/j.1432-2277.2008.00726.x. [DOI] [PubMed] [Google Scholar]
- 7.del Rio ML, Lucas CL, Buhler L, Rayat G, Rodriguez-Barbosa JI. HVEM/LIGHT/BTLA/CD160 cosignaling pathways as targets for immune regulation. J Leukoc Biol. 2010;87:223–235. doi: 10.1189/jlb.0809590. [DOI] [PubMed] [Google Scholar]
- 8.Croft M. Co-stimulatory members of the TNFR family: keys to effective T-cell immunity? Nat Rev Immunol. 2003;3:609–620. doi: 10.1038/nri1148. [DOI] [PubMed] [Google Scholar]
- 9.Hsu H, Solovyev I, Colombero A, Elliott R, Kelley M, Boyle WJ. ATAR, a novel tumor necrosis factor receptor family member, signals through TRAF2 and TRAF5. J Biol Chem. 1997;272:13471–13474. doi: 10.1074/jbc.272.21.13471. [DOI] [PubMed] [Google Scholar]
- 10.Steinberg MW, Turovskaya O, Shaikh RB, Kim G, McCole DF, Pfeffer K, Murphy KM, Ware CF, Kronenberg M. A crucial role for HVEM and BTLA in preventing intestinal inflammation. J Exp Med. 2008;205:1463–1476. doi: 10.1084/jem.20071160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gavrieli M, Watanabe N, Loftin SK, Murphy TL, Murphy KM. Characterization of phosphotyrosine binding motifs in the cytoplasmic domain of B and T lymphocyte attenuator required for association with protein tyrosine phosphatases SHP-1 and SHP-2. Biochem Biophys Res Commun. 2003;312:1236–1243. doi: 10.1016/j.bbrc.2003.11.070. [DOI] [PubMed] [Google Scholar]
- 12.Vendel AC, Calemine-Fenaux J, Izrael-Tomasevic A, Chauhan V, Arnott D, Eaton DL. B and T lymphocyte attenuator regulates B cell receptor signaling by targeting Syk and BLNK. J Immunol. 2009;182:1509–1517. doi: 10.4049/jimmunol.182.3.1509. [DOI] [PubMed] [Google Scholar]
- 13.Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, Hurchla MA, Zimmerman N, Sim J, Zang X, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol. 2003;4:670–679. doi: 10.1038/ni944. [DOI] [PubMed] [Google Scholar]
- 14.Hurchla MA, Sedy JR, Gavrieli M, Drake CG, Murphy TL, Murphy KM. B and T lymphocyte attenuator exhibits structural and expression polymorphisms and is highly induced in anergic CD4+ T cells. J Immunol. 2005;174:3377–3385. doi: 10.4049/jimmunol.174.6.3377. [DOI] [PubMed] [Google Scholar]
- 15.Compaan DM, Gonzalez LC, Tom I, Loyet KM, Eaton D, Hymowitz SG. Attenuating lymphocyte activity: the crystal structure of the BTLA-HVEM complex. J Biol Chem. 2005;280:39553–39561. doi: 10.1074/jbc.M507629200. [DOI] [PubMed] [Google Scholar]
- 16.Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell. 2001;104:487–501. doi: 10.1016/s0092-8674(01)00237-9. [DOI] [PubMed] [Google Scholar]
- 17.Cai G, Anumanthan A, Brown JA, Greenfield EA, Zhu B, Freeman GJ. CD160 inhibits activation of human CD4+ T cells through interaction with herpesvirus entry mediator. Nat Immunol. 2008;9:176–185. doi: 10.1038/ni1554. [DOI] [PubMed] [Google Scholar]
- 18.Gonzalez LC, Loyet KM, Calemine-Fenaux J, Chauhan V, Wranik B, Ouyang W, Eaton DL. A coreceptor interaction between the CD28 and TNF receptor family members B and T lymphocyte attenuator and herpesvirus entry mediator. Proc Natl Acad Sci USA. 2005;102:1116–1121. doi: 10.1073/pnas.0409071102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheung TC, Humphreys IR, Potter KG, Norris PS, Shumway HM, Tran BR, Patterson G, Jean-Jacques R, Yoon M, Spear PG, et al. Evolutionarily divergent herpesviruses modulate T cell activation by targeting the herpesvirus entry mediator cosignaling pathway. Proc Natl Acad Sci USA. 2005;102:13218–13223. doi: 10.1073/pnas.0506172102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sedy JR, Gavrieli M, Potter KG, Hurchla MA, Lindsley RC, Hildner K, Scheu S, Pfeffer K, Ware CF, Murphy TL, Murphy KM. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat Immunol. 2005;6:90–98. doi: 10.1038/ni1144. [DOI] [PubMed] [Google Scholar]
- 21.Mauri DN, Ebner R, Montgomery RI, Kochel KD, Cheung TC, Yu GL, Ruben S, Murphy M, Eisenberg RJ, Cohen GH, et al. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity. 1998;8:21–30. doi: 10.1016/s1074-7613(00)80455-0. [DOI] [PubMed] [Google Scholar]
- 22.Cheung TC, Steinberg MW, Oborne LM, Macauley MG, Fukuyama S, Sanjo H, D’Souza C, Norris PS, Pfeffer K, Murphy KM, et al. Unconventional ligand activation of herpesvirus entry mediator signals cell survival. Proc Natl Acad Sci USA. 2009;106:6244–6249. doi: 10.1073/pnas.0902115106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ware CF, Sedý JR. TNF superfamily networks: bidirectional and interference pathways of the herpesvirus entry mediator (TNFSF14) Curr Opin Immunol. 2011;23:627–631. doi: 10.1016/j.coi.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.del Rio ML, Kaye J, Rodriguez-Barbosa JI. Detection of protein on BTLAlow cells and in vivo y-mediated down-modulation of BTLA on lymphoid and myeloid cells of C57BL/6 and BALB/c BTLA allelic variants. Immunobiology. 2010;215:570–578. doi: 10.1016/j.imbio.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 25.Ito T, Iwamoto K, Tsuji I, Tsubouchi H, Omae H, Sato T, Ohba H, Kurokawa T, Taniyama Y, Shintani Y. Trimerization of murine TNF ligand family member LIGHT increases the cytotoxic activity against the FM3A mammary carcinoma cell line. Appl Microbiol Biotechnol. 2011;90:1691–1699. doi: 10.1007/s00253-011-3168-8. [DOI] [PubMed] [Google Scholar]
- 26.Katsel PL, Greenstein RJ. Eukaryotic gene transfer with liposomes: effect of differences in lipid structure. Biotechnol Annu Rev. 2000;5:197–220. doi: 10.1016/s1387-2656(00)05036-5. [DOI] [PubMed] [Google Scholar]
- 27.del Rio ML, Penuelas-Rivas G, Dominguez-Perles R, Ramirez P, Parrilla P, Rodriguez-Barbosa JI. Antibody-mediated signaling through PD-1 costimulates T cells and enhances CD28-dependent proliferation. Eur J Immunol. 2005;35:3545–3560. doi: 10.1002/eji.200535232. [DOI] [PubMed] [Google Scholar]
- 28.del Rio ML, Rodriguez-Barbosa JI, Bölter J, Ballmaier M, Dittrich-Breiholz O, Kracht M, Jung S, Förster R. CX3CR1+ c-kit+ bone marrow cells give rise to CD103+ and CD103- dendritic cells with distinct functional properties. J Immunol. 2008;181:6178–6188. doi: 10.4049/jimmunol.181.9.6178. [DOI] [PubMed] [Google Scholar]
- 29.Betts MR, Brenchley JM, Price DA, De Rosa SC, Douek DC, Roederer M, Koup RA. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 30.Vikingsson A, Pederson K, Muller D. Enumeration of IFN-gamma producing lymphocytes by flow cytometry and correlation with quantitative measurement of IFN-gamma. J Immunol Methods. 1994;173:219–228. doi: 10.1016/0022-1759(94)90300-x. [DOI] [PubMed] [Google Scholar]
- 31.Oehen S, Brduscha-Riem K, Oxenius A, Odermatt B. A simple method for evaluating the rejection of grafted spleen cells by flow cytometry and tracing adoptively transferred cells by light microscopy. J Immunol Methods. 1997;207:33–42. doi: 10.1016/s0022-1759(97)00089-6. [DOI] [PubMed] [Google Scholar]
- 32.Brehm MA, Daniels KA, Ortaldo JR, Welsh RM. Rapid conversion of effector mechanisms from NK to T cells during virus-induced lysis of allogeneic implants in vivo. J Immunol. 2005;174:6663–6671. doi: 10.4049/jimmunol.174.11.6663. [DOI] [PubMed] [Google Scholar]
- 33.Hale G, Clark M, Waldmann H. Therapeutic potential of rat monoclonal antibodies: isotype specificity of antibody-dependent cell-mediated cytotoxicity with human lymphocytes. J Immunol. 1985;134:3056–3061. [PubMed] [Google Scholar]
- 34.Watts TH, Gommerman JL. The LIGHT and DARC sides of herpesvirus entry mediator. Proc Natl Acad Sci USA. 2005;102:13365–13366. doi: 10.1073/pnas.0506707102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nelson CA, Fremont MD, Sedy JR, Norris PS, Ware CF, Murphy KM, Fremont DH. Structural determinants of herpesvirus entry mediator recognition by murine B and T lymphocyte attenuator. J Immunol. 2008;180:940–947. doi: 10.4049/jimmunol.180.2.940. [DOI] [PubMed] [Google Scholar]
- 36.Morel Y, Schiano de Colella JM, Harrop J, Deen KC, Holmes SD, Wattam TA, Khandekar SS, Truneh A, Sweet RW, Gastaut JA, et al. Reciprocal expression of the TNF family receptor herpes virus entry mediator and its ligand LIGHT on activated T cells: LIGHT down-regulates its own receptor. J Immunol. 2000;165:4397–4404. doi: 10.4049/jimmunol.165.8.4397. [DOI] [PubMed] [Google Scholar]
- 37.Murphy TL, Murphy KM. Slow down and survive: enigmatic immunoregulation by BTLA and HVEM. Annu Rev Immunol. 2010;28:389–411. doi: 10.1146/annurev-immunol-030409-101202. [DOI] [PubMed] [Google Scholar]
- 38.Tschetter JR, Mozes E, Shearer GM. Progression from acute to chronic disease in a murine parent-into-F1 model of graft-versus-host disease. J Immunol. 2000;165:5987–5994. doi: 10.4049/jimmunol.165.10.5987. [DOI] [PubMed] [Google Scholar]
- 39.Pulaiev RA, Puliaeva IA, Ryan AE, Via CS. The parent-into-F1 model of graft-vs-host disease as a model of in vivo T cell function and immunomodulation. Curr Med Chem Immunol Endocr Metab Agents. 2005;5:575–583. doi: 10.2174/156801305774962204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ferrara JL, Reddy P. Pathophysiology of graft-versus-host disease. Semin Hematol. 2006;43:3–10. doi: 10.1053/j.seminhematol.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 41.Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol. 2009;9:271–285. doi: 10.1038/nri2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hehlgans T, Pfeffer K. The intriguing biology of the tumour necrosis factor/tumour necrosis factor receptor superfamily: players, rules and the games. Immunology. 2005;115:1–20. doi: 10.1111/j.1365-2567.2005.02143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu Y, Flies AS, Flies DB, Zhu G, Anand S, Flies SJ, Xu H, Anders RA, Hancock WW, Chen L, Tamada K. Selective targeting of the LIGHT-HVEM costimulatory system for the treatment of graft-versus-host disease. Blood. 2007;109:4097–4104. doi: 10.1182/blood-2006-09-047332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Albring JC, Sandau MM, Rapaport AS, Edelson BT, Satpathy A, Mashayekhi M, Lathrop SK, Hsieh CS, Stelljes M, Colonna M, et al. Targeting of B and T lymphocyte associated (BTLA) prevents graft-versus-host disease without global immunosuppression. J Exp Med. 2010;207:2551–2559. doi: 10.1084/jem.20102017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sakoda Y, Park JJ, Zhao Y, Kuramasu A, Geng D, Liu Y, Davila E, Tamada K. Dichotomous regulation of GVHD through bidirectional functions of the BTLA-HVEM pathway. Blood. 2011;117:2506–2514. doi: 10.1182/blood-2010-08-301325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ye Q, Fraser CC, Gao W, Wang L, Busfield SJ, Wang C, Qiu Y, Coyle AJ, Gutierrez-Ramos JC, Hancock WW. Modulation of LIGHT-HVEM costimulation prolongs cardiac allograft survival. J Exp Med. 2002;195:795–800. doi: 10.1084/jem.20012088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scheu S, Alferink J, Pötzel T, Barchet W, Kalinke U, Pfeffer K. Targeted disruption of LIGHT causes defects in costimulatory T cell activation and reveals cooperation with lymphotoxin beta in mesenteric lymph node genesis. J Exp Med. 2002;195:1613–1624. doi: 10.1084/jem.20020215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wan X, Zhang J, Luo H, Shi G, Kapnik E, Kim S, Kanakaraj P, Wu J. A TNF family member LIGHT transduces costimulatory signals into human T cells. J Immunol. 2002;169:6813–6821. doi: 10.4049/jimmunol.169.12.6813. [DOI] [PubMed] [Google Scholar]
- 49.Tamada K, Ni J, Zhu G, Fiscella M, Teng B, van Deursen JM, Chen L. Cutting edge: selective impairment of CD8+ T cell function in mice lacking the TNF superfamily member LIGHT. J Immunol. 2002;168:4832–4835. doi: 10.4049/jimmunol.168.10.4832. [DOI] [PubMed] [Google Scholar]
- 50.Shi G, Luo H, Wan X, Salcedo TW, Zhang J, Wu J. Mouse T cells receive costimulatory signals from LIGHT, a TNF family member. Blood. 2002;100:3279–3286. doi: 10.1182/blood-2002-05-1404. [DOI] [PubMed] [Google Scholar]
- 51.Cheung TC, Oborne LM, Steinberg MW, Macauley MG, Fukuyama S, Sanjo H, D’Souza C, Norris PS, Pfeffer K, Murphy KM, et al. T cell intrinsic heterodimeric complexes between HVEM and BTLA determine receptivity to the surrounding microenvironment. J Immunol. 2009;183:7286–7296. doi: 10.4049/jimmunol.0902490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hurchla MA, Sedy JR, Murphy KM. Unexpected role of B and T lymphocyte attenuator in sustaining cell survival during chronic allostimulation. J Immunol. 2007;178:6073–6082. doi: 10.4049/jimmunol.178.10.6073. [DOI] [PubMed] [Google Scholar]
- 53.Sykes M, Spitzer TR. Non-myeloblative induction of mixed hematopoietic chimerism: application to transplantation tolerance and hematologic malignancies in experimental and clinical studies. Cancer Treat Res. 2002;110:79–99. doi: 10.1007/978-1-4615-0919-6_4. [DOI] [PubMed] [Google Scholar]
- 54.Hakim F, Fowler DH, Shearer GM, Gress RE. Animal models of acute and chronic graft-versus-host disease. Curr Protoc Immunol. 2001;Chapter 4(Unit 4.3) doi: 10.1002/0471142735.im0403s27. [DOI] [PubMed] [Google Scholar]
- 55.Sprent J, Surh CD. Normal T cell homeostasis: the conversion of naive cells into memory-phenotype cells. Nat Immunol. 2011;12:478–484. doi: 10.1038/ni.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hickman SP, Turka LA. Homeostatic T cell proliferation as a barrier to T cell tolerance. Philos Trans R Soc Lond B Biol Sci. 2005;360:1713–1721. doi: 10.1098/rstb.2005.1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu Z, Bensinger SJ, Zhang J, Chen C, Yuan X, Huang X, Markmann JF, Kassaee A, Rosengard BR, Hancock WW, et al. Homeostatic proliferation is a barrier to transplantation tolerance. Nat Med. 2004;10:87–92. doi: 10.1038/nm965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schroeder MA, DiPersio JF. Mouse models of graft-versus-host disease: advances and limitations. Dis Model Mech. 2011;4:318–333. doi: 10.1242/dmm.006668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gleichmann E, Gleichmann H. Pathogenesis of graft-versus-host reactions (GVHR) and GVH-like diseases. J Invest Dermatol. 1985;85:115s–120s. doi: 10.1111/1523-1747.ep12275619. [DOI] [PubMed] [Google Scholar]
- 60.Ferrara JL, Levine JE, Reddy P, Holler E. Graft-versus-host disease. Lancet. 2009;373:1550–1561. doi: 10.1016/S0140-6736(09)60237-3. [DOI] [PMC free article] [PubMed] [Google Scholar]