

Abstract
RNA is a tool used in many fields, from molecular and cellular biology to medicine and nanotechnology. For most of these uses, the integrity of RNA is required and must be maintained during storage. Even though freezing is currently the storage method of choice, the increasing number of samples to be stored and the costly use of a cold chain have highlighted the need for room temperature preservation methods. Here, we report a new room temperature technology that consists in drying RNA samples in the presence of a stabilizer in stainless steel minicapsules. These air- and water-tight capsules isolate RNA from the atmosphere and maintain an anhydrous and anoxic environment. Through the evaluation of RNA integrity over time at room temperature or 90 °C, we identified atmospheric humidity as a major deleterious factor. The degradation rate dependence in temperature fitted an Arrhenius model, with an activation energy of 28.5 kcal/mol and an extrapolated room temperature degradation rate of 3.2 10−13/nt/s (95% confidence interval: 2.3–4.2/nt/s). In these conditions, it is expected that an RNA molecule will be subjected to 0.7–1.3 cut every 1000 nucleotides per century. In addition, we showed that stored RNA is compatible for further analyses, such as reverse transcription-quantitative PCR. No significant change in the Cq values was observed over a simulated period of several decades. At last, our data are consistent with a sequence-independent degradation rate of RNA in the solid state.
Keywords: RNA storage, stability, room temperature, dry state, degradation, anhydrous and anoxic atmosphere
Introduction
RNA is a tool used in many fields, from molecular and cellular biology to medicine and nanotechnology. For instance, RNA populations (whole or subsets) are quantitatively analyzed for studying cellular functions, elucidating normal or pathological mechanisms or seeking molecular signatures for diagnostic purposes (microarrays, RNAseq). In addition, specific RNA species are used as clinical standards1, 2 or as agents for controlling gene expression (siRNA, ribozymes). For reviews, see Baginsky et al,3 Malone et al,4 Wang et al5.
For most of these uses, integrity of RNA is required and must be maintained during storage. However, this molecule can be affected by multiple degradation reactions. First, it is very sensitive to oxidation by reactive oxygen species. In vivo, they are produced by respiration.6 Outside the cell, they can be generated by mechanisms generally involving metallic ions.7, 8 Oxidation could also result from attacks by ozone, an atmospheric pollutant that rapidly reacts with RNA either in solution or in the solid state.9 Degradation can also occur through the activity of some metallic complexes catalyzing the hydrolytic cleavage of the phosphodiester bond or by contaminating nucleases.
However, the main degradative event is the spontaneous cleavage of the phosphodiester linkage through transesterification resulting from a nucleophilic attack of the phosphorus atom by the neighboring 2′OH. A large variety of agents such as specific acids and bases as well as Brønsted acids and base acting as catalysts can be involved. For reviews, see Emilsson et al10 and Oivanen et al.11 RNase A and some ribozymes share this mechanism.12 Water is involved, for instance, by providing hydroxyl or hydronium ions or by allowing proton transfer. As expected, dehydration of RNA strongly inhibits its degradation.13 However, partial rehydration by atmospheric water restores the initial instability while still in the solid state.13
Another characteristic of the reaction is that it is highly dependent on the geometry of the molecule. Indeed, in the transition state, the 2′ oxygen, the phosphorus and a negatively charged oxygen of the phosphate group must be ‘in line'. This structural requirement leads up to 10 000-fold rate variations depending on the local secondary and tertiary structures of the molecule.14, 15
In order to prevent degradation, RNA samples are generally stored frozen at −20 °C or −80 °C or under liquid nitrogen. However, even at a low temperature, RNA retains some reactivity. It has been shown, for instance, that ribonucleases are still active at −20 °C on frozen RNA.16 In addition, the activity of some ribozymes is still significant at −70 °C13 and can even be enhanced by freezing.13, 17 More importantly, the increase in the number of samples that need to be stored by up to hundreds of thousands in biorepositories, biobanks and biological resource centers leads to problems of space, costs, maintenance and security.18 Shipping of RNA samples, usually done in dry ice, is costly and can be challenging for air transportation (regulations, limited weight, long distance travel, and so on). Obviously, an effective room temperature storage and shipping procedure is needed.
To answer this need, several methods for room temperature storage of purified RNA procedures have been proposed. Some companies offer commercial solutions in which RNA is dehydrated in the presence of hydrophilic additives, claimed to protect RNA by entrapment in matrices.19 However, being hydrophilic, they cannot be expected to protect the sample against atmospheric water during storage. Procedures using RNA adsorption on insoluble solid matrixes have also been described. Whatman's FTA paper binds nucleic acids and inactivates ribonucleases but the amounts to be stored and retrieved are low and the storage time is limited.20 Another procedure uses adsorption of the sample on a solid substrate in the presence of a reducing agent and storage in an organic solvent.21 However, no validation of this procedure has yet been published. At last, a method involving a chemical protection of the 2′ OH- is promising, but it necessitates an additional deprotection step and awaits further validation.22 In addition, although all these procedures claim to be efficient in storing RNA, none of them gave a quantitative estimation of the RNA lifetime under the conditions they used. To provide an optimal RNA protection at ambient temperature, we extended to RNA a procedure previously validated for the storage of DNA.23, 24 In this process, RNA is thoroughly dried in the presence of an additive and kept in airtight and waterproof stainless steel minicapsules under an anhydrous and anoxic atmosphere.
Here we showed that (1) efficient RNA storage required full protection from the atmosphere; (2) the room temperature degradation rates of a set of RNA species (mRNAs as well as rRNAs) were proportional to their lengths, suggesting that in the solid state most RNA molecules degraded independently of their sequence; and (3) our procedure made the RNA degradation rate slow enough to allow reliable gene expression studies after long-term storage. Most importantly, we gave a quantitative estimation of the RNA lifetime when RNA was stored protected from the atmosphere.
Materials and methods
RNA sources and preparation
All the glassware and solutions were RNAse-free. A 3313 nucleotide-long HPLC-purified mRNA, coding for yeast β-galactosidase, was purchased from Mitoprod (Bordeaux, France). Human total RNA was prepared from HeLa cells. Pellets of 5 × 106 cells were resuspended in 1 ml of lysis buffer (4 ℳ guanidine thiocyanate, 1% β-mercaptoethanol, 40 mℳ sodium acetate, 0.17 % sarkosyl) and extracted by an equal volume of phenol:chloroform:isoamyl alcohol (250:49:1) and then precipitated by adding 1 volume of isopropyl alcohol and 0.1 volume of sodium acetate (2 ℳ, pH 4) to the aqueous phase. The RNA pellet was washed with 70% ethanol and resuspended in RNAse-free water. To obtain different RNA preparations, bacterial total RNA was extracted from Escherichia coli cells following the same protocol but with one or two extraction steps with the phenol:chloroform:isoamyl alcohol (250:49:1) solution. The overall quality of these various RNA preparations was the same. To increase the variability among the samples, RNA was resuspended in either water, TE buffer (10 mℳ Tris-HCl pH 8 and 1 mℳ EDTA) or Tris buffer (10 mℳ Tris-HCl pH 8). RNA quality and quantity were evaluated by UV measurements (spectrophotometer, SmartSPECTM 3000, Bio-rad, Marnes-la-Coquette, France) and by agarose gel electrophoresis. All RNA samples were stored frozen at −80 °C before being processed.
RNA encapsulation
Preliminary studies (not shown here) indicated, first, that optimal RNA stability was reached by using an Imagene proprietary stabilizer and, second, that RNA retrieval after treatment and storage was quantitative. In each experiment, 2 μg of RNA was mixed with the stabilization solution and deposited in a 0.2 ml glass insert contained in a stainless steel minicapsule. The solution was then thoroughly vacuum-dried. The minicapsules were then laser-welded under anhydrous argon according to the process developed by Imagene company.24 When needed, capsules were opened and RNA was rehydrated by adding 20 or 30 μl of water, followed by 10-min incubation on ice.
Accelerated degradation studies
Minicapsules were heated at temperatures ranging from 50 to 130 °C in an oven. For studying the effect of moist air exposure, a set of minicapsules were opened and placed in an atmosphere of approximately 50% relative humidity inside closed glass bottles as previously described in.23, 24 At each time point, the capsules were quickly removed from the oven and stored at −20 °C until analysis. The t0 time was set at 10 min to allow the establishment of temperature and hydration equilibriums.
Analyses by capillary electrophoresis
Total RNA samples (200 ng) were heat denatured for 2 min at 70 °C and run with an RNA 6000 nano kit on a Bioanalyzer (Agilent Technologies, Les Ulis, France).
Agarose gel electrophoresis and quantitative analysis
Five hundred nanograms of RNA samples were analyzed using agarose gel electrophoresis as it gives a quantitative measurement of fluorescence signals. RNA was heat denatured for 5 min at 70 °C or 75 °C before being run on a 1.5% agarose gel at 50 V in TAE 0.5X (40 mℳ Tris acetate, 1 mℳ EDTA). The gels were stained for 1 h with ethidium bromide (500 ng/μl) and photographed with a digital imaging device under UV light (Quantum ST4 1000, Vilber Lourmat, Marne-la-vallée, France). The images were analyzed with the Bio1D image analysis software (Vilber Lourmat). RNA bands of interest (28S for human total RNA, 23S for bacterial rRNA and mRNA) were identified and their fluorescence was recorded. As fluorescence intensities are proportional to RNA mass in these conditions, the proportion of intact RNA was calculated by normalizing fluorescence intensities to the fluorescence intensity at t0 time. Proportions were normalized to represent the proportion of intact phosphodiester bond by taking into account the size of the molecule of interest (28S=5070 nt, 23S=2904 nt, mRNA=3313 nt).
Statistical analysis, estimation of degradation rates and the Arrhenius model
Proportions of intact phosphodiester bonds in the studied RNA species were plotted against incubation time. The degradation kinetic constant for a given temperature, kT, was determined by fitting the data points to an exponential decrease, where the proportion of intact phosphodiester bonds is 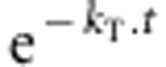 with T being the temperature in K and t being the heating time in seconds.
with T being the temperature in K and t being the heating time in seconds.
Estimation of the lognkT confidence intervals was performed with the R software linear regression tool.
To construct the Arrhenius plot of RNA degradation in the minicapsules, two methods were used. First, the kT values were estimated by exponential curve fitting and used on the Arrhenius plot, log kT=f(1/T). The k25 °C can then be extrapolated through linear regression. The second method used a personal R program allowing the determination of confidence intervals on the extrapolated values of kT by fitting all the data points of the kinetics at the same time23 (Supplementary Data S1).
Reverse transcription and quantitative PCR experiments on RNA heated at 90 °C to accelerate aging
Capsules were heated at 90 °C for up to 30 days and stored at −20 °C until use.
After rehydration, the equivalent of 700 ng of total RNA was converted into cDNA either using (dT)18 (Fermentas, Dominique DUTSCHER SAS, Brumath, France) for reverse transcription initiation at the 3′ end of mRNAs (Figure 3) or the ‘verso cDNA synthesis' kit (Abgene, Dominique DUTSCHER SAS) with random hexamers (Figure 4). The reaction mixture was incubated for 5 min at 25 °C, for 45 min at 42 °C and then for 2 min at 95 °C. A fivefold-diluted cDNA solution of 2 μl was used for quantitative PCR (qPCR) with the ‘maxima SYBRGreen qPCR master mix ROX solution' provided in the kit (Fermentas). Each 25 μl reaction volume contained 250 nℳ of each forward and reverse primer (Table 1) and 5 μl of a 100-fold diluted ROX solution in addition to cDNA. After 10 min at 95 °C, the 40 cycles (15 s at 95 °C, 30 s at 60 °C and 30 s at 72 °C) were carried out on the Agilent MX3000p machine. No template control and no reverse transcriptase controls were included on each plate of qPCR. qPCR reactions were set up in duplicate. The Cq values were calculated using the MXPro software (amplification-based threshold, data treated individually). Amplicon targets in transcripts from eight genes – GAPDH, PSMB6, TUBA1B, PPIE, TBP, EEF1a1, β2M and 18S (GenBank accession numbers are reported in Table 1) – were used. The transcripts are expressed at different levels in HeLa cells: from highly abundant (18S) to very low (TBP) via intermediate levels. Amplicon lengths varied from 75 pb to 221 pb (Table 1).
Table 1. Gene identification and oligonucleotide sequences.
| GenBank | Amplicon | ||
|---|---|---|---|
| Gene | transcript ID | length (bp) | Oligonucleotides sequences |
| TBP | NM_003194 | 90 | For: cacgaaccacggcactgatt |
| Rev: ttttcttgctgccagtctggac | |||
| EEF1A1 | NM_001402 | 221 | For: ctggagccaagtgctaacatg |
| Rev: actggtgttctcaaacccgg | |||
| B2M | NM_004048 | 177 | For: actacactgaattcacccc |
| Rev: tctgcttgcttgctttttaa | |||
| 18S | NR_003286 | 112 | For: cctggataccgcagctagga |
| Rev: gcggcgcaatacgaatgcccc | |||
| GAPDH | NM_002046 | 120 | For: ttggtatcgtggaaggactcatg |
| Rev: gggctctccagaacatcatcc | |||
| PPIE | NM_001195007 | 75 | For: gcgttcattccttttggaga |
| Rev: aaatcctcggtgcttttctg | |||
| TUBA1B | NM_006082 | 107 | For: ttacctcgactcttagcttgtcg |
| Rev: ggatggagatgcactcacg | |||
| PSMB6 | NM_002798 | 116 | For: gataccgggaagacctgatg |
| Rev: aatggcaaaggactgcctta |
Size dependency of RNA degradation rate in the minicapsules
Reverse transcription-qPCR (RT-qPCR) reactions were set up using (dT)18 for the reverse transcription of aged or control mRNA as described in the above paragraph. The Cq for each reaction was plotted as a function of the incubation time at 90 °C. The values of k were calculated from the linear relationship of Cq=f(t90 °C) as shown below. At the time, t:
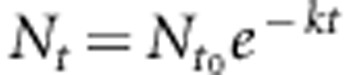 |
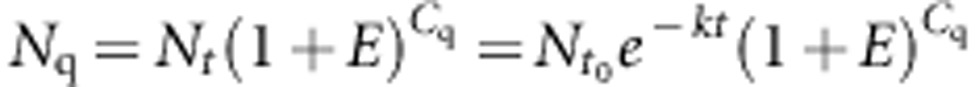 |
 |
where 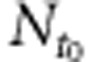 , Nt and Nq are the copy numbers of the considered amplicon, respectively, at the beginning, at time t and at the threshold of the degradation kinetic. Nq is constant for a given run of qPCR.
, Nt and Nq are the copy numbers of the considered amplicon, respectively, at the beginning, at time t and at the threshold of the degradation kinetic. Nq is constant for a given run of qPCR.
E signifies PCR efficiency. According to the literature and our present work, E is independent of the degradation state and can be considered a constant for a given amplicon (see Discussion).
Therefore, 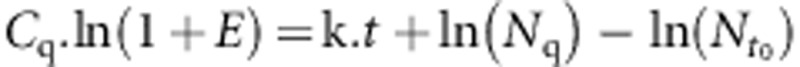 and
and
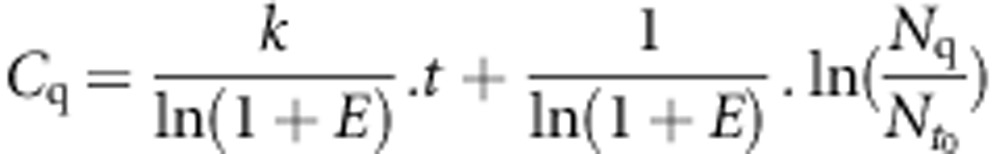 |
As E,  and Nq are constants, Cq values plotted as a function of time must be fitted by a straight line, with a slope being (k/(ln(1+E)).
and Nq are constants, Cq values plotted as a function of time must be fitted by a straight line, with a slope being (k/(ln(1+E)).
Results
Degradation of solid-state RNA in the presence or absence of a moist atmosphere
We first evaluated the stability of two RNA species at room temperature. First, encapsulated β-galactosidase mRNA samples were left for up to 6 months at room temperature and run on an electrophoresis gel (Figure 1a). On the basis of the measurements of the fluorescence intensity of the bands, one can conclude that no significant degradation occurred over this time period (Figure 1a). Thereafter, we compared the degradation kinetics of rRNA when fully protected from air and when exposed to air. As seen in Figure 1b, RNA exposed to air exhibited a clear degradation: after 92 weeks at room temperature, no intact 28S rRNA molecule could be seen and the RIN value dropped from 7.3 to 2.0. In contrast, when protected from air, the RIN number slightly dropped from 7.2 to 6.8 after 23 months at room temperature.
Figure 1.

RNA degradation at room temperature in the presence or absence of a moist atmosphere. β-Galactosidase mRNA samples (a) and total RNA (b–d) were dried and encapsulated under anoxic and anhydrous atmosphere. After incubation at room temperature, at −20 °C or at 90 °C, aliquots corresponding to 500 ng of the initial RNA amounts were denatured for 3 min at 75 °C and run on agarose gel. (a) Degradation of HPLC-purified β-galactosidase mRNA. The proportion of undegraded mRNA in minicapsules stored at −20 °C is given under each lane (the experiments were performed in triplicate, but a single representative gel is shown). ‘−80 °C': RNA stored at −80 °C for 24 weeks. ‘−20 °C': RNA in minicapsules stored at −20 °C for 24 weeks. (b) Degradation of total RNA at room temperature. Half of the capsules were opened and exposed to air. The ratios of the fluorescence intensities of 28S rRNA and 18S rRNA as well as the RIN values are given for each sample under the respective lanes. (c) Total RNA degradation at 90 °C in the absence of air (minicapsule conditions). Total RNA samples were dried and stored under anoxic and anhydrous atmosphere in minicapsules. They were heated at 90 °C under 50% relative humidity (RH). Two kinetics were run for 8 h and 240 h. Five hundred nanograms of total RNA samples were analyzed on electrophoresis gels. The proportion of intact 28S molecule, measured by the Bio1D software, is indicated for each time point (the experiments were performed in triplicate; a single representative gel is shown). (d) Same as in (c), but with RNA exposed to air under 50% RH (the experiments were performed in triplicate; a single representative gel is shown).
In order to quantify the degradation rate difference of RNA preserved in the presence or absence of a moist atmosphere, RNA aging was accelerated by heating. Degradation of total RNA was performed at 90 °C either in closed capsules or in opened capsules, and placed in a 50% relative humidity atmosphere (relative humidity control was necessary to maintain RNA in the same hydration state as it is at room temperature).
Figures 1c and d show that, after 8 h of exposure at 50% relative humidity, the amount of 28S rRNA band was no longer measurable, whereas, when protected from air, about 90% of the 28S band was still present. The degradation rate constants at 90 °C, k90 °C, as determined by exponential curve fitting were 7.7 × 10−10/nt/s and 1.9 × 10−8/nt/s for RNA protected or unprotected from moist air, respectively. This corresponds to a 25-fold increase in the degradation rate constant at 90 °C.
Determination of the room temperature chain-breaking rate of air-protected solid-state RNA
As the RNA degradation rate at room temperature in the minicapsules was slow, it was not possible to directly measure it over the time of the study. However, it can be estimated by using the Arrhenius model on data drawn from kinetics run at different temperatures. In order to obtain a wide variety of samples, we used RNA samples that differed in their origin (bacteria and human cell lines) and the nature of the resuspension buffer used at the end of extraction (10 mℳ Tris-HCl pH 7, 10 mℳ Tris-HCl 1 mM EDTA pH 8 and RNAse-free water pH 7.2). Seventy-two kinetics were run at temperatures ranging from 50 to 130 °C (not shown). The proportion of two intact RNA species (human 28S rRNA and E. coli 23S rRNA in total RNA samples) was monitored over time. Supplementary Figure 1, Supplementary Material, is a representative example of these kinetics.
R software was used for determining the strand-breaking rate constant kT from each of the kinetics. The kT values and the confidence intervals for all RNA species (28S and 23S) were averaged. The log10 of these values gave a straight line when plotted as a function of 1/T (K−1) (Figure 3). The temperature dependency of the RNA strand-breaking rates followed the Arrhenius model. No discrepancies were found between the 28S and the 23S strand-breaking rate constants. Thus, the degradation rate constant at room temperature in the absence of both atmospheric water and oxygen could be determined by linear extrapolation and was estimated to 3.2 10−13/nt/s at 25 °C with a 95% confidence interval of (2.3–4.2) 10−13/nt/s. This experiment also confirmed the decrease in stability induced by exposure to moisture (Figure 3, drop data point).
Size dependency of RNA degradation rate in the minicapsules
The degradation rate constants at 90 °C were calculated for five mRNA species by linear regression of the plots of Cq values as a function of time (data not shown). These rate constants appeared to be proportional to the distance between the 5′ of the terminal poly(A) and the 5′ end of the amplicon (Figure 2) (y= 6.10−10 * (size in nt), r2= 0.84). This suggests that, in the minicapsules, the RNA degradation rate depends mainly on its size and can be considered as being independent of its sequence and of the presence of local structures.
Figure 2.

Degradation rates as a function of RNA length on several mRNA species. Total RNA samples in minicapsules were heated at 90 °C to accelerate aging. Reverse transcription reactions were set up using (dT)18 oligonucleotides. The qPCR reactions were run using a SybrGreen fluorescent dye and transcript-specific oligonucleotides of TBP, β2M, GAPDH, TUBA1B and PSMB6 as described in Materials and Methods (cf Table 1). For each time point, the Cq value was measured and plotted as a function of time t. The relationship Cq=f(t) was linear for each mRNA species (not shown) and allowed the calculation of the degradation rate constants at 90 °C, k. k was plotted as a function of the distance between the 3′ end of the mRNA species (+18 nucleotides) and the 5′ end of the forward primer. The straight line is a linear fit through the data points.
At last, the normalized calculated k values for each mRNA species were reported on the Arrhenius graph (Figure 3, small circles). These values are in agreement with the other data summarized on the Arrhenius graph.
Figure 3.

Temperature dependence of the degradation rate constant of RNA stored in the minicapsules plotted according to the Arrhenius model. Degradation rates (k) of 23S rRNA resuspended after purification in water (open squares), Tris-HCl-EDTA (open diamonds) or Tris-HCl only (open circles) and those of 28S rRNA resuspended in water (closed diamond) were determined at temperatures ranging from 50 to 130 °C with R software (28S=5070 nt 23S=2904 nt, mRNA=3313 nt). Degradation rates of five mRNA species were calculated through RT-qPCR reactions (Figure 2). Log10(k) value was plotted as a function of 1/T. Confidence interval (95%) was calculated with R software as described in Methods. As a comparison, the log10(k) corresponding to the 23S rRNA incubated in opened minicapsules under 50% RH at 90 °C (drop) was also plotted.
QPCR validation experiments on minicapsule-stored RNA
As RT-qPCR measurements are widely used, in particular for gene expression analysis, it was necessary to run this type of experiment on aged RNA samples. Encapsulated human total RNA was heated at 90 °C for increasing periods of times. The rehydrated RNA samples were submitted to a randomly primed reverse transcription, and qPCR reactions were run on eight RNA species that have very different levels of expression in HeLa cells. As controls, RT-qPCR reactions for TUBA1B and 18S amplicons were run on RNA solutions stored at −80 °C. Figure 4 shows either no significant changes or a slight increase in the Cq values for EEF1a1 and β2M qPCR reactions.
Figure 4.

Evolution of Cq as a function of time at 90 °C. RT-qPCR reactions and Cq calculations were performed as described in Materials and Methods on amplicons from the following transcripts: TUBA1B (open diamonds), PSMB6 (open squares), PPIE (closed circles), TBP (open circles), EEF1a1 (open triangles), β2M (closed squares), GAPDH (closed diamonds) and 18S rRNA (closed triangles). The slopes of the linear regressions are indicated for each amplicon.
The PCR efficiencies of the qPCR reactions remained stable over the kinetics for each amplicon (SDs of the mean efficiency for each amplicon were between 0.01 and 0.17 and did not correlate with the degradation evolution over time). These data agree with previous studies showing no change in PCR efficiency with RNA degradation.25
No difference in Cq values was observed between the control solution stored at −80 °C and the encapsulated RNA (TUBA1B: Cq= 20.55 and Cq=20.54, and 18S rRNA: Cq= 7.7+/−0.15, n= 4). These data as well as others (independent evaluations, to be published) suggest that there is no bias in RT-qPCR outcome between RNA stored in the minicapsules and RNA stored in solution at −80 °C.
In conclusion, RNA remained compatible with analyses by RT-qPCR when preserved in minicapsules (see Discussion).
Discussion
Air- and moisture-tight containers are necessary for reliable room temperature storage of RNA
Our results (Figures 1 and 2) agree and extend previous observations made on a ribozyme13 showing that almost complete water removal strongly reduces the rate of RNA phosphodiester bond cleavage. This is to be expected as water has a direct role in the transesterification reaction, but reduction in degradation rate can also be due to a loss of molecular mobility induced by the solid state.26, 27, 28 However, if that is well established for pharmaceuticals, there is no direct evidence for nucleic acids.
The highly dehydrated state must be maintained as moisture exposure increases the degradation rate through partial rehydration.29 We also observed that the presence of an additive further increased RNA stability (not shown here) but provided no protection against air exposure. This sensitivity to air exposure precludes the use of usual laboratory plastic tubes for room temperature RNA storage as these tubes are not moisture tight. In contrast, welded-sealed stainless steel minicapsules maintain an anhydrous and anoxic atmosphere over time.
Estimation of the degradation rate at room temperature of encapsulated RNA
Through linear extrapolation of the Arrhenius plot (Figure 3), we estimated the degradation rate constant of RNA stored in the minicapsules at 25 °C to be 3.2 10−13/nt/s (95% confidence interval: 2.3–4.2/nt/s). In these conditions, it is expected that an RNA molecule will be subjected to 0.7–1.3 cut every 1000 nucleotides per century. Accordingly, the β-galactosidase mRNA and the 28S rRNA are expected to undergo less than 10% of degradation over a 24-month period. This extrapolation agrees with the results of Figure 1. The latter are to be compared with the results of a recent paper from Brisco and Morley who found that, in solution, RNA molecules undergo between 0.7 and 7.6 cut per 1000 nucleotides per 30 min at 90 °C.30 This corresponds to a degradation rate that is about 1000-fold faster than that in the minicapsules at the same temperature.
In these storage conditions, the activation energy of the strand-breaking reaction is 28.5 kcal/mol. This value is quite close to the activation energy of the base-catalyzed cleavage in the solution of a 22-nucleotide-long RNA molecule, which was found to be 29 kcal.31 This suggests that the degradation mechanism is the same as in solution and that lowering the molecular mobility may not be involved in degradation inhibition.
The linearity of the Arrhenius plot (Figure 3) over a wide range of temperatures (50–130 °C) is a good indication that no other parameter apart from temperature (in particular, structural changes) is significantly involved in the determination of the dehydrated RNA degradation rate.31 This idea is supported by the data in Figure 2 and by spectroscopic studies showing that dehydration had the same effect as heating in destroying the secondary and tertiary structure.29 This is important as it is known that RNA degradation rates are very heterogeneous in solution, mainly because of the variety of local RNA conformation.15 The coherence between the rates of degradation of eight different mRNA and rRNA species suggests that, in the minicapsules, RNA may degrade independently of its sequence. In addition, the fact that the temperature dependency of the degradation rate follows the Arrhenius model validates heating as a way of simulating extended periods of time.
RT-qPCR on aged RNA. Implication for gene expression measurements
Reverse transcription coupled to qPCR is widely used for gene expression measurements (ie, the measure of the copy number of specific transcripts). It has been shown to be sensitive even to moderate degradation.25, 32, 33, 34 A loose correlation has been seen between Cq increase and target size. For instance, short amplicons (70–250 bp) are more or less independent of a slight degradation of the RNA quality, whereas products over 400 bp are strongly dependent.33 In the most currently used procedure (random priming for the reverse transcription step), the loss of measured copy number is less due to fragmentation of the sequence to be amplified than due to a decrease in the priming efficiency of this target. The decrease upon fragmentation is not directly dependent on the length of the amplicon and is variable among species. In some cases, it has even been seen that degradation could raise the efficiency of the reverse transcription reaction.34, 35 This is probably due to suppression of the inhibitory effect of the RNA secondary structure.36
According to our Arrhenius model, the last point of the kinetics at 90 °C simulates a 160-year storage period at room temperature. Even up to this equivalent of 160-year storage, the Cq increase is not significant (Figure 4). Tentative linear regressions showed low linear regression coefficients and low slopes with a maximum slope of 0.01 Cq value per year relative to the β2M transcript. According to this value, the maximum Cq increase in 10 years at room temperature should be 0.1 Cq, corresponding to a loss of 7% in copy number. The minimum slope (a=0.002 for TUBA1B and PSMB6) would result in less than 1% copy number difference after 10 years of storage at room temperature. The PCR efficiency did not change along the kinetics and we saw some dependence on the length of the amplicon (Table 1), which is in agreement with previous studies.25, 33
These losses are acceptable for most gene expression measurements. They could be minimized by reducing the size of the amplicon and by using priming oligonucleotides complementary of sequences immediately downstream of the target as done by Briscoe and Morley.30 At last, some currently emerging gene expression techniques eliminate the reverse transcription step. In this case, it will be advantageous to have a storage procedure that allows a degradation rate similar for all RNA sequences.
In conclusion, the process described here allows the following: (1) reliable long-term RNA storage of molecular biology and clinical standards or patient samples; (2) quantitative recovery (Supplementary Figure 2); (3) economically sustainable archiving and shipping by avoiding the maintenance and alarm costs of freezers and by a 10-fold decrease in storage space due to the capsule size, for instance; and (4) an ideal green alternative to −80 °C freezers and dry-ice shipping without reliance on chain cold chain from storage to shipping.
Acknowledgments
We thank D Coudy for help and discussions, the ENSTBB, Marc Bonneu for discussions and making available a qPCR machine, Pr. Merlio and Y Drissi for supplying oligonucleotides and Integragen for sharing their Bioanalyzer. This work was supported by the ‘Conseil Régional d'Aquitaine', ‘FEDER', ‘Genopole' and ‘Oséo Innovation'
S Tuffet is CEO and shareholder of Imagene Company; M Colotte, A-L Fabre, A Luis are employees of Imagene Company; J Bonnet, is a shareholder, has consulted for Imagene Company and received compensation.
Footnotes
Supplementary Information accompanies this paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary Material
References
- Madej RM, Davis J, Holden MJ, Kwang S, Labourier E, Schneider GJ. International standards and reference materials for quantitative molecular infectious disease testing. J Mol Diagn. 2010;12:133–143. doi: 10.2353/jmoldx.2010.090067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White HE, Matejtschuk P, Rigsby P, et al. Establishment of the first World Health Organization International Genetic Reference Panel for quantitation of BCR-ABL mRNA. Blood. 2010;116:e111–e117. doi: 10.1182/blood-2010-06-291641. [DOI] [PubMed] [Google Scholar]
- Baginsky S, Hennig L, Zimmermann P, Gruissem W. Gene expression analysis, proteomics, and network discovery. Plant Physiol. 2010;152:402–410. doi: 10.1104/pp.109.150433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone J, Oliver B. Microarrays, deep sequencing and the true measure of the transcriptome. BMC Biol. 2011;9:34. doi: 10.1186/1741-7007-9-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen HE, Specht E, Broedbaek K, et al. RNA modifications by oxidation: a novel disease mechanism. Free Radic Biol Med. 2012;52:1353–1361. doi: 10.1016/j.freeradbiomed.2012.01.009. [DOI] [PubMed] [Google Scholar]
- Bruskov VI, Malakhova LV, Masalimov ZK, Chernikov AV. Heat-induced formation of reactive oxygen species and 8-oxoguanine, a biomarker of damage to DNA. Nucleic Acids Res. 2002;30:1354–1363. doi: 10.1093/nar/30.6.1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RK, Xu Z, Bohannon KE, Wang B, Bruner MW, Volkin DB. Evaluation of degradation pathways for plasmid DNA in pharmaceutical formulations via accelerated stability studies. J Pharm Sci. 2000;89:76–87. doi: 10.1002/(SICI)1520-6017(200001)89:1<76::AID-JPS8>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Cataldo F. Ozone degradation of ribonucleic acid (RNA) Polym Degrad Stabil. 2005;89:274–281. [Google Scholar]
- Emilsson GM, Nakamura S, Roth A, Breaker RR. Ribozyme speed limits. RNA. 2003;9:907–918. doi: 10.1261/rna.5680603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oivanen M, Kuusela S, Lonnberg H. Kinetics and mechanisms for the cleavage and isomerization of the phosphodiester bonds of RNA by bronsted acids and bases. Chem Rev. 1998;98:961–990. doi: 10.1021/cr960425x. [DOI] [PubMed] [Google Scholar]
- Perreault DM, Anslyn EV. Unifying the current data on the mechanism of cleavage–transesterification of RNA. Angew Chem Int Ed. 1997;36:432–450. [Google Scholar]
- Seyhan AA, Burke JM. Mg2+-independent hairpin ribozyme catalysis in hydrated RNA films. RNA. 2000;6:189–198. doi: 10.1017/s1355838200991441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soukup GA, Breaker RR. Relationship between internucleotide linkage geometry and the stability of RNA. RNA. 1999;5:1308–1325. doi: 10.1017/s1355838299990891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaukinen U, Lyytikainen S, Mikkola S, Lonnberg H. The reactivity of phosphodiester bonds within linear single-stranded oligoribonucleotides is strongly dependent on the base sequence. Nucl Acids Res. 2002;30:468–474. doi: 10.1093/nar/30.2.468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S, Huang Y, van Huystee RB. Improved plant RNA stability in storage. Anal Biochem. 2004;326:122–124. doi: 10.1016/j.ab.2003.10.026. [DOI] [PubMed] [Google Scholar]
- Kazakov SA, Balatskaya SV, Johnston BH. Ligation of the hairpin ribozyme in cis induced by freezing and dehydration. RNA. 2006;12:446–456. doi: 10.1261/rna.2123506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M. Biorepositories: building better biobanks. Nature. 2012;486:141–146. doi: 10.1038/486141a. [DOI] [PubMed] [Google Scholar]
- Wan E, Akana M, Pons J, et al. Green technologies for room temperature nucleic acid storage. Curr Issues Mol Biol. 2009;12:135–142. [PMC free article] [PubMed] [Google Scholar]
- Natarajan P, Trinh T, Mertz L, Goldsborough M, Fox DK. Paper-based archiving of mammalian and plant samples for RNA analysis. Biotechniques. 2000;29:1328–1333. doi: 10.2144/00296pf01. [DOI] [PubMed] [Google Scholar]
- Novoradovskaya N, Basehore L, Scott BramanJC.Stabilization of nucleic acids on solid supports2009. WO 2009/029433 A2.
- Bates M, Goldsborough A.RNA sample stabilization in the presence of a transitional metal2011. WO 2011/008553 A1.
- Bonnet J, Colotte M, Coudy D, et al. Chain and conformation stability of solid-state DNA: implications for room temperature storage. Nucleic Acids Res. 2010;38:1531–1546. doi: 10.1093/nar/gkp1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colotte M, Coudy D, Tuffet S, Bonnet J. Adverse effect of air exposure on the stability of dna stored at room temperature. Biopreserv Biobank. 2011;9:47–50. doi: 10.1089/bio.2010.0028. [DOI] [PubMed] [Google Scholar]
- Fleige S, Pfaffl M. RNA integrity and the effect on the real-time qRT-PCR performance. Mol Aspects Med. 2006;27:126–139. doi: 10.1016/j.mam.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Byrn SR, Xu W, Newman AW. Chemical reactivity in solid-state pharmaceuticals: formulation implications. Adv Drug Deliv Rev. 2001;48:115–136. doi: 10.1016/s0169-409x(01)00102-8. [DOI] [PubMed] [Google Scholar]
- Shalaev EY, Zografi G. How does residual water affect the solid-state degradation of drugs in the amorphous state. J Pharm Sci. 1996;85:1137–1141. doi: 10.1021/js960257o. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, Suryanarayanan R. Local mobility in amorphous pharmaceuticals—characterization and implications on stability. J Pharm Sci. 2009;98:2935–2953. doi: 10.1002/jps.21728. [DOI] [PubMed] [Google Scholar]
- Falk M. The ultraviolet spectra of ribonucleic acids in solid state and in solution. Rev Canadienne Chim. 1965;43:3151–3159. [Google Scholar]
- Brisco MJ, Morley AA. Quantification of RNA integrity and its use for measurement of transcript number. Nucleic Acids Res. 2012;40:e144. doi: 10.1093/nar/gks588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Breaker RR. Kinetics of RNA degradation by specific base catalysis of transesterification involving the 2'-hydroxyl group. J Am Chem Soc. 1999;121:5364–5372. [Google Scholar]
- Imbeaud S, Graudens E, Boulanger V, et al. Towards standardization of RNA quality assessment using user-independent classifiers of microcapillary electrophoresis traces. Nucl Acids Res. 2005;33:e56. doi: 10.1093/nar/gni054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleige S, Walf V, Huch S, Prgomet C, Sehm J, Pfaffl MW. Comparison of relative mRNA quantification models and the impact of RNA integrity in quantitative real-time RT-PCR. Biotechnol Lett. 2006;28:1601–1613. doi: 10.1007/s10529-006-9127-2. [DOI] [PubMed] [Google Scholar]
- Vermeulen J, De Preter K, Lefever S, et al. Measurable impact of RNA quality on gene expression results from quantitative PCR. Nucleic Acids Res. 2011;39:e63. doi: 10.1093/nar/gkr065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Meth. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Das M, Harvey I, Chu LL, Sinha M, Pelletier J. Full-length cDNAs: more than just reaching the ends. Physiol Genomics. 2001;6:57–80. doi: 10.1152/physiolgenomics.2001.6.2.57. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.