

Abstract
Background and aim
Impaired fatty liver regeneration has already been reported in many genetic modification models. However, in diet-induced simple hepatic steatosis, which showed similar phenotype with clinical pathology, whether liver regeneration is impaired or not remains unclear. In this study, we evaluated liver regeneration in mice with diet-induced simple hepatic steatosis, and focused on excess lipid accumulation occurring during liver regeneration.
Methods
Mice were fed high fat diet (HFD) or control diet for 9–10 weeks. We analyzed intrahepatic lipid accumulation, DNA replication, and various signaling pathways including cell proliferation and ER stress during liver regeneration after partial hepatectomy. In addition, some of mice were pretreated with tauroursodeoxycholic acid (TUDCA), a chemical chaperone which alleviates ER stress, and then we estimated TUDCA effects on liver regeneration.
Results
The peak of hepatocyte BrdU incorporation, the expression of proliferation cell nuclear antigen (PCNA) protein, and the expressions of cell cycle-related genes were observed in delayed time in HFD mice. The expression of phosphorylated Erk1/2 was also delayed in HFD mice. The amounts of liver triglyceride were at least twofold higher in HFD mice at each time point. Intrahepatic palmitic acid was increased especially in HFD mice. ER stress induced during liver regeneration was significantly higher in HFD mice. In HFD mice, pretreatment with TUDCA reduced ER stress and resulted in improvement of delayed liver regeneration.
Conclusion
In simple hepatic steatosis, lipid overloading occurring during liver regeneration might be caused ER stress and results in delayed hepatocyte DNA replication.
Keywords: Fatty liver, Liver regeneration, Lipotoxicity, ER stress
Introduction
Non-alcoholic fatty liver disease (NAFLD) is a hepatic component of metabolic syndrome and is closely associated with insulin resistance and dyslipidemia [1]. Recently, the prevalence of NAFLD has reached up to 20–30 % of the healthy population [2, 3], and NAFLD is a very common problem in the world. Nowadays, it is estimated that more than 20 % of the patients who plan for liver resection have various degrees of fatty liver [4]. Liver resection is the most curative treatment option for patients with malignant hepatic tumors. Postoperative mortality was decreased by advances in hepatic surgery technique, improvements in patient selection criteria, and accurate perioperative management. However, postoperative complications still remain as important problems in patients with fatty liver [5–7]. It is clinically significant to elucidate the difference between fatty liver regeneration and normal liver regeneration.
The liver has a remarkable capacity to recover from injury. Liver regeneration is highly orchestrated by the activation of multiple pathways, such as cytokines, growth factors, intracellular signaling events, transcription factors, and metabolic networks [8–10]. Fatty liver might be considered as one of major risk factors for impaired liver regeneration [11–16], but others have mentioned that mild fat infiltration has no influence on liver regeneration [17–19]. Many studies have demonstrated that fatty liver regeneration is based on genetically modified models, which are considerably influenced by their own genetic abnormalities on the hepatocyte proliferation process, or on a methionine–choline deficient diet-induced fatty liver model which showed various degrees of inflammation without obesity. These models significantly differed from clinical pathology. Recently, several studies have reported about impaired liver regeneration in diet-induced fatty liver models which were similar in clinical conditions [11, 12], but the regeneration process of fatty liver has not been fully elucidated.
Intrahepatic lipid accumulation occurring from the early regeneration phase in partial hepatectomy (PHx) rodent models seemed to be an essential process [20]. The most important source of lipids that accumulated in the regenerating liver was considered to be mainly free fatty acids (FFAs) supplied from adipose tissue, indeed de novo hepatic fatty acid synthesis also has been reported [21–24]. Lipids were consumed as energy for hepatocyte DNA replication and materials for phospholipids synthesis [25, 26]. However, the influence of excess lipid accumulation during liver regeneration is not clearly understood. Therefore, in this study, we evaluated the relation between liver regeneration and excess lipid accumulation in mice with high fat diet (HFD)-induced simple hepatic steatosis.
Materials and methods
Animals
Eight-week-old male C57BL6/J mice were purchased from Clea Japan (Tokyo, Japan). Mice were housed in a temperature- and humidity-controlled environment with 12/12 h light/dark cycle. Mice were divided into two groups. The control group (CD) was fed a standard mice chow (MF: Oriental Yeast, Osaka, Japan), and the other group (HFD) was fed a high-fat diet (HFD60: Oriental Yeast) ad libitum for 9–10 weeks. Two-thirds PHx, according to Higgins and Anderson [27], was performed at the same time of the day. Four to eight mice were killed at several time points after PHx. Mice received intraperitoneal injection of bromodeoxyuridine (BrdU) (100 μg/g body weight, Sigma Aldrich, Tokyo, Japan) 4 h before killing for evaluation of hepatocyte DNA replication. Moreover, some mice received intravenous injection of tauroursodeoxycholic acid (TUDCA) (75 μg/g body weight, Sigma Aldrich) 1 h before PHx to alleviate endoplasmic reticulum (ER) stress. Blood was collected aseptically from the inferior vena cave and centrifuged (2,000 g, 10 min, 4 °C) and plasma was collected. The remnant liver was removed en bloc and weighed. The liver was either fixed with 10 % buffered formaldehyde or embedded in compound and frozen at −80 °C for histological examination, or immediately frozen in liquid nitrogen and stored at −80 °C for protein, mRNA, and lipid extraction. The experimental protocols and animal maintenance procedures used in this study were approved by the Ethics Review Committee for Animal Experimentation of Osaka University Graduate School of Medicine.
Analysis procedures
The concentrations of plasma alanine aminotransferase (ALT), triglycerides (TG), total cholesterol (TC), and glucose were measured using the transaminase CII-test Wako kit, TG E-test Wako kit, TC E-test Wako kit, and glucose CII-test Wako kit (Wako Pure Chemical Industries, Tokyo, Japan). The concentration of plasma FFAs was measured using the NESCAUTO NEFA Kit-U (Alfresa-phama, Osaka, Japan), insulin was measured using the insulin ELISA kit (Morinaga Institute of Biological Science, Yokohama, Japan), total bilirubin was measured by the bilirubin oxidase method, and albumin was measured by BCG method (Oriental Yeast).
Measurement of hepatic lipid contents
We performed oil red O staining on liver sections of unfixed freshly frozen tissue to assessed hepatic lipid contents visually. Total lipids were extracted from the liver as described previously [28]. Hepatic TG contents and components of hepatic FFAs were measured by using TG E-test Wako kit and Gas-chromatograph (SRL Inc, Tokyo, Japan).
Immunohistological analysis
For detection of hepatocyte DNA replication, liver sections were subjected to BrdU immunohistochemical staining using BrdU IHC Kit (Kamiya Biomedical Company, WA, USA). In each section, we counted the number of BrdU positive nuclear cells and calculated the BrdU labeling index (percentage of BrdU-positive cell). To assess ER stress, we also demonstrated immunohistochemical staining on liver sections using anti-BiP/glucose-regulated protein of 78-kDa (GRP78) antibody (Cell Signaling Technology Inc., Tokyo, Japan) and anti-spliced form of X-box binding protein 1 (sXBP-1) antibody (Santa Cruz Biotechnology Inc., CA, USA). These sections were heated under pressure with Target Retrieval Solution (pH9) (DakoCytomation, Kyoto, Japan) before incubation with primary antibody. We used goat anti-rabbit secondary antibody and avidin–biotin complex (VECTASTAIN ABC Rabbit IgG Kit, Vector Laboratories, Inc., CA, USA). These sections were visualized by diaminobenzidine tetrahydrochloride (DAB Kit, Vector Laboratories) and haematoxylin (Wako Pure Chemical Industries).
Western blotting
Total extracts prepared from liver tissues were separated on SDS-PAGE and transferred onto PVDF membrane as described previously. We used primary antibodies specific to proliferation cell nuclear antigen (PCNA) (DakoCytomation), phospho-Met (Thy1234/1235), c-Met, phospho-epidermal growth factor receptor (EGFR) (Tyr1068), total EGFR, phospho-Erk1/2 (Thr202/Tyr204), total Erk1/2, phospho-Akt (Ser473), total Akt, GRP78 (Cell Signaling Technology Inc.), sXBP-1 (Santa Cruz Biotechnology Inc.), and GAPDH (Trevigen, MD, USA). Immunoreactive bands were visualized on film by using ECL detection reagent (GE Healthcare, WI, USA), and quantitative densitometric analysis of each band was performed using Fluor Chem IS-8000 (Alpha Innotech Corp., CA, USA). Additionally, total extracts prepared from BNL-Cl2 cells treated for 10 h with or without tunicamycin (5 μg/ml, Sigma Aldrich), a chemical inducer of ER stress, were used as a positive or negative control of sXBP-1.
Determination of gene expression levels
Total RNA was extracted from whole livers with QIAshredder and an RNeasy Mini Kit according to the instructions provided by the manufacturer (Qiagen, Hilden, Germany), and then transcribed into complementary DNA with a ReverTra Ace qPCR RT Kit (Toyobo, Osaka, Japan). Quantitative real-time RT-PCR was performed with a THUNDERBIRD SYBR qPCR Mix (Toyobo) using specific primers on a LightCycler according to the instructions provided by the manufacturer (Roche Diagnostics, IN, UAS). The Quantitect gene assay kit was used for analysis of murine cyclin D1 (QT12443), cyclin E2 (QT12448), Foxm1 (QT14235), cyclin A2 (QT12428), cyclin B1 (QT268697), GRP78 (QT14828), activating transcription factor 6 (ATF6) (QT226641), inositol-requiring enzyme 1α (IRE1α) (QT78943), PKR-like ER kinase (PERK) (QT13666), CCAAT/enhancer-binding protein homologous protein (CHOP) (QT13198), and GAPDH (QT14433) (Qiagen). For analysis of sXBP-1, we used the customized primer (sense: CTGAGTCCGAATCAGGTGCAG; antisense: GTCCATGGGAAGATGTTCTGG). The mRNA expression levels were normalized relative to GAPDH mRNA expression level and expressed in arbitrary units.
Statistical analysis
Statistical analysis was conducted using JMP 9.0 software (SAS Institute Inc., NC, USA). Continuous variables were expressed as mean ± standard error (SE) by analysis of variance (ANOVA). ANOVA for the groups was performed by Wilcoxon test. Statistical significance was defined as p < 0.05.
Results
High fat diet induced simple hepatic steatosis and metabolic disorders
HFD mice significantly gained body weight, but not resulted in increasing liver weight (Table 1). Although micro- and macrovesicular lipid accumulation in hepatocyte were clearly visible by oil red O staining in HFD mice liver (Fig. 3a), there was no sign of inflammatory change by hematoxylin–eosin staining (data not shown). Moreover, there was no significant difference in plasma ALT levels in both groups. Plasma TC, TG, FFA, glucose, and insulin levels were significantly higher in HFD mice than in CD mice (Table 1). In this study, HFD mice showed dyslipidemia and insulin resistance.
Table 1.
Physiological and biochemical characteristics
| Control | HFD | |
|---|---|---|
| Body weight (g) | 27.2 ± 0.5 | 34.2 ± 0.1* |
| Liver weight (g) | 1.17 ± 0.02 | 1.09 ± 0.02 |
| ALT (IU/l) | 16.3 ± 2.7 | 17.9 ± 2.2 |
| TG (mg/dl) | 83.8 ± 8.9 | 149.6 ± 11.4* |
| TC (mg/dl) | 89.8 ± 11.8 | 133.4 ± 9.1* |
| FFA (mEq/l) | 700.0 ± 139.2 | 1301.3 ± 106.3* |
| Blood glucose (mg/dl) | 112.2 ± 7.7 | 143.1 ± 9.1* |
| IRI (ng/ml) | 0.32 ± 0.1 | 1.47 ± 0.4* |
Data were mean ± SE
* p < 0.05 vs. Control by ANOVA and Wilcoxon test
Fig. 3.
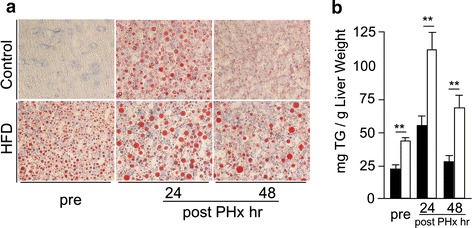
Lipid accumulation during liver regeneration. a Oil red O staining (original magnification ×400). b Hepatic tryglyceride contents (black squares Control, white squares HFD, n = 5–8; mean ± SE, **p < 0.01 HFD mice vs. CD mice in each time point by ANOVA and Wilcoxon test)
Liver regeneration was delayed in mice with simple hepatic steatosis
At 36 h after PHx the BrdU positive hepatocytes were rapidly increased in CD mice, but there were few in HFD mice. At 48 h after PHx in HFD the BrdU positive hepatocytes were increased and BrdU labeling index of HFD mice was similar to that of CD mice at 36 h after PHx (Fig. 1a). The expression of PCNA protein was also observed in delayed time in HFD mice compared with in CD mice (Fig. 1b). Next, we investigated the expression of cell cycle-regulated genes, such as cyclin D1, cyclin E2, Foxm1, cyclin A2, and cyclin B1 (Fig 1c). The mRNA levels of cyclin D1 and cyclin E2 were slightly higher in HFD mice than in CD mice before PHx, but the expression levels of these were similar in both groups during liver regeneration. The mRNA levels of Foxm1, cyclin A2, and cyclin B1 were significantly lower at 36 h after PHx in HFD mice than in CD mice. These results indicated that the remnant liver of HFD mice showed delayed hepatocyte DNA replication and cell cycle progression.
Fig. 1.
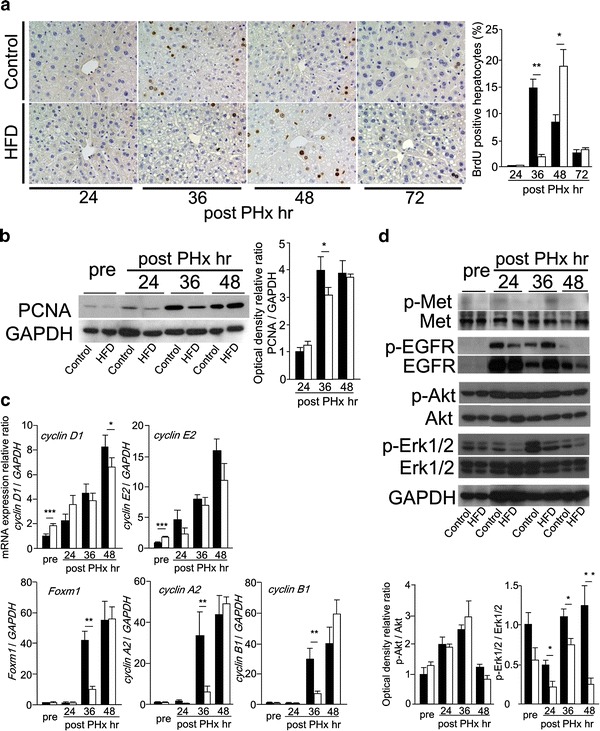
Delayed liver regeneration after PHx in simple fatty liver. a BrdU immunohistochemical staining (original magnification ×200), and BrdU labeling index. b The expression of PCNA protein detected by Western blotting. c The mRNA levels of cell-cycle related genes, cyclin D1, cyclin E2, Foxm1, cyclin A2, and cyclin B1 measured by real-time RT-PCR. d The activations of Akt and Erk1/2 evaluated by Western blotting (black squares Control, white squares HFD, n = 5–8; mean ± SE, *p < 0.05, **p < 0.01, and ***p < 0.005 HFD mice vs. CD mice in each time point by ANOVA and Wilcoxon test)
Growth factor receptors and downstream pathways
Among several growth factor receptors, phosphorylation of EGFR was detected from 24 h after PHx in both groups (Fig. 1d). We observed its downstream pathways such as Akt and Erk1/2 (Fig. 1d). Akt was well phosphorylated during liver regeneration, and there were no differences between both groups. Although phosphorylation of Erk1/2 was detected from 24 h and lasted to 48 h after PHx in CD mice, it could not be detected in HFD mice at 24 h after PHx. Phosphorylation of Erk1/2 during liver regeneration was delayed in HFD mice compared with in CD mice.
Liver volume and liver function examinations
We calculated liver mass regeneration ratio at several time points (Fig. 2a). Liver mass regeneration ratio in HFD mice at 48 h after PHx was slightly higher compared with in CD mice, but it was similar in both groups at the early phase (within 3 days after PHx). At a later phase (such as 6–8 days after PHx), the liver mass regeneration ratio of HFD mice was significantly lower than that of CD mice. At the early phase, plasma ALT levels were dramatically increased in both groups, and were significantly higher in HFD mice than in CD mice from 12 to 36 h after PHx. In contrast, plasma total bilirubin levels were slightly increased, but there was no significant difference in both groups. Plasma albumin levels showed no remarkable change in both groups before and after PHx (Fig. 2b).
Fig. 2.
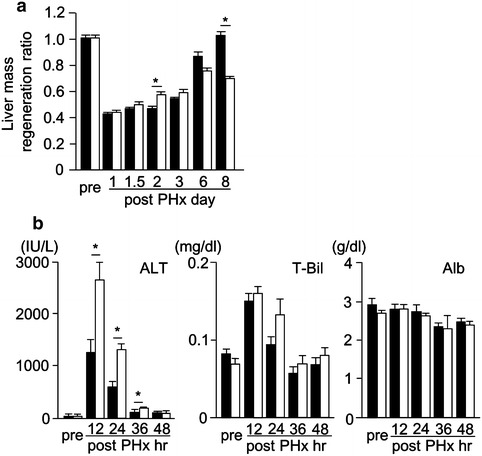
Liver mass regeneration ratio and serum liver function examinations. a Liver mass regeneration ratio during liver regeneration. b The change of plasma ALT, total bilirubin (T-Bil), and albumin (Alb) levels during liver regeneration (black squares Control, white squares HFD, n = 5–8; mean ± SE, *p < 0.05 HFD mice vs. CD mice in each time point by ANOVA and Wilcoxon test)
Lipid accumulation and FFA components during liver regeneration after PHx
Lipid accumulation into hepatocytes was shown during liver regeneration in both groups. Micro- and macrovesicular lipid accumulation were shown at 24 h after PHx in both groups by oil red O staining (Fig. 3a). In HFD mice, the amounts of liver TG were at least twofold higher than in CD mice at each time point (Fig. 3b). This significant increase in hepatic lipid contents of HFD mice after PHx might affect the liver mass regeneration ratio at the early phase. In each group, hepatic lipid extracts from the same time points were mixed together for measurement of FFA components, and the results were shown in Table 2. Among the FFAs, the amounts of palmitic acid and oleic acid were dramatically increased during liver regeneration in HFD mice compared with in CD mice (Table 2).
Table 2.
The comportment of FFAs of liver lipid extracts
| FFAs | Control | HFD | ||||
|---|---|---|---|---|---|---|
| Pre-PHx | 24 h after PHx | 48 h after PHx | Pre-PHx | 24 h after PHx | 48 h after PHx | |
| C12:0 | 0.056 | 0.080 | 0.056 | 0.063 | 0.148 | 0.100 |
| C14:0 | 0.404 | 0.504 | 0.262 | 0.349 | 1.202 | 0.766 |
| C16:0 | 9.842 | 17.652 | 10.684 | 15.465 | 33.478 | 29.782 |
| C16:1 | 0.652 | 2.496 | 0.730 | 1.216 | 5.702 | 3.492 |
| C18:0 | 6.964 | 7.972 | 7.816 | 7.976 | 10.566 | 10.394 |
| C18:1 ω9 | 4.266 | 13.600 | 5.602 | 15.061 | 46.372 | 38.826 |
| C18:2 ω6 | 7.164 | 22.450 | 10.430 | 7.398 | 22.942 | 18.284 |
| C18:3 ω6 | 0.092 | 0.204 | 0.128 | 0.171 | 0.350 | 0.396 |
| C18:3 ω3 | 0.192 | 0.946 | 0.230 | 0.252 | 1.440 | 0.922 |
| C20:0 | 0.164 | 0.236 | 0.168 | 0.240 | 0.276 | 0.354 |
| C20:1 ω9 | 0.146 | 0.418 | 0.246 | 0.368 | 0.890 | 0.858 |
| C20:2 ω6 | 0.084 | 0.224 | 0.120 | 0.130 | 0.348 | 0.258 |
| C20:3 ω9 | 0.022 | 0.020 | 0.000 | 0.085 | 0.132 | 0.116 |
| C20:3 ω6 | 0.504 | 0.626 | 0.516 | 0.500 | 0.836 | 0.814 |
| C20:4 ω6 | 3.182 | 3.300 | 2.972 | 5.225 | 6.200 | 5.252 |
| C20:5 ω3 | 0.476 | 0.700 | 0.370 | 0.106 | 0.530 | 0.296 |
| C22:0 | 0.188 | 0.198 | 0.156 | 0.236 | 0.204 | 0.218 |
| C22:4 ω6 | 0.050 | 0.140 | 0.088 | 0.248 | 0.614 | 0.484 |
| C22:5 ω3 | 0.268 | 0.686 | 0.356 | 0.286 | 0.832 | 0.750 |
| C22:6 ω3 | 0.120 | 0.116 | 0.116 | 0.059 | 0.080 | 0.104 |
| C24:0 | 3.912 | 6.900 | 5.268 | 3.814 | 5.406 | 6.776 |
| C24:1 ω9 | 0.160 | 0.168 | 0.162 | 0.081 | 0.092 | 0.110 |
Values are expressed as mg FFA/g LW. In each group, hepatic lipid extracts from same time points were mixed together for measurement of FFA components. C16:0; palmitic acid, C18:1; oleic acid
ER stress was occurred during liver regeneration in mice with simple hepatic steatosis
Recently, some studies have reported that palmitic acid induced ER stress in a dose dependent manner in vitro [29, 30]. In this study, the amount of intrahepatic palmitic acid was increased during liver regeneration (Table 2). We investigated ER stress during liver regeneration. At first we observed the mRNA levels of ER stress related genes, such as GRP78, IRE1α, ATF6, PERK, sXBP-1, and CHOP (Fig. 4a). Before PHx, the mRNA level of sXBP-1 in HFD mice was significantly higher than in CD mice, and the other mRNA levels showed no significant difference in both groups. In this study, we suggested that simple hepatic steatosis itself did not induce ER stress. The mRNA level of GRP78 was increased at 12 h after PHx and subsequent time points in both groups, and was significantly higher in HFD mice than in CD mice. Among three unfolded protein response (UPR) pathways, the mRNA level of PERK was increased in only HFD mice, and the others were not increased in both groups. Additionally, the mRNA level of sXBP-1 was remarkably increased from 12 to 24 h after PHx in both groups, and was significantly higher in HFD mice than in CD mice. The mRNA level of CHOP was also increased in both groups, and further increased in HFD mice compared with in CD mice. Next, we investigated the protein expressions of GRP78 and sXBP-1 during liver regeneration. The expression of GRP78 protein was increased from 12 to 24 h after PHx in both groups, and was significantly higher in HFD mice than in CD mice at 24 h after PHx (Fig. 4b). Immunohistochemical analysis also confirmed the increased expression of GRP78 protein in HFD mice compared with in CD mice at 24 h after PHx (Fig. 4b). By Western blotting, the expression of sXBP-1 protein was detectable only in HFD mice at 24 h after PHx (Fig. 4c). Immunohistochemical staining demonstrated that the number of sXBP-1 positive nuclear cells was increased in HFD mice compared with in CD mice at 24 h after PHx (Fig. 4c).
Fig. 4.
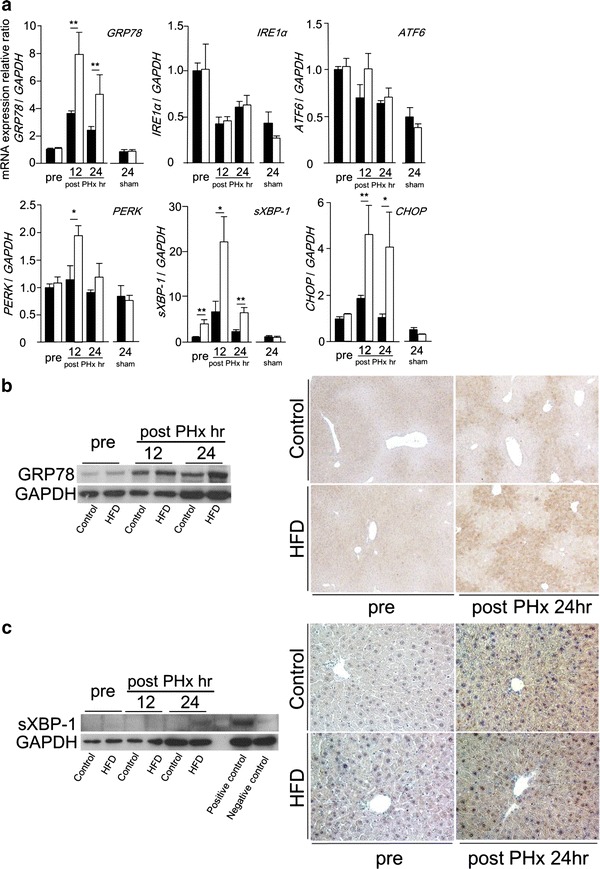
Enhanced ER stress was observed in simple fatty liver during liver regeneration. a The mRNA levels of ER stress-related genes, GRP78, IRE1α, ATF6, PERK, sXBP-1, and CHOP measured by real-time RT-PCR. b The expression of GRP78 protein detected by Western blotting and immunohistochemical staining (original magnification ×100). c The expression of sXBP-1 protein detected by Western blotting and immunohistochemical staining (original magnification ×200) (black squares Control, white squares HFD, n = 5–8; mean ± SE, *p < 0.05 and **p < 0.01 HFD mice vs. CD mice in each time point by ANOVA and Wilcoxon test)
The effects of TUDCA pretreatment for liver regeneration in mice with simple hepatic steatosis
We established a hypothesis that delayed liver regeneration shown in HFD mice is related with ER stress. To evaluate the influence of ER stress, both groups received intravenous injection of TUDCA 1 h before PHx. The mRNA levels of sXBP-1 and CHOP were significantly reduced in HFD mice with TUDCA pretreatment at 24 h after PHx (Fig. 5a). The mRNA level of GRP78 at 24 h after PHx showed no change with or without TUDCA pretreatment in both groups (Fig. 5a). However, the expression of GRP78 protein was remarkably reduced at 24 h after PHx in both groups with TUDCA pretreatment (Fig. 5b). The expression of sXBP-1 protein was not detectable in HFD mice with TUDCA pretreatment at 24 h after PHx (Fig. 5c). Phosphorylated Erk1/2 was detected at 24 h after PHx in HFD mice with TUDCA pretreatment (Fig. 6a). At 36 h after PHx, the BrdU labeling index and the expression of PCNA protein in HFD mice with TUDCA pretreatment were reached up to similar degree of those in CD mice at same time point (Fig. 6b, c). TUDCA pretreatment improved Foxm1 and cyclin A2 gene expressions in HFD mice at 36 h after PHx (Fig. 6d). TUDCA prevented ER stress and improved delayed liver regeneration in HFD mice. These results suggest that ER stress might be an important risk factor of delayed liver regeneration in fatty liver.
Fig. 5.
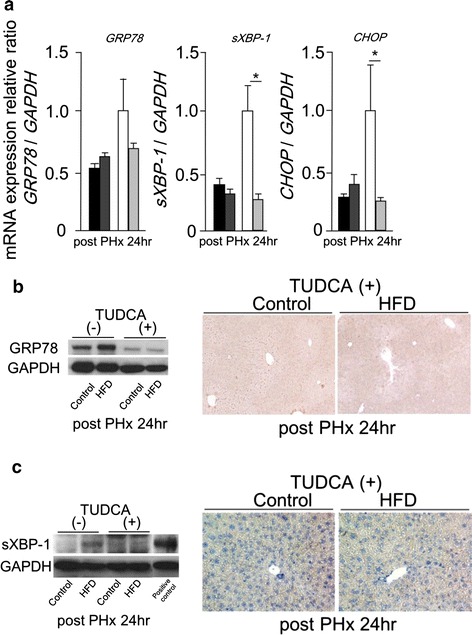
TUDCA pretreatment prevented ER stress induces during liver regeneratin in simple fatty liver. a The mRNA levels of GRP78, sXBP-1, and CHOP at 24 h after PHx with or without TUDCA pretreatment measured by real-time RT-PCR. b The expression of GRP78 protein at 24 h after PHx with or without TUDCA pretreatment detected by Western blotting and immunohistochemical staining (original magnification ×100). c The expression of sXBP-1 protein at 24 h after PHx with or without TUDCA pretreatment detected by Western blotting and immunohistochemical staining (original magnification ×200) (black squares Control, dark gray squares Control with TUDCA, white squares HFD, light gray squares HFD with TUDCA, n = 4–6; mean ± SE, *p < 0.05 mice with TUDCA pretreatment vs. mice without TUDCA pretreatment fed the same diet at each time point by ANOVA and Wilcoxon test)
Fig. 6.
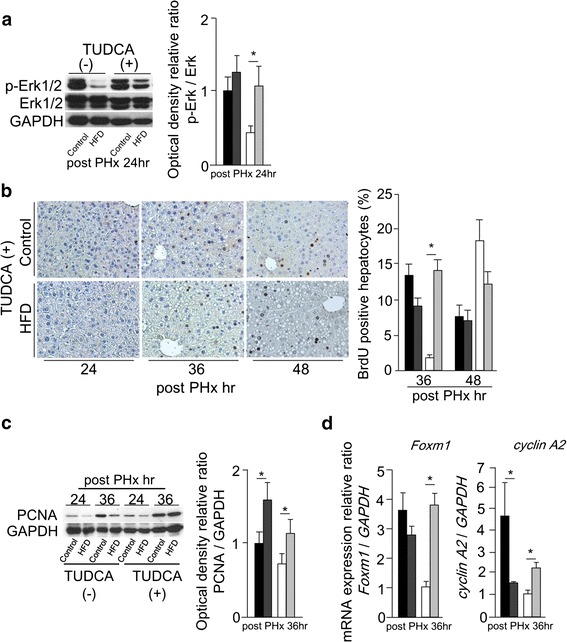
TUDCA pretreatment improved delayed liver regeneration in simple fatty liver. a The activation of Erk1/2 at 24 h after PHx with or without TUDCA pretreatment evaluated by Western blotting. b BrdU immunohistochemical staining (original magnification ×200) with TUDCA pretreatment, and BrdU labeling index at 36 and 48 h after PHx with or without TUDCA pretreatment. c The expression of PCNA protein at 24 and 36 h after PHx with or without TUDCA pretreatment detected by Western blotting, densitometric analysis was performed at 36 h after PHx. d The mRNA levels of Foxm1 and cyclin A2 at 36 h after PHx with or without TUDCA pretreatment measured by real-time RT-PCR (black squares Control, dark gray squares Control with TUDCA, white squares HFD, light gray squares HFD with TUDCA, n = 4–6; mean ± SE, *p < 0.05 mice with TUDCA pretreatment vs. mice without TUDCA pretreatment fed same diet in each time point by ANOVA and Wilcoxon test)
Discussion
We demonstrated delayed liver regeneration in mice with HFD-induced simple hepatic steatosis. In this model, the expression levels of hepatic ER stress markers before PHx were similar to those of non-steatotic liver in control mice. We thought this model was suitable to evaluate the association of ER stress with excess lipid accumulation occurring during liver regeneration.
Many previous studies which reported impaired fatty liver regeneration have used models induced by genetic alteration. For instance, ob/ob mice and KK-Ay mice failed to induce cell progression G1 phase to S phase because of alteration in innate immune response and abnormal Janus kinase (JAK)-signal transducers and activators of transcription (STAT) signaling [14, 15]. Additionally, several studies have reported impaired liver regeneration in diet-induced fatty liver models [11, 12, 19]. One of them, fed HFD, reported that IkappaB kinase beta (IKKβ) overexpression and high leptin level resulted in G1 arrest [12]. Most of them failed in cyclin D1 expression and showed severe mortality. However, in this study, the mRNA expressions of G1 related-genes, such as cyclin D1 and E2, were similar in both groups. Additionally, both groups showed high survival rate (date not shown). Therefore, we suggested that delayed hepatocyte DNA replication in mice with simple hepatic steatosis was induced by another factors, which have not been reported previously.
ER is the cellular organelle where proteins and lipids are synthesized and modified. ER stress is caused by various insults, such as oxidative stress, chemical toxicity, viral infection, and metabolic disorders. The UPR, induced by ER stress, is mediated by three primary signal transducer molecules: IRE1α, ATF6, and PERK [31]. It has been reported that NAFLD patients have shown various degrees of UPR [32], and a rat model fed saturated FFAs rich diet also showed hepatic ER stress [33]. To evaluate the influence of lipid overaccumulation for liver regeneration, we at first analyzed the change of intrahepatic FFA components during liver regeneration. Interestingly, intrahepatic FFA components increasing during liver regeneration were similar in both groups, not influenced by their nutritional conditions. Among FFAs, the amount of intrahepatic palmitic acid was dramatically increased during liver regeneration, especially in mice with simple hepatic steatosis. Several studies have reported the changes of FFA components after PHx. These authors described the roles of unsaturated FFAs as signal transducers, but did not mention saturated FFAs [34, 35]. Recently, many studies have reported the cytotoxic effects of saturated FFAs, such as stearic acid and palmitic acid, and the cytoprotective effects of unsaturated FFAs, such as oleic acids and linoleic acid. In in vitro studies, some authors demonstrated that saturated FFAs induced ER stress in a dose-dependent manner and promoted cell death, mainly by apoptosis, and others reported that unsaturated FFAs protected cell from saturated FFAs-induced lipotoxicity by lipid droplets formation [29, 30]. In this study, the mRNA expressions of GRP78, a major ER chaperone, and of UPR pathways were not different in both groups before PHx. During liver regeneration, the expression of GRP78 protein was increased significantly higher in mice with simple hepatic steatosis. The mRNA overexpressions of sXBP-1 and CHOP, downstream molecule of UPR, were detected especially in mice with simple hepatic steatosis. sXBP-1 protein was detected in mice with simple hepatic steatosis only at 24 h after PHx. These results suggested that various levels of ER stress was induced not only in fatty liver but also in normal liver during liver regeneration after PHx, however the trigger level might be higher in fatty liver than in normal liver. We also obtained c-jun NH2 terminal kinase (JNK) activity and TUNEL staining, but there was no evidence of phosphorylation of JNK and TUNEL positive cell in both groups (data not shown). In this study, the trigger level would not be enough to induce hepatocyte apoptosis even after PHx of simple fatty liver. We suggested that intrahepatic palmitic acid accumulation occurring during liver regeneration might be overloaded with simple hepatic steatosis, and result in induction of ER stress.
TUDCA, a hydrophilic bile acid, causes concentration-dependent decreases in intracellular calcium, and acts as a chemical chaperone to enhance protein folding and protect cells against ER stress [36]. Recently, many studies have reported that TUDCA has been approved for clinical use as a protective agent in various diseases, for instance, improved insulin resistance by reduced ER stress [37, 38]. In the ischemia/reperfusion liver injury model, severe ER stress was induced and resulted in hepatocyte apoptosis, TUDCA pretreatment improved liver damage [39, 40]. To investigate whether ER stress induced during liver regeneration had influence on hepatocyte DNA replication or not, we also performed PHx with TUDCA pretreatment. As a result, TUDCA abolished ER stress induced during liver regeneration in mice with simple hepatic steatosis and ameliorated hepatocyte DNA replication remarkably. Moreover, delayed phosphorylation of Erk1/2 and the expression of Foxm1 were also improved by TUDCA pretreatment. Therefore, we suggested that Erk1/2 and Foxm1 might be key molecules influenced by ER stress in this model.
Erk1/2, a member of the mitogen-activated protein kinase (MAPK) family, is known as an essential molecule in growth factor signaling pathways, and regulates G1/S transition [41]. Erk1/2 also down-regulates anti-proliferative genes including CHOP and Gadd45α during G1 phase, and sustained activation of Erk1/2 is needed for successful S phase entry [42, 43]. Akt and MAPK pathways are considered major cell proliferation and survival signaling cascades. Recently, several studies reported a cross-talk relation between these pathways under ER stress induced by drugs in vitro [44, 45], but these cascades during liver regeneration under ER stress were not clearly understood. It was also reported that Foxm1 activation, which is related to regulations of DNA replication and mitosis [46, 47], might be regulated by Erk1/2 [48]. However, the mechanism of Foxm1 regulation was not fully understood. In addition, Foxm1 was up-regulated in several stress conditions such as oxidative stress and hypoxia [49, 50], but there was no report discussed Foxm1 expression under ER stress. In this study, unfortunately, we could not make direct effect of ER stress on Foxm1 activation clear. Further investigation is needed to make mechanism of ER stress induced delayed liver regeneration clear.
In conclusion, we demonstrated that simple hepatic steatosis itself might be an important risk factor of delayed liver regeneration. ER stress induced during liver regeneration resulted in delayed hepatocyte DNA replication in fatty liver. Intrahepatic lipid accumulation occurred during liver regeneration might be an important trigger of ER stress. We suggested that management of ER stress might have a possibility of improvement on delayed fatty liver regeneration.
Conflict of interest
None.
Abbreviations
- NAFLD
Non-alcoholic fatty liver disease
- PHx
Partial hepatectomy
- FFAs
Free fatty acids
- BrdU
Bromodeoxyuridine
- TUDCA
Tauroursodeoxycholic acid
- ER
Endoplasmic reticulum
- ALT
Alanine aminotransferase
- TG
Triglycerides
- TC
Total cholesterol
- GRP78
Glucose-regulated protein of 78-kDa
- sXBP-1
Spliced form of X-box binding protein 1
- PCNA
Proliferation cell nuclear antigen
- EGFR
Epidermal growth factor receptor
- ATF6
Activating transcription factor 6
- IRE1α
Inositol-requiring enzyme 1α
- PERK
PKR-like ER kinase
- CHOP
CCAAT/enhancer-binding protein homologous protein
- UPR
Unfolded protein response
- JAK
Janus kinase
- STAT
Signal transducers and activators of transcription
- IKKβ
IkappaB kinase beta
- JNK
c-Jun NH2 terminal kinase
- MAPK
Mitogen-activated protein kinase
References
- 1.Speliotes E, Massaro J, Hoffmann U, Vasan R, Meigs J, Sahani D, et al. Fatty liver is associated with dyslipidemia and dysglycemia independent of visceral fat: The framingham heart study. Hepatology. 2010. [DOI] [PMC free article] [PubMed]
- 2.Chitturi S, Farrell G, Hashimoto E, Saibara T, Lau G, Sollano J, et al. Non-alcoholic fatty liver disease in the Asia-Pacific region: definitions and overview of proposed guidelines. J Gastroenterol Hepatol. 2007;22(6):778–787. doi: 10.1111/j.1440-1746.2007.05001.x. [DOI] [PubMed] [Google Scholar]
- 3.Adams L, Lymp J, St Sauver J, Sanderson S, Lindor K, Feldstein A, et al. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129(1):113–121. doi: 10.1053/j.gastro.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 4.Bernuau J, Rueff B, Benhamou JP. Fulminant and subfulminant liver failure: definitions and causes. Semin Liver Dis. 1986;6(2):97–106. doi: 10.1055/s-2008-1040593. [DOI] [PubMed] [Google Scholar]
- 5.Guglielmi A, Ruzzenente A, Conci S, Valdegamberi A, Iacono C. How much remnant is enough in liver resection? Dig Surg. 2012;29(1):6–17. doi: 10.1159/000335713. [DOI] [PubMed] [Google Scholar]
- 6.van den Broek MA, Olde Damink SW, Dejong CH, Lang H, Malagó M, Jalan R, et al. Liver failure after partial hepatic resection: definition, pathophysiology, risk factors and treatment. Liver Int. 2008;28(6):767–780. doi: 10.1111/j.1478-3231.2008.01777.x. [DOI] [PubMed] [Google Scholar]
- 7.de Meijer VE, Kalish BT, Puder M, Ijzermans JN. Systematic review and meta-analysis of steatosis as a risk factor in major hepatic resection. Br J Surg. 2010;97(9):1331–1339. doi: 10.1002/bjs.7194. [DOI] [PubMed] [Google Scholar]
- 8.Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology. 2006;43(2 Suppl 1):S45–S53. doi: 10.1002/hep.20969. [DOI] [PubMed] [Google Scholar]
- 9.Kiso S, Kawata S, Tamura S, Inui Y, Yoshida Y, Sawai Y, et al. Liver regeneration in heparin-binding EGF-like growth factor transgenic mice after partial hepatectomy. Gastroenterology. 2003;124(3):701–707. doi: 10.1053/gast.2003.50097. [DOI] [PubMed] [Google Scholar]
- 10.Kiso S, Kawata S, Tamura S, Higashiyama S, Ito N, Tsushima H, et al. Role of heparin-binding epidermal growth factor-like growth factor as a hepatotrophic factor in rat liver regeneration after partial hepatectomy. Hepatology. 1995;22(5):1584–1590. [PubMed] [Google Scholar]
- 11.Tanoue S, Uto H, Kumamoto R, Arima S, Hashimoto S, Nasu Y, et al. Liver regeneration after partial hepatectomy in rat is more impaired in a steatotic liver induced by dietary fructose compared to dietary fat. Biochem Biophys Res Commun. 2011;407(1):163–168. doi: 10.1016/j.bbrc.2011.02.131. [DOI] [PubMed] [Google Scholar]
- 12.DeAngelis RA, Markiewski MM, Taub R, Lambris JD. A high-fat diet impairs liver regeneration in C57BL/6 mice through overexpression of the NF-kappaB inhibitor, IkappaBalpha. Hepatology. 2005;42(5):1148–1157. doi: 10.1002/hep.20879. [DOI] [PubMed] [Google Scholar]
- 13.Yamauchi H, Uetsuka K, Okada T, Nakayama H, Doi K. Impaired liver regeneration after partial hepatectomy in db/db mice. Exp Toxicol Pathol. 2003;54(4):281–6. [DOI] [PubMed]
- 14.Yang SQ, Lin HZ, Mandal AK, Huang J, Diehl AM. Disrupted signaling and inhibited regeneration in obese mice with fatty livers: implications for nonalcoholic fatty liver disease pathophysiology. Hepatology. 2001;34(4 Pt 1):694–706. doi: 10.1053/jhep.2001.28054. [DOI] [PubMed] [Google Scholar]
- 15.Aoyama T, Ikejima K, Kon K, Okumura K, Arai K, Watanabe S. Pioglitazone promotes survival and prevents hepatic regeneration failure after partial hepatectomy in obese and diabetic KK-A(y) mice. Hepatology. 2009;49(5):1636–1644. doi: 10.1002/hep.22828. [DOI] [PubMed] [Google Scholar]
- 16.Gazit V, Weymann A, Hartman E, Finck BN, Hruz PW, Tzekov A, et al. Liver regeneration is impaired in lipodystrophic fatty liver dystrophy mice. Hepatology. 2010;52(6):2109–2117. doi: 10.1002/hep.23920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rao MS, Papreddy K, Abecassis M, Hashimoto T. Regeneration of liver with marked fatty change following partial hepatectomy in rats. Dig Dis Sci. 2001;46(9):1821–1826. doi: 10.1023/A:1010654908938. [DOI] [PubMed] [Google Scholar]
- 18.Picard C, Lambotte L, Starkel P, Sempoux C, Saliez A, Van den Berge V, et al. Steatosis is not sufficient to cause an impaired regenerative response after partial hepatectomy in rats. J Hepatol. 2002;36(5):645–652. doi: 10.1016/S0168-8278(02)00038-7. [DOI] [PubMed] [Google Scholar]
- 19.Veteläinen R, van Vliet AK, van Gulik TM. Severe steatosis increases hepatocellular injury and impairs liver regeneration in a rat model of partial hepatectomy. Ann Surg. 2007;245(1):44–50. doi: 10.1097/01.sla.0000225253.84501.0e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Delahunty TJ, Rubinstein D. Accumulation and release of triglycerides by rat liver following partial hepatectomy. J Lipid Res. 1970;11(6):536–543. [PubMed] [Google Scholar]
- 21.Newberry EP, Kennedy SM, Xie Y, Luo J, Stanley SE, Semenkovich CF, et al. Altered hepatic triglyceride content after partial hepatectomy without impaired liver regeneration in multiple murine genetic models. Hepatology. 2008;48(4):1097–1105. doi: 10.1002/hep.22473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glende EA, Morgan WS. Alteration in liver lipid and lipid fatty acid composition after partial hepatectomy in the rat. Exp Mol Pathol. 1968;8(2):190–200. doi: 10.1016/0014-4800(68)90015-4. [DOI] [PubMed] [Google Scholar]
- 23.Rudnick DA, Davidson NO. Functional relationships between lipid metabolism and liver regeneration. Int J Hepatol. 2012;2012:549241. doi: 10.1155/2012/549241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gove CD, Hems DA. Fatty acid synthesis in the regenerating liver of the rat. Biochem J. 1978;170(1):1–8. doi: 10.1042/bj1700001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farrell GC. Probing Prometheus: fat fueling the fire? Hepatology. 2004;40(6):1252–1255. doi: 10.1002/hep.20522. [DOI] [PubMed] [Google Scholar]
- 26.Delgado-Coello B, Briones-Orta MA, Macías-Silva M, Mas-Oliva J. Cholesterol: recapitulation of its active role during liver regeneration. Liver Int. 2011;31(9):1271–1284. doi: 10.1111/j.1478-3231.2011.02542.x. [DOI] [PubMed] [Google Scholar]
- 27.Higgins GM, Anderson RM. Experimental pathology of the liver. 1. Restoration of the liver of the white rat following partial surgical removal. Arch Pathol. 1931;12:186–189. [Google Scholar]
- 28.Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957;226(1):497–509. [PubMed] [Google Scholar]
- 29.Mantzaris MD, Tsianos EV, Galaris D. Interruption of triacylglycerol synthesis in the endoplasmic reticulum is the initiating event for saturated fatty acid-induced lipotoxicity in liver cells. FEBS J. 2011;278(3):519–530. doi: 10.1111/j.1742-4658.2010.07972.x. [DOI] [PubMed] [Google Scholar]
- 30.Listenberger LL, Han X, Lewis SE, Cases S, Farese RV, Ory DS, et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci USA. 2003;100(6):3077–3082. doi: 10.1073/pnas.0630588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110(10):1389–1398. doi: 10.1172/JCI0216886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Puri P, Mirshahi F, Cheung O, Natarajan R, Maher JW, Kellum JM, et al. Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology. 2008;134(2):568–576. doi: 10.1053/j.gastro.2007.10.039. [DOI] [PubMed] [Google Scholar]
- 33.Wang D, Wei Y, Pagliassotti MJ. Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology. 2006;147(2):943–951. doi: 10.1210/en.2005-0570. [DOI] [PubMed] [Google Scholar]
- 34.Kishino T, Tanno M, Yamada H, Saito S, Matsumoto S. Changes in liver fatty acid unsaturation after partial hepatectomy in the rat. Lipids. 2000;35(4):445–452. doi: 10.1007/s11745-000-543-9. [DOI] [PubMed] [Google Scholar]
- 35.Domitrovic R, Milin C, Radosevic-Stasic B. Liver fatty acid and element changes after partial hepatectomy in mice fed olive oil- and corn oil-enriched diets. Biol Trace Elem Res. 2006;109(1):61–74. doi: 10.1385/BTER:109:1:061. [DOI] [PubMed] [Google Scholar]
- 36.Xie Q, Khaoustov VI, Chung CC, Sohn J, Krishnan B, Lewis DE, et al. Effect of tauroursodeoxycholic acid on endoplasmic reticulum stress-induced caspase-12 activation. Hepatology. 2002;36(3):592–601. doi: 10.1053/jhep.2002.35441. [DOI] [PubMed] [Google Scholar]
- 37.Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313(5790):1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kars M, Yang L, Gregor MF, Mohammed BS, Pietka TA, Finck BN, et al. Tauroursodeoxycholic acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes. 2010;59(8):1899–1905. doi: 10.2337/db10-0308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ben Mosbah I, Alfany-Fernández I, Martel C, Zaouali MA, Bintanel-Morcillo M, Rimola A, et al. Endoplasmic reticulum stress inhibition protects steatotic and non-steatotic livers in partial hepatectomy under ischemia–reperfusion. Cell Death Dis. 2010;1:e52. [DOI] [PMC free article] [PubMed]
- 40.Anderson CD, Upadhya G, Conzen KD, Jia J, Brunt EM, Tiriveedhi V, et al. Endoplasmic reticulum stress is a mediator of posttransplant injury in severely steatotic liver allografts. Liver Transpl. 2011;17(2):189–200. doi: 10.1002/lt.22220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Meloche S, Pouysségur J. The ERK1/2 mitogen-activated protein kinase pathway as a master regulator of the G1- to S-phase transition. Oncogene. 2007;26(22):3227–3239. doi: 10.1038/sj.onc.1210414. [DOI] [PubMed] [Google Scholar]
- 42.Yamamoto T, Ebisuya M, Ashida F, Okamoto K, Yonehara S, Nishida E. Continuous ERK activation downregulates antiproliferative genes throughout G1 phase to allow cell-cycle progression. Curr Biol. 2006;16(12):1171–1182. doi: 10.1016/j.cub.2006.04.044. [DOI] [PubMed] [Google Scholar]
- 43.Villanueva J, Yung Y, Walker JL, Assoian RK. ERK activity and G1 phase progression: identifying dispensable versus essential activities and primary versus secondary targets. Mol Biol Cell. 2007;18(4):1457–1463. doi: 10.1091/mbc.E06-10-0908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hu P, Han Z, Couvillon AD, Exton JH. Critical role of endogenous Akt/IAPs and MEK1/ERK pathways in counteracting endoplasmic reticulum stress-induced cell death. J Biol Chem. 2004;279(47):49420–49429. doi: 10.1074/jbc.M407700200. [DOI] [PubMed] [Google Scholar]
- 45.Dai R, Chen R, Li H. Cross-talk between PI3 K/Akt and MEK/ERK pathways mediates endoplasmic reticulum stress-induced cell cycle progression and cell death in human hepatocellular carcinoma cells. Int J Oncol. 2009;34(6):1749–1757. doi: 10.3892/ijo_00000306. [DOI] [PubMed] [Google Scholar]
- 46.Wang X, Kiyokawa H, Dennewitz MB, Costa RH. The Forkhead Box m1b transcription factor is essential for hepatocyte DNA replication and mitosis during mouse liver regeneration. Proc Natl Acad Sci USA. 2002;99(26):16881–16886. doi: 10.1073/pnas.252570299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Korver W, Roose J, Clevers H. The winged-helix transcription factor Trident is expressed in cycling cells. Nucleic Acids Res. 1997;25(9):1715–1719. doi: 10.1093/nar/25.9.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma RY, Tong TH, Cheung AM, Tsang AC, Leung WY, Yao KM. Raf/MEK/MAPK signaling stimulates the nuclear translocation and transactivating activity of FOXM1c. J Cell Sci. 2005;118(Pt 4):795–806. doi: 10.1242/jcs.01657. [DOI] [PubMed] [Google Scholar]
- 49.Park HJ, Carr JR, Wang Z, Nogueira V, Hay N, Tyner AL, et al. FoxM1, a critical regulator of oxidative stress during oncogenesis. EMBO J. 2009;28(19):2908–2918. doi: 10.1038/emboj.2009.239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xia L, Mo P, Huang W, Zhang L, Wang Y, Zhu H, et al. The TNF-α/ROS/HIF-1-induced upregulation of foxmi expression promotes HCC proliferation and resistance to apoptosis. Carcinogenesis. 2012. [DOI] [PubMed]