

Abstract
An expanded CUG repeat transcript (CUGexp) is the causative agent of myotonic dystrophy type 1 (DM1) by sequestering muscleblind-like 1 protein (MBNL1), a regulator of alternative splicing. Based on a ligand (1) that was previously reported to be active in an in vitro assay, we present the synthesis of a small library containing ten dimeric ligands (4-13) that differ in length, composition and attachment point of the linking chain. The oligoamino linkers gave a greater gain in affinity for CUG RNA and were more effective when compared to oligoether linkers. The most potent in vitro ligand (9) was shown to be aqueous-soluble and both cell- and nucleus-permeable displaying almost complete dispersion of MBNL1 ribonuclear foci in a DM1 cell model. Direct evidence for the bioactivity of 9 was observed in its ability to disperse ribonuclear foci in individual live DM1 model cells using time-lapse confocal fluorescence microscopy.
Introduction
Myotonic dystrophy type 1 (DM1) is the most common form of adult muscular dystrophy and is a disease for which there is currently no treatment, only palliative therapy.1 DM1 is a trinucleotide expansion disease (TRED) that is caused by an aberrant expansion of a CTG repeat sequence in the 3'-untranslated region of the dystrophia myotonica protein kinase (DMPK) gene.2 The CTGexp leads to a CUGexp transcript that has a unique secondary structure, consisting of repetitive UU mismatches and CG base pairs.3,4 This toxic CUGexp transcript sequesters all three paralogs of human MBNL proteins including MBNL1, a key regulator of the alternative splicing process.5-7 Thus, although DM1 has a complex pathogenesis, it is known to be an RNA-gain-of-function disease with the high affinity MBNL1·CUGexp interaction playing a major role.8-10 Another proposed mechanism involves the increased steady-state level of CUG-binding protein 1 (CUG-BP1) as a result of the presence of CUGexp.11-14 The CUGexp RNA is a validated drug target,15-17 having been successfully targeted by several oligonucleotides including synthetic short interfering RNAs,18 a morpholino antisense oligonucleotide (ASO),19 2’-O-(2-methoxyethyl) ASO,15 a D-amino acid hexapeptide,20 and several small molecules including pentamidine,21 benzo[g]quinolone-based heterocycles,22 a Hoechst derivative (H1),23 a modularly assembled Hoechst 33258,24,25 and a triaminotriazine-acridine conjugate, reported by our laboratory (ligand 1).26
Ligand 1 was reported as a highly selective, albeit moderate (CUG)12·MBNL1 inhibitor (IC50 = 46 μM) in an in vitro assay.26 The hydrogen bonding recognition unit, the triaminotriazine ring was found to be essential for recognition and inhibition of the (CUG)12·MBNL1 interaction because acridine derivatives lacking this unit did not exhibit inhibition potency in an in vitro assay.26 However, ligand 1 had two shortcomings: modest inhibition potency and poor cell permeability. Recently a conjugate of ligand 1 containing a oligoamine-derivative side chain was found to be cell-permeable and bioactive.27
Considering the repeating nature of CUGexp, one logical approach to increasing the affinity of 1 for CUGexp is through the generation of multivalent ligands.28,29 The multivalent effect has proven useful to increase the binding affinity and selectivity of other ligands toward a wide variety of multivalent targets including CUGexp.30-38 The increase in affinity of multivalent ligands arises from the thermodynamic advantage inherent in a cooperative binding system.29,39 Upon binding of the first module, the overall entropy of the ligand·CUGexp complex is significantly lowered by having the second binding module localized in the vicinity of its binding site.28 However, in nearly all cases examined, the dimeric binding constant rarely approaches the very high level expected based on ΔGdimer >> 2ΔGmonomer because of entropic and enthalpic costs involved in bivalent binding.28 In particular, conformational rigidity can cause spatial mismatch and diminish the binding enthalpy of the second module, whereas conformational flexibility raises the entropic cost for the binding of the second module. Thus, it is essential to have the right linker to maximize the multivalent effect as both rigidity and flexibility can potentially diminish this effect.
Another advantage of multivalent ligands is their potential to become cell-permeable, by taking advantage of the polarity of appropriate linkers. However, development of bioactive multivalent ligands have some obstacles such as their large size and molecular weight reducing their “drugability.”38 Indeed, a tetrameric and pentameric Hoechst 33528 ligand were developed successfully as highly effective inhibitors of the MBNL1·CUGexp interaction in vitro but both were found to be insoluble and cell-impermeable.25 The use of dimeric ligands is an attractive pathway that is becoming more prevalent in drug discovery efforts40-45 and has the added advantage of more moderate molecular weights. This approach seemed particularly well suited to ligand 1 (vide infra). To accomplish this goal with limited structural knowledge of the ligand-binding mode,46,47 a small library of dimeric ligands was created with a range of chains linking two units analogous to 1.
Results and Discussion
Rational Design and Synthesis of Dimeric Ligands
The design of a dimeric CUGexp-binding ligand requires the following: (1) a monomeric ligand with at least a modest affinity and selectivity for CUGexp, (2) an appropriate handle for the attachment of the linker to the monomeric ligand, that would not interfere with the ligand·CUGexp interaction, and (3) an appropriate linker to connect the two monomers so that each module can bind to CUGexp. From the previous studies in our lab, it was determined that 1 is a highly selective and modest monomeric inhibitor for (CUG)12·MBNL1 (IC50 = 46 μM) with over 50 fold selectivity for CUG repeat relative to a random duplex.26 Ligand 1 was designed to serve as a “stacked intercalator” with the acridine unit inserting between the GC base pair and the U-triaminotriazine-U base triplet (i.e., the acridine stacked on the triaminotriazine).47
The covalent attachment of 1 to a linker required an appropriately positioned, reactive functional group. Possible sites for this group on 1 were the acridine ring, the triaminotriazine recognition unit, or the linking chain between these two components. Modification of the recognition unit was considered less desirable because of the potential to block key hydrogen bonding functionality involved in the UU mismatch recognition. However, various bis-acridine intercalators spanning two or more base pairs were synthesized and studied previously.48-52 The most straightforward synthesis involved an acridine ring containing a carboxylic acid group that could be amidated with a linker diamine. Placing the carboxylic acid group in either the 1- or 3-position of the acridine ring is challenging because the synthetic route proceeds through an inseparable mixture of both derivatives.53
The remaining two positions of the acridine ring, i.e., the 2- and 4-positions, were considered more tractable from a synthetic standpoint. Importantly, it was found that the chloro- and methoxy-groups in 1 could be replaced with a 2- or 4-carboxamido group (see 2 and 3 in Figure 1, respectively) without reducing its affinity for CUG repeats or its inhibition of the MBNL1·CUG complex. Although modeling was not carried out, it appeared that dimeric ligands interconnected at the 2- and 4-positions would have the linker and triaminotriazine recognition units located on the same or the opposite sides of the acridine unit, thereby likely requiring a non-threading or threading54 mechanism of binding, respectively. This simple analysis suggested that these two isomeric dimers would significantly increase the diversity of our library, especially when considering the ultimate RNA-ligand complex.
Figure 1.

Library of synthesized monomeric (1-3) and dimeric (4-13) ligands.
Linker units with terminal amino groups were selected as they would form a stable amide bond with the carboxylic acid handle on the acridine ring.55,56 Without a firm structural data for the binding mode, the linker chain should be flexible enough to allow bivalent binding. As noted above, interconnection can result in an “all or nothing” effect,57 whereby an appropriately designed rigid linker puts the second binding module at the UU mismatch site but an inappropriate linker greatly diminishes the binding of one or possibly even both modules. The polarity of the linker is another important consideration as aqueous solubility issues may arise from use of a polymethylene chain. Because the original monomeric ligand is minimally aqueous-soluble, both polyamide and alkyl chains were ruled out as potential linkers. Thus, all of the diamine linkers used in this study contained either oligoether or oligoamino groups. Oligoethers were either oligoethylene and oligopropylene glycols which are attractive moieties for drug delivery, because of their flexibility and polarity.58 Oligoamino groups introduce positive charge to the dimeric ligand, therefore increasing its aqueous-solubility and affinity to the RNA polyphosphate backbone.59 Moreover, cells have a polyamine transporting system (PTS) that potentially makes such conjugates cell-permeable.60
The heteroatom-rich linkers used here possess various lengths from 10 to 21 atoms and were designed to span at least two central GC base-pairs according to the nearest-neighbor exclusion principle,61 thereby allowing the two triaminotriazine recognition units to bind minimally to consecutive UU mismatches. The actual linkers used were either commercially available or synthesized as shown in Schemes 3-8 in the Supporting Information. The two resulting series of dimeric ligands that were synthesized from these linkers and evaluated are shown in Figure 2.
Figure 2.

General synthetic scheme for dimeric ligands
Optical Melting Studies
It has been suggested that MBNL1 displays a preference for single stranded (ss) CUGexp and thus destabilizes the double stranded (ds) CUGexp stem loop upon binding.62,63 If correct, this model suggests that ligands capable of stabilizing the ds form of CUGexp may prove to be the most effective inhibitors of the MBNL1·CUGexp interaction. The increase in (CUG)12 melting temperature upon ligand binding correlates both with (CUG)12 stem loop stability and ligand binding strength. Therefore, we studied the effect of ligands on the Tm of (CUG)12, the thermal denaturation study of (CUG)12 being carried out in the presence of one equivalent of select ligands. Monophasic melting curves were obtained in each case with ΔTm values shown in Table 1. Ligands 4, 5 and 6 were not fully soluble in the buffer used and a ΔTm could not be obtained. For the monomeric ligand 2, the ΔTm was 1.8 °C. For the oligoether-linker dimeric ligands, 7 and 8, ΔTm value was 2.7 and 1.8 °C, respectively. The values of the monomeric and oligoether-linker dimeric ligands are similar, suggesting that 7 and 8 are binding to (CUG)12 only with one of their binding modules. This can be explained by the possible coil-like conformation of oligoethylene glycol linkers, preventing the desired distance between binding modules.64 Oligopropylene glycol linker in 6 was designed to avoid this coil-like conformation, however it was aqueous-insoluble.
Table 1.
ΔTm value of (CUG)12 upon adding ligand in a 1:1 ratio.a
| Ligand | Solution | ΔTm(°C) |
|---|---|---|
| 2 | 5% DMSO | 1.8 ± 0.61 |
| 7 | 5% DMSO | 2.7 ± 0.4 |
| 4 | N.S. | ______ |
| 12 | 5% DMSO | N.D. |
| 13 | 5% DMSO | N.D. |
| 5 | N.S. | ______ |
| 8 | 5% DMSO | 1.8 ± 0.2 |
| 6 | N.S. | ______ |
| 9 | Aqueous | 9.3 ± 0.6 |
| 10 | Aqueous | 9.6 ± 1.3 |
| 11 | Aqueous | 8.4 ± 2.1 |
Each value is mean ± s.d. of three independent experiments.
N.S. – not sufficiently soluble.
The dimeric ligands 9, 10 and 11 containing oligoamino linkers had much higher ΔTm values of 9.3, 9.6, and 8.4 °C, respectively. This finding suggests that 9, 10, and 11, unlike 7 and 8, have a more optimized linker to bind to (CUG)12 and are better able to stabilize the ds form of (CUG)12 than the corresponding monomer. These results suggest that the composition of the linker is more important than the number of atoms it contains. For example, 7 contains the same number of atoms in its linker as does 11, yet the latter gives a >3-fold higher ΔTm value. Overall, the thermal denaturation data suggest that ligands 9, 10 and 11 have the greatest potential given their higher ΔTm values, but more quantitative binding data was sought to corroborate these preliminary results. Ligands 2 and 3 (data not shown) showed similar binding affinity and inhibition potency so 2, was used as the representative monomeric ligand throughout this study.
Steady State Fluorescence Studies
To measure the binding affinity of the ligands to CUGexp, a steady state fluorescence titration method using 5’-TAMRA-(CUG)6 -3’ was utilized.46 It is possible that binding of a ligand to a UU mismatch, close to 5’-TAMRA, could change the structure from ss to ds, and thus lead to quenching of the 5’-TAMRA by the 3’-G through photoinduced electron transfer.65,66 Ligands were titrated into TAMRA-(CUG)6 solution and the TAMRA fluorescence intensity was observed to gradually decrease with increasing ligand concentration as a result of fluorophore quenching by the bound ligand. A plot of normalized fluorescence intensity versus concentration of each ligand yielded a binding isotherm and KD value (Figure 3) that showed a trend paralleling that of the ΔTm study.
Figure 3.

a) Fluorescence titrations of TAMRA-(CUG)6 with ligands. Comparison of normalized fluorescence intensity change of TAMRA-(CUG)6 in the presence of increasing concentrations of ligands. TAMRA was excited at 560 nm and its emission was recorded at 590 nm. Error bars represent mean ± s.d. of three independent experiments. b) KD was derived by fitting the Fluorescence intensities at different concentrations of ligands into the following equation: where [L] is the concentration of each ligand and Fmax and F0 are the maximum and minimum of normalized fluorescence intensities, respectively.
Two of the oligoether-linker dimeric ligands, 7 and 12, gave a KD value close to the monomeric ligand 2. On the other hand, dimeric ligands, 11, 10 and 9, showed a 14, 17 and 206- fold decrease in KD compared to 2, respectively. Although 9 and 10 have the same number of atoms in the linker group, the latter has an additional amino group, yet it is ligand 9 that is a 10-fold tighter binder. The origin of this difference is difficult to identify with certainty without knowing the exact protonation state and preferred conformation of the linking chain. There may also be a role for a specific placement of the amino groups in the linker of 9 for a favorable electrostatic interaction with the polyphosphate backbone of (CUG)6.59
Inhibition of MBNL1·CUG Interaction Using a SPR-Based Biosensor
We recently described a simple SPR-based method capable of directly measuring MBNL1 complexation of (CUG)12 in real time under equilibrium conditions and in a label-free format. Further, we were able to use this technique to quantify the inhibition potency of selected ligands.27 To rule out non-specific inhibition due to aggregation and non-selective RNA binding, the assays were performed in the presence of both Tween-20 and an excess of competitor tRNA.67 As described previously, biotinylated (CUG)12 was immobilized on a streptavidin coated biosensor chip and incubated with different concentrations of each ligand to reach a steady state response unit (RU) over 150 s. The response to the binding of each ligand was found to be negligible in comparison to MBNL1 binding, so the effect of the ligand could be ignored. The method involved successive injections of a 0.65 μM solution of MBNL1 and enough ligand to keep its concentration identical to that of pre-incubation with the varying responses correlating with ligand concentration. Because the SPR signal directly reflects the binding of MBNL1 to the biotinylated (CUG)12, the differences in the response curves are a result of inhibition by the ligand. The maximum RU at 150 s was recorded for each ligand concentration and converted to the % (CUG)12 bound by MBNL1, with all values normalized to that measured in the absence of each ligand. Fitting the data points in the plot of % (CUG)12 bound by MBNL1 versus increasing concentrations of each ligand (Figure 4a) gave an apparent IC50 value for each ligand (Figure 4b).
Figure 4.

a) Fitting the response unit data in the plot of % (CUG)12 bound by MBNL1 versus increasing concentrations of each ligand to a dose-response curve by SPR assay. b) IC50 values and bivalent effects were derived. Error bars represent mean ± s.d. of three independent experiments.
Normalized IC50 values can be calculated by dividing the actual IC50 value by the number of binding modules in the ligand. The ratio between normalized IC50 values of the representative monomeric ligand, 2, and the dimeric ligand is called bivalent effect. The IC50 values (Figure 4b) show that more than the linker length, its composition is critical for an optimal bivalent effect. The oligoether-linker dimeric ligands, 13, 8, 12 and 7, show a small bivalent effect of 3, 4, 5 and 8, respectively, whereas oligoamino-linker dimeric ligands, 9, 10, 11, show a much bigger bivalent effect of 133, 24 and 29, respectively.
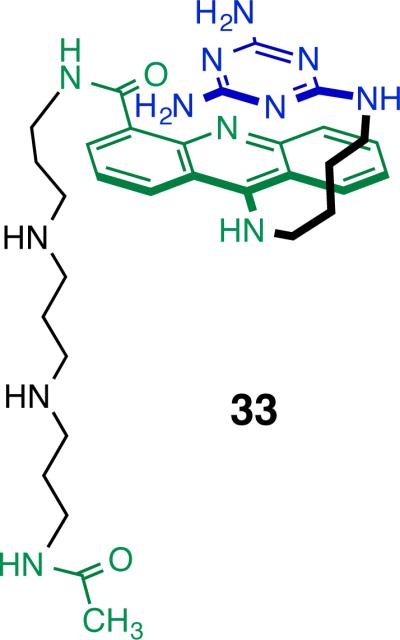
Do the increases in affinity for dimeric ligands 9, 10, 11 originate in bivalent binding of the acridine-triaminotriazine units? Caution must be exercised in interpreting simple increases in KD or improvements in IC50 values, because the linker chain may also contribute directly to binding and inhibition. Indeed, the recently reported monomeric ligand 33 containing an analogous oligoamine side chain to that in 9 gave ΔTm = 2.5 °C and IC50 = 15 ± 2 μM using the same RNA substrate and analytical methods.27 In comparison, the monomeric ligand 2 studied here gave ΔTm = 1.8 ± 0.6 °C, KD = 66 ± 1 μM and IC50 = 293 ± 19 μM. These ΔTm values are nearly the same but the improvement in the IC50 value suggests that the oligoamine does provide additional binding and inhibitory strength. Perhaps the best evidence ligand 9 is a bivalent binder is the increased melting temperature it affords (ΔTm = 9.3 ± 0.6 °C) which is significantly above that for 33 (ΔTm = 2.5 °C). The significant melting temperature increase for 9 relative to 2 and 33 is attributed in large part to the intercalation of the second acridine unit consistent with a bivalent interaction.
These in vitro experiments demonstrate that 9 with ΔTm = 9.3 ± 0.6 °C, KD = 0.32 ± 0.02 μM and IC50 = 1.1 ± 0.1 μM, is the optimal dimeric ligand. Thus, 9 binds to (CUG)12 with a high affinity, stabilizing the stem loop structure and inhibiting the (CUG)12·MBNL1 interaction selectively. The evidence collected is consistent with the improved binding affinity and inhibition potency of 9 resulting from a bivalent binding mode with a positive contribution from oligoamine linker. All of the in vitro experiments were performed in 1X PBS buffer, which was chosen because it most closely mimics physiological conditions.
Bioactivity of 9 in a DM1 Cell Model
Following the successful in vitro studies described above, the optimized dimeric ligand 9 was examined in a cell-based assay. Dimeric ligands 5 and 11 and the corresponding monomeric ligand 2 served as controls. Spermine was studied as a negative control because its structure is analogous to the linker in 9. The cytotoxicity of 2 and 9 in HeLa cells and examined and both showed less than 20% cytotoxicity at concentrations up to 75 μM by a sulforhodamine B assay in 24 h.68 The cellular and nuclear penetration of ligands could be monitored by the inherent fluorescence of the acridine unit. It was found that 5 and 9 are cell-permeable and nucleus-permeable whereas 2 and 11 are not (Figure 5, column 2).
Figure 5.

Confocal fluorescent images show CUGexp foci in DM1 model cells are present in rows 2-6, no ligand treatment, treated with spermine (negative control), 2, 9 and 5, respectively. CUGexp foci are not present in negative control cells, row 1, as well as row 7 where DM1 cell model is treated with 9, for 36 h. Corresponding fluorophores in columns 1-3 are TO-PRO-3, Acridine ring and Cy3-(CAG)10 FISH probe, respectively.
It is a distinguishing characteristic of DM1 cells that MBNL1 aggregates with CUGexp in nuclear foci.69 To visualize the effect of these ligands on DM1 ribonuclear foci, confocal microscopy was applied to a DM1 cell model, HeLa cells that are transfected with a plasmid containing a truncated DMPK mini-gene with CTG960.70 As a negative control cell, HeLa cells were transfected with a truncated DMPK-CTG0 (i.e., no CTG repeat) containing plasmid.70 To label (CUG)960 foci, Cy3-(CAG)10 was used as a fluorescence in situ hybridization (FISH) probe. TO-PRO-3 was used to stain the nucleus. This particular dye was chosen because it does not interfere with the fluorescence of the acridine ring of the ligands and or that from Cy3 used in the Cy3-(CAG)10 FISH probe.
DM1 model cells were treated with three different concentrations (20, 35 and 50 μM) of ligands 2, 5, 11 and 9 for 36 h, followed by a fluorescence in situ hybridization (FISH) study with the Cy3-(CAG)10 probe that labels CUG960 nuclear foci. Over a hundred cells were counted and classified as having foci or no foci for each. Representative images for each sample are shown in Figures 1-14 in the Supporting Information. The fraction of cells with (CUG)960 foci was measured at various concentrations of each of ligand (Figure 6). Ligands 2, 5, 11 and spermine showed no significant foci dispersion in any concentration (p-value > 0.05). However for 9, partial foci dispersion was observed at 20 μM and 35 μM (p-value < 0.009) and full foci dispersion was observed at 50 μM concentration.
Figure 6.

Plot of CUGexp foci-containing cell fraction at various concentrations of ligands. It is noteworthy that data for ligand 5 (50 μM) was not measurable due to solubility issues. These data are gathered from counting more than one hundred cells. The cells were not counted in a blinded manner, but loss of foci was very apparent. Cells with a single foci were not counted as containing nuclear foci. The error bars represent mean ± standard error of at least three independent experiments. The symbol indicates a p-value < 0.009, otherwise giving a p-value > 0.05.
MBNL1 Foci Dispersion in Live DM1 Cell model by 9
Although the reduction in the foci-containing cells upon treatment with 9 was statistically significant, this approach provides only indirect evidence of the foci dispersion. With fixed-cell microscopy the dispersion of ribonuclear foci upon addition of 9 is not directly observed over time in the same cell. Thus, the effect of 9 in a live DM1 cell model was investigated using time-lapse confocal microscopy analogous to the method described recently.27
The DM1 cell model consisted of HeLa cells transfected with the same plasmid used above with a truncated DMPK-CTG960 minigene and also a plasmid containing a GFP-MBNL1 minigene, to track the nuclear foci by monitoring the GFP tag on MBNL1.70 To monitor ligand uptake and MBNL1 foci dispersion in real time, the model DM1 cells were incubated with 9 at 50 μM and individual live cells were examined by confocal microscopy over time. The observation made at t = 0 was just prior to addition of 9 and MBNL1 nuclear foci were clearly present (Figure 7a, t = 0). It was necessary to use a Petri dish with an imprinted 500 μm grid to obtain the absolute position of each individual cell. This way each cell could be relocated following the incubation interval using differential interference contrast (DIC) microscopy. DIC images of 9-treated and untreated DM1 model cells corresponding to cells in Figures 7a and 7b, used to relocate each individual cell, are shown in Figures 15 and 16 in the Supporting Information.
Figure 7.

Live cell microscopy demonstrates a direct evidence for MBNL1 foci dispersion with 9. a) Live DM1 cell model are treated with 9 (50 μM) at t = 0, immediately after the first image is taken. Fluorescence of 9, confirms its penetration to the nucleus. MBNL1 nuclear foci are gradually dispersing over time in two cells. b) Two live cell show stability of foci in DM1 cell model, in the absence of 9, over the period of 10 h. Each box shows 120 μM X 120 μM.
Over time, ligand 9 was observed to penetrate the cellular and nuclear membrane and the intense foci were fully dispersed with the green fluorescence of MBNL1 spread throughout the nucleus (Figure 7a, t = 3, 6, and 10 h). To validate this observation, a Z-stacked image of 1 μm-separated slices from the whole 9-treated DM1 model cell was obtained. This confirmed that upon treatment with 9 at 50 μM, almost all the foci are dispersed over the entire nucleus at t = 10 h (Supplementary Movie 1 and Supplementary Figure 17). As a negative control, to rule out spontaneous MBNL1 foci dispersion over time, untreated DM1 model cells were studied in the same way. The presence of MBNL1 foci at all time points confirmed the stability of the foci (Figure 7b). A Z-stacked image of 1 μm-separated slices from the whole untreated DM1 model confirmed the presence of multiple MBNL1 foci at various Z planes of the cell (Supplementary Movie 2 and Supplementary Figure 18). It is noteworthy that the increase in cell size and number of MBNL1 foci is caused by natural growth of live cells and continuous expression of GFP-MBNL1 in real time. These results provide direct evidence of the ability of 9 to penetrate the cellular and nuclear membrane and disperse almost all of the MBNL1·CUGexp aggregates over a 10 h period.
Conclusion
We report here an approach to targeting CUGexp using a dimeric ligand based on previously reported in vitro active ligand 1.26 Limited by a firm structural knowledge on the ligand·CUGexp complex,46,47 we designed and synthesized a small library of dimeric ligands to bind to neighboring CUG sites. This library of 10 dimeric ligands varied in composition, length and attachment point of linker. The in vitro activity of ligands was studied by three methods: (1) optical melting to measure the stabilization level of ds (CUG)12, (2) steady state fluorescence to measure the binding affinity to (CUG)6 and (3) SPR to measure inhibition of the (CUG)12·MBNL1 complex.
The bivalent effect was found to depend more on the composition of the linker than its length. The dimeric ligands containing oligoamines as linkers were the most potent and they exhibited excellent water solubility. Among the four dimeric ligands with oligoamine linkers, 9 was the most potent ligand in in vitro assays. In comparison to monomeric ligand 2 it had a 206-fold greater affinity to (CUG)6 and 266-fold greater inhibition potency in inhibiting formation of the (CUG)12·MBNL1 complex. Comparison with previously reported monomeric ligand 3327 supports the formation of a bivalent complex with a portion of the gain in affinity and inhibition potency originating in the polyamine-derivative linker of 9. In DM1 cell model, 9 was bioactive as partial foci dispersion was observed at 20 and 35 μM (p-value < 0.009) and full foci dispersion was observed at 50 μM concentration. To obtain direct evidence of its bioactivity, ligand 9 was examined by time-lapse confocal fluorescence microscopy and gradual MBNL1 foci dispersion was observed in individual live DM1 model cells at 50 μM over a 10 h period. To validate that foci are dispersed over the entire nucleus, a Z-stacked image containing 1 μm-separated slices from the whole DM1 model cell was obtained. These positive results suggest that dimeric ligand 9 is a good candidate for further development. Indeed, the next step will be to measure downstream effects of 9 on splicing as well as its toxicity in animals, positive results in these two areas pointing toward future studies in animal models of DM1. These efforts are underway and will be reported in due course.
Experimental Section
General Synthetic Procedure for Compounds 18-27
A round-bottom flask, equipped with a stir bar, was charged with 14 or 15 (1 equivalent) and freshly distilled thionyl chloride (16 equiv.). Synthetic schemes for Compounds 14 and 15 are shown in Scheme 2 of the Supporting Information. A catalytic amount of DMF was added and the mixture heated gently under reflux at 70 °C, stirring until homogeneous and then for 2 h. The excess thionyl chloride was distilled off and the last traces were removed azeotropically via co-evaporation with DCM (3 × 50 mL). The residue was left under vacuum (minimally) for 1 h to afford the crude intermediate as a yellow powder. The crude intermediate was dissolved in anhydrous DCM. Anhydrous triethylamine was added to the solution until the pH was 11 and it was cooled to 0 °C. The corresponding diamine (0.45 equiv.), compounds 34-43 shown in Supporting Information, or methylamine (1.1 equiv) was added and the solution was stirred at 0 °C for 2 h and slowly warmed to room temperature overnight. The solvent was removed by rotary evaporation and the crude mixture was purified via flash chromatography (SiO2; CH2Cl2:MeOH, generally from 98:2 to 95:5) to yield 16-27 as a yellow solid. The yield of each compound is shown in the Supporting Information.
General Synthetic Procedure for Compounds 4, 6-8, 12, 13, 29-32
A round-bottom flask, equipped with a stir bar, was charged with one of compounds 18-27 (0.5 equivalent) or 16-17 (1 equiv.) and 28 (1.1 equivalent). Synthetic scheme for compounds 28 is shown in Scheme 1 of Supporting Information. DIPEA (1.1 equivalent) and anhydrous DMF (25 mL) were added. The solution was heated at 80 °C for 6 h. The solvent was removed by rotary evaporation and the product purified via flash chromatography (basic alumina; DCM:methanol:NH4OH, generally from 95:4.9:0.1 to 90:9.5:0.5) to yield the corresponding compound, 2-4, 6-8, 12-13, or 29-32 as a yellow solid. The yield of each compound is shown in the Supporting Information.
General Synthetic Procedure for Compounds 5, 9-11
A round-bottom flask, equipped with a stir bar, was charged with one of compounds 29-32 (1 equivalent). Anhydrous TFA (30 mL) and anhydrous DCM (70 mL) were added and stirred at room temperature for 6 h. The solvents were removed to yield compounds 5, 9-11 as a yellow solid in quantitative yield.
Compound Purification and Characterization
Following chromatographic purification, compounds were observed as single spots on thin layer chromatography plates. The key monomeric and dimeric ligands were estimated to be ≥95% based on the 1H NMR spectra that showed single homogeneous compounds as judged by the single set of peaks. This estimate was confirmed quantitatively for control ligand 2 and dimeric ligands 5-7, 9, and 11 using HPLC (LC-MS) with traces shown in the Supporting Information. HPLC showed ligand 9, which was studied in cell culture, to be ca. 99% pure and its elemental analysis (CHNF) showed it to be ca. a hexa-ammonium ion trifluoroacetate salt. See Supporting Information for additional details.
Bioassays, Methods, and Origin of Biological Materials
The MBNL1N protein expression and purification procedures and origin of CTG0 and CTG960 plasmids and RNAs were described previously.27 The optical melting experiments, surface plasmon resonance (SPR) analysis, and steady state fluorescence-based binding assays followed directly from the previously reported.27
FISH (Fluorescence In Situ Hybridization)
A total of ca. 120,000 HeLa cells were seeded in each well of a 6-well plate on coverslips. After a day, the cells were transfected with 500 ng DMPK−CTG0 or DMPK−CTG960 plasmid, as described previously.27 Ligands were added to wells at different concentrations (20, 35 and 50 μM). After 36 h, cells were fixed and processed and slides were imaged and analyzed, as described previously. The following table indicates the excitation filters used in these experiments.
| Fluorophore | Component | Excitation wavelength (nm) |
|---|---|---|
| Acridine | Ligands | 405 |
| Cy3 | CUG960 | 555 |
| TO-PRO-3 | Nucleus | 639 |
The p-values were calculated using two-tailed Student t test by Microsoft® Excel 2010.
Live Cell Imaging
Cells were prepared as described previously.27 Ligand 9 was added to give a final concentration of 50 μM. Live-cell, time-lapse images were taken before addition of 9 as well as at 3, 6 and 10 h time points, as described previously.
Supplementary Material
ACKNOWLEDGEMENT
Support of this work by the National Institutes of Health (R01AR058361 to S.C.Z. and A.M.B.) is gratefully acknowledged.
ABBREVIATIONS USED
- ASO
antisense oligonucleotide
- CUG-BP1
CUG-binding protein 1; myotonic dystrophy type 1
- DM1
dystrophia myotonica protein kinase
- ds
double stranded
- DMPK
dystrophia myotonica protein kinase
- FISH
fluorescence in situ hybridization
- MBNL1
muscleblind-like 1 protein
- PTS
polyamine transporting system
- RU
response unit
- ss
single stranded
- SPR
surface plasmon resonance
- TAMRA
carboxytetramethylrhodamine
- Tm
melting temperature
- TRED
trinucleotide expansion disease
Footnotes
Present Addresses Anne M. Baranger: University of California, Berkeley Yuan Fu: Cornell University
The authors declare no competing financial interest.
Supporting Information
Experimental procedures, full characterization data for the compounds, supplementary confocal microscopy images and movies. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Foff EP, Mahadevan MS. Therapeutics development in myotonic dystrophy type 1. Muscle Nerve. 2011;44:160–169. doi: 10.1002/mus.22090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, Timmers AM, Hauswirth WW, Swanson MS. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302:1978–1980. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- 3.Mooers BHM, Logue JS, Berglund JA. The structural basis of myotonic dystrophy from the crystal structure of CUG repeats. Proc. Natl. Acad. Sci. U. S. A. 2005;102:16626–16631. doi: 10.1073/pnas.0505873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kiliszek A, Kierzek R, Krzyzosiak WJ, Rypniewski W. Structural insights into CUG repeats containing the ‘stretched U-U wobble’: implications for myotonic dystrophy. Nucleic Acids Res. 2009;37:4149–4156. doi: 10.1093/nar/gkp350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grammatikakis I, Goo YH, Echeverria GV, Cooper TA. Identification of MBNL1 and MBNL3 domains required for splicing activation and repression. Nucleic Acids Res. 2011;39:2769–2780. doi: 10.1093/nar/gkq1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kanadia RN, Shin J, Yuan Y, Beattie SG, Wheeler TM, Thornton CA, Swanson MS. Reversal of RNA missplicing and myotonia after muscleblind overexpression in a mouse poly(CUG) model for myotonic dystrophy. Proc. Natl. Acad. Sci. U. S. A. 2006;103:11748–11753. doi: 10.1073/pnas.0604970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O'Rourke JR, Swanson MS. Mechanisms of RNA-mediated disease. J. Biol. Chem. 2009;284:7419–7423. doi: 10.1074/jbc.R800025200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mulders SA, van Engelen BG, Wieringa B, Wansink DG. Molecular therapy in myotonic dystrophy: focus on RNA gain-of-function. Hum. Mol. Genet. 2010;19:R90–97. doi: 10.1093/hmg/ddq161. [DOI] [PubMed] [Google Scholar]
- 9.Lee JE, Cooper TA. Pathogenic mechanisms of myotonic dystrophy. Biochem. Soc. Trans. 2009;37:1281–1286. doi: 10.1042/BST0371281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cooper TA. A reversal of misfortune for myotonic dystrophy? New Engl. J. Med. 2006;355:1825–1827. doi: 10.1056/NEJMcibr064708. [DOI] [PubMed] [Google Scholar]
- 11.Ward AJ, Rimer M, Killian JM, Dowling JJ, Cooper TA. CUGBP1 overexpression in mouse skeletal muscle reproduces features of myotonic dystrophy type 1. Hum. Mol. Genet. 2010;19:3614–3622. doi: 10.1093/hmg/ddq277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang L, Lee JE, Wilusz J, Wilusz CJ. The RNA-binding protein CUGBP1 regulates stability of tumor necrosis factor mRNA in muscle cells: implications for myotonic dystrophy. J. Biol. Chem. 2008;283:22457–22463. doi: 10.1074/jbc.M802803200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kuyumcu-Martinez NM, Wang GS, Cooper TA. Increased steady-state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC-mediated hyperphosphorylation. Mol. Cell. 2007;28:68–78. doi: 10.1016/j.molcel.2007.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jones K, Jin B, Iakova P, Huichalaf C, Sarkar P, Schneider-Gold C, Schoser B, Meola G, Shyu AB, Timchenko N, Timchenko L. RNA Foci, CUGBP1, and ZNF9 are the primary targets of the mutant CUG and CCUG repeats expanded in myotonic dystrophies type 1 and type 2. Amer. J. Pathol. 2011;179:2475–2489. doi: 10.1016/j.ajpath.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wheeler TM, Leger AJ, Pandey SK, MacLeod AR, Nakamori M, Cheng SH, Wentworth BM, Bennett CF, Thornton CA. Targeting nuclear RNA for in vivo correction of myotonic dystrophy. Nature. 2012;488:111–115. doi: 10.1038/nature11362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cooper T. a. Chemical reversal of the RNA gain of function in myotonic dystrophy. Proc. Natl. Acad. Sci. U. S. A. 2009;106:18433–18434. doi: 10.1073/pnas.0910643106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee JE, Bennett CF, Cooper TA. RNase H-mediated degradation of toxic RNA in myotonic dystrophy type 1. Proc. Natl. Acad. Sci. U. S. A. 2012;109:4221–4226. doi: 10.1073/pnas.1117019109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sobczak K, Wheeler TM, Wang W, Thornton CA. RNA interference targeting CUG repeats in a mouse model of myotonic dystrophy. Mol. Ther. 2013;21:380–387. doi: 10.1038/mt.2012.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wheeler TM, Sobczak K, Lueck JD, Osborne RJ, Lin X, Dirksen RT, Thornton CA. Reversal of RNA dominance by displacement of protein sequestered on triplet repeat RNA. Science. 2009;325:336–339. doi: 10.1126/science.1173110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.García-López A, Llamusí B, Orzáez M, Pérez-Payá E, Artero RD. In vivo discovery of a peptide that prevents CUG-RNA hairpin formation and reverses RNA toxicity in myotonic dystrophy models. Proc. Natl. Acad. Sci. U. S. A. 2011;108:11866–11871. doi: 10.1073/pnas.1018213108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Warf MB, Nakamori M, Matthys CM, Thornton CA, Berglund JA. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc. Natl. Acad. Sci. U. S. A. 2009;106:18551–18556. doi: 10.1073/pnas.0903234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ofori LO, Hoskins J, Nakamori M, Thornton CA, Miller BL. From dynamic combinatorial ‘hit’ to lead: in vitro and in vivo activity of compounds targeting the pathogenic RNAs that cause myotonic dystrophy. Nucleic Acids Res. 2012;40:6380–6390. doi: 10.1093/nar/gks298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parkesh R, Childs-Disney JL, Nakamori M, Kumar A, Wang E, Wang T, Hoskins J, Tran T, Housman D, Thornton CA, Disney MD. Design of a bioactive small molecule that targets the myotonic dystrophy type 1 RNA via an RNA motif-ligand database and chemical similarity searching. J. Am. Chem. Soc. 2012;134:4731–4742. doi: 10.1021/ja210088v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pushechnikov A, Lee MM, Childs-Disney JL, Sobczak K, French JM, Thornton CA, Disney MD. Rational design of ligands targeting triplet repeating transcripts that cause RNA dominant disease: application to myotonic muscular dystrophy type 1 and spinocerebellar ataxia type 3. J. Am. Chem. Soc. 2009;131:9767–9779. doi: 10.1021/ja9020149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Childs-Disney JL, Hoskins J, Rzuczek SG, Thornton CA, Disney MD. Rationally designed small molecules targeting the RNA that causes myotonic dystrophy type 1 are potently bioactive. ACS Chem. Biol. 2012;7:856–862. doi: 10.1021/cb200408a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arambula JF, Ramisetty SR, Baranger AM, Zimmerman SC. A simple ligand that selectively targets CUG trinucleotide repeats and inhibits MBNL protein binding. Proc. Natl. Acad. Sci. U. S. A. 2009;106:16068–16073. doi: 10.1073/pnas.0901824106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jahromi AH, Nguyen L, Fu Y, Miller KA, Baranger AM, Zimmerman SC. A Novel CUG(exp)·MBNL1 Inhibitor with Therapeutic Potential for Myotonic Dystrophy Type 1. ACS Chem. Biol. 2013;8:1037–1043. doi: 10.1021/cb400046u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krishnamurthy VM, Estroff LA, Whitesides GM. Multivalency in Ligand Design, in Fragment-based Approaches in Drug Discovery. In: Jahnke W, Erlanson DA, editors. Wiley-VCH Verlag GmbH & Co.; KGaA, Weinheim, FRG: 2006. doi: 10.1002/3527608761.ch2. [Google Scholar]
- 29.Mammen M, Choi SK, Whitesides GM. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angew. Chem. Int. Ed. 1998;37:2755–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 30.Lees WJ, Spaltenstein A, Kingery-Wood JE, Whitesides GM. Polyacrylamides bearing pendant alpha-sialoside groups strongly inhibit agglutination of erythrocytes by influenza A virus: multivalency and steric stabilization of particulate biological systems. J. Med. Chem. 1994;37:3419–3433. doi: 10.1021/jm00046a027. [DOI] [PubMed] [Google Scholar]
- 31.Jervis PJ, Moulis M, Jukes JP, Ghadbane H, Cox LR, Cerundolo V, Besra GS. Towards multivalent CD1d ligands: synthesis and biological activity of homodimeric alpha-galactosyl ceramide analogues. Carbohydr. Res. 2012;356:152–162. doi: 10.1016/j.carres.2012.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sucheck SJ, Wong AL, Koeller KM, Boehr DD, Draker K.-a., Sears P, Wright GD, Wong C-H. Design of Bifunctional Antibiotics that Target Bacterial rRNA and Inhibit Resistance-Causing Enzymes. J. Am. Chem. Soc. 2000;122:5230–5231. [Google Scholar]
- 33.Tamiz AP, Zhang J, Zhang M, Wang CZ, Johnson KM, Kozikowski AP. Application of the Bivalent Ligand Approach to the Design of Novel Dimeric Serotonin Reuptake Inhibitors. J. Am. Chem. Soc. 2000;122:5393–5394. [Google Scholar]
- 34.Gestwicki JE, Cairo CW, Strong LE, Oetjen K. a., Kiessling LL. Influencing receptor-ligand binding mechanisms with multivalent ligand architecture. J. Am. Chem. Soc. 2002;124:14922–14933. doi: 10.1021/ja027184x. [DOI] [PubMed] [Google Scholar]
- 35.Agnelli F, Sucheck SJ, Marby KA, Rabuka D, Yao SL, Sears PS, Liang FS, Wong CH. Dimeric aminoglycosides as antibiotics. Angew. Chem. Int. Ed. Engl. 2004;43:1562–1566. doi: 10.1002/anie.200353225. [DOI] [PubMed] [Google Scholar]
- 36.Lee MM, Pushechnikov A, Disney MD. Rational and modular design of potent ligands targeting the RNA that causes myotonic dystrophy 2. ACS Chem. Biol. 2009;4:345–355. doi: 10.1021/cb900025w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Childs-Disney JL, Tsitovich PB, Disney MD. Using modularly assembled ligands to bind RNA internal loops separated by different distances. ChemBioChem. 2011;12:2143–2146. doi: 10.1002/cbic.201100298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shonberg J, Scammells PJ, Capuano B. Design strategies for bivalent ligands targeting GPCRs. ChemMedChem. 2011;6:963–974. doi: 10.1002/cmdc.201100101. [DOI] [PubMed] [Google Scholar]
- 39.Kitov PI, Bundle DR. On the nature of the multivalency effect: a thermodynamic model. J. Am. Chem. Soc. 2003;125:16271–16284. doi: 10.1021/ja038223n. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Y, Gilliam A, Maitra R, Damaj MI, Tajuba JM, Seltzman HH, Thomas BF. Synthesis and biological evaluation of bivalent ligands for the cannabinoid 1 receptor. J. Med. Chem. 2010;53:7048–7060. doi: 10.1021/jm1006676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Birnkammer T, Spickenreither A, Brunskole I, Lopuch M, Kagermeier N, Bernhardt G, Dove S, Seifert R, Elz S, Buschauer A. The bivalent ligand approach leads to highly potent and selective acylguanidine-type histamine H(2) receptor agonists. J. Med. Chem. 2012;55:1147–1160. doi: 10.1021/jm201128q. [DOI] [PubMed] [Google Scholar]
- 42.Kühhorn J, Götz A, Hübner H, Thompson D, Whistler J, Gmeiner P. Development of a bivalent dopamine D(2) receptor agonist. J. Med. Chem. 2011;54:7911–7919. doi: 10.1021/jm2009919. [DOI] [PubMed] [Google Scholar]
- 43.Kühhorn J, Hübner H, Gmeiner P. Bivalent dopamine D2 receptor ligands: synthesis and binding properties. J. Med. Chem. 2011;54:4896–4903. doi: 10.1021/jm2004859. [DOI] [PubMed] [Google Scholar]
- 44.Peng Y, Sun H, Lu J, Liu L, Cai Q, Shen R, Yang CY, Yi H, Wang S. Bivalent Smac mimetics with a diazabicyclic core as highly potent antagonists of XIAP and cIAP1/2 and novel anticancer agents. J. Med. Chem. 2012;55:106–114. doi: 10.1021/jm201072x. [DOI] [PubMed] [Google Scholar]
- 45.Liu J, Brahimi F, Saragovi HU, Burgess K. Bivalent diketopiperazine-based tropomysin receptor kinase C (TrkC) antagonists. J. Med. Chem. 2010;53:5044–5048. doi: 10.1021/jm100148d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haghighat Jahromi A, Honda M, Zimmerman SC, Spies M. Single-molecule study of the CUG repeat-MBNL1 interaction and its inhibition by small molecules. Nucleic Acids Res. 2013;41:6687–6697. doi: 10.1093/nar/gkt330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wong CH, Richardson SL, Ho YJ, Lucas AM, Tuccinardi T, Baranger AM, Zimmerman SC. Investigating the binding mode of an inhibitor of the MBNL1.RNA complex in myotonic dystrophy type 1 (DM1) leads to the unexpected discovery of a DNA-selective binder. ChemBioChem. 2012;13:2505–2509. doi: 10.1002/cbic.201200602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wakelin LPG. Polyfunctional DNA intercalating agents. Med. Res. Rev. 1986;6:275–340. doi: 10.1002/med.2610060303. [DOI] [PubMed] [Google Scholar]
- 49.Zimmerman SC, Lamberson CR, Cory M, Fairley TA. Topologically Constrained Bifunctional Intercalators - DNA Intercalation by a Macrocyclic Bisacridine. J. Am. Chem. Soc. 1989;111:6805–6809. [Google Scholar]
- 50.Veal JM, Li Y, Zimmerman SC, Lamberson CR, Cory M, Zon G, Wilson WD. Interaction of a macrocyclic bisacridine with DNA. Biochemistry. 1990;29:10918–10927. doi: 10.1021/bi00501a009. [DOI] [PubMed] [Google Scholar]
- 51.Denny WA. Acridine derivatives as chemotherapeutic agents. Curr. Med. Chem. 2002;9:1655–1665. doi: 10.2174/0929867023369277. [DOI] [PubMed] [Google Scholar]
- 52.Fechter EJ, Olenyuk B, Dervan PB. Design of a sequence-specific DNA bisintercalator. Angew. Chem. Int. Ed. Engl. 2004;43:3591–3594. doi: 10.1002/anie.200454231. [DOI] [PubMed] [Google Scholar]
- 53.Goodell JR, Madhok AA, Hiasa H, Ferguson DM. Synthesis and evaluation of acridine- and acridone-based anti-herpes agents with topoisomerase activity. Bioorg. Med. Chem. 2006;14:5467–5480. doi: 10.1016/j.bmc.2006.04.044. [DOI] [PubMed] [Google Scholar]
- 54.Rhoden Smith A, Iverson BL. Threading polyintercalators with extremely slow dissociation rates and extended DNA binding sites. J. Am. Chem. Soc. 2013;135:12783–12789. doi: 10.1021/ja4057344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Antonini I, Polucci P, Magnano A, Gatto B, Palumbo M, Menta E, Pescalli N, Martelli S. Design, synthesis, and biological properties of new bis(acridine-4-carboxamides) as anticancer agents. J. Med. Chem. 2003;46:3109–3115. doi: 10.1021/jm030820x. [DOI] [PubMed] [Google Scholar]
- 56.Caffrey CR, Steverding D, Swenerton RK, Kelly B, Walshe D, Debnath A, Zhou YM, Doyle PS, Fafarman AT, Zorn JA, Land KM, Beauchene J, Schreiber K, Moll H, Ponte-Sucre A, Schirmeister T, Saravanamuthu A, Fairlamb AH, Cohen FE, McKerrow JH, Weisman JL, May BC. Bis-acridines as lead antiparasitic agents: structure-activity analysis of a discrete compound library in vitro. Antimicrob. Agents Chemother. 2007;51:2164–2172. doi: 10.1128/AAC.01418-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bobrovnik SA. The influence of rigid or flexible linkage between two ligands on the effective affinity and avidity for reversible interactions with bivalent receptors. J. Mol. Recognit. 2007;20:253–262. doi: 10.1002/jmr.836. [DOI] [PubMed] [Google Scholar]
- 58.Knuf EC, Jiang J-K, Gin MS. Preparation of discrete oligoethers: synthesis of pentabutylene glycol and hexapropylene glycol by two complementary methods. J. Org. Chem. 2003;68:9166–9169. doi: 10.1021/jo034728k. [DOI] [PubMed] [Google Scholar]
- 59.Fischer W, Brissault B, Prévost S, Kopaczynska M, Andreou I, Janosch A, Gradzielski M, Haag R. Synthesis of linear polyamines with different amine spacings and their ability to form dsDNA/siRNA complexes suitable for transfection. Macromol. Biosci. 2010;10:1073–1083. doi: 10.1002/mabi.201000082. [DOI] [PubMed] [Google Scholar]
- 60.Delcros JG, Tomasi S, Carrington S, Martin B, Renault J, Blagbrough IS, Uriac P. Effect of spermine conjugation on the cytotoxicity and cellular transport of acridine. J. Med. Chem. 2002;45:5098–5111. doi: 10.1021/jm020843w. [DOI] [PubMed] [Google Scholar]
- 61.Crothers DM. Calculation of binding isotherms for heterogenous polymers. Biopolymers. 1968;6:575–584. doi: 10.1002/bip.1968.360060411. [DOI] [PubMed] [Google Scholar]
- 62.Laurent FX, Sureau A, Klein AF, Trouslard F, Gasnier E, Furling D, Marie J. New function for the RNA helicase p68/DDX5 as a modifier of MBNL1 activity on expanded CUG repeats. Nucleic Acids Res. 2012;40:3159–3171. doi: 10.1093/nar/gkr1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fu Y, Ramisetty SR, Hussain N, Baranger AM. MBNL1-RNA recognition: contributions of MBNL1 sequence and RNA conformation. ChemBioChem. 2012;13:112–119. doi: 10.1002/cbic.201100487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alessi ML, Norman AI, Knowlton SE, Ho DL, Greer SC. Helical and coil conformations of poly(ethylene glycol) in isobutyric acid and water. Macromolecules. 2005;38:9333–9340. [Google Scholar]
- 65.Vámosi G, Gohlke C, Clegg RM. Fluorescence characteristics of 5-carboxytetramethylrhodamine linked covalently to the 5′ end of oligonucleotides: multiple conformers of single-stranded and double-stranded dye-DNA complexes. Biophys. J. 1996;71:972–994. doi: 10.1016/S0006-3495(96)79300-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Qu P, Chen XD, Zhou XX, Li X, Zhao XS. Fluorescence quenching of TMR by guanosine in oligonucleotides. Sci. China, Ser. B. 2009;52:1653–1659. [Google Scholar]
- 67.Feng BY, Shoichet BK. A detergent-based assay for the detection of promiscuous inhibitors. Nat. Protoc. 2006;1:550–553. doi: 10.1038/nprot.2006.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc. 2006;1:1112–1116. doi: 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- 69.Echeverria GV, Cooper TA. RNA-binding proteins in microsatellite expansion disorders: mediators of RNA toxicity. Brain Res. 2012;1462:100–111. doi: 10.1016/j.brainres.2012.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ho TH, Savkur RS, Poulos MG, Mancini MA, Swanson MS, Cooper TA. Colocalization of muscleblind with RNA foci is separable from mis-regulation of alternative splicing in myotonic dystrophy. J. Cell Sci. 2005;118:2923–2933. doi: 10.1242/jcs.02404. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.