

Abstract
The objective of this study was to elucidate the effect of VDAC1 on Alzheimer’s disease (AD)-related genes, mitochondrial activity, and synaptic viability. Recent knockout studies of VDAC1 revealed that homozygote VDAC1 knockout (VDAC1−/−) mice exhibited disrupted learning and synaptic plasticity, and in contrast, VDAC1+/− mice appeared normal in terms of lifespan, fertility, and viability relative to wild-type mice. However, the effects of reduced VDAC1 on mitochondrial/synaptic genes and mitochondrial function in AD-affected neurons are not well understood. In the present study, we characterized mitochondrial/synaptic and AD-related genes and mitochondrial function in VDAC1+/− mice and VDAC1+/+ mice. We found reduced mRNA levels in the AD-related genes, including AβPP, Tau, PS1, PS2, and BACE1; increased levels of the mitochondrial fusion genes Mfn1, Mfn2; reduced levels of the fission genes Drp1 and Fis1; and reduced levels of the mitochondrial permeability transition pore genes VDAC1, ANT, and CypD in VDAC1+/− mice relative to VDAC1+/+ mice. Hexokinase 1 and 2 were significantly upregulated in the VDAC+/− mice. The synaptic genes synaptophysin, synapsin 1 and 2, synaptobrevin 1 and 2, neurogranin, and PSD95 were also upregulated in the VDAC1+/− mice. Free radical production and lipid peroxidation levels were reduced in the VDAC1+/− mice, and cytochrome oxidase activity and ATP levels were elevated, indicating enhanced mitochondrial function in the VDAC1+/− mice. These findings suggest that reduced VDAC1 expression, such as that we found in the VDAC1+/− mice, may be beneficial to synaptic activity, may improve function, and may protect against toxicities of AD-related genes.
Introduction
Alzheimer’s disease (AD) is a late-onset, neurodegenerative disease, characterized by a progressive decline of memory and cognitive functions, and changes in behavior and personality [1]. Intraneuronal amyloid beta (Aβ) and Aβ deposits early in the disease process, and intracellular hyperphosphorylated tau and neurofibrillary tangles (NFTs) later in the disease process were found in postmortem brains from AD patients [2–4]. AD has been also associated with the loss of synapses and synaptic dysfunction, inflammatory responses, and abnormalities in the structure and function of mitochondria [5–6].
Recent research revealed that mitochondrial dysfunction and synaptic damage are early events in AD pathogenesis [7–11]. Several studies found increased free radical production, lipid peroxidation, oxidative DNA and protein damage reduced ATP production, and cytochrome oxidase activity in postmortem brains from AD patients, compared to postmortem brains from control subjects, suggesting the presence of mitochondrial dysfunction in AD pathogenesis[12–17]. The mechanistic link between mitochondria and AD pathogenesis has only recently been established [4,11,18]. Using biochemical, molecular, and electron microscopy studies, and postmortem brains from AD patients and AβPP mice, we [8,19,20,21] and others [12, 22–25] found Aβ associated with mitochondria and mitochondrial dysfunction. Further, recent research also revealed that increased mitochondrial fission genes, Drp1, Fis1 and decreased fusion gene, Mfn1, Mfn2 and Opa1 in AD postmortem brains, brain tissues from AD transgenic mice and cells that express/produce Aβ, indicating that the presence of abnormal mitochondrial dynamics in AD progression and pathogenesis [10,20, 26,27].
Mitochondria are the powerhouses of the cell, responsible for cell survival and cell death. They perform several important functions, including ATP production, intracellular calcium regulation, free radical production, and scavenging [28]. Mitochondria consist of two biolipid membranes, the inner and outer. and the inner mitochondrial membrane houses the electron transport chain, tricarboxylic acid, and beta-oxidation. The inner membrane provides a highly efficient barrier to the flow of ions. The outer mitochondrial membrane is basically porous and allows the passage of low molecular-weight substances between the cytosol and the mitochondrial intermembrane space. The mitochondrial permeability transition (MPT) pores are formed by 3 important proteins: voltage-dependent anion channel 1 (VDAC1), the protein in the outer membrane of mitochondria; adenine nucleotide translocator (ANT), the protein in the inner membrane of mitochondria, and the matrix protein cyclophilin D (CypD). The metabolites and nuclear-encoded mitochondrial proteins pass through the MPT pores, and VDAC1 regulates the mitochondrial pore gating [29–32]. Recent research also revealed that increased levels of MPT pore forming proteins, VDAC1, ANT and CypD in AD postmortem brains and AD transgenic mice, indicating that mitochondrial pore gating may be impaired in AD neurons [33–35].
VDACs, also referred as porins, reported to perform several important functions in the cell, including maintaining synaptic plasticity through mitochondrial permeability transition pore regulating the shape and structure of mitochondria; regulating the interaction of hexokinase with mitochondria; and regulating apoptosis signaling [36–37]. In studies aimed at elucidating the normal function of VDAC proteins, researchers generated VDAC1 and VDAC3 heterozygote embryonic stem cells to obtain mouse chimeras and to breed hetero- and homozygote knockout mice. Mutant and wild-type alleles of the VDAC3 locus were transmitted in the expected Mendelian ratios, but VDAC1−/− mice were born in fewer-than-expected numbers, suggesting partial embryonic lethality of VDAC1−/− particularly between embryonic days 10.5 to 11. 5 [38]. The surviving VDAC1−/− mice were fertile, but they were mildly retarded in growth. However, the VDAC1 and VDAC3 heterozygote knockout (VDAC1+/− and VDAC3+/−) mice were fertile and had a normal lifespan.
Recently, our laboratory reported that in postmortem AD brains and AβPP transgenic mouse brains, Aβ (monomers and oligomers) and phosphorylated tau interacted with VDAC1. These interactions increased with disease progression, suggesting that Aβ and phosphorylated tau, in combination, may block the transport of organelles between mitochondria and the cytoplasm, possibly causing defects in oxidative phosphorylation and mitochondrial ATP synthesis.
It is unclear whether partial reduction of VDAC1 expression reduces interaction of Aβ and phosphorylated tau with VDAC1 [39]. If partial reduction of VDAC1 does reduce mitochondrial dysfunction and synaptic deficiencies and reduce the interaction of Aβ and phosphorylated tau with VDAC1, a key question is how much VDAC1 is minimally sufficient to inhibit the interaction between VDAC1 and Aβ, and between VDAC1 and phosphorylated tau, yet can maintain synaptic activity, mitochondrial function, and neuronal survival in brains of AD transgenic mice? To address this question, we determined mitochondrial activity in VDAC1+/− mice and VDAC1+/+ mice, focusing on mitochondrial structural gene expressions, electron transport chain genes, mitochondrial dynamics in the inner membrane and matrix genes of mitochondria, and mitochondrial function and GTPase Drp1 enzymatic activity. We also identified mRNA levels of AD-related genes and synaptic activity of synaptic and dendritic proteins.
Materials and Methods
VDAC1 mice and brain tissues
Brain tissues from VDAC1 heterozygote knockout (VDAC1+/−) mice and wild-type VDAC1+/+ mice (control) [38] were used to determine the effects of reduced VDAC1 on mitochondrial, synaptic and AD gene expression and mitochondrial function. The VDAC1+/+ and VDAC1+/− mice were housed at Baylor College of Medicine, and the Baylor College of Medicine Institutional Animal Care and Use Committee approved all procedures for animal care according to guidelines set forth by the National Institutes of Health. We genotyped all the mice for the VDAC1 gene, using DNA prepared from tail biopsies of 2- to 3-week-old pups from VDAC+/− mice, following Weeber et al. [38].
Real-time RT-PCR quantification of mRNA expression of peroxiredoxins, ETC, and neuroprotective genes
Using the reagent TriZol (Invitrogen, Grand Island, NY), total RNA was isolated from VDAC+/− (n=4) and VDAC+/+ (n=4) mice. Using primer express Software (Applied Biosystems, Foster City, CA), the oligonucleotide primers were designed for the housekeeping genes, β-actin and GAPDH; mitochondrial-encoded ETC genes ([Complex I, NADH dehydrogenase 3–(ND3) & NADH dehydrogenase 6 (ND6]), Complex III – CytB, Complex IV – COX1 & 2, and Complex V – ATP6); the mitochondrial structural fission genes Drp1 and Fis1; the mitochondrial structural fusion genes MFN1, MFN2, Opa1, VDAC1, ANT; and the mitochondrial matrix genes CypD and BcLxL; hexokinase 1 and hexokinase 2; the synaptic genes synaptophysin, PSD95, synapsin 1, synapsin 2, synaptobrevin 1 and 2, GAP43, synaptopodin, and neurogranin; and the AD-related genes APP, Tau, PS1, PS2, BACE1, GSK3α, and GSK3β. The primer sequences and amplicon sizes are listed in Table 1.
Table 1.
Summary of real-time RT-PCR oligonucleotide primers used in measuring mRNA expression in mitochondrial genes, Alzheimer’s disease-related genes, and synaptic genes in VDAC1+/− mice and VDAC1+/+ mice
| Genes | DNA Sequence (5′-3′) | PCR Product Size |
|---|---|---|
| Mitochondrial Structural Genes | ||
| Drp1 | Forward Primer GCGCTGATCCCGCGTCAT | 54 |
| Reverse Primer CCGCACCCACTGTGTTGA | ||
| Fis1 | Forward Primer GCCCCTGCTACTGGACCAT | 62 |
| Reverse Primer CCCTGAAAGCCTCACACTAAGG | ||
| MFN1 | Forward Primer TCTCCAAGCCCAACATCTTCA | 62 |
| Reverse Primer ACTCCGGCTCCGAAGCA | ||
| MFN2 | Forward Primer ACAGCCTCAGCCGACAGCAT | 56 |
| Reverse Primer TGCCGAAGGAGCAGACCTT | ||
| Opa1 | Forward Primer TGGGCTGCAGAGGATGGT | 59 |
| Reverse Primer CCTGATGTCACGGTGTTGATG | ||
| VDCA1 | Forward Primer CTCCCACATACGCCGATCTT | 58 |
| Reverse Primer GCCGTAGCCCTTGGTGAAG | ||
| ANT | Forward Primer CACCCATCGAGAGGGTCAA | 54 |
| Reverse Primer ATTTGCTTGCTGGCATGCT | ||
| Cyclophilin D | Forward Primer CAGCCAAGCCCTCCAACTC | 58 |
| Reverse Primer GCCGATGTCCACGTCAAAG | ||
| BcLXL | Forward Primer GAGCCATTGAGTGAGGTGCTTT | 60 |
| Reverse Primer TCCCAAGCAGCCTGAATTTC | ||
| Mitochondrial-encoded Electron Transport Chain Genes | ||
| ND3-Complex I | Forward Primer TGTACTCAGAAAAAGCAAATCCATATG | 73 |
| Reverse Primer AATAATAGAAATGTAATTGCTACCAAGAAAAA | ||
| ND6-Complex I | Forward Primer TTGATGGTTTGGGAGATTGGTT | 75 |
| Reverse Primer TGCCGCTACCCCAATCC | ||
| CytB-Complex III | Forward Primer TTATCGCGGCCCTAGCAA | 75 |
| Reverse Primer TAATCCTGTTGGGTTGTTTGATCC | ||
| COX 1-Complex IV | Forward Primer GAAGAGACAGTGTTTCATGTGGTGT | 73 |
| Reverse Primer TCCTGGGCCTTTCAGGAATA | ||
| COX2-Complex IV | Forward Primer CATCCCAGGCCGACTAAATC | 74 |
| Reverse Primer TTTCAGAGCATTGGCCATAGAA | ||
| ATP6-Complex V | Forward Primer TGTGGAAGGAAGTGGGCAA | 72 |
| Reverse Primer CCACTATGAGCTGGAGCCGT | ||
| AD-related Genes | ||
| APP | Forward Primer AACATGATCAGTGAGCCCAGAA | 59 |
| Reverse Primer CGTCAGCGAAGGCATGAGA | ||
| PS1 | Forward Primer ATCGGCCTGTGCCTTACATT | 55 |
| Reverse Primer GCTGGCAACGCTTTCTTGA | ||
| PS2 | Forward Primer AGACCATGCTCGCATTCATG | 62 |
| Reverse Primer AGGACGTCCGCTCATCACA | ||
| Tau | Forward Primer CCCCCGCCGATGATG | 79 |
| Reverse Primer AGTCACGTCTTCAGCAGTTGGA | ||
| BACE1 | Forward Primer TCCGGCTCAGAACTACAGTGTAAT | 58 |
| Reverse Primer TGCGGCGTTTTCATGGT | ||
| Hexokinase 1 | Forward Primer GACCCGAGGCATCTTCGA | 62 |
| Reverse Primer AGCAGCGCTAATCGGTCACT | ||
| Hexokinase 2 | Forward Primer CGCCGGATTGGAACAGAA | 75 |
| Reverse Primer CCCGTCGCTAACTTCACTCACT | ||
| GSK-3β | Forward Primer TGTTGGGCCCTGAGGTAGAT | 82 |
| Reverse Primer CCTAGGGTACCAGCTCAAAACAA | ||
| Synaptic Genes | ||
| Synaptophysin | Forward Primer 5′ CATTCAGGCTGCACCAAGTG 3′ | 59 |
| Reverse Primer 5′ TGGTAGTGCCCCCTTTAACG 3′ | ||
| PSD95 | Forward Primer 5′ GGACATTCAGGCGCACAAG 3′ | 58 |
| Reverse Primer 5′ TCCCGTAGAGGTGGCTGTTG 3′ | ||
| Synapsin 1 | Forward Primer 5′ TGAGGACATCAGTGTCGGGTAA 3′ | 64 |
| Reverse Primer 5′ GGCAATCTGCTCAAGCATAGC 3′ | ||
| Synapsin 2 | Forward Primer 5′ TCCCACTCATTGAGCAGACATACT 3′ | 63 |
| Reverse Primer 5′ GGGAACGTAGGAAGCGTAAGC 3′ | ||
| Synaptobrevin 1 | Forward Primer 5′ TGCTGCCAAGCTAAAAAGGAA 3′ | 68 |
| Reverse Primer 5′ CAGATAGCTCCCAGCATGATCA 3′ | ||
| Synaptobrevin 2 | Forward Primer 5′ CGGAAGAGTCAGTCTCCATTGG 3′ | 64 |
| Reverse Primer 5′ 5′ CACCTGCAGATAATGTCGTGCTA 3′ | ||
| Neurogranin | Forward Primer 5′ CCTCAACACCGGCAATGG 3′ | 61 |
| Reverse Primer 5′ AATATCGTCGTCTGGCTTGGA 3′ | ||
| GAP43 | Forward Primer 5′ CTGAGGAGGAGAAAGACGCTGTA 3′ | 57 |
| Reverse Primer 5′ TCCTGTCGGGCACTTTCC 3′ | ||
| Synaptopodin | Forward Primer 5′ TCCTGCGCCCTGAACCTA 3′ | 70 |
| Reverse Primer 5′ GACGGGCGACAGAGCATAGA 3′ | ||
| Housekeeping Genes | ||
| Beta Actin | Forward Primer ACGGCCAGGTCATCACTATTC | 65 |
| Reverse Primer AGGAAGGCTGGAAAAGAGCC | ||
| GAPDH | Forward Primer TTCCCGTTCAGCTCTGGG | 59 |
| Reverse Primer CCCTGCATCCACTGGTGC | ||
mRNA expression of the genes mentioned above were measured using SYBR-Green chemistry-based quantitative real-time RT-PCR, as previously described in Gutala and Reddy and Reddy et al [40–41]. Briefly, 2 μg of DNAse-treated total RNA was used as the starting material, to which was added 1 μl of oligo (dT), 1 μl of 10 mM dNTPs, 4 μl of 5× first strand buffer, 2 μl of 0.1 M DTT, and 1 μl RNAse out. First, the reagents RNA, dT, and dNTPs were mixed, then heated at 65°C for 5 min, and finally chilled on ice until the remaining components were added. The samples were incubated at 42°C for 2 min, and then 1 μl of Superscript III (40 U/μl) was added. The samples were then incubated at 42°C for 50 min, at which time the reaction was inactivated by heating it at 70°C for 15 min.
Quantitative real-time PCR amplification reactions were performed using RNA from the VDAC1+/+ and VDAC+/− mice in an ABI Prism 7900 sequence detection system (Applied Biosystems), in a 25-μl volume of total reaction mixture. The reaction mixture consisted of 1× PCR buffer containing SYBR-Green; 3 mM MgCl2; 100 nm of each primer; 200 nm of dATP, dGTP, and dCTP each; 400 nm of dUTP; 0.01 U/μl of AmpErase UNG; and 0.05 U/μl of AmpliTaq Gold. A 20 ng cDNA template was added to each reaction mixture.
The CT-values of β-actin and the GAPDH were tested to determine the unregulated endogenous reference gene in VDAC1+/+ and VDAC+/− mice. In the latter case, the CT-value was similar in the VDAC1+/+ and VDAC+/− mice. The CT-value is an important quantitative parameter in real-time PCR analysis [20, 40–41]. All RT-PCR reactions were carried out in triplicate, with no template control. The PCR conditions were: 50°C for 2 min and 95°C for 10 min, followed by 40 cycles at 95°C for 15 sec and at 60°C for 1 min. The fluorescent spectra were recorded during the elongation phase of each PCR cycle. To distinguish specific amplicons from non-specific amplifications, a dissociation curve was generated. The CT-values were calculated with sequence-detection system software V1.7 (Applied Biosystems) and an automatic setting of base line, which was the average value of PCR, cycles 3–15, plus CT generated 10 times its standard deviation. The amplification plots and CT-values were exported from the exponential phase of PCR directly into a Microsoft Excel worksheet for further analysis.
The mRNA transcript level was normalized against β-actin and the GAPDH at each dilution. The standard curve was the normalized mRNA transcript level, plotted against the log-value of the input cDNA concentration at each dilution. To compare β-actin, GAPDH, and neuroprotective markers, relative quantification was performed according to the CT method (Applied Biosystems; [20,40–41]. Briefly, the comparative CT method involved averaging triplicate samples which were taken as the CT values for β-actin, GAPDH, and mitochondrial, synaptic, and AD-related genes. β-actin normalization was used in the present study because β-actin CT values were similar for the mitochondrial, synaptic, and AD-related genes in the VDAC1+/+ and VDAC+/− mice. The ΔCT-value was obtained by subtracting the average β-actin CT value from the average CT-value for the mitochondrial, synaptic, and AD-related genes. The ΔCT of the VDAC+/+ mice was used as the calibrator. Fold change was calculated according to the formula 2−(ΔΔCT), where ΔΔCT is the difference between ΔCT and the ΔCT calibrator value. To determine the statistical significance of mRNA expression between VDAC1+/− and VDAC1+/+, the difference in CT value between VDAC1+/− and VDAC1+/+ was used in relation to the normalization of β-actin, and statistical significance was calculated using one-way ANOVA.
Immunoblotting analysis of mitochondrial permeability transition pore proteins
To determine, if reduced VDAC1 levels in influence MPT pore proteins, VDAC1, ANT and CypD, we performed immunoblotting analyses of protein lysates from VDAC+/+ mice (n=4) and VDAC1+/− mice (n=4). Twenty μg protein lysates were resolved on a 4–12% Nu-PAGE gel (Invitrogen, Grand Island, NY). Theseresolved proteins were transferred to PVDF (Novax Inc, San Diego, CA) and then incubated with a blocking buffer (5% dry milk dissolved in a TBST buffer) for 1 h at room temperature. The nylon membranes were incubated overnight with primaryantibodies of VDAC1 (1:400 rabbit polyclonal, Abcam, Cambridge, MA), ANT (1:200, Mouse Monoclonal, Pierce Biotechnology, INC. Rockford, Il) CypD (1:300, mouse monoclonal, Bellerica, MA) and beta actin (1:500, mouse monoclonal, Sigma-Aldrich, St Luis, MO).
The membranes were washed with a TBST buffer 3 times at 10-min intervals and then incubated for 2 h withappropriate secondary antibodies, followed by 3 additional washes at 10-min intervals. The APP, Tau and VDAC1 proteins weredetected with the Supersignal West Pico chemiluminescent reagent (Thermo Scientific). Scanned images of the exposed X-ray film were analyzed with ImageJ to determine relative band intensity. Quantification was performed on western blots of protein lysates from VDAC1+/+ mice and VDAC1+/− mice using densitometry analysis. Comparisons were made between VDAC1+/+ mice and VDAC1+/− mice.
Mitochondrial Function
H2O2 production
The production of H2O2 in tissues from the cerebral cortex and the cerebellum of VDAC1+/− (n=4) and VDAC1+/+ (n=4) mice were measured with an Amplex® Red H2O2 Assay Kit (Molecular Probes), as previously described [21,42]. Briefly, the production of H2O2 was measured in isolated mitochondria from the cerebral cortex and cerebellum of the VDAC1+/− and VDAC1+/+ mice. A BCA Protein Assay Kit (Pierce Biotechnology) was used to measure protein concentration in a reaction mixture that contained mitochondrial proteins (μg/μl), Amplex Red reagents (50 μM), horseradish peroxidase(0.1 U/ml), and a reaction buffer (1X). The mixture was incubated at room temperature for 30 min, followed by spectrophotometer readings of fluorescence (570 nm). H2O2 production was then determined, using a standard curve equation expressed in nmol/μg mitochondrial protein.
Cytochrome oxidase activity
Cytochrome oxidase activity was measured in mitochondria isolated from the cerebral cortex and cerebellum of VDAC1+/− (n=4) and VDAC1+/+ (n=4) mice, as described elsewhere [21,42]. Enzyme activity was assayed spectrophotometrically with a Sigma Kit (Sigma-Aldrich) following manufacturer’s instructions. Briefly, 2 μg mitochondrial protein was added to 1.1 ml of a reaction solution containing 50 μl 0.22 mM ferricytochrome c fully reduced by sodium hydrosulphide, Tris-HCl at pH 7.0, and 120 mM potassium chloride. The absorbance of wavelength at 550 mM (or its decrease) was recorded in 1-min reactions, at 10-sec intervals. Cytochrome c oxidase activity was measured according to the following formula: mU/mg total mitochondrial protein = (A/min sample − [A/min blank] × 1.1 mL × 21.84). The protein concentrations were determined, following the BCA method.
ATP levels
The levels of ATP were measured in mitochondria that were isolated from cerebral cortex and cerebellum tissues of VDAC1+/− (n=4) and VDAC1+/+ (n=4) mice, with an ATP determination kit (Molecular Probes) [43]. This bioluminescence assay was based on the reaction of ATP with recombinant firefly luciferase and its substract luciferin. Luciferase catalyzes the formation of light from ATP and luciferin. Luciferin is the emitted light that is linearly related to the concentration of ATP, which was measured with a luminometer. ATP was measured from mitochondrial pellets, using a standard curve method.
Lipid peroxidation assay
Lipid peroxidates are unstable indicators of oxidative stress in neurons [44]. 4-hydroxy-2-nonenol (HNE) is the final product of lipid peroxidation. HNE was measured in the cerebral cortex and cerebellum tissues of VDAC1+/− (n=4) and VDAC1+/+ (n=4) mice, with an HNE-His ELISA Kit (Cell BioLabs, Inc. [San Diego, CA]). Briefly, freshly prepared protein was added to a 96-well protein binding plate and incubated overnight at 4°C. It was then washed 3 times with a wash buffer. The washed protein and the anti-HNE-His antibody were added to wells, incubated for 2 h at room temperature, and then washed 3 times. The samples were then incubated with a secondary antibody that was conjugated with peroxidase for 2 h at room temperature. They were then incubated with an enzyme substrate. Optical density was measured to quantify the level of HNE.
Fission-linked GTPase enzymatic activity
Using a Novus Biological calorimetric kit (Littleton, CO), GTPase Drp1 enzymatic activity was measured in the cerebral cortex and cerebellum tissues from VDAC1+/+ (n=4) and VDAC1+/− (n=4) mice, following GTPase assay methods described in Shirendeb et al. [45–46]. The enzymatic activity was based on Drp1 hydrolyzing GTP to GDP and to inorganic phosphorous (Pi). GTPase activity was measured, based on the amount of Pi that the GTP produced. By adding the ColorLock Gold (orange) substrate to the Pi generated from GTP, GTP activity was assessed, based on the inorganic complex solution (green). Calorimetric measurements (green) were read in the wavelength range of 650 nm. GTPase Drp1 activity in cerebral cortex and cerebellum tissues were compared from the VDAC1+/− and VDAC1+/+ mice.
Results
mRNA expression of mitochondrial structural genes
Using mRNA prepared from 2-month-old VDAC1+/− and VDAC1+/+ (wild-type) mice, we determined the effects of reduced VDAC1 on 9 genes: Drp1 and Fis1 (fission); Mfn1, Mfn2, and Opa1 (fusion); VDAC1 (outermembrane); ANT (innermembrane); and CypD and BclXL (matrix genes) (Table 2). We also measured hexokinases 1 and 2, in order to understand the effects of reduced VDAC1 on hexokinases in mice.
Table 2.
mRNA Fold Changes of Mitochondrial, Synaptic and Alzheimer’s Disease-Related Genes in 2-month-old VDAC +/− Mice Relative to VDAC +/+ Mice.
| Genes | mRNA Fold Changes | P Values | |
|---|---|---|---|
| AD-related Genes | APP | −1.3* | 0.01 |
| Tau | −1.4* | 0.03 | |
| BACE 1 | −1.2 | 0.3 | |
| Presenilin 1 | −1.2 | 0.2 | |
| Presenilin 2 | −1.4* | 0.04 | |
| GSK-3α | −1.2 | 0.2 | |
| GSK-3β | −1.3* | 0.04 | |
| Synaptic Genes | Synaptophysin | 1.5* | 0.01 |
| PSD-95 | 1.3 | 0.4 | |
| Synapsin 1 | 1.6* | 0.01 | |
| Synapsin 2 | 1.8* | 0.03 | |
| Synaptobrevin 1 | 1.3* | 0.04 | |
| Synaptobrevin 2 | 1.3* | 0.02 | |
| Neurogranin | 1.7** | 0.003 | |
| GAP43 | 1 | 0.7 | |
| Synaptopodin | 1.2 | 0.18 | |
| Mitochondrial Dynamics Genes | DRP1 | −1.2 | 0.14 |
| FIS1 | −1.2 | 0.35 | |
| Mfn1 | 1.5* | 0.04 | |
| Mfn2 | 1.2 | 0.07 | |
| OPA1 | 1.2 | 0.71 | |
| Cyclophylin D | −1.4* | 0.01 | |
| VDAC | −1.7* | 0.02 | |
| ANT | −1.6* | 0.03 | |
| BCL-XL | 1.0 | 0.83 | |
| Hexokinase 1 | 1.4* | 0.01 | |
| Hexokinase 2 | 1.4* | 0.02 | |
| Mitochondrial-encoded Electron Transport Chain Genes | ND3-Complex I | 1.0 | 0.95 |
| ND6-Complex I | 1.1 | 0.3 | |
| CytB-Complex III | −1.2 | 0.09 | |
| COX1-Complex IV | −1.3* | 0.02 | |
| COX2-Complex IV | −1.2 | 0.06 | |
| ATP6-Complex V | 1.2 | 0.14 | |
In the VDAC1+/− mice compared to the VDAC+/+ mice, mRNA expression levels were decreased in Drp1, by 1.2 fold; and in Fis1, by 1.2 fold, but not significantly so. In contrast, the mRNA expression levels of Mfn1 increased significantly (1.5 fold, P=0.04), as did Mfn2 (1.2 fold, P=0.07) and Opa1 (1.2 fold, P=0.71) in the VDAC1+/− mice compared to the VDAC+/+ mice (Table 3). Thus, the reduction of VADC1 corresponded to a reduction in fission, in the VDAC1+/− mice. Interestingly, mitochondrial permeability transition pore encoded genes were significantly down-regulated in the VDAC1+/− mice relative to the VDAC1+/+ mice: VDAC1, by 1.7 fold (P=02); ANT, by 1.8 fold (P=0.03), and CypD, by 1.4 fold (P=0.01). To understand the involvement of hexokinases in mitochondrial ATP link in the VDAC1+/− mice, we also measured mRNA levels of hexokinase 1 and 2 in the mice. Interestingly, we found significantly increased levels of mRNA for hexokinase 1 (1.3 fold, P=0.01) and hexokinase 2 (1.4 fold, P=0.02) in the VDAC1+/− mice relative to the VDAC1+/+ mice.
mRNA expression of electron transport chain genes
To determine the effects of reduced VDAC1 on mRNA levels of electron transport chain genes, we measured mRNA expression in Complex I (ND3, ND6), Complex III (CytB), Complex IV (COX1 and COX2), and Complex V (ATP6), using cortical tissues from VDAC1+/− and VDAC+/+ mice.
As shown in Table 2, decreased levels of mRNA expressions were found in Complex III (CytB, 1.2 fold, P=0.09) and Complex IV (COX1, 1.2 fold, P=0.02; COX2, 1.3 fold, P =0.06) in the VDAC1+/− mice relative to VDAC1+/+ mice. We also found slightly increased mRNA expression of Complex V ATPase 6 (by 1.2 fold, [but not significant]) in VDAC1+/− mice relative to VDAC+/+ mice.
mRNA expression of AD-related genes
To determine the effects of reduced VDAC1 on AD-related genes, we measured levels of APP, Tau, PS1, PS2, BACE1, GSKβα, and GSKβα using cerebral cortex tissues from VDAC1+/− and VDAC1 +/+ mice.
As shown in Table 2, levels of mRNA expression were significantly decreased for APP (1.3 fold, P=0.01), tau (1.4 fold, P=0.03), BACE1 (1.2 fold, P=0.3 [not significant]), PS1 (1.2 fold, P=0.2 [not significant]), and PS2 (1.4 fold, P=0.04). These results indicate that reduced VDAC1 may be beneficial to AD mice, because reduced expression of AD related genes inhibit apoptotic cell death. We also measured mRNA levels of GSK3α and GSK3β in the VDAC1+/− mice relative to the VDAC+/+ mice and found decreased levels of mRNA for both GSK3β (by 1.3 fold, P=0.04) and GSK3α (1.3 fold, P=0.2 [not significant]).
mRNA expression of synaptic genes
We studied the effects of partially reduced VDAC1 on the synaptic genes synaptophysin, PSD95, synapsin 1, synapsin 2, synaptobrevin 1, synaptobrevin 2, neurogranin, GAP43, and synaptopodin, using mRNA prepared from 2-month-old VDAC1+/− and VDAC1+/+ mice (Table 2).
mRNA expressions were significantly increased for synaptophysin (1.5 fold, P=0.01), synapsin 1 (1.6 fold, P=0.01), synapsin 2 (1.8 fold, P=0.03), synaptobrevin 1 (1.3 fold, P=0.04), synaptobrevin 2 (1.3 fold, P=0.02), and neurogranin (1.7 fold, P=0.003) (Table 2). The levels of mRNA expression were increased, but not significantly, for synaptopodin (1.2 fold, P=0.18) and PSD95 (1.3 fold, P=0.4). mRNA levels were unchanged for GAP43. These findings suggest that reduced VDAC1 is beneficial for maintaining synaptic activity in neurons in these mice.
Immunoblotting analysis of mitochondrial permeability transition pore proteins
To determine, if reduced VDAC1 levels influence MPT pore proteins, VDAC1, ANT and CypD, we conducted immunoblotting analysis of VDAC1, ANT and CypD proteins using protein lysates from VDAC+/+ mice (n=4) and VDAC1+/− mice (n=4). As shown Fig. 1, we found significantly decreased protein levels for VDAC1 (P=0.001) and CypD (P=0.001) in VDAC+/− mice relative to VDAC1+/+ mice, indicating that partial reduction of VDAC1 in mice, reduce protein levels ofVDAC1 and CypD. However, we did not find significant reduction of protein levels for ANT in VDAC1+/− mice relative to VDAC1+/+ mice.
Figure 1.
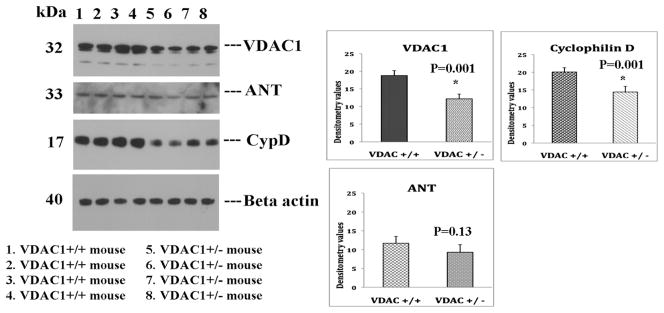
Immunoblotting analysis of mitochondrial permeability transition pore proteins in VDAC1+/+ mice (n=4) and VDAC1+/− (n=4) mice.
Mitochondrial function
To determine whether a reduction in VDAC1 affects mitochondrial function in the VDAC1+/− mice, we characterized mitochondrial function by measuring H2O2 production, cytochrome oxidase activity, lipid peroxidation, and ATP production in cerebral cortex tissues from the VDAC1+/− and VDAC1+/+ mice. We also measured mitochondrial fission-linked GTPase enzymatic activity in the VDAC1+/− and VDAC+/+ mice to determine whether reduced VDAC1 affects mitochondrial fragmentation and mitochondrial dynamics.
H2O2 production
Significantly decreased levels of H2O2 were found in the cerebral cortex tissues from the VDAC1+/− mice relative to the VDAC1 +/+ mice (P<0.05) (Fig. 2A). These findings suggest that reduced VDAC1 influences H2O2 levels in the cortex.
Figure 2.
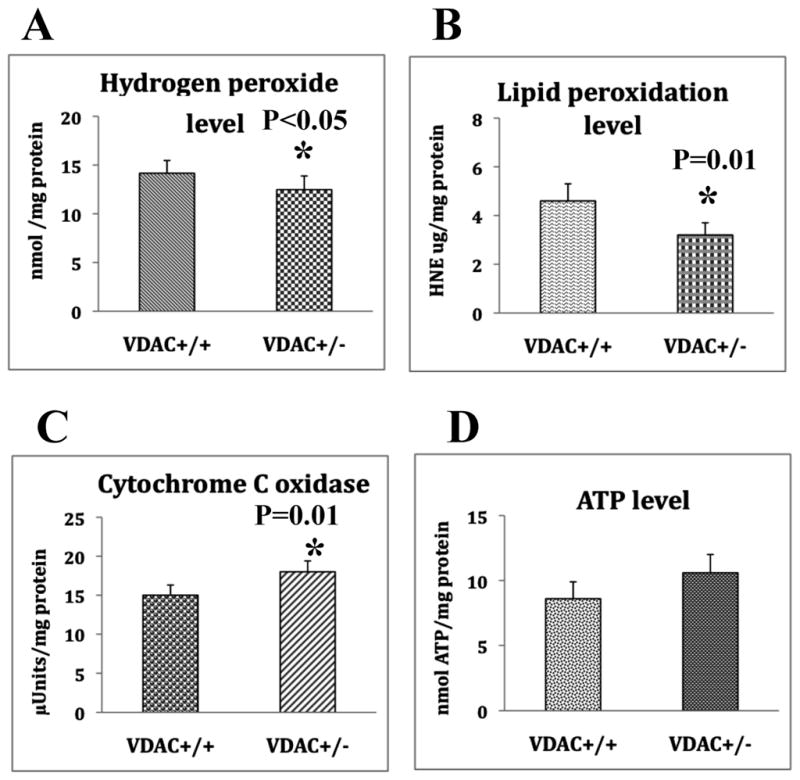
Mitochondrial functional parameters in VDAC1+/+mice (n=4) and VDAC+/− mice (n=4). A represents hydrogen peroxide production, significantly decreased in VDAC1+/− mice relative to VDAC+/+ mice. B. Represents lipid peroxidation levels, significantly decreased in VDAC1+/− mice relative to VDAC+/+ mice. C. Represents cytochrome c oxidase activity, significantly increased in VDAC1+/− mice relative to VDAC+/+ mice. D. Represents ATP levels, significantly increased in VDAC1+/− mice relative to VDAC+/+ mice.
Lipid peroxidation
4-Hydroxy-nonenol, a marker for lipid peroxidation levels, were significantly reduced (P=0.01) in the cerebral cortex tissues from the VDAC1+/− mice relative to the VDAC1+/+ mice (Fig. 2B), indicating that reduced VDAC1 corresponded to reduced lipid peroxidation in the VDAC+/− mice and may likely have some influence on the reduction of lipid peroxidation.
Cytochrome oxidase activity
Cytochrome oxidase activity, an indicator in Complex IV of oxidative phosphorylation, was significantly increased (P=0.01) in the cerebral cortex from the VDAC1+/− mice relative to the VDAC1 +/+ mice (Fig. 2C), indicating that reducedVDAC1 may enhance cytochrome c oxidase activity.
ATP production
Mitochondrial ATP levels were increased in VDAC1+/− mice relative to VDAC1+/+ mice, but not significantly so (Fig. 2D), indicating that reduced VDAC1 may beneficial physiologically to VDAC1+/− mice since ATP levels did not significantly rise.
GTPase Drp1 enzymatic activity
As shown in Fig. 3, GTPase Drp1 enzymatic activity was significantly reduced in the cerebral cortex tissues from both the VDAC1+/− and the VDAC1+/+ mice, indicating that reduced VDAC1 may reduce mitochondrial fragmentation in VDAC+/− mice.
Figure 3.
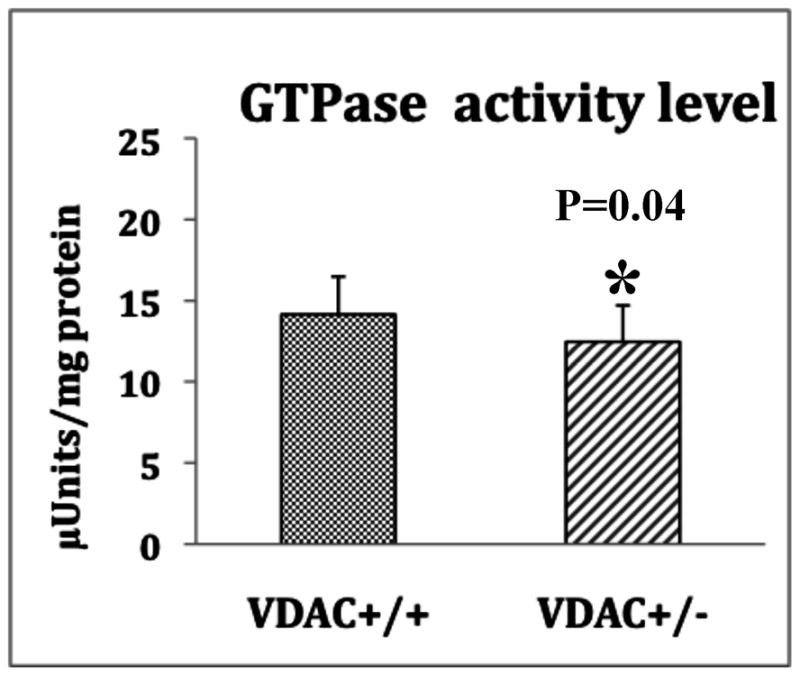
GTPase Drp1 activity in VDAC1+/+ (n=4) and VDAC+/− mice (n=4). Significantly decreased reduced in VDAC1+/− mice relative to VDAC+/+ mice.
Discussion
The purpose of this study was to characterize mitochondrial and synaptic genes and mitochondrial function in VDAC1 heterozygote knockout (VDAC1+/−) mice. This characterization is critical because homozygote VDAC1 knockout (or VDAC1−/−) mice have shown disrupted learning and synaptic plasticity [33], but it was unknown whether problems would be present if VDAC1 were partially reduced. If such problems are not present or are reduced in the VDAC1+/− mice, this result would point to a possible beneficial impact of partially reduced VDAC1 on a disease state such as AD [39] and ALS [47].
The VDAC1+/− mice were normal in terms of lifespan, fertility, and viability, and phenotypically similar to the VDAC1+/+ mice. The VDAC1+/− mice also exhibited reduced mRNA levels of AD related-genes (including BACE1, APP, Tau, PS1, and PS2) and reduced levels of Drp1 and Fis1, ANT, and CypD. These mice exhibited increased levels of mitochondrial fusion genes (Mfn1 and Mfn2) and hexokinase 1 and 2, and their synaptic genes, synaptophysin, synapsin 1 and 2, synaptobrevin 1 and 2, neurogranin, and PSD95 were upregulated. Further, the VDAC1+/− mice exhibited enhanced mitochondrial function.
Taken together, these findings suggest that partial reduction of VDAC1 does not affect mitochondrial and synaptic viability and may be beneficial to the maintenance of mitochondrial function and synaptic activity. Additional studies are needed to further evaluate reduced VDAC1 as a possible therapeutic approach to reduce VDAC1 in persons with AD.
Mitochondrial function in VDAC1 heterozygote knockout mice
One of the objectives of our study was to better understand the link between reduced VDAC1 and mitochondrial function in VDAC+/− mice. We also found H2O2 and lipid peroxidation levels significantly reduced in VDAC+/− mice compared to the VDAC+/+ mice, and cytochrome c oxidase and mitochondrial ATP significantly increased. This combination may be related to reduced VDAC1 since increased VDAC1 expression has been inked to mitochondrial dysfunction and apoptotic cell death [48–52].
Decreased levels of mRNA expressions were found in Complex III and Complex IV in the VDAC1+/− mice relative to VDAC1+/+ mice. Interestingly, an increased level of cytochrome c oxidase activity was observed in VDAC1+/− mice. However, there was no direct correlation between mRNA levels and cytochrome c oxidase activity. It is possible that increased cytochrome oxidase activity may be linked to reduced VDAC1 in VDAC1+/− mice. Further research is needed to determine the direct link between VDAC1 levels and cytochrome c oxidase activity, and how much VDAC1 mRNA expression is necessary to maintain oxidative phosphorylation/mitochondrial function, including cytochrome oxidase activity and ATP production in normal and disease state is an important area of investigation.
We found progressively increased defective mitochondrial function (increased free radicals, lipid peroxidation fission-linked GTPase enzymatic activity and reduced cytochrome oxidase activity and ATP) in AβPP transgenic mice that also showed progressively increased VDAC1 levels [46]. Other researchers found results consistent with our previous and present findings. Increased VDAC1 was found to induce apoptotic cell death in humans and rodents [50–52], and the overexpression of N-terminal VDAC1 stimulated the oligomerization of VDAC1 [44], leading to the release of apoptotic factors, including cytochrome c, Smac/Diablo, and apoptosis-inducing factor and the subsequent apoptosis in cells/mice. All of these findings support the possibility that increased VDAC1 is linked to mitochondrial dysfunction and apoptotic cell death.
Further, the significantly reduced levels of fission-linked GTPase activity in VDAC1+/− mice that we found may be related to the reduction of VDAC1 deficiency in these mice. If so, this finding may suggest that an increase in VDAC1 expression may be related to an increase in GTPase activity, mitochondrial fragmentation, and apoptosis. This possibility is supported by studies using postmortem brain tissues from AD and Huntington disease patients and AD and Huntington transgenic mice, which found increased GTPase activity, excessive mitochondrial fragmentation [21,45,46,53]. The correspondences among increased GTPas activity, mitochondrial fragmentation, and reduced VDAC1 is supported by our real-time RT-PCR data on mitochondrial dynamics: that reduced VDAC1 corresponds to increased mRNA levels of Mfn1 and Mfn2 and decreased mRNA levels of Drp1 and Fis1 in the VDAC1+/− mice.
mRNA levels of AD, mitochondrial and synaptic genes in heterozygote VDAC1 knockout mice
Mitochondrial porins are involved in multiple cellular functions, including the regulation of mitochondrial ATP and calcium flux, learning, and synaptic plasticity [38]. Homozygote VDAC1 knockout mice did show disrupted spatial learning, deficits in long and short-term synaptic plasticity, and problems with fertility. However, heterozygote knockout VDAC1 mice were viable and fertile, and had a normal lifespan, without any learning or synaptic plasticity problems. However, there have been no published reports of investigations into synapses and mitochondria, none on AD-related genes and their expressions. Interestingly, our finding that reduced VDAC1 did not affect mRNA expressions of electron transport chain suggests that reduced VDAC1 may reduce mitochondrial fragmentation, and maintain mitochondrial pore opening and closure.
Our findings of significantly reduced mRNA levels of AD-related genes in the VDAC1+/− mice suggest that a reduction in VDAC1 in early disease progression may suppress the expression of AD-related genes, and this suppression may in turn inhibit apoptotic cell death in neurons. Because the overexpression of APP, PS1, PS2, Tau and BACE1 promotes apoptotic cell death via mitochondria. Therefore, reduced VDAC1 may be beneficial to slow AD progression.
The significantly increased mRNA levels that we found for the synaptic genes (Table 2) in the VDAC1+/− mice suggest that reduced VDAC1 is beneficial for synaptic activity in neurons from mice. Findings from our current study support our previous study using AβPP transgenic mice, in which increased VDAC1 levels correlated with reduced synaptic and mitochondrial activity at different stages in disease progression [20,21,46]. Our current findings also suggest that reduced VDAC1 is beneficial to neurons not only in a diseased state but also to synaptic activity in general. Needed is additional research into the extent that reduced VDAC1 can protect synaptic or neuronal damage in the presence of Aβ and phosphorylated tau.
In summary, we found that VDAC1+/− mice, noteworthy for their reduced levels of VDAC1 in comparison to VDAC1+/+ mice, showed improved mitochondrial function and synaptic activity and reduced expressions of several AD related genes.
Acknowledgments
This research was supported by NIH grants AG028072, AG042178, and RR000163, and a grant from the Medical Research Foundation of Oregon.
References
- 1.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 2.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 3.LaFerla FM, Green KN, Oddo S. Intracellular amyloid-beta in Alzheimer’s disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 4.Reddy PH, Beal MF. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol Med. 2008;14:45–53. doi: 10.1016/j.molmed.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reddy PH, Tripathi R, Troung Q, Tirumala K, Reddy TP, Anekonda V, Shirendeb UP, Calkins MJ, Reddy AP, Mao P, Manczak M. Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer’s disease: Implications to mitochondria-targeted antioxidant therapeutics. Biochim Biophys Acta. 2012;1822:639–649. doi: 10.1016/j.bbadis.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swerdlow RH. Brain aging, Alzheimer’s disease, and mitochondria. Biochim Biophys Acta. 2011;1812:1630–1639. doi: 10.1016/j.bbadis.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reddy PH, McWeeney S, Park BS, Manczak M, Gutala RV, Partovi D, Jung Y, Yau V, Searles R, Mori M, Quinn J. Gene expression profiles of transcripts in amyloid precursor protein transgenic mice: up-regulation of mitochondrial metabolism and apoptotic genes is an early cellular change in Alzheimer’s disease. Hum Mol Genet. 2004;13:1225–1240. doi: 10.1093/hmg/ddh140. [DOI] [PubMed] [Google Scholar]
- 8.Manczak M, Park BS, Jung Y, Reddy PH. Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: implications for early mitochondrial dysfunction and oxidative damage. Neuromolecular Med. 2004;5:147–162. doi: 10.1385/NMM:5:2:147. [DOI] [PubMed] [Google Scholar]
- 9.Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc Natl Acad Sci U S A. 2010;107:18670–18675. doi: 10.1073/pnas.1006586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Calkins MJ, Manczak M, Mao P, Shirendeb UP, Reddy PH. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum Mol Genet. 2011;20:4515–4529. doi: 10.1093/hmg/ddr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu X, Perry G, Smith MA, Wang X. Abnormal Mitochondrial Dynamics in the Pathogenesis of Alzheimer’s Disease. J Alzheimers Dis. 2013;33:S253–S562. doi: 10.3233/JAD-2012-129005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J Neurosci. 2006;26:9057–9068. doi: 10.1523/JNEUROSCI.1469-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Parker WD, Jr, Filley CM, Parks JK. Cytochrome oxidase deficiency in Alzheimer’s disease. Neurology. 1990;40:1302–1303. doi: 10.1212/wnl.40.8.1302. [DOI] [PubMed] [Google Scholar]
- 14.Maurer I, Zierz S, Möller HJ. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol Aging. 2000;21:455–462. doi: 10.1016/s0197-4580(00)00112-3. [DOI] [PubMed] [Google Scholar]
- 15.Smith MA, Perry G, Richey PL, Sayre LM, Anderson VE, Beal MF, Kowall N. Oxidative damage in Alzheimer’s. Nature. 1996;382:120–121. doi: 10.1038/382120b0. [DOI] [PubMed] [Google Scholar]
- 16.Wang J, Xiong S, Xie C, Markesber WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J Neurochem. 2005;93:953–962. doi: 10.1111/j.1471-4159.2005.03053.x. [DOI] [PubMed] [Google Scholar]
- 17.Sultana R, Boyd-Kimball D, Cai J, Pierce WM, Klein JB, Merchant M, Butterfield DA. Proteomics analysis of the Alzheimer’s disease hippocampal proteome. J Alzheimers Dis. 2007;11:153–164. doi: 10.3233/jad-2007-11203. [DOI] [PubMed] [Google Scholar]
- 18.Du H, Guo L, Yan SS. Synaptic mitochondrial pathology in Alzheimer’s disease. Antioxid Redox Signal. 2012;11:1467–1475. doi: 10.1089/ars.2011.4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manczak M, Anekonda TS, Henson E, Park BS, Quinn J, Reddy PH. Mitochondria are a direct site of A beta accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum Mol Genet. 2006;15:1437–1449. doi: 10.1093/hmg/ddl066. [DOI] [PubMed] [Google Scholar]
- 20.Manczak M, Calkins MJ, Reddy PH. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum Mol Genet. 2011;20:2495–2509. doi: 10.1093/hmg/ddr139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manczak M, Reddy PH. Abnormal interaction of VDAC1 with amyloid beta and phosphorylated tau in Alzheimer’s disease neurons cause mitochondrial dysfunction. Hum Mol Genet. 2012;21:5131–5146. doi: 10.1093/hmg/dds360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2009;106:14670–14675. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Caspersen C, Wang CN, Yao J, Sosunov A, Chen X, Lustbader JW, Xu HW, Stern D, McKhann G, Yan SS. Mitochondrial Abeta: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005;19:2040–1. doi: 10.1096/fj.05-3735fje. [DOI] [PubMed] [Google Scholar]
- 24.Devi L, Ohno M. Mitochondrial dysfunction and accumulation of the β-secretase-cleaved C-terminal fragment of APP in Alzheimer’s disease transgenic mice. Neurobiol Dis. 2012;45:417–424. doi: 10.1016/j.nbd.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crouch IJ, Blake R, Duce JA, Ciccotosto CD, Li QX, Barnham KJ, Curtain CC, Cherny RA, Cappai R, Dyrks T, Masters CL, Trounce IA. Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-beta1–42. J Neurosci. 2005;25:672–679. doi: 10.1523/JNEUROSCI.4276-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, Casadesus G, Zhu X. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A. 2008;105:19318–19323. doi: 10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, Zhu X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci. 2009;29:9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reddy PH, Reddy TP. Mitochondria as a therapeutic target for aging and neurodegenerative diseases. Curr Alzheimer Res. 2011;8:393–409. doi: 10.2174/156720511795745401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hodge T, Colombini M. Regulation of metabolite flux through voltage-gating of VDAC channels. J Membr Biol. 157:271–279. doi: 10.1007/s002329900235. [DOI] [PubMed] [Google Scholar]
- 30.Rostovtseva T, Colombini M. VDAC channels mediate and gate the flow of ATP: implications for the regulation of mitochondrial function. Biophys J. 72:1954–1962. doi: 10.1016/S0006-3495(97)78841-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–555. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Colombini M. VDAC structure, selectivity, and dynamics. Biochim Biophys Acta. 2012;1818:1457–1465. doi: 10.1016/j.bbamem.2011.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med. 2008;14:1097–1105. doi: 10.1038/nm.1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Du H, Guo L, Wu X, Sosunov AA, McKhann GM, Chen JX, Yan SS. Cyclophilin D deficiency rescues Aβ-impaired PKA/CREB signaling and alleviates synaptic degeneration. Biochim Biophys Acta. 2013 Mar 16; doi: 10.1016/j.bbadis.2013.03.004. S0925-4439(13)00078-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo L, Du H, Yan S, Wu X, McKhann GM, Chen JX, Yan SS. Cyclophilin D deficiency rescues axonal mitochondrial transport in Alzheimer’s neurons. PLoS One. 2013;8:e54914. doi: 10.1371/journal.pone.0054914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raghavan A, Sheiko T, Graham BH, Craigen WJ. Voltage-dependant anion channels: Novel insights into isoform function through genetic models. Biochim Biophys Acta. 2012;1818:1477–1485. doi: 10.1016/j.bbamem.2011.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shimizu S, Narita M, Tsujimoto Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature. 1999;399:483–487. doi: 10.1038/20959. [DOI] [PubMed] [Google Scholar]
- 38.Weeber EJ, Levy M, Sampson MJ, Anflous K, Armstrong DL, Brown SE, Sweatt JD, Craigen WJ. The role of mitochondrial porins and the permeability transition pore in learning and synaptic plasticity. J Biol Chem. 2002;277:18891–18897. doi: 10.1074/jbc.M201649200. [DOI] [PubMed] [Google Scholar]
- 39.Reddy PH. Is the mitochondrial outermembrane protein VDAC1 therapeutic target for Alzheimer’s disease? Biochim Biophys Acta. 2013;1832:67–75. doi: 10.1016/j.bbadis.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gutala RV, Reddy PH. The use of real-time PCR analysis in a gene expression study of Alzheimer’s disease post-mortem brains. J Neurosci Methods. 2004;132:101–107. doi: 10.1016/j.jneumeth.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 41.Reddy TP, Manczak M, Calkins MJ, Mao P, Reddy AP, Shirendeb U, Park B, Reddy PH. Toxicity of neurons treated with herbicides and neuroprotection by mitochondria-targeted antioxidant SS 31. Int J Environ Res Public Health. 2011;8:203–221. doi: 10.3390/ijerph8010203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Manczak M, Mao P, Calkins MJ, Cornea A, Reddy AP, Murphy MP, Szeto HH, Park B, Reddy PH. Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in Alzheimer’s disease neurons. J Alzheimers Dis. 2010;20:S609–S631. doi: 10.3233/JAD-2010-100564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Drew B, Leeuwenburgh C. Method for measuring ATP production in isolated mitochondria: ATP production in brain and liver mitochondria of Fischer-344 rats with age and caloric restriction. Am J Physiol Regul Integr Comp Physiol. 2003;285:R1259–R1267. doi: 10.1152/ajpregu.00264.2003. [DOI] [PubMed] [Google Scholar]
- 44.Hall ED, Bosken JM. Measurement of oxygen radicals and lipid peroxidation in neural tissues. Curr Protoc Neurosci. 2009;Chapter 7(Unit 7. 17):1–51. doi: 10.1002/0471142301.ns0717s48. [DOI] [PubMed] [Google Scholar]
- 45.Shirendeb UP, Calkins MJ, Manczak M, Anekonda V, Dufour B, McBride JL, Mao P, Reddy PH. Mutant huntingtin’s interaction with mitochondrial protein Drp1 impairs mitochondrial biogenesis and causes defective axonal transport and synaptic degeneration in Huntington’s disease. Hum Mol Genet. 2012;21:406–420. doi: 10.1093/hmg/ddr475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Manczak M, Reddy PH. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum Mol Genet. 2012;21:2538–2547. doi: 10.1093/hmg/dds072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Israelson A, Arbel N, Da Cruz S, Ilieva H, Yamanaka K, Shoshan-Barmatz V, Cleveland DW. Misfolded mutant SOD1 directly inhibits VDAC1 conductance in a mouse model of inherited ALS. Neuron. 2010;67:575–587. doi: 10.1016/j.neuron.2010.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shoshan-Barmatz V, Keinan N, Zaid H. Uncovering the role of VDAC in the regulation of cell life and death. J Bioenerg Biomembr. 2008;40:183–191. doi: 10.1007/s10863-008-9147-9. [DOI] [PubMed] [Google Scholar]
- 49.Shoshan-Barmatz V, De Pinto V, Zweckstetter M, Raviv Z, Keinan N, Arbel N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol Aspects Med. 31:227–285. doi: 10.1016/j.mam.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 50.Abu-Hamad S, Zaid H, Israelson A, Nahon E, Shoshan-Barmatz V. Hexokinase-I protection against apoptotic cell death is mediated via interaction with the voltage-dependent anion channel-1: mapping the site of binding. J Biol Chem. 2008;283:13482–13490. doi: 10.1074/jbc.M708216200. [DOI] [PubMed] [Google Scholar]
- 51.Ghosh T, Pandey N, Maitra A, Brahmachari SK, Pillai B. A role for voltage-dependent anion channel Vdac1 in polyglutamine-mediated neuronal cell death. PLoS One. 2007;2:e1170. doi: 10.1371/journal.pone.0001170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zaid H, Abu-Hamad S, Israelson A, Nathan I, Shoshan-Barmatz V. The voltage-dependent anion channel-1 modulates apoptotic cell death. Cell Death Differ. 2005;12:751–760. doi: 10.1038/sj.cdd.4401599. [DOI] [PubMed] [Google Scholar]
- 53.Shirendeb U, Reddy AP, Manczak M, Calkins MJ, Mao P, Tagle DA, Reddy PH. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: implications for selective neuronal damage. Hum Mol Genet. 2011;20:1438–1455. doi: 10.1093/hmg/ddr024. [DOI] [PMC free article] [PubMed] [Google Scholar]