

Abstract
Periodate oxidation followed by borohydride reduction converts the well-known antithrombotics heparin and low-molecular weight heparins (LMWHs) into their “glycol-split” (gs) derivatives of the “reduced oxyheparin” (RO) type, some of which are currently being developed as potential anti-cancer and anti-inflammatory drugs. Whereas the structure of gs-heparins has been recently studied, details of the more complex and more bioavailable gs-LMWHs have not been yet reported. We obtained RO derivatives of the three most common LMWHs (tinzaparin, enoxaparin, and dalteparin) and studied their structures by two-dimensional nuclear magnetic resonance (2D NMR) spectroscopy and ion-pair reversed-phase high-performance liquid chromatography (IPRP-HPLC) coupled with electrospray ionization mass spectrometry (ESI-MS). The LC-MS analysis was extended to their heparinase-generated oligosaccharides. The combined NMR/LC-MS analysis of RO-LMWHs provided evidence for glycol-splitting-induced transformations mainly involving internal nonsulfated glucuronic and iduronic acid residues (including partial hydrolysis with formation of “remnants”), and for the hydrolysis of the gs uronic acid residues when formed at the non-reducing ends (mainly, in RO-dalteparin). Evidence for minor modifications, such as ring contraction of some dalteparin internal aminosugar residues was also obtained. Unexpectedly, the N-sulfated 1,6-anhydro-mannosamine residues at the enoxaparin reducing end were found to be susceptible to the periodate oxidation. In addition, in tinzaparin and enoxaparin the borohydride reduction converts the hemiacetalic aminosugars at the reducing end to alditols. Typical LC-MS signatures of RO-derivatives of individual LMWH both before and after digestion with heparinases included oligosaccharides generated from the original antithrombin-binding and “linkage” regions.
Keywords: Low-molecular weight heparins, Glycol-splitting, Nuclear Magnetic Resonance, Liquid chromatography, Mass spectrometry
Introduction
Low-molecular weight heparins (LMWHs), obtained by chemical or enzymatic depolymerisation of heparin, are widely used as antithrombotic drugs [1]. They have characteristic terminal residues [2], but their linear internal sequences, like heparin, are primarily 1,4-linked trisulfated disaccharide (TSD) repeating units, containing 2-O-sulfated iduronic acid (I2S) and N-sulfate-glucosamine-6-O-sulfate (ANS,6S). These regular “fully sulfated” regions are interspersed by undersulfated sequences bearing nonsulfated iduronic (I) and, in a somewhat higher proportion, glucuronic (G) acids, which are usually preceded by N-acetylated glucosamine residues (ANAc). The specific pentasaccharide sequence N-acetyl-D-glucosamine-6-O-sulfate α1→4 D-glucuronic acid β1→4 D-glucosamine N,3-O,6-O-trisulfate α1→4 L-iduronic acid 2-O-sulfate α1→4 D-glucosamine N,6-O-disulfate (ANAc,6S-G-ANS,3S,6S-I2S-ANS,6S), present only in some of the chains [3], constitutes the antithrombin-binding region (ATBR), essential for a high anticoagulant and antithrombotic activity of both unfractionated heparins and LMWHs.
In the last decade, several new therapeutic uses of heparins, LMWHs, and their derivatives have been envisaged in various fields [4], especially in inflammation and cancer [5–7]. To reduce risks of bleeding in non-anticoagulant/non-antithrombotic uses of heparins and LMWHs, removal or inactivation of their ATBR is required. Although removal of the ATBR-containing chains could be achieved exploiting their interaction with AT [8], a more practical way of reducing anticoagulant properties, without impairing other biological activities, is periodate oxidation of heparins [9,10]. Periodate oxidation is a classical reaction in carbohydrate chemistry that has been widely applied to determine vicinal carbons bearing unsubstituted hydroxyl and/or amino groups [11]. When applied for heparins, periodate oxidation mainly leads to the splitting of the C(2)-C(3) bond of nonsulfated uronic acids, including the essential glucuronic acid within the ATBR [12,13]. The primary products of the glycol-splitting reaction (the polyaldehydic oxy-heparins) are usually stabilized by borohydride reduction, leading to reduced oxy-heparins (RO-heparins) [9,13]. Under controlled conditions, periodate oxidation can be performed with limited cleavage of glycosidic bonds, i.e., without significant depolymerisation at the level of glycol-split (gs) residues.
The glycol-splitting reaction usually does not impair the biological activities of heparin chains that are not critically dependent on the intact structure of the AT-binding sequence. In fact, most of the literature data indicate that the glycol-splitting gives rise to a higher protein-binding activity of gs-heparins than their parent GAGs [14], most probably due to the increased local mobility of gs residues, which act as flexible joints along the polysaccharide chains, thus, facilitating interactions with the heparin/HS binding sites of proteins [7,9]. A number of non-anticoagulant gs-heparins, differing for the molecular weight and the extent of glycol-splitting, are being considered as potential antilipemic, platelet interaction-inhibiting, antithrombotic, antiangiogenic, antimetastatic, antimalarial, labor-reducing in obstetrics, and anticancer drugs [7,9,14–21]. Some of these potential drugs under development (such as, M402 [21]) were obtained from LMWHs.
The structures of a heparin [22] and a glycol-split heparin chain are depicted in Fig. 1, which also includes the “linkage region” (LR, structure 1c), i.e., the nonsulfated sequence G-Gal-Gal-Xyl, which links the carbohydrate chains with the core peptide chain of its natural proteoglycan precursor (“macromolecular heparin”) through a serine (Ser) residue [23]. The LR sequence is usually present in commercial heparins [23], however, its content and structure depends on the strength of oxidative “bleaching” treatment. Usually a few percents of intact or partially modified LR remain in heparins and in some LMWHs. G and Xyl residues, bearing vicinal hydroxyl groups, can be split. (Fig. 1d).
Fig. 1.

Simplified formula of a representative ATBR-containing chain of porcine mucosal heparin (a) and the corresponding glycol-split derivative (b) obtained by periodate oxidation and borohydride reduction of nonsulfated glucuronic (G/G′) and iduronic (I) residues, to generate gsG/gsG′, and gsI [13]
LR = linkage region [23] at the “reducing” end (RE). The biochemical formula of ATBR is indicated in structure (a), starting with a residue I that is not part of the pentasaccharidic active site, but most often precedes it. Structure (a) is adapted from [22]; a major modification involves the ANS,6S residue at the non-reducing end (NRE), assumed to be the consequence of cleavage (by an endo-β-D-glucosidase) of a G-ANS6S glycosidic bond in the chains of the heparin proteoglycan precursor (“macromolecular heparin”). The structure of the “full” linkage region LR is reported in (c) and (d) for heparin and RO-heparin, respectively, where GLR is the G residue in the LR, Gal1 and Gal2 are two galactose residues, Xyl is xylose, Ser is serine [23]. The two glycol-split residues (gsGLR and gsXyl) in the LR of gs-heparin were not previously described (see text). Circled residues in the unmodified heparin structure (a) and (c) highlight unsubstituted diol groups susceptible of glycol-splitting
The structures of LMWHs are even more complex than those of their parent heparins, since both chemical and enzymatic depolymerisation of the original polysaccharide modifies the heparin sequences, at least at the site of chain cleavage, making each LMWH structurally (Fig. 2), and, to some extent biologically, different from each other [24]. Glycol-splitting and reduction reactions are expected to introduce further local modifications in LMWHs.
Fig. 2.

Simplified structures of tinzaparin, enoxaparin, and dalteparin
Internal sequences are indicated as substantially the same as in non-depolymerized heparin. The end groups susceptible of being modified by periodate oxidation or borohydride reduction are circled
Pharmaceutical development of drugs of this type requires setting up appropriate analytical methods to control and guarantee preparation reproducibility and to establish correlations between structure and biological activity. Structural characterization of heparin species largely relies on NMR and/or LC-MS analysis, with the latter most often performed on fragments [25]. Significant advances in the analysis of heparin/heparan sulfate di- and oligosaccharides have been achieved by coupling ion-pair reversed-phase high-performance liquid chromatography (IPRP-HPLC) with electrospray ionization mass spectrometry (ESI-MS) [13,26–30]. However, as with heparins, one major problem in the analysis of gs-heparins is associated with their structural microheterogeneity, which can be unravelled only through careful cleavage of their chains and identification of the generated di- and oligosaccharide fragments. Glycol-splitting followed by enzymatic digestion with a cocktail of heparinases (heparin lyases I, II and/or III) has been recently used for structural characterization of heparins of different origins [13]. Heparin lyases preferentially cleave glycosidic bonds of heparin unmodified sequences, “skipping” disaccharide units containing gs-residues and thus generating oligosaccharides with incorporated gsG/gsI residues.
In the present study we obtained RO-derivatives of the three most common commercially available LMWHs (tinzaparin, enoxaparin, and dalteparin) and characterized their structures by heteronuclear single-quantum coherence (HSQC) NMR spectroscopy and IPRP-HPLC coupled with electrospray ionization quadrupole time-of-flight mass spectrometry (ESI-Q-TOF-MS). The resistance of oligosaccharides containing gs residues to heparinases was exploited to unravel the problem of the structural microheterogeneity and to obtain more structural information on unmodified heparin sequences.
Experimental
Reagents and starting materials
Low-molecular weight heparins were samples of commercial formulated or bulk materials of: tinzaparin sodium (Innohep, LEO Pharma, Denmark), enoxaparin sodium (Clexane, Sanofi Aventis, Italy) and dalteparin sodium (Fragmin, Pfizer, Italy), N-acetyl heparosan is a gift of K. Jann [31], synthetic pentasaccharide Arixtra® (fondaparinux, GSK, France). Heparin lyases I (EC4.2.2.7), II and III (EC4.2.2.8) were purchased from Grampian Enzymes, UK. Sodium periodate (>99%), dibutylamine (>99.5%), methanol (LC-MS grade), acetonitrile (LC-MS grade), acetic acid (glacial, 99.9%), formic acid (98–100%), ammonium chloride (> 99.5%) were purchased from Sigma-Aldrich, sodium borohydride (95%), from Riedel-de Haën, sodium acetate, from Merck, calcium acetate (>97%), from BDH, ethylenediaminetetraacetic acid (EDTA, >98%), from Fluka. Deionized (conductivity less than 0.06 μS) and filtered (Millipore filter 0.22 μm) water was used for sample dilution and for mobile phases.
Preparation of RO-derivatives
Glycol-split derivatives of tinzaparin, enoxaparin, dalteparin and N-acetyl heparosan (K5 polysaccharide) were prepared using the following procedure: about 40 – 50 mg of each sample were dissolved in 1.5 ml of H2O, and 1.5 ml of 0.2 M NaIO4 were added. The reaction mixture was left for 20 h at 4°C in the dark under stirring to avoid unspecific oxidation. The excess of periodate was neutralized by adding 200 μl of ethylene glycol maintaining the stirring for a further hour at 4°C. Solid sodium borohydride (~ 70 mg) was added in portions to the reaction mixture and after 16 h at 4°C in the dark under stirring, the pH value was adjusted under stirring at 4°C to 7 with 5% (v/v) acetic acid. The solutions containing the gs-derivatives were desalted on a Sephadex G-10 column (80 x 2.5 cm) using water – ethanol 9:1 (v/v) as a mobile phase at 2.6 ml/min, monitoring the absorbance at 210 nm (Cary 50 UV-Vis spectrophotometer, Varian). After freeze drying, RO-samples were obtained with yields in the range of 85–92 %.
Molecular weight determination
Molecular weight determinations were performed by HP-SEC-TDA on a Viscotek (Houston, Texas) instrument equipped with a VE1121 pump, Rheodyne valve (100 μl), and triple detector array 302 equipped with refraction index (RI), viscometer, and light-scattering (90° and 7°) systems, as described in [13]. Weight-average mean molecular mass (MW) for RO-tinzaparin, RO-enoxaparin and RO-dalteparin were found to be 7.2, 4.7 and 6.4 kDa, respectively, somewhat lower than Mw values obtained for the parent LMWHs (7.9, 5.2 and 7.5 kDa, respectively).
Digestion with heparinases
The original and RO-LMWHs (100 μg) were enzymatically depolymerised by heparin lyases I, II, III. The substrate (5–10 mg) was dissolved in water to obtain a 20 mg/ml solution. then 149 μl of solution containing 50 mM sodium acetate and 5 mM calcium acetate, and 3 μl of heparin lyases mixture (1 μl of each lyase, 2 mU/μl enzyme solution) were added to 5 μl of the substrate solution. The reaction was stirred at 37°C (Termo shaker TS-100 Biosan) for 24 h, then, stopped by adding 3 μl of 3% formic acid. Each sample was diluted two times with water and analyzed by LC-MS.
RO-enoxaparin was digested with heparinases I, II and III also in a preparative scale for further fractionation and detailed studies by LC-MS and NMR: 175 μl of 2 mU/μl solutions of each enzyme were added to a RO-enoxaparin solution (37 mg in 50 ml of a buffer containing 50 mM sodium acetate buffer and 5 mM calcium acetate). The digestion was carried out for 24 h at 37°C, and after the second addition of the enzymes (175 μl of 2 mU/μl solutions of each enzyme), the reaction was maintained for further 24 h at 37°C, then, stopped by adding 1 ml of 3% formic acid. The reaction mixture was filtered using Millipore filter 3 μm and concentrated up to 2 ml for SEC fractionation (see next section) and further detailed LC-MS and NMR analysis.
Preparative size-exclusion chromatography
HPLC system (Bioline Knauer S1050) was used for the size fractionation of the heparinase-digested RO-enoxaparin. 2 ml of the concentrated solution (see the previous section) were loaded onto the column (Superdex S30, 84 x 2 cm) and eluted with 0.25 M ammonium chloride at a flow-rate 5.0 ml/min. Based on the detection at 210 nm, seven fractions were obtained and, then, desalted on the Biofox 40/100 SEC Agarose column (89 x 3 cm) using water – ethanol 9:1 (v/v) as a mobile phase delivered at a flow-rate 15 ml/min. After concentration and freeze-drying each gs-oligosaccharide fraction was analyzed by 2D NMR and LC-MS.
Sodium borohydride reduction of the digested RO-enoxaparin fractions
The reduction of the SEC fractions of the digested RO-enoxaparin (see the previous section) were performed by adding of 5 μl of 30 mg/ml solution of sodium borohydride into an aliquot (50 μl) of 0.5 mg/ml solution of each fraction. The mixture was left at room temperature for 4 h and, then, analyzed by LC-MS.
NMR analysis
The samples (5 – 8 mg for LMWHs and RO-LMWHs, 1 – 2 mg for the isolated fractions of heparinase digest of RO-enoxaparin) were dissolved in 500 μl of D2O, added of 100 μl of 10 mM solution of EDTA in D2O, then freeze dried. 500 μl of D2O were added to each lyophilized sample and the procedure of lyophilization was repeated. Spectra of the original and RO-LMWHs were recorded at 25°C on a Bruker Avance 500 MHz spectrometer (Karlsruhe, Germany), except for the fractions of the heparinase-digested RO-enoxaparin for which the NMR spectra were acquired on a Bruker Avance 600 MHz spectrometer (Karlsruhe, Germany). Both instruments were equipped with 5-mm TCI cryoprobe. Integration of peak volumes in the 2D-HSQC spectra was made using standard Bruker TOPSPIN 3.0 software. The relative content of monosaccharide residues was calculated from the corresponding anomeric cross-peaks identified by HSQC following the procedure previously applied to heparins [32] and to LMWHs [2]. HSQC spectra were obtained in phase-sensitivity enhanced pure-absorption mode with decoupling in the acquisition period (Bruker puse program hsqcetgpsisp.2). Spectra were recorded using a spectral width of 8 ppm and 80 ppm in the proton and carbon dimensions, respectively. The carrier frequencies for proton and carbon were 4.7 ppm and 80 ppm. Spectra were acquired into a time domain of 1024 complex points, using from 16 to 32 scans for each of 320 increments. The repetition time and the acquisition time were set at 2s and 0.178s, respectively, with a total measurements ranging from 3h:05min up to 6h:10min. The matrix size of 1024 × 320 data points was zero lled to 4096 × 2048 by application of a squared cosine function prior to Fourier transformation. 2D-TOCSY spectra was acquired using 32 scans per series of 2048 × 512 data points and a mixing time of 80ms. A zero lling in F1 (4096 × 2048) and a shifted (π/3) squared cosine function was applied prior to Fourier transformation.
LC-MS analysis of the unmodified and RO-LMWHs and their corresponding heparinase digests
LC-MS analysis of all samples was performed on an Ultimate 3000 HPLC-UV system (Dionex) coupled to an ESI-QTOF mass spectrometer MicrOTOF-Q (Bruker Daltonics, Germany). Ion-pair reversed-phase separation was carried out on a Kinetex-C18 column (2.1 x 100 mm, ODS 2.6 μm, 100 Å, Phenomenex) with Security Guard Cartridges Gemini C18 (4 x 2.0 mm, Phenomenex). A binary solvent system was used for gradient elution.
LC-MS analysis of the heparinase-digested original and RO-LMWHs was performed as previously described in [13]. For profiling the intact unmodified LMWHs and RO-LMWHs, methanol in the mobile phases, used for the analysis of the enzymatic digests [13], was substituted by acetonitrile. This substitution led to a decrease in column backpressure and, consequently, permitted an increase in the flow-rate (from 100 up to 250 μl/min), which resulted in a decrease of total analysis time, and, more important, improved chromatographic performance. Thus, for profiling the non-digested original and RO-LMWHs, mobile phases A (10 mM dibutylamine, 10 mM acetic acid in water – acetonitrile (9:1)) and B (10 mM dibutylamine and 10 mM acetic acid in acetonitrile), delivered at 0.25 ml/min, were used. Because enoxaparin, tinzaparin and dalteparin are characterized by a different molecular weight distribution, slightly different multistep gradients were used for their profiling (Table 1). Under the applied conditions the retention times vary in a range 1.0 – 1.5 %.
Table 1.
Gradient used and the LMWH quantity loaded for the LC-MS profiling of the LMWHs and their RO-derivatives
| Enoxaparin and RO-enoxaparin | Tinzaparin and RO-tinzaparin | Dalteparin and RO-dalteparin | |||
|---|---|---|---|---|---|
|
| |||||
| Multistep gradient
| |||||
| Time (min) | % Eluent B | Time (min) | % Eluent B | Time (min) | % Eluent B |
|
| |||||
| 0 | 10 | ||||
| 0 | 0 | 0 | 5 | 4 | 10 |
| 5 | 0 | 5 | 5 | 15 | 18 |
| 130 | 35 | 125 | 35 | 30 | 18 |
| 140 | 70 | 130 | 70 | 115 | 35 |
| 145 | 70 | 135 | 70 | 133 | 70 |
| 148 | 0 | 138 | 5 | 138 | 70 |
| 170 | 0 | 160 | 5 | 140 | 10 |
| 160 | 10 | ||||
|
| |||||
| Sample quantity loaded, μg | |||||
|
| |||||
| 15 | 15 | 30 | 30 | 30 | 30 |
|
| |||||
| Corresponding Figure
| |||||
|
| |||||
| Fig. 4 and ESM Fig. S5 | ESM Fig. S6 | ESM Fig. S7 | |||
LC conditions: column C18 100 x 2.1 mm, 2.6 μm; eluents A and B – 10 mM DBA and 10 mM CH3COOH in H2O-CH3CN = 9:1 (v/v) and CH3CN, respectively; flow rate – 0.25 ml/min
The MS spectrometric conditions were as follows: ESI in negative ion mode, drying gas temperature +180°C, drying gas flow-rate 7.0 l/min, nebulizer pressure 0.9 bar, capillary voltage +3.2 kV. The mass spectra of the oligosaccharides were acquired in a scan mode (m/z scan range 200 – 2000). The fragmentation of the ion with m/z 488.5, present in the digested RO-enoxaparin, was performed by applying LC-ESI-MS2 method. Collision-induced dissociation (CID) was used, the isolation window was set at 5 Da and the collision energy was −20 eV.
Nomenclature and structure identification
In the present work the abbreviation RO is used for describing the samples obtained by glycol-splitting followed by the reduction to highlight this latter step, while gs is used to indicate the split units within RO-LMWHs chains.
The oligosaccharide identification was based on the MS data. Their formula was only accepted if the m/z of the candidate analyte matched with the theoretical value within 5 ppm for oligosaccharides up to dp 8. For higher chain length oligosaccharides an error within 10 ppm was accepted. The comparison of the experimentally obtained isotope pattern with the theoretical one was also used to confirm the assigned structure. For each peak a “first-level structure” was assigned using MS data and considering the chemical nature and treatments of the starting material. The nomenclature previously applied for the heparinase-digested RO-heparins [13] was used. The abbreviation system is similar to that of Henriksen et al [33] and includes the number of monosaccharide residues, sulfate groups, and N-acetyl groups. In our case, a number and the symbol gs were also added to indicate the number of the glycol-split residues. “Remnants” of the hydrolyzed internal gs residues were indicated by the symbol R (see Fig. 1e), and those from non-reducing end as R(Unr), respectively. Symbols ΔU, U and A indicate a 4,5-unsaturated uronic acid, a saturated one and a glucosamine unit, respectively, at the NRE. Symbol “ol” indicates the alditol form of terminal units at the RE; symbols aM.ol and Rc indicates 2,5-anhydromannitol and contracted ring residues typical for dalteparin. Symbols 1,6aA and 1,6aM were used for the NMR cross peaks of 1,6-anhydroglucosamine and 1,6-anhydromannosamine units typical for enoxaparin; while for LC-MS data 1,6aA was used for both epimers because it was not possible to distinguish them from MS data.
Results
NMR analysis of RO-LMWHs
In order to detect and quantify monosaccharidic residues and units of LMWHs affected by the glycol-splitting reaction, RO-LMWHs were analyzed by 2D-NMR spectroscopy, using the 1H/13C HSQC approach previously developed for the structural characterization of unmodified heparins [32] and LMWHs [2]. The HSQC spectra of heparin species consist of a characteristic series of spots (cross peaks) over the typical range of monodimensional 1H and 13C spectra, the most informative for the current purpose usually being in the region of the anomeric (H-1/C-1) protons and carbons (5.6–5.4 / 94–108 ppm, including the small region of unsaturated 1H and 13C at 6.0-5.8/108–111.ppm), which is regarded as the “structural reporter” region of the spectra. As indicated in Fig. 3 for tinzaparin (blue spots) and RO-tinzaparin (red spots), signals for typical residues are located in subregions of the spectrum. Disappearance of cross peaks associated with residues known to be susceptible to periodate/borohydride reactions as indicated in Fig. 1 and 2, and appearance of new ones, with slight perturbation of cross peaks, clearly attributable to residues that are unaffected by these reactions, are evident in Fig. 3, Fig. S1 and S2 (see Electronic Supplementary Material,). Chemical shifts associated with gs-residues in different heparin-related compounds (heparins, LMWHs and K5PS) are listed in Table 2. Table S1 (see Electronic Supplementary Material) reports the relative content of typical residues for the three prepared RO-LMWH samples measured from the signal volumes [2,32].
Fig. 3.

Anomeric (a) and “ring” (b) regions of the 1H/13C HSQC NMR spectra of RO-tinzaparin (red spots) superimposed on the spectrum of the corresponding unmodified LMWH (blue spots) Symbols for typical end-group signals are framed with rectangles
A – glucosamine, A* – N-sulfate-glucosamine-3-O-sulfate, ΔU2S – 4,5-unsaturated 2-O-sulfated uronic acid, I/G – iduronic and glucuronic acids, GLR – glucuronic acid of the linkage region sequence, gsI/gsG – glycol-split iduronic and glucuronic acids, gsU – glycol-split uronic acids, Gal – galactose, Xyl – xylose, Ser - serine
Table 2.
1H and 13C chemical shifts in the anomeric region characteristic for the RO-LMWHs
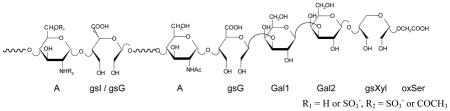 R1 = H or SO3−, R2 = SO3− or COCH3 | ||
|---|---|---|
| RESIDUE | 1H, ppm | 13C, ppm |
| Aminosugars | ||
| ANAc-(gsG)a | 5.09 | 99.0 |
| ANAc-(gsI)b | 5.12 | 98.0 |
| ANS-(gsI)c | 5.39 | 99.8 |
| ANS-(R)d | 5.34 | 99.2 |
| Glycol-split uronic acids | ||
| gsG-(ANAc)a | 4.71 | 106.7 |
| gsI-(ANAc)b | 4.94 | 106.9 |
| gsI-(ANS)c | 4.98 | 106.9 |
| gsG (RO-heparinse and RO-tinzaparin) | 4.87 | 106.4 |
| gsG (RO-LMWHs)f | 4.80 | 106.5 |
| gsIe | 4.98 | 106.9 |
| gsU-(A*)g | 4.90 | 103.2 |
| Residues of the gsLR | ||
| gsG-(Gal1) = gsGLR | 4.92 | 107.6 |
| Gal1 | 4.66 | 106.9 |
| Gal2 | 4.61 | 105.0 |
| gsXyl | 4.75 | 105.6 |
assignment consistent with the 2D NMR spectrum of RO-K5 (RO-derivative of N-acetylheparosan), spectrum not shown
assignment consistent with the 2D NMR spectrum of RO-derivative of N-acetylated bovine lung heparin
published in [14]
R – remnant generated by hydrolysis of glycol-split uronic acids; data obtained from 2D NMR spectrum of the fraction of heparinase-digested RO-enoxaparin, containing ΔU2S-ANS6S and ΔU2S-ANS6S-R
reported in [13] for RO-heparins
present work
from NMR analysis of RO-dalteparin (this cross peak correlates with CH2OH in the TOCSY spectrum indicating that it is a glycol-split uronic acid); A* = ANS3S(6S)
As expected, for all the three LMWHs the strongest signals are those that are less affected by glycol-splitting, i.e., those of the internal “regular” sequences of the trisulfated disaccharide (TSD) I2S-ANS6S [32] Distinct spots are associated with G-, gsG- and ANAc-containing disaccharide sequences (5.0-4.5/108-104 ppm). The H-1/C-1 signals most typical for the gs residues (at 4.98/106.9 ppm and at 4.80-4.87/106.4 ppm) are at positions similar to those that we recently identified for gsI and gsG, respectively, in RO-heparins [13,14]. Notably, cross-peak G-(A*) at 4.62/103.9 ppm, taken as a marker of the ATBR [34], is common to the three unmodified LMWHs but disappears upon glycol-splitting. The corresponding gsG-(A*) signal (at 4.90/103.2) was identified in RO-LMWHs. Such an assignment was confirmed by TOCSY experiments (see Electronic Supplementary Material, Fig. S3), indicating a correlation of the signal with CH2OH signals of a gsU residues. Moreover, cross peaks with similar chemical shifts were found in the spectrum of the RO-derivative of the synthetic pentasaccharide AGA*IA (4.94/103.4 ppm) and in that (4.98/103.1 ppm) of the RO-derivative of a heparin fraction enriched in ATBR-containing chains obtained by affinity chromatography [13] (data not shown). Signal gsG-(A*) is clearly observed only in the spectrum of RO-dalteparin (see Electronic Supplementary Material, Fig. S2,), but can be also detected in the expanded scale spectrum of the other RO-LMWHs. It is thought that different contents of the glycol-split ATBR disaccharide largely reflect different contents of G-(A*) in the original LMWHs as reported in the Table S1 (see Electronic Supplementary Material). Together with these signals from “internal” gsI and gsG, at least one cross peak (at 4.92/107.6), typical for RO-heparin species containing the linkage region, can be assigned to the glycol-split G residue (gsGLR) preceding the first galactose residue (Gal1). As in the spectra of the parent LMWHs, the gsGLR signal intensity indicates that the content of LR in RO-tinzaparin is consistently higher than in RO-enoxaparin, while that in RO-dalteparin is very low, the corresponding signal not being observed in spectrum (see Electronic Supplementary Material, Fig. S2).
NMR data (Fig. 3) show that not only internal sequences but also several end-groups are susceptible to glycol-splitting and/or reduction. For example, the cross-peaks of both α and β anomers of aminosugars and uronic acids at the RE typical for unmodified tinzaparin (ANSα, ANSβ, Fig. 3) and enoxaparin (ANSα, ANSβ MNSα, MNSβ, I2Sα, I2Sβ, see Fig. S1 of the Electronic Supplementary Material) disappeared after the RO-reaction. The resulting RE-alditols are not detectable in the complex “ring region” of the NMR spectra due to the signal overlapping with the free primary hydroxyl groups at C-6 of glucosamine and those at C-2 and C-3 of gs-uronic acids (Fig. 3, Fig. S1). The enoxaparin cross-peaks of H4/C4 ΔU and those of H1/C1 in 1,6aM are among those that disappear upon periodate oxidation/borohydride reduction (See Discussion). The NMR spectrum of dalteparin confirms its relative structural homogeneity, with a 2-O-sulfated iduronic acid at the NRE and a 2,5-anhydromannitol (aM.ol) at the RE, this latter residue well identifiable in the “ring region” of the NMR spectrum (see Electronic Supplementary Material, Fig. S2). A week signal at 5.45/104.5 ppm in both dalteparin and RO-dalteparin was recognized and assigned for the first time to an internal glucosamine contracted ring (Rc) residue using homonuclear (1D-COSY and 1D-TOCSY) and heteronuclear (edited-HSQC and HSQC-TOCSY) experiments (Electronic Supplementary Material Fig. S4). Particularly, the intra-residue connections were obtained starting from the 2-C-hydroxymethyl signal at 2.53 ppm, not affected by signal overlapping (Electronic Supplementary Material Fig. S4). The nitrous acid heparin treatment such as that used to obtain dalteparin has been previously reported to cause also deamination of some internal glucosamine residues to give by ring contraction a 2-deoxy-2-C-hydroxymethyl pentofuranosidic residues [35–37]. The found chemical shift of the hexocyclic CH2 group (H2a/b) is in agreement with the value reported previously for methyl-2,5-dideoxy-2-C-(hydroxymethyl)-α-D- xylo-pentofuranoside [36].
Direct LC-MS analysis of RO-LMWHs
To obtain compositional profiles of the RO-LMWHs, their samples were analyzed directly by the LC-MS method in comparison with those of the original unmodified LMWHs. Although a detailed analysis of unmodified LMWHs is outside the scope of this work, some general features of their HPLC-MS chromatograms are described here for comparison with the corresponding RO-derivatives (Fig. 4 and Fig. S5–S7 of Electronic Supplementary Material).
Fig. 4.

LC-MS profiles of enoxaparin (a), RO-enoxaparin (c) and the corresponding expanded chromatograms (b, d)
LC conditions: column C18 100 x 2.1 mm,.2.6 μm; eluents A and B – 10 mM DBA and 10 mM CH3COOH in H2O-CH3CN = 9:1 (v/v) and CH3CN, respectively; gradient: 0 min – 0%B, 5 min – 0%B, 130 min – 35%B, 140 min – 70%B, 145 min – 70%B, 148 min – 0%B, 170 min – 0%B; flow rate – 0.25 ml/min
Oligosaccharides from dp2 up to 22 (ΔU22,32,0, see Electronic Supplementary Material Table S2) were observed in tinzaparin, from dp 2 up to dp 20 (ΔU20,28,0, Fig. 4, see also Electronic Supplementary Material Table S2) in enoxaparin and from dp 4 up to dp 22 (U22,31,0-aM.ol and U22,32,0-aM.ol, see Electronic Supplementary Material Table S2) in dalteparin. Comparable results were reported using pentylamine acetate as ion pair reagent for the UPLC separation of tinzaparin oligosaccharides [26] and in an earlier SEC-MS study of Henriksen [33] oligosaccharides up to dp 18 were detected in tinzaparin.
The present LC-MS method highlights both the structural heterogeneity of the analyzed samples and their different molecular weight distributions. For example, while the LC-MS chromatogram of enoxaparin appers highly heterogeneous (Fig. 4), that of dalteparin (Electronic Supplementary Material, Fig. S7), a LMWH known to be extensively purified by ion-exchange chromatography [34], reflects a lower heterogeneity and a lower content of dp4 and dp6 fractions with respect to tinzaparin (Electronic Supplementary Material, Fig. S6) and enoxaparin (Fig. 4, Electronic Supplementary Material Fig. S5). The highly-sulfated even-numbered oligosaccharides are prevalent in all three LMWHs (Electronic Supplementary Material, Fig. S5–S7). N-Acetyl groups were observed more frequently within longer chains (Electronic Supplementary Material, Fig. S5–S7).
Peculiar residues typical for each LMWH are clearly observed in their LC-MS profiles. In tinzaparin and enoxaparin LC-MS chromatograms the major components absorb at 232 nm due to the presence of 4,5-unsaturated uronic acids (ΔU2S/ΔU) at the NRE (Fig. 2) formed as a result of the depolymerisation by β-elimination reaction (LC-UV chromatograms not shown), while analogs bearing saturated uronic acids were found only in low amounts. The enoxaparin LC-MS profile appears more complex due to the presence of oligosaccharides bearing at the RE either hemiacetalic or 1,6-anhydro (1,6aA) forms differing from each olther by 18 Da (Fig. 4, Fig. S5). In the LC-MS profile of dalteparin some oligosaccharides containing both aM.ol and Rc residues, detectable due to a mass decrease of 15 Da in comparison with the oligosaccharides bearing only aM.ol residues, have been found (Electronic Supplementary Material, Fig. S7 and Table S2), confirming the ring contracted residues observed in NMR spectra.
Among the minor components, oligosaccharides with an odd number of residues and a glucosamine at the NRE are detectable (Electronic Supplementary Material, Fig. S5–S7) in all three LMWHs, suggesting that some of the original heparin chains terminate with glucosamine units, as previously reported [33,38,39]. Odd-numbered oligosaccharides starting and terminating with uronic acid residues are minor components of tinzaparin (such as ΔU9,10,1, Electronic Supplementary Material Fig. S6 and Table S2) and in dalteparin (U5,6,0 and U5,7,0, see Electronic Supplementary Material Fig. S7 and Table S2), but are well evident in the enoxaparin (ΔU5,6,0 and ΔU5,7,0, Fig. 4, see also Electronic Supplementary Material, Fig. S5 and Table S2).
Comparison of LC-MS profiles of RO-LMWHs with the corresponding LMWHs (Electronic Supplementary Material Fig S5–S7) provides information on changes in mass values of individual oligosaccharides upon periodate oxidation and borohydride reduction at sites predicted in Fig. 2. A 2 Da mass increase is expected for each internal glycol-split residue with respect to the corresponding unmodified oligosaccharides. The possibility to obtain more structural information by differentiating isomers through RO-modification is also worth noting. For example, after glycol-splitting the enoxaparin tetrasaccharide ΔU4,5,0 gives rise to at least two tetrasaccharides containing internal 2-O-sulfated uronic acid ΔU4,5,0-ol and a third one containing a nonsulfated uronic acid ΔU4,5,0,1gs-ol (Electronic Supplementary Material Fig. S5 and Table S2). As particular cases, several gs-ATBR containing oligosaccharides were found (for example, ΔU8,10,1,2gs-ol and ΔU10,13,1,2gs-ol, etc, Fig. 4, see also Electronic Supplementary Material Table S2). The possibility to distinguish isomeric oligosaccharides can be shown also for RO-tinzaparin and RO-dalteparin (Electronic Supplementary Material Fig. S6,S7).
LC-MS analysis revealed also the modifications induced by periodate oxidation/sodium borohydrate reduction in the end-groups, confirming the NMR data. The most evident effect of the reduction step is formation of alditol residues at the RE (involving a 2 Da mass increase) for tinzaparin and enoxaparin oligosaccharides originally bearing aminosugars in hemiacetalic form (Fig. 4, Electronic Supplementary Material Fig. S5, S6). The behaviour of some minor short oligosaccharides of enoxaparin such as ΔU2,3,0 and ΔU3,4,0 appearing in RO-enoxaparin as ΔU2,3,0-ol and ΔU3,4,0-ol, respectively (Fig. 4), clearly confirms these data. The presence of residues at the RE susceptible to reduction was used for the structural assignment of oligosaccharides in RO-LMWHs: the “first” increment of “2H” was assigned to the reduced RE residue (either glucosamine or uronic acid). A further increase of the mass value of 2 or 4 Da were assigned to oligosaccharides (in the alditol form) with 1 or 2 gs residues, respectively (for example, ΔU4,5,0,1gs-ol and ΔU8,10,1,2gs-ol in RO-enoxaparin, Fig. 4). Because the dalteparin RE residues are exclusively constituted by aM.ol (Fig. 2), not susceptible to glycol-splitting nor to borohydride reduction, the gs-oligosaccharides present in RO-dalteparin are easier to be assigned on the MS basis, because each increment of 2Da should indicate only the presence of one gs-residue.
On the other hand, LC-MS showed that practically all the NRE residues of RO-dalteparin were susceptible to the glycol-splitting and to further chain cleavage (as illustrated in Fig. 5). In the case of the 2-O-sulfated uronic acid at the NRE, more represented in dalteparin, the bond between two vicinal OH-groups at C-3 and C-4 can be actually split, while in the case of the nonsulfated uronic acid at the NRE, simultaneous oxidation of the three vicinal OH-groups at C-2, C-3 and C-4 is likely to occur. Further loss of the formed remnants at the NRE (gsI2S(nr) and R(Unr), respectively, Fig. 5) seems to easily occur. In fact, a relatively large number of RO-dalteparin odd-numbered oligosaccharides start with a glucosamine (see Electronic Supplementary Material Fig. S7). The presence of some remnant-bearing structures was useful for the identification of LMWH oligosaccharides having a saturated uronic acid at the NRE and for distinguishing isomers. For example, the broad U8,11,0-aM.ol peak in the dalteparin LC-MS chromatogram can be explained by the presence of at least two isomers, regular one (constituted by TSD units) and another one having G-A* unit at the NRE that were identified by LC/MS analysis of RO-dalteparin (see Electronic Supplementary Material, Fig S7). The presence of G-A*-bearing oligosaccharides is in agreement with the NMR results showing a relatively high contents of A*, G-(A*) and Gnr residues (see Electronic Supplementary Material Table S1).
Fig. 5.

Terminal residues at the non-reducing end (NRE) and at the reducing end (RE) confirmed or found for unmodified and RO-LMWHs
NOTE: Since structures have been confirmed or elucidated by MS spectroscopy, the anionic groups (COOH, SO3H) are represented in their undissociated form
Hydrolytic loss of the remnants formed at the NRE were also observed in enoxaparin, which contains low amounts of oligosaccharides starting with glucuronic acid at the NRE (see Electronic Supplementary Material Table S1). For example, R(Unr)-A3,5,0-ol (Fig. 4, see also Electronic Supplementary Material Fig. S5) and R(Unr)-A3,5,0-1,6aA (Electronic Supplementary Material Fig. S5 and Table S2) were observed in RO-enoxaparin and the last one can be structurally correlated with a tetrasaccharide G-ANS3S6S-U2S-ANS1.6aA, isolated from the enoxaparin dp4 fraction (unpublished data of our group). A further remnant loss at the NRE is reflected, especially in RO-enoxaparin, by the presence of the trisaccharides A3,5,0-ol and A3,6,0-ol, this latter bearing an ANS3S6S residue.
For species starting with glucosamine at the NRE splitting between C-3 and C-4 is also possible. However, it seems that A5,8,0-1,6aA in enoxaparin (Fig. 4) and A5,8,0-aM.ol in dalteparin (see Electronic Supplementary Material Fig. S7) are resistant to the oxidation because the corresponding analytes were found unchanged in the corresponding RO-derivative chromatograms. Since such components are minor and difficult to characterize, in order to evaluate the susceptibility to glycol-splitting of terminal NRE glucosamine residues the RO-reaction was performed also on a model compound, synthetic pentasaccharide ANS,6S-G-ANS,3S,6S-I2S-A-OMeNS,6S (Arixtra®). This experiment showed that an excess of periodate should be used for completing the splitting of both internal glucuronic acid and glucosamine at the NRE (data not shown). The resistance of the mentioned above oligosaccharides to the periodate oxidation may be also explained by the presence of the 3-O-sulfated glucosamine at the NRE of some heparin chains [39]. However, such species may be also formed by the cleavage of the terminal uronic acids, which originally are present in dalteparin and enoxaparin (as described above).
The hypothesis that the formation of the oligosaccharides starting with glucosamine at the NRE as a result of an internal gs uronic acids hydrolysis could not be confirmed because no corresponding remnant (R, Fig. 1e)) bearing oligosaccharides were found. However, to verify if the developed LC-MS method permits to detect such species a RO-tinzaparin sample was prepared using HCl instead of acetic acid, for neutralizing the NaBH4-reaction mixture. The NMR spectrum of the HCl treated preparation (not shown) showed a lower amount of generated gs residues than in the preparation treated with acetic acid, supporting the hypothesis that partial hydrolysis occurred. Indeed, a number of remnant (R) bearing species was observed (Electronic Supplementary Material Fig. S6), indicating that the present analytical conditions permit to observe and monitor such a hydrolysis process.
Elution order
For oligosaccharides with the same dp, the higher the sulfation degree, the higher the retention time RT (for example, RT (ΔU6,7,0) < RT (ΔU6,8,0) < RT (ΔU6,9,0) in enoxaparin Fig. 4). The presence of a N-acetyl group causes a slight increase of the retention time (RT (ΔU6,7,0) < RT (ΔU6,7,1)) that indicates that not only electrostatic but also hydrophobic interactions between analytes and stationary phase can take place. Unexpectedly, the retention time of ΔU16,21,0 is somewhat lower that that of ΔU14,21,0 in tinzaparin (Electronic Supplementary Material Fig. S6) and enoxaparin (Electronic Supplementary Material Fig. S5). This finding may be explained by the fact that the longer the oligosaccharide chains, the less the influence of additional disaccharide units and the more crucial the influence of the number of anionic groups per chain. Enoxaparin oligosaccharides with a 1,6aA-residues are eluted after their analoges bearing hemiacetalic aminosugars (RT ΔU6,8,0 < RT ΔU6,8,0-1.6aA). Unsaturation in the NRE uronic acid increases the RT (RT (U6,9,0) < RT (ΔU6,9,0)), probably due to the higher acidity of the uronic acid carboxy-group when conjugated with a double bond, leading to a stronger interaction with dibutylammonium cation. The same fact may take place in the case of the odd enoxaparin oligosaccharides starting with ΔU(2S), which shows higher RT than oligosaccharides starting with a glucosamine residue (RT (A3,4,0) < RT (ΔU3,4,0)); moreover, ΔU3,4,0, bearing two carboxyl groups, have more anion centres able to interact with dibutylammonium cation. Oligosaccharides in alditol form are eluted after their unmodified analogs (RT (ΔU6,8,0) < RT (ΔU6,8,0-ol)), and this is probably caused by the higher flexibility of the reduced open form with respect to that of the hemiacetalic one that facilitates the interactions between sulfate groups and dibutylamine on the stationary phase. The presence of an uronic acid remnant increases the retention time (e.g., in RO-tinzaparin (RT (ΔU8,12,0-ol) < RT (ΔU8,12,0-R)), probably, due to the additional charge of the remnant carboxyl group (R) present at the RE (Fig. 1e).
LC-MS profiling of heparinase-digested RO-LMWHs
The main limitation in LC-MS profiling of LMWHs and their RO-derivatives is that only the major species can be separated and identified; in fact, component overlapping in terms of retention times and mass values, makes difficult to characterize the minor components. This problem becomes more crucial for higher oligosaccharides, especially when unmodified oligosaccharides and glycol-split ones have similar retention times. The gradient optimization for LMWHs is limited by the total analysis time due to the wide range of the species to be profiled, from dp 2 up to at least dp 22.
In order to obtain more informative data, all RO-LMWHs were also analyzed after exhaustive enzymatic digestion with a mixture of heparinases I, II and III which leads to concentration of sequences containing gs-residues and to the identification of peculiar structures otherwise difficult to be characterized. The LC-MS profiles of heparinase-digested RO-LMWHs (Fig. 6, and v Fig. S8, S9) are definitely simpler than those of the corresponding non-digested samples (Fig. 4, Fig. S5–S7). Like those of similarly digested RO-heparins [13], they essentially consist of disaccharide, tetrasaccharide, hexasaccharide fragments and very low amounts of some octasaccharides present only in the digest of RO-dalteparin (see Electronic Supplementary Material Fig. S9). The major disaccharide ΔU2,3,0 (ΔU2S-ANS6S), the minor disaccharide ΔU2,2,0 (ΔU2S-ANS) and several tetrasaccharides resistant to the enzymatic cleavage (such as, ΔU4,6,0-ol in RO-tinzaparin, ΔU4,5,0-1,6aA in RO-enoxaparin, ΔU4,5,0-aM.ol in RO-dalteparin) are also present in the corresponding digest of LMWHs (not shown), characterizing the regular sequences not containing nonsulfated uronic acids.
Fig. 6.
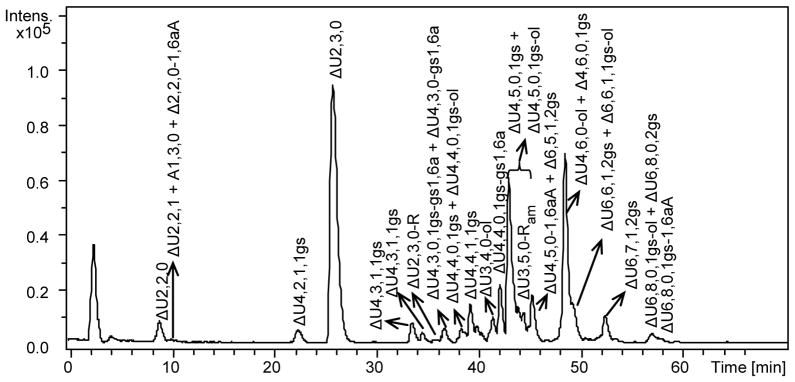
LC-MS chromatogram of the heparinase-digest of RO-enoxaparin
LC conditions: column C18 100 x 2.1 mm, 2.6 μm; eluents A and B – 10 mM DBA and 10 mM CH3COOH in H2O-CH3OH = 9:1 (v/v) and CH3OH, respectively; 0 min – 17% B, 10 min – 17%B, 30 – 42%, 50 min – 50% B, 65 min – 90%, 75 min – 90%B, 76 min – 17% B, 95 min – 17 %B; flow rate – 0.1 ml/min; injection volume – 5 μl.
Sodium borohydride reduction was used to distinguish between internal gs-unit and alditol end residue. For example, the double-charged ion with m/z 497.02 could correspond to both ΔU4,4,0,1gs and ΔU4,4,0-ol. After sodium borohydride reduction two peaks with m/z values were observed: a) m/z 498.02, indicating that this oligosaccharide containing one internal gs-unit (gsU), corresponds to the structure ΔUS-ANS6(S)-gsU-ANS6(S), where only one of the two sulfate groups within parenthesis is present; b) unmodified m/z 497.02, which should have an internal 2-O-sulfated uronic acid (US) and corresponds to ΔUS-ANS-US-A.olNS structure. The additional NaBH4 reduction of the RO-enoxaparin digest proved also the structure assigned to the minor tetrasaccharides terminating with an uronic acid remnant (ΔU4,4,1,1gs-R, m/z 577.03 for [M-2H]2− ion form, and ΔU4,5,0,1gs-R, m/z 596.01 for [M-2H]2− ion form), for which the m/z values remained constant after the additional reduction
The fragments derived from enzymatic digestion of RO-LMWHs differ from those of unmodified LMWHs especially in their hexasaccharide components, which are practically absent in the heparinase digests of non-gs LMWHs (not shown). It is important to note that while the species containing two gs residues are seldom observable in the profiles of the intact RO-LMWHs (Fig. 4 and Electronic Supplementary Material Fig, S5–S7), some of these species can be detected in the chromatograms of all the RO-LMWHs digests (for example, ΔU6,7,1,2gs, Fig. 6).
As expected, the digested RO-tinzaparin also consists of even-numbered oligosaccharides terminating at the RE with both hexosamine formed during the enzymatic cleavage and some with alditols generated by the RO-reaction (see Electronic Supplementary Material Fig. S8). Notably, no disaccharide in alditol form was detected (see Discussion).
The LC-MS profile of RO-enoxaparin enzymatic digest (Fig. 6), is definitely more complex, especially as regards end residues, than the corresponding profiles of RO-tinzaparin and RO-dalteparin digests, making structural assignments more difficult. Since nonsulfated uronic acids of RO-LMWH chains are split and cannot be recognized by the heparinases, the 4,5-unsaturated uronic acid residues were assumed to be always 2-O-sulfated for the oligosaccharides present in the RO-LMWH digests. Furthermore, to verify if the observed mass increment of 2 Da was caused by the generation of one internal gs residue or by reduction of terminal hemiacetal, the RO-enoxaparin digest was further treated with sodium borohydride (profiles not shown). Species susceptible to this reduction (i.e., those converted to alditol forms) are identified with oligosaccharides originally terminating with reducing hexosamines and consequently bearing internal gs, while the reduction resistant oligosaccharides were considered to already have an alditol group at the RE. This approach was used to distinguish ΔU4,6,0-ol (Fig. 6) and ΔU4,6,0,1gs (not shown in the chromatogram); this minor tetrasaccharide, contains a N-sulfated-glucosamine-3,6-O-disulfate residue and is likely to be one of the markers of the sulfated ATBR (I2S-ANS6S-G-ANS3S6S-I2S-ANS6S), previously observed in the enzymatic digest of RO-heparin from bovine lung [13].
The LC-MS chromatogram of the heparinase-digest of RO-dalteparin (see Electronic Supplementary Material Fig. S9) is less complex than those of the other two RO-LMWH digests, reflecting in part a rather homogeneous internal structure of the original LMWH and the fact that practically all chains in the original dalteparin terminate at the RE with aM.ol groups, resistant to the glycol-splitting. Notably, the RO-dalteparin digests contain also octasaccharides ΔU8,9,1,1gs-aM.ol and ΔU8,9,1,2gs-aM.ol (both in traces), indicating that all the glycosidic bonds of these oligosaccharides are resistant to cleavage by heparinases (see Discussion).
RO-induced modifications of peculiar end-groups (linkage region, mannosamine, 1,6-anhydromannosamine)
The LC-MS chromatogram of tinzaparin reveals the presence of species containing intact linkage region such as ΔU4,3,1-LR, ΔU4,4,1-LR, ΔU8,6,2-LR, ΔU8,9,1-LR, ΔU10,11,1-LR and ΔU10,11,2-LR (see Electronic Supplementary Material Fig. S6).
As expected and illustrated in Fig. 1, the glucuronic acid and xylose units of the LR are susceptible to glycol-splitting, and, depending on the reaction conditions the LR can generate two forms, such as the one shown in Fig. 1d (gsLR) or further hydrolyzed at the level of gs-xylose, to give the serine lacking fragment gsG-Gal-Gal-CH(CH2OH)2 (gsLR(2)).
The LC-MS analysis of the oligosaccharides containing the gsLR/gsLR(2) in the RO-tinzaparin digest allowed the characterization of the environment near the LR. Of note, ΔU2,1,1-gsLR(2) is formed in trace amount, which indicates that the first uronic acid preceeding GLR is almost always nonsulfated. Different tetrasaccharides containing one gs residue linked to the gsLR(2), such as ΔU4,3,1,1gs-gsLR(2), ΔU4,4,1,1gs-gsLR(2) and ΔU4,2,1,1gs-gsLR(2), were detected. The first level structure of the former corresponds to the previously observed heparin oligosaccharides ΔU2S-ANS6S-G/I-ANAc-LR [23]. Only one specie containing two acetyl groups (ΔU4,1,2,1gs-gsLR(2)) was observed, but in very low amounts, indicating that, when the second uronic acid after GLR is 2-O-sulfated, the second glucosamine residue is almost always N-sulfated and not N-acetylated. Moreover, the presence of ΔU4,4,1,1gs-gsLR(2) indicates that the first N-acetylglucosamine can also be 6-O-sulfated, as also found in heparin [23]. Among higher oligosaccharides, ΔU6,3,2,2gs-gsLR(2) and ΔU6,2,2,2gs-gsLR(2) were the most evident, while a mono-N-acetylated analog (ΔU6,3,1,2gs-gsLR(2)) was observed in trace. Notably, no species with three N-acetylglucosamines were found. Considering the known biosynthesis pathway of heparin [40], these data could indicate that when the second uronic acid after GLR is nonsulfated, the following glucosamine is N-acetylated. Octasaccharide chains with three gs units linked to the gsLR(2) were also found, even if in traces (Electronic Supplementary Material Table S2). Combining these data with the results obtained by the LC-MS analysis of the intact RO-tinzaparin, it is likely that the N-acetylated domain in the closest proximity of LR of the parent heparin should be very short (i.e., di- or tetrasaccharide sequences).
Several gsLR-bearing oligosaccharides with the sulfation/acetylation pattern similar to that observed in tinzaparin were found also in enoxaparin after SEC-fractionating its digested RO-derivative (Electronic Supplementary Material Fig. S10). No oligosaccharides bearing 2-O-phosphorylated xylose [41] were observed. It would probably be possible to detect these particular LR-bearing oligosaccharides after previous isolation of the LR-containing fraction.
In the LC-MS chromatogram of the digested RO-enoxaparin, an oligosaccharide with m/z 507.476(2−) attributed to a ΔU3,5,0- aminosugar remnant (ΔU3,5,0-Ram) structure was observed (neutral molecular formula C22H35N1O34S5, theoretical m/z 507.475) (Fig. 5,6). The formation of this trisaccharide, also found in RO-tinzaparin and some RO-heparins [13], as a trace component, may be explained by the oxidation of glucosamines bearing free NH2-groups, which can be present in heparin. The same explanation does not hold for RO-enoxaparin, where ΔU3,5,0-Ram is present in relatively high quantity. Notably, 2D NMR spectrum of the parent enoxaparin did not show cross peaks of glucosamines with free NH2. We suggest that the generation of ΔU3,5,0-Ram in RO-enoxaparin could result from an oxidation of the N-sulfated mannosamine residue (Fig. 5,6).
Other unknown structures were observed (m/z 448.5, 449.5 and 488.5) in the heparinase-digest of RO-enoxaparin. The LC-MS data show that these analytes are likely to have the same backbone but differ by number of sulfate-, acetyl groups, and gs residues (Electronic Supplementary Material Table S3). The fact that all the three species absorb at 232 nm indicates the presence of an unsaturated uronic acid at the NRE. Useful information was obtained especially by a subsequent SEC-fractionation. LC-MS analysis showed that the SEC-fraction eluted between tri- and tetrasaccharides pools is enriched in compounds of the unknown structures (m/z 448.5, 449.5 and 488.5, see Electronic Supplementary Material Fig. S10), that is in agreement with the hypothesized neutral formula (with an error less than 5 ppm) (see Electronic Supplementary Material Table S3). These species were also found to be resistant to borohydride reduction, suggesting that they do not have hemiacetalic monosaccharide at the RE.
Two different structures with the same neutral formula were found to correspond to the observed unusual m/z values: the first one with the same backbone as ΔU3,5,0-Ram but containing N-acetylated glucosamine, and the second structure bearing a split 1,6-anhydro-aminosugar at the RE (gs1,6a, Fig. 5 and Electronic Supplementary Material Table S4). A subsequent MS/MS experiment indicated that the main observed fragments could be formed from both backbone types (either an acetylated trisaccharide with an aminosugar remnant, or a tetrasaccharide with gs1,6a) (see Electronic Supplementary Material Fig. S11). Several minor signals may be more informative. For example, m/z 247,494 may correspond to a disaccharide unit with two sulfate groups, suggesting that the only glucosamine is sulfated but not acetylated, ruling out the structure with a remnant and a N-acetyl substituent.
Moreover, the HSQC spectrum of the SEC fraction enriched in structurally unknown compounds did not show cross-peaks corresponding to the N-acetyl-glucosamine-6-O-sulfate linked to 2-O-sulfated-iduronic acid (ANAc6S-(I2S) C1-H 5.15/96.6 ppm, C2-H 4,03/56.2 ppm) [42]. This cross-peak should have been observed if the unknown compound with m/z 488.5 had the acetylated structure ΔU2S-ANAc6S-I2S-Ram. Notably, the ANAc6S-(I2S) unit is rarely present within heparin chains. Therefore, all experimental data, i.e.: i) absorbance at 232 nm; ii) resistance under the reductive conditions; iii) accurate m/z value; iv) correspondence of the experimental isotopic pattern with theoretic one; v) resistance to the reduction and to the enzymatic cleavage; and vi) NMR data, confirm that the structure with the split 1,6-anhydromannosamine is the most probable.
Sequences with three subsequent glycol-split residues
The sequences with three glycol-split uronic acids in contiguous disaccharide units were previously observed in trace amounts in the heparinase-digest of the RO-derivative of pig mucosal heparin [13]. This indicates that sequences with three subsequent nonsulfated uronic acid units are present within heparin chains. A similar result was obtained for the RO-tinzaparin where octasaccharides with three internal gs units and glycol-split LR sequence were found (described above). A fraction containing highly sulfated mono-N-acetylated octasaccharides with three gs units (ΔU8,7,1,3gs, ΔU8,8,1,3gs, ΔU8,9,1,3gs) was isolated from the RO-enoxaparin digest by SEC fractionation (see Electronic Supplementary Material Fig. S10). Borohydride reduction revealed that octasaccharides with both three gs-units (ΔU8,7,1,3gs, ΔU8,8,1,3gs, ΔU8,9,1,3gs) and two gs-units and reduced aminosugar at the RE (ΔU8,7,1,2gs-ol, ΔU8,8,1,2gs-ol, ΔU8,9,1,2gs-ol, ΔU8,10,1,2gs-ol) were present. The cross-peak of the α-N-sulfated glucosamine (ANSα), observed in the 2D NMR spectrum of the digest before reductive treatment, confirms the presence of octasaccharides with three gs units and hemiacetalic N-sulfated glucosamine at the RE. It is worth noting that in the case of the octasaccharides with two gs residues their resistance to the enzymatic cleavage may be explained by the position of the 2-O-sulfated uronic acid (US) that is likely to be close to the alditol unit in the proposed structure ΔUS-A-gsU-A-gsU-A-US-A-ol.
Discussion
Modifications generated in LMWHs by the glycol-splitting periodate oxidation followed by a “stabilizing” borohydride reaction mainly involve internal nonsulfated uronic acid residues and end-residues susceptible to splitting and/or reduction. These structural modifications change both NMR spectra and LC-MS profiles of the LMWHs, providing complementary and characteristic fingerprints of both LMWHs and their RO-derivatives. Thus, the combined NMR/LC-MS analysis can allow to characterize the individual RO-LMWH, compare different RO-samples and provide structural information on the starting material. Whereas the HSQC spectra are currently exploited for quantitative evaluation of individual monosaccharide and disaccharide units in heparins [32] and LMWHs [2], a note of caution is necessary when dealing with gs-derivatives, since the local flexibility of gs residues may affect their relaxation times and scalar coupling values. The reported quantitative data (Online Resource 3), though compatible with our overall conclusions, should accordingly be taken as provisional, until a deeper study is made using well-defined model compounds.
The HSQC NMR spectra and the LC-MS profiles of RO-LMWHs also retain some features characteristic of the LMWH production method, partial depolymerisation with heparinase I (tinzaparin), base-catalyzed β-elimination (enoxaparin), and nitrous acid depolymerisation (dalteparin). All of these depolymerisation methods cleave only glycosidic bonds between an aminosugar and the adjacent uronic acid that leads to the formation of even-numbered oligosaccharides (Fig. 2). Surprisingly, RO-dalteparin has been shown to contain numerous odd-numbered oligosaccharides (see Electronic Supplementary Material Fig. S7), probably due to the glycol-splitting of the dalteparin terminal uronic acid residues bearing 3,4-diol groups and the further hydrolysis of the generated remnants (Fig. 5). Together with the observed high content of odd-numbered oligosaccharides, the occurrence of occasional ring contracted residues diversifies RO-dalteparin from the other RO-LMWHs.
Interestingly, RO-enoxaparin was the only one, among the RO-LMWHs of the present study, showing a LC-MS profile less complex than that of the parent LMWH (Fig. 4). This can be reasonably explained by the presence of end residues ΔU2S at the NRE, and N-sulfated 1,6aA groups at the RE, resistant to glycol-splitting. Minor ΔU residues from one side, as well as the above mentioned 1,6aM on the other side, may both contribute to some chain shortening caused by their oxidation. While, the glycol-splitting of the ΔU was expected, the oxidation of the N-sulfated 1,6aM residues not bearing two vicinal hydroxyls was quite unexpected.
Hydrolytic cleavage of glycosydic bonds (especially those of the gsU-residues), as well as partial cleavage and eventual hydrolysis at the level of terminal gs residues should be considered as possible events under the chosen conditions of RO-reaction. For example, addition of both sodium borohydride and strong acids (for example, hydrochloric acid) used for the pH adjustment, may represent critical steps since it can locally generate high basic or acidic conditions [12,13]. We observed that the acidic hydrolytic effect can be decreased by using mild acids (i.e. acetic acid). Although under our experimental conditions the conversion yields of the parent LMWHs to the corresponding RO-derivatives are quite high (>85%), the decrease (~20%) observed in their average MW (see Experimental) indicates some chain shortening with respect the parent LMWHs. Remnant bearing species, markers of such a chain cleavage, can be detected by the developed LC-MS method.
The plurality of species observed in the LC-MS chromatograms of RO-LMWHs suggests that some minor components may result from side-reactions such as hydrolysis at the level of internal gs residues (with generation of shorter chains terminating with remnant residues), oxidation of some end groups, and/or further cleavage of the generated remnants. In view of a further optimization of the RO-reaction conditions that could limit the impact of side-reactions, the present LC-MS method represents an useful and reproducible tool for fingerprinting RO-LMWH.
On the other hand, the glycol-splitting-induced local mobility is likely to exert an influence on retention times in LC-MS profiles of components of RO-LMWHs, allowing separation of some isomeric oligosaccharides otherwise difficult to achieve.
LC-MS analysis of heparinase digests provide additional structural information on oligosaccharide components of RO-LMWHs, and, indirectly, on the parent compounds. Since heparin lyases recognize the “regular” sequences of heparin but not residues introduced by the depolymerisation reactions [24,43,44] nor glycol-split residues [13], incubation of RO-LMWHs with heparinases generated fragments one or more disaccharide unit(s) longer than the corresponding ones detected in the LMWH digests, with incorporated gs-units, providing information about the length of the undersulfated heparin sequences. The combination of glycol-splitting and enzymatic digestion demonstrated that the sequences containing up to three subsequent nonsulfated uronic acids are present within heparin chains.
The specificity of the heparinases can be exploited for confirming the structure of terminal residues at the RE (such as 1,6aA in RO-enoxaparin and aM.ol in RO-dalteparin) and some sequences bearing them. As implied in previous reports on enoxaparin and dalteparin in different contexts [24,43,44], these residues at the RE are not recognized by heparinases and tetrasaccharide fragments are generated instead of disaccharides as would be the case if the end residues were unmodified glucosamines. The present study suggests that also the terminal alditols (as observed for practically all species of RO-tinzaparin, and for a number of chains in RO-enoxaparin) exert some inhibition of heparinases, with preferential cleavage of glycosidic bonds one disaccharide unit further towards the NRE. In fact, LC-MS peaks attributable to alditol disaccharides were never observed in the enzymatic digests, indicating that the heparinase-generated RO-LMWH fragment terminating with an alditol cannot be shorter than a trisaccharide (for example, ΔU3,4,0-ol).
An additional inhibition exerted by glycol-split residues, first observed by us on RO-derivatives of full-length heparins [13], permitted to characterize the heparin sequences near the linkage region by the first time, and to identify the ATBR-containg sequences with different sulfation/acetylation pattern. These ATBR-derived fragments can be more easily unraveled because the glycosidic bond of 3-O-sulfated glucosamine (A*) is preferentially cleaved by heparinases and the A*-containing oligosaccharides terminate with a “truncated” N,3,6-trisulfated glucosamine [45]. The ATBR-derived fragments accordingly terminate at the RE with a G-A* disaccharide; moreover, since both the bond connecting glucosamine to the gsG-A* unit [46] and that of the gs-residue (gsI) linked to the next sequence (A-gsG-A*) [13] are not cleaved by heparinases, most of these typical fragments have the minimum size of a hexasaccharide. The hexasaccharide ΔU6,7,1,2gs (in the case of dalteparin it could be also incorporated in the minor octasaccharide ΔU8,9,1,2gs-aM.ol) was observed in the LC-MS profiles of all the RO-LMWH digests. Tetrasaccharide ΔU4,6,0,1gs (generated from the highly sulfated ATBR variant [13]) was also found in trace amount in all RO-LMWHs digests.
Conclusions
This work provides previously unavailable criteria for structural analysis of gs-LMWHs as well as a complement to current methods for characterization of LMWHs. In particular, combined 2D NMR and LC-MS analysis permits the detection of the major internal modifications introduced in LMWH chains by periodate oxidation/borohydride reduction. Modifications of end residues susceptible to glycol-splitting and /or reduction can also be detected by this approach. In addition, the present analysis of RO-LMWHs and their heparinase digests provides an informative profiling complementary to that currently obtained for unmodified LMWHs, and a better characterization of isomeric oligosaccharides. The combination of glycol-splitting and enzymatic digestion provides more informative data also on the starting material, first of all, about the length of the sequences containing subsequent nonsulfated uronic acids and the structure of terminal sequences in RO-LMWHs. In particular, the N-acetylated domain in the closest proximity to the LR was characterized using this approach, providing information on the heparin biosynthesis. ATBR-containing oligosaccharides can be identified by the LC-MS analysis of RO-LMWHs and their heparinase-digests. Additionally, the NMR/LC-MS approach permits monitoring of the efficiency of conversion of nonsulfated uronic acids to the corresponding gs residues as well as the extent of side reactions occurring under different experimental conditions such as cleavage of remnants of terminal gs residues and hydrolysis of internal ones.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Health (grant number R01-CA138535) and The Italian Association for Cancer Research (AIRC, grant number IG10569).
References
- 1.Weitz JI. Low-molecular weight heparins. N Engl J Med. 1997;337:688–698. doi: 10.1056/NEJM199709043371007. [DOI] [PubMed] [Google Scholar]
- 2.Guerrini M, Guglieri S, Naggi A, Sasisekharan R, Torri G. Low molecular weight heparins: structural differentiation by bidimentional nuclear magnetic resonance spectroscopy. Semin Thromb Hemost. 2007;33:478–487. doi: 10.1055/s-2007-982078. and references therein. [DOI] [PubMed] [Google Scholar]
- 3.Casu B. In: Chemistry and Biology of Heparin and Heparan Sulfate. Garg HG, Linhardt RJ, Hales CA, editors. Elsevier; Amsterdam: 2005. and references therein. [Google Scholar]
- 4.Lever R, Page CP. Novel drug development opportunities for heparin. Nat Rev Drug Discov. 2002;1:140–148. doi: 10.1038/nrd724. [DOI] [PubMed] [Google Scholar]
- 5.Fuster MM, Esko JD. The sweet and sour of cancer: glycans as novel therapeutic agents. Nat Rev Cancer. 2005;5:526–542. doi: 10.1038/nrc1649. [DOI] [PubMed] [Google Scholar]
- 6.Casu B, Vlodavsky I, Sanderson RD. Non-anticoagulant heparins and inhibition of cancer. Pathophysiol Haemost Thromb. 2007;36:195–203. doi: 10.1159/000175157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Casu B, Naggi A, Torri G. Heparin-derived heparan sulfate mimics to modulate heparan sulfate-protein interaction in inflammation and cancer. Matrix Biology. 2010;29:442–452. doi: 10.1016/j.matbio.2010.04.003. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Höök M, Björk I, Hopwood J, Lindahl U. Anticoagulant activity of heparin: separation of high-activity and low-activity heparin species by affinity chromatography on immobilized antithrombin. FEBS Lett. 1976;66:90–93. doi: 10.1016/0014-5793(76)80592-3. [DOI] [PubMed] [Google Scholar]
- 9.Naggi A. In: Chemistry and Biology of Heparin and Heparan Sulfate. Garg HG, Linhardt RJ, Hales CA, editors. Elsevier; Amsterdam: 2005. and references therein. [Google Scholar]
- 10.Mousa SA, Linhardt R, Francis JL, Amirkhosravi A. Anti-metastatic effect of a non-anticoagulant low-molecular-weight heparin versus the standard low-molecular-weight heparin, enoxaparin. Thromb Haemost. 2006;96:816–821. doi: 10.1160/th06-05-0289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perlin AS. Glycol-cleavage oxidation. Advances Carbohydr Chem Biochem. 2008;60:183–250. doi: 10.1016/S0065-2318(06)60005-X. [DOI] [PubMed] [Google Scholar]
- 12.Islam T, Butler M, Sikkander SA, Toida T, Linhardt RJ. Further evidence that periodate cleavage of heparin occurs primarily through the antithrombin binding site. Carbohydr Res. 2002;337:2239–2243. doi: 10.1016/s0008-6215(02)00229-x. [DOI] [PubMed] [Google Scholar]
- 13.Alekseeva A, Casu B, Torri G, Pierro S, Naggi A. Profiling glycol-split heparins by high-performance liquid chromatography/mass spectrometric analysis of their heparinase-generated oligosaccharides. Anal Biochem. 2013;434:112–122. doi: 10.1016/j.ab.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Casu B, Guerrini M, Naggi A, Perez M, Torri G, Ribatti D, Carminati P, Giannini G, Penco S, Pisano C, Belleri M, Resnati M, Presta M. Short heparin sequences spaced by glycol-split uronate residues are antagonists of fibroblast growth factor 2 and angiogenesis inhibitors. Biochemistry. 2002;41:10519–10528. doi: 10.1021/bi020118n. [DOI] [PubMed] [Google Scholar]
- 15.Naggi A, Casu B, Perez M, Torri G, Cassinelli G, Penco S, Pisano C, Giannini G, Ishai-Michaeli R, Vlodavsky I. Modulation of the heparanase-inhibiting activity of heparin through selective desulfation, graded N-acetylation, and glycol-splitting. J Biol Chem. 2005;280:12103–12113. doi: 10.1074/jbc.M414217200. [DOI] [PubMed] [Google Scholar]
- 16.Hostetter N, Naggi A, Torri G, Ishai-Michaeli R, Casu B, Vlodavsky I, Borsig L. P-selectin and heparanase-dependent antimetastatic activity of non-anticoagulant heparins. FASEB J. 2007;21:3562–3572. doi: 10.1096/fj.07-8450com. [DOI] [PubMed] [Google Scholar]
- 17.Leitgeb AM, Blomqvist K, Cho-Ngwa F, Samje M, Nde P, Titanji V, Wahlgrene M. Low anticoagulant heparin disrupts Plasmodium falciparum rosettes in fresh clinical isolates. Am J Trop Med Hyg. 2011;84:390–396. doi: 10.4269/ajtmh.2011.10-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ekman-Ordeberg G, Hellgren M, Åkerud A, Andersson E, Dubicke A, Sennström M, Byström B, Tzortzatos G, Gomez MF, Edlund1 M, Lindahl U, Malmström A Low molecular weight heparin stimulates myometrial contractility and cervical remodeling in vitro. Acta Obstetr et Gynecol Scand. 2009;88:984–989. doi: 10.1080/00016340903176818. [DOI] [PubMed] [Google Scholar]
- 19.Vogt AM, Pettersson F, Moll K, Johnsson C, Normark J, Ribacke U, Egwang TG, Ekre H-P, Spilmann D, Chen Q, Wahlgren M. Release of sequestered malaria parasites upon injection of a glycosaminoglycan. PLoS Pathogens. 2006;2:0853–0863. doi: 10.1371/journal.ppat.0020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weitz JI, Young E, Johnston M, Stafford AR, Fredenburgh JC, Hirsh J. Vasoflux, a new anticoagulant with a novel mechanism of action circulation. Circulation. 1999;99:682–689. doi: 10.1161/01.cir.99.5.682. [DOI] [PubMed] [Google Scholar]
- 21.Zhou H, Roy S, Cochran E, Zouaoui R, Chu CL, Duffner J, Zhao G, Smith S, Galcheva-Gargova Z, Karlgren J, Dussault N, Kwan RYQ, Moy E, Barnes M, Long A, Hohan C, Qi YW, Shriver Z, Ganguly T, Schultes B, Venkataraman G, Kishimoto TK. M402, a novel heparan sulfate mimetic, targets multiple pathways in tumor progression and metastasis. PLosONE. 2011;6:e21106. doi: 10.1371/journal.pone.0021106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiao Z, Tappen BR, Ly M, Zhao W, Canova LP, Guan H, Linhardt RJ. Heparin mapping using heparin lyases and the generation of a novel low molecular weight heparin. J Med Chem. 2011;54:603–610. doi: 10.1021/jm101381k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iacomini M, Casu B, Guerrini M, Naggi A, Pirola A, Torri G. “Linkage region” sequence of heparin and heparan sulfate: Detection and quantification by Nuclear Magnetic Resonance spectroscopy. Anal Biochem. 1999;274:50–58. doi: 10.1006/abio.1999.4230. and references therein. [DOI] [PubMed] [Google Scholar]
- 24.Linhardt RJ, Loganathan D, Al-Hakim A, Wang H-M, Walenga JM, Hoppensteadt D, Fareed J. Oligosaccharide mapping of low-molecular weight heparins: structure and activity differences. J Med Chem. 1990;33:1639–1645. doi: 10.1021/jm00168a017. [DOI] [PubMed] [Google Scholar]
- 25.Capila I, Gunay NS, Shriver Z, Venkataraman G. In: Chemistry and Biology of Heparin and Heparan Sulfate. Garg HG, Linhardt RJ, Hales CA, editors. Elsevier; Amsterdam: 2005. [Google Scholar]
- 26.Langeslay DJ, Urso E, Gardini C, Naggi A, Torri G, Larive CK. Reversed-phase ion-pair ultra-high-performance-liquid chromatography-mass spectrometry for fingerprinting low-molecular-weight heparins. J Chromatogr A. 2013;1292:201–210. doi: 10.1016/j.chroma.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 27.Korir AK, Limtiaco JF, Gutierrez SM, Larive CK. Ultraperformance ion-pair liquid chromatography coupled with electrospray time-of flight mass spectrometry for compositional profiling and quantification of heparin and heparan sulphate. Anal Chem. 2008;80:1297–1306. doi: 10.1021/ac702235u. [DOI] [PubMed] [Google Scholar]
- 28.Kuberan B, Lech M, Zhang L, Wu LZ, Beeler DL, Rosenberg R. Analysis of heparan sulfate oligosaccharides with ion pair-reverse phase capillary high performance liquid chromatography-microelectrospray ionization time-of-flight mass spectrometry. J Am Chem Soc. 2002;124:8707–8718. doi: 10.1021/ja0178867. [DOI] [PubMed] [Google Scholar]
- 29.Doneanu CE, Chen W, Geber JC. Analysis of oligosaccharides derived from heparin by ion-pair reverse-phase chromatography/mass spectrometry. Anal Chem. 2009;81:3485–3499. doi: 10.1021/ac802770r. [DOI] [PubMed] [Google Scholar]
- 30.Wang B, Buhse LF, Al-Hakim A, Boyne MT, II, Keire DA. Characterization of currently marketed heparin products: analysis of heparin digests by RPIP-UHPLC-QTOF-MS. J Pharm Biomed Anal. 2012;67–68:42–50. doi: 10.1016/j.jpba.2012.04.033. [DOI] [PubMed] [Google Scholar]
- 31.Vann WF, Schmidt MA, Jann B, Jann K. The Structure of the Capsular Polysaccharide (K5 Antigenn) of Urinary-Tract-Infective Escherichia coli 010:K5:H4. Eur J Biochem. 1981;116:359–364. doi: 10.1111/j.1432-1033.1981.tb05343.x. [DOI] [PubMed] [Google Scholar]
- 32.Guerrini M, Naggi A, Guglieri S, Santarsiero R, Torri G. Complex glycosaminoglycans: profiling substitution patterns by two-dimensional nuclear magnetic resonance spectroscopy. Anal Biochem. 2005;337:35–47. doi: 10.1016/j.ab.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 33.Henriksen J, Ringborg LH, Roepstorrf P. On-line size-exclusion chromatography/mass spectrometry of low molecular mass heparin. J Mass Spec. 2004;39:1305–1312. doi: 10.1002/jms.723. [DOI] [PubMed] [Google Scholar]
- 34.Bisio A, Vecchietti D, Citterio L, Guerrini M, Raman R, Bertini S, Eisele G, Naggi A, Sasisekharan R. Structural features of low-molecular weight heparins affecting their affinity to antithrombin. Thromb Haemost. 2009;102:865–873. doi: 10.1160/TH09-02-0081. [DOI] [PubMed] [Google Scholar]
- 35.Shively JE, Conrad HE. Stoichiometry of the Nitrous Acid Deaminative Cleavage of Model Amino Sugar Glycosides and Glycosaminoglycuronans. Biochem. 1970;9:33–43. doi: 10.1021/bi00803a005. [DOI] [PubMed] [Google Scholar]
- 36.Baer HH, Astles DJ, Chin H-C, Siemsen L. The formation of branched-chain deoxypentofuranosides by ring contraction in the reductive desulfonyloxylation of hexopyranoside p-toluenesulfonates. Can J Chem. 1985;63:432–439. [Google Scholar]
- 37.Horton D, Philips KD. The nitrous acid deamination of glycosides and acetates of 2-amino-2-deoxy-D-glucose. Carbohyd Res. 1973;30:367–374. [Google Scholar]
- 38.Thanawiroon C, Rice KG, Toida T, Linhardt RJ. Liquid chromatography/mass spectrometry sequencing approach for highly sulfated heparin-derived oligosaccharides. J Biol Chem. 2004;279:2608–2615. doi: 10.1074/jbc.M304772200. [DOI] [PubMed] [Google Scholar]
- 39.Rudd TR, Macchi E, Muzi L, Ferro M, Gaudesi D, Torri G, Casu B, Guerrini M, Yates EA. Unravelling structural information from complex mixtures utilising correlation spectroscopy applied to HSQC spectra. Anal Chem. 2013 doi: 10.1021/ac4014379. [DOI] [PubMed] [Google Scholar]
- 40.Lindahl U, Feingold DS, Roden L. Biosynthesis of heparin. Trends Biochem Sci. 1986;11:221–225. [Google Scholar]
- 41.Tone Y, Pedersen LC, Yamamoto T, Izumikawa T, Kitagawa H, Nishihara J, Tamura J, Negishi M, Sugahara K. 2-O-Phosporylation of xylose and 6-O-sulfation of galactose in the protein linkage region of glucosaminoglycans influence the glucuronyltransferase-I activity involved in the linkage region synthesis. JBC. 2008;283:16801–16807. doi: 10.1074/jbc.M709556200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yates EA, Santini F, Guerrini M, Naggi A, Torri G, Casu B. 1H and 13C NMR spectral assignments of the major sequences of twelve systematically modified heparin derivatives. Carbohydr Res. 1996;294:15–27. doi: 10.1016/s0008-6215(96)90611-4. [DOI] [PubMed] [Google Scholar]
- 43.Viskov C, Mourier P. US Pat Appl US 2004/0265943 A1 Method for quantitatively determining specific groups constituting heparins of low molecular weight heparins. 2004
- 44.Ozug J, Wudyka S, Gunay NS, Beccati D, Lansing J, Wang J, Capila I, Shriver Z, Kaundinya GV. Structural elucidation of the tetrasaccharide pool in enoxaparin sodium. Anal Bioanal Chem. 2012;403:2733–2744. doi: 10.1007/s00216-012-6045-0. [DOI] [PubMed] [Google Scholar]
- 45.Xiao Z, Zhao W, Yang B, Zhang Z, Guan H, Linhardt RJ. Heparinase 1 selectivity for the 3,6-di-O-sulfo-2-deoxy-2-sulfamido-α-D-glucopyranose (1,4) 2-O-sulfo-αL-idopyranosyluronic acid (GlcNS3S6S-IdoA2S) linkages. Glycobiol. 2011;21:13–22. doi: 10.1093/glycob/cwq123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shriver Z, Sundaram M, Venkataraman G, Fareed J, Linhardt R, Biemann K, Sasisekharan R. Cleavage of the antithrombin III binding site by heparinases and its implication in the generation of low-molecular weight heparin. Proc Natl Acad Sci USA. 2000;97:10365–10370. doi: 10.1073/pnas.97.19.10365. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.