

Summary
Haemophilia replacement factors, both plasma-derived and recombinant, are in relatively short supply and are high-cost products. This has stymied the study and development of alternative methods of administration of haemophilia therapy even in the most economically advanced countries, owing to the large amounts of material needed because bioabsorption and bioavailability of haemophilic factors can be less than 10% when using non-intravenous routes of delivery. There is therefore a need to increase access to therapy worldwide by decreasing the cost and increasing the abundance so that therapy can be achieved through simplified, alternative delivery methods. Transgenic livestock have been used to produce haemophilic factors in milk. Only the pig mammary gland has been shown to carry out the post-translational processing necessary to enable both the biological activity and long circulation half-life needed for therapeutic glycoproteins. Furthermore, the large amounts of recombinant protein that can be produced from pig milk make feasible the use of alternative delivery methods such as oral, intratracheal, subcutaneous, and intramuscular administration.
Keywords: factor IX, haemophilia, transgenic animal
Introduction
The haemophilias are congenital disorders characterized by frequent bleeding episodes, especially into the joints and muscles, that cause severe arthritis and crippling. Untreated severe haemophilia patients rarely survive past adolescence. The most common haemophilias are caused by congenital deficiency in the clotting proteins factor (F) VIII (haemophilia A) or FIX (haemophilia B), which occur in about 1 : 5000 and 1 : 23 000 male children, respectively. These patients are currently treated with replacement therapy using intravenous administration of purified FVIII or FIX produced by purification from donor plasma or in cell-culture bioreactors. According to a 1995 audit in the state of California, the average healthcare cost for haemophilia patients receiving what is now considered suboptimal care was US$140 000 per patient per year of life [1], of which the overwhelming majority represents the direct cost of the clotting factor concentrate. Replacement of clotting factor after a haemorrhage is recognized as a suboptimal treatment, as some damage still ensues. Optimal therapy consists of regular replacement of the clotting factor to prevent bleeding episodes. Some of the most urgent problems that complicate the clinical situation include:
optimal care of patients in the developed countries is not possible because of the high cost and insufficient supply of clotting factors for replacement therapy;
there is currently no access to clotting factors for replacement therapy for 80% of the world’s haemophiliacs;
clotting factor shortages occur and are exacerbated by raw material shortages (i.e. plasma) and production-plant shutdowns;
there is a lack of affordable and adequate amounts of clotting factors for alternative routes of administration, such as oral dosage, which would circumvent the complications and difficulties of intravenous therapy.
The high cost of purified haemophilic factor proteins severely limits treatment in the most developed countries to therapy needed when bleeding occurs (episodic treatment) [2], and regrettably, little to no treatment for 80% of the world’s haemophilia A and B patients, who live in developing countries [3]. Episodic therapy, compared with preventative or prophylactic therapy, is suboptimal for severe haemophilia, but still requires on average at least 50 000 international units (IU) of FIX or more per patient per year [4] at an average wholesale price of US$1.18 per IU [5]. The limited supply of donor plasma, the low productivity of cell culture bioreactors, and high development costs of about US$800 000 000 for a new therapeutic product [6] contribute to the high cost of haemophilic factor proteins. As a result, the limited supply of either plasma or recombinant factors precludes the broad use of prophylactic therapy, which is now regarded as the most effective treatment to reduce the number of bleeding episodes and prevent joint damage [7,8].
This paper discusses innovative bioengineering efforts that would provide effective and affordable supplies of replacement clotting factors and thus make possible methods of administration other than intravenous injection to treat haemophilia. FIX is used as a detailed example, but it is recognized that current research into production and alternative methods of delivery for FVIII closely parallel that of FIX.
Molecular difficulties and ramifications in the production of haemophilic factors
Several fundamental biochemical constraints are encountered in the production of recombinant human FIX that make it a scarce protein therapeutic. These constraints lie not only in the relationship between the complexity of FIX function and its structure, but also in the limited productivity that is inherent to animal cells cultured on a large scale. The biological activity of FIX requires a complex molecular structure that contains post-translational modifications. Figure 1 shows the salient molecular features of FIX. FIX is a member of the vitamin K-dependent family of proteins distinguished by the post-translational γ-carboxylation of glutamate (Glu) to γ-carboxyglutamate (Gla). This post-translational modification requires vitamin K as a cofactor and the FIX propeptide as the binding site for the carboxylase enzyme complex [9]. After the N-terminal glutamates are γ-carboxylated, the pro-peptide is removed. Both of these molecular processing steps are required for biological activity. In terms of a therapeutic product specification, FIX products must have at least 6 γ-carboxyglutamates per molecule to be functional [10]. The complexity of these post-translational modifications necessitates production of biologically active FIX in mammalian cells. However, very few mammalian cells are capable of making these post-translational modifications at rates sufficient for commercial-scale production.
Fig. 1.

FIX amino acid sequence, indicating the propeptide (Thr-18–Arg1), post-translational modifications in the “Gladomain” (Tyr1–Glu40), and the activation peptide (Ala146–Arg180).
Because of the rate limitations in post-translational modification, animal cells will produce a mixture of FIX molecules. Purification processing is then used to enrich the FIX mixture with species having the most desirable therapeutic properties. BeneFix® manufacture provides a good illustration. BeneFix® is the only recombinant human FIX product approved by the United States Food and Drug Administration (USFDA), and it is produced in Chinese hamster ovary cells. The source material collected from genetically engineered cell culture is widely diverse in the extent and variety of post-translational modification of the FIX molecules; nevertheless, subsequent purification processes yield a highly efficacious and safe therapeutic. The problem of having a mixture of biologically active and inactive FIX populations was solved by designing a purification process consisting of four chromatographic steps designed to purify and select for γ-carboxylated recombinant FIX [11]. Briefly, highly purified BeneFix® contains a mixture of subpopulations with 10, 11 and 12 γ-carboxyglutamates [12]. This is consistent with the reported limitations in the ability of Chinese hamster ovary cells to completely γ-carboxylate FIX [10]. Another salient feature of BeneFix® is that, unlike plasma-derived FIX products, it contains essentially no phosphorylation at Ser158, and only 15% sulphation of Tyr155 [12]. Pharmacokinetic studies in humans indicate that this lack of phosphorylation and sulphation may lead to a modestly lower initial recovery of activity in plasma after intravenous infusion for BeneFix®, compared with plasma-derived FIX [13].
Other post-translational modifications made to FIX include β-hydroxylation of Asp63, two N-linked glycosylation sites at Asn157 and Asn167 within the activation peptide, and four O-linked glycosylation sites at Ser53, Ser61, Thr159, and Thr169 (Fig. 1). The specific contributions of these post-translational modifications to FIX biological activity have not yet been determined. However, the presence of terminal sialic acid moieties on N-linked oligosaccharides has been shown to significantly improve the circulation half-life of therapeutic proteins [14]. Plasma-derived FIX has complex N-linked oligosaccharides that are tri-and tetra-antennary and terminated with sialic acid (N-acetylneuraminic acid) [15]. N-linked oligosaccharides of BeneFix® are similar to plasma-derived FIX [12].
The production of FVIII by genetically engineered animal cells is even more highly constrained by molecular complexity than is FIX [16]. For example, FVIII is limited in its rate of secretion, which is related to its post-translational structure. Post-translational structures arising from proteolytic processing, sulphation, and glycosylation each contribute to FVIII biological function. Some of these rate limitations can be ameliorated by molecular engineering of the primary structure of the FVIII molecule while still maintaining or even improving the function and stability [17]. However, even with these improvements, the rate of synthesis and secretion of FVIII by any animal cell is arguably one of the most difficult tasks that has been encountered in recombinant protein expression. Thus, other improvements in production technology must be made to make FVIII abundant and less costly, such as increasing the concentration of cells in the bioreactor.
Large-scale production characteristics of haemophilic factors made using plasma, cell culture or transgenic mammary tissue
The primary cause of the current scarcity of haemophilic factors is that only low concentrations of FVIII and FIX occur in human plasma, and only limited concentrations can be produced in the bioreactor culture of genetically engineered animal cells. FVIII is found in plasma at about 0.2 μg mL−1 and FIX at only 5 μg mL−1. The exact concentrations of FVIII made in animal cell bioreactors at production scale are not published by manufacturers, but are likely to be much less than 10 μg mL−1 accumulated after about every 24–48 h of production phase. FIX is also likely to be produced at similarly low concentrations by large-scale bioreactors. The low concentration of recombinant haemophilic factor protein made by current animal-cell culture technology is due to two process limitations: a low concentration of cells of about 5 × 106 cells mL−1 of reactor volume and low production of protein made by each animal cell. While on a per cell basis, recombinant protein production has been increased by genetic engineering of both the protein and the cell itself, very little progress has been made in raising the concentration of cells that can be cultured at manufacturing scale.
The resultant low concentration of these haemophilic factors in the bioreactor dictates that downstream processing must both concentrate and purify the final product at very high cost [18]. Typically, the overall production costs will be on the order of about US$10 000 or more per gramme of recombinant protein. For FVIII and FIX, this translates to a direct manufacturing cost on order of US$0.10 per unit. More importantly, the exceedingly large production volumes needed to satisfy clinical demands require very costly investments in manufacturing facilities. Specifically, a large portion of the cost is associated with both establishing and maintaining the validated status of large-scale processes needed to consistently produce material that will meet federal regulatory requirements [6,19]. The limitations in current manufacturing capacity have a serious impact on haemophilia therapy. For example, even though about 90% of patients in the USA receive recombinant FIX therapy, only 13% of the children with haemophilia B in the USA receive prophylaxis [20].
The projected process volumes needed to satisfy the estimated clinical demand for prophylactic intravenous therapy for FIX in the USA translate to large capital investment costs in new manufacturing facilities [19]. Figure 2 compares the estimated amounts of FIX source materials needed to meet the demand for prophylactic intravenous therapy for the estimated 3000 patients at a level of 200 000 IU patient−1 year−1. To satisfy this demand using plasma-derived FIX would require processing over 1 million litres of plasma per year. A comparable amount of recombinant FIX produced using industry-standard cell-culture bioreactors would require 600 000 L of supernatant per year and greater than US$50 000 000 capital investment. In contrast to both plasma and animal cell culture, only 60 genetically engineered pigs that produce FIX in their milk would be needed to supply prophylactic intravenous treatment to the 3000 patients in the USA.
Fig. 2.
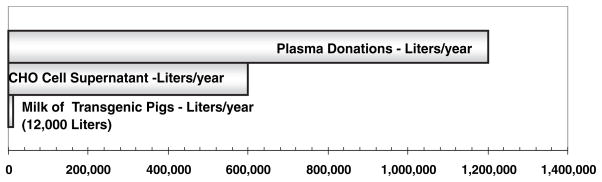
Volumes of source materials for FIX required to meet the needs for prophylactic therapy of all haemophilia B patients in the USA (3000 patients @200 000 IU per patient per year). The amount of FIX in each source is – plasma (1 IU mL−1); CHO cell bioreactor (2 IU mL−1) and transgenic pig milk (100 IU mL−1, Lindsay et al. 2004). A 50% purification yield for each source is assumed. The milk of 60 pigs (12 000 L year−1) could supply the entire amount of FIX needed for prophylaxis in the USA.
Transgenic animal production of haemophilic factors
The milk of transgenic livestock can be a prodigious and expedient source of complex therapeutic proteins such as recombinant haemophilic factors relative to cell culture bioreactors [21]. When considered as potential bioreactors, mammary tissues can produce high concentrations of secreted proteins, owing to the high cell density of the gland, with about 109 cells mL−1 of tissue. This cell density is 2–3 orders of magnitude greater than that possible in mammalian cell-culture production technology used to make complex recombinant therapeutic proteins. The result is remarkable protein production rates of about 1–15 g L−1h−1 in milk. The specific choice of making haemophilic factors in the pig rather than other livestock is based in part upon the combination of high volume of milk that can be harvested per day (1–3 L) and the quality of post-translational processing of complex proteins by pig mammary cells relative to other animal cells. A transgenic pig typically yields about 0.4–1.25 L per milking. Pigs can be milked up to five times per day over two 50-day lactations per year, and so the total milk yield from the average sow is 100–300 L year−1 [21]. Even more important than the consideration of milk volume, however, is the fact that the mammary epithelial cells of the pig are unique among livestock in making the complex post-translational modifications needed for FIX [22] and FVIII [23] biological activity. From a broad biochemical and physiological perspective, the pig is more closely related to humans than the ruminants, which include the cow, sheep, and goat. As an example of the differences between ruminant and pig biochemistry, recombinant FIX production in sheep was unsuccessful, with only low levels of biologically active FIX being produced: less than 0.5 ng of biologically active FIX could be recovered from each millilitre of milk [24]. The sheep was even less productive than donor plasma at 0.0001 U mL−1 of milk. In contrast, recombinant human FIX has been produced at levels up to 100 U mL−1 of milk in transgenic pigs [22], and proper proteolytic cleavage of recombinant FVIII to a mature two-chain form has been demonstrated in the milk [23]. With respect to N-linked glycans that are needed for long circulation half-life in therapeutic proteins [14], endogenous porcine milk glycoproteins such as lactoferrin contain complex glycans terminated with N-acetylneuraminic acid (sialic acid) [25]. In contrast, ruminant lactoferrins contain undesirable high-mannose glycans [25], and recombinant antithrombin III produced in transgenic goats also contained high-mannose glycans [26]. Thus, ruminants are probably unable to make a recombinant FIX or FVIII with the glycosylation needed for a long circulation half-life.
The safety requirements for USFDA approval of a transgenic animal supply of recombinant haemophilic factors
The regulatory concerns for production of haemophilic factors derived from transgenic animals have been largely delineated. In recognition of the feasibility of this production method, the USFDA has issued a document, Points to Consider in the Manufacture and Testing of Therapeutic Products for Human Use Derived from Transgenic Animals. Importantly, pioneering work on the safety and efficacy of recombinant antithrombin III produced in the milk of transgenic animals has been demonstrated in clinical trials by Genzyme Transgenics Corporation [27]. To reduce risks of disease transmission, multiple barriers (chromatography, solvent/detergent treatment, and nanofiltration) can be placed in the purification process to provide logs of pathogen removal and/or inactivation of common classes of lipid-enveloped and non-lipid-enveloped viruses. Maintenance of specific pathogen-free production herds is also an important requirement. Many of these steps are currently in place for many human plasma-derived FIX products, animal cell-derived products such as BeneFix®, and animal products such as heparin and porcine FVIII (Hyate:C®). The process technology for implementing these steps with transgenic animal-derived products has been demonstrated to be directly transferable [28].
Clinical experience with the porcine-derived FVIII product Hyate:C® is instructive for assessing and reducing the risks involved in zoonosis from porcine derived therapeutic protein products. Hyate:C® is a product alternative used to treat haemophilia A patients who develop an immune response to plasma-derived or recombinant human FVIII. These patients produce antibodies (inhibitors) that neutralize the activity of these FVIII products. Hyate:C® was originally produced using largely unregulated slaughterhouse pigs, and had been given to patients since 1984 without any viral-inactivation steps in the purification process. In 1996, detection of porcine parvovirus (PPV) DNA in multiple batches halted production. In a comprehensive retrospective study of haemophiliacs receiving Hyate:C® prior to 1996, it was concluded there was no evidence of transmission of PV, encephalomyocarditis virus, or porcine respiratory and reproductive syndrome virus to patients receiving Hyate:C® [29]. Thus, the threat of the most common pathogens that occur in pigs has been evaluated for a haemophilia therapy derived specifically from pigs having exposure to these diseases. Another example of a safety concern for parenteral therapeutics is transmissible spongiform encephalopathies such as bovine spongiform encephalopathy (BSE). It has been recently shown that swine are resistant to nutritional challenges of highly infective tissue homogenates from BSE-infected cattle [30]. In addition, specific pathogen-free transgenic production herds can be fed only plant-derived diets.
Alternative delivery routes for FVIII and FIX enabled by abundant supply
Alternative delivery methods that increase the quality of life for haemophilia patients might be made possible if an abundance of haemophilic factors were available. While these alternatives to intravenous infusion increase the ease of administration, they decrease the bioavailability, and therefore their application is limited by the lack of abundance of haemophilic factors. For example, aerosolization of FIX may provide an alternative, needle-free therapeutic option for delivery of FIX across the epithelial cell layer in the lungs [31]. This recent study demonstrated that bioavailabilities of 11% (relative to intravenous administration) were obtained in haemophilia B dogs using 200 IU kg−1-bodyweight intratracheal dosing regimen. The pharmacokinetic results were consistent with a slow release from a deposition site in the lungs. This intratracheal administration produced therapeutic levels of both antigen and activity, and the activity was detected through 72 h post-administration. In addition, there has been a group of studies to determine the absorption rate and bioavailability of plasma-derived FIX administered through other extravascular routes: subcutaneous, intramuscular, and intraperitoneal [32,33]. While all of these routes resulted in measurable FIX activity, the subcutaneous and intramuscular injections resulted in bioavailabilities of 63.5% and 82.8%, respectively, in the haemophilia B dog model [32]. Subcutaneous administration of high concentrations of up to 4000 IU mL−1 resulted in bioavailabilities of 35% in cynomolgus monkeys and 46% in haemophilia B dogs [33]. Importantly, while all of these extravascular delivery routes are less efficient than intravenous, they provide a combination of simplicity of administration and sustained FIX activity. These advantages may be greatest when applied to factor prophylaxis in children, for whom intravenous access is most troublesome, the level of physical activity (and coincident risk of bleeding) greatest, and yet the total dose requirements are smaller, as they are based on bodyweight. Thus, if an abundant and economical source of FIX were available, then the advantage of clinical simplicity becomes a more important and feasible consideration.
Oral dosage formulations
An oral dosage form of FVIII or FIX may provide the most facile and convenient delivery, especially in children, in whom intravenous delivery is problematic. Oral dosage is feasible only when abundance and cost are favourable, and low patient plasma levels are needed to achieve effective therapy. In general, low bioavailability of oral protein dosage forms arise from degradation within the gastrointestinal environment and the resistance of high molecular weight proteins to transport through the mucus gel layer and epithelia and into the bloodstream. The inefficiency due to the gastrointestinal environment can be mitigated by the following: modifying the pH of the stomach; competitively inhibiting pancreatic proteases with inhibitors such as soybean trypsin inhibitor, aprotinin, or casein; and high dosage regimens. A low efficiency of FVIII or FIX absorption will be less cost-intensive in children and infants compared with adults because total body weight is low, and therefore total units needed to reach therapeutic levels is lower. For example, in the case of FIX and FVIII, only about 5% of normal plasma levels are needed to prevent spontaneous bleeding and chronic recurrent arthropathy. This translates to only about 250 and 12 ng mL−1 of plasma for FIX and FVIII, respectively. These levels have been achieved in human and dog plasma using liposome and enzyme-inhibitor formulations of oral dosages of human plasma-derived FVIII and FIX [34,35].
Hemker et al. [34] formulated plasma-derived FVIII in liposomes, administered a dose of 290 U to a haemophilia A adult patient, and obtained greater than 5% FVIII activity in plasma for 50 h after dosing. The profile is consistent with a slow absorption process resulting in a broad plasma-level response. A broad plateau of 5% activity or greater sustained over more than 48 h is desirable. Horikoshi et al. [35] also used a liposomal formulation, administered a dose of 14 U of plasma-derived FIX kg−1 bodyweight in a 9.2-kg dog, and measured 125% of normal FIX activity by activated partial throboplastin time. In both these FVIII and FIX oral-dosage studies, however, sufficient numbers of animal or human subjects were not investigated to assess the variability in bioavailability that would be expected using oral dosage. It is likely that at that time, the supply of plasma-derived materials precluded the effective parametric study of appropriate formulations to determine optimum bioavailability.
Conclusions
The relationship of the structure and function of FVIII and FIX requires they be made in a mammalian cell. The pig mammary epithelial cell can make the post-translational modifications needed for a functional, recombinant human FIX with desirable pharmacokinetic properties. The high cell density of the mammary gland enables prodigious concentrations of FIX to be made in the milk. The low milk volumes required would enable a rapid scale-up to manufacturing an abundant supply of FIX that would meet clinical demand for prophylactic treatment that is not currently being met by conventional animal cell bioreactor technology. Recognition of the potential for transgenic animal production of haemophilic factors was recently confirmed by the Medical and Scientific Advisory Committee (MASAC) of the United States National Haemophilia Foundation. They state in their Recommendations [36] that manufacturers should strive to make coagulation products less expensive and that methods such as the use of transgenic animals would increase supply and availability. It is also clear that alternative delivery methods that dramatically improve the ease of treatment and quality of patient life, particularly for children, will be feasible if an abundance of haemophilic factors becomes available. This may apply even more strongly to locations where patient care and compliance are more challenging due to economic conditions.
References
- 1.Globe DR, Curtis RG, Koerper MA. Utilization of care in haemophilia: a resource-based method for cost analysis from the Haemophilia Utilization Group Study (HUGS) Haemophilia. 2004;10 (Suppl 1):63–70. doi: 10.1111/j.1355-0691.2004.00881.x. [DOI] [PubMed] [Google Scholar]
- 2.Fischer K, Van Der Bom JG, Molho P, et al. Prophylactic versus on-demand treatment strategies for severe haemophilia: a comparison of costs and long-term outcome. Haemophilia. 2002;8:745–52. doi: 10.1046/j.1365-2516.2002.00695.x. [DOI] [PubMed] [Google Scholar]
- 3.Mannucci PM. Hemophilia: treatment options in the twenty first century. J Thromb Haemost. 2003;1:1349–55. doi: 10.1046/j.1538-7836.2003.00262.x. [DOI] [PubMed] [Google Scholar]
- 4.Linden JV, Kolakoski MH, Du Lima JEP, Lipton RA. Factor concentrate usage in persons with hemophilia in New York State. Transfusion. 2003;43:470–5. doi: 10.1046/j.1537-2995.2003.00343.x. [DOI] [PubMed] [Google Scholar]
- 5.Drug Topics Red Book. Montvale, NJ: Medical Economics; 2003. [Google Scholar]
- 6.Frantz S. Why are clinical costs so high? Nat Rev Drug Discov. 2003;2:851–2. [Google Scholar]
- 7.Miners AH, Sabin CA, Tolley KH, Lee CA. Primary prophylaxis for individuals with severe haemophilia: how many hospital visits could treatment prevent? J Intern Med. 2000;247:493–9. doi: 10.1046/j.1365-2796.2000.00633.x. [DOI] [PubMed] [Google Scholar]
- 8.Panicker J, Warrier I, Thomas R, Lusher JM. The overall effectiveness of prophylaxis in severe haemophilia. Haemophilia. 2003;9:272–8. doi: 10.1046/j.1365-2516.2003.00757.x. [DOI] [PubMed] [Google Scholar]
- 9.Suttie JW. Synthesis of vitamin K-dependent proteins. FASEB J. 1993;7:445–52. doi: 10.1096/fasebj.7.5.8462786. [DOI] [PubMed] [Google Scholar]
- 10.Kaufman RJ, Wasley LC, Furie BC, Furie B, Shoemaker CB. Expression, purification, and characterization of recombinant gamma-carboxylated factor IX synthesized in Chinese hamster ovary cells. J Biol Chem. 1986;261:9622–8. [PubMed] [Google Scholar]
- 11.Harrison S, Adamson S, Bonam D, et al. The manufacturing process for recombinant factor IX. Semin Hematol. 1998;35 (Suppl 2):4–10. [PubMed] [Google Scholar]
- 12.Bond M, Jankowski M, Patel H, et al. Biochemical characterization of recombinant factor IX. Semin Hematol. 1998;35 (Suppl 2):11–7. [PubMed] [Google Scholar]
- 13.Ewenstein BM, Joist JH, Shapiro AD, et al. Pharmacokinetic analysis of plasma-derived and recombinant F IX concentrates in previously treated patients with moderate or severe hemophilia B. Transfusion. 2002;42:190–7. doi: 10.1046/j.1537-2995.2002.00039.x. [DOI] [PubMed] [Google Scholar]
- 14.Varki A, Cummings R, Jeffrey Esko, Freeze H, Hart G, Marth J. Essentials of Glycobiology. Plainview (NY): Cold Spring Harbor Laboratory Press; 1999. [PubMed] [Google Scholar]
- 15.Makino Y, Omichi K, Kuraya N, et al. Structural analysis of N-linked sugar chains of human blood clotting factor IX. J Biochem (Tokyo) 2000;128:175–80. doi: 10.1093/oxfordjournals.jbchem.a022738. [DOI] [PubMed] [Google Scholar]
- 16.Miao HZ, Sirachainan N, Palmer L, et al. Bioengineering of coagulation factor VIII for improved secretion. Blood. 2004;103:3412–9. doi: 10.1182/blood-2003-10-3591. [DOI] [PubMed] [Google Scholar]
- 17.Tendulkar R, Pipe SW, Miao H, Kaufman RJ. Asparagine-linked glycosylation sites within the B domain of coagulation Factor VIII improve secretion efficiency. Blood. 2001;98:705. [Google Scholar]
- 18.Garber K. rFactor VIII deficit questioned. Nat Biotechnol. 2000;18:1133. doi: 10.1038/81098. [DOI] [PubMed] [Google Scholar]
- 19.Garber K. Biotech industry faces new bottleneck. Nat Biotechnol. 2001;19:184–5. doi: 10.1038/85559. [DOI] [PubMed] [Google Scholar]
- 20.Blanchette VS, McCready M, Achonu C, Abdolell M, Rivard G, Manco-Johnson MJ. A survey of factor prophylaxis in boys with haemophilia followed in North American haemophilia treatment centres. Haemophilia. 2003;9 (Suppl 1):19–26. doi: 10.1046/j.1365-2516.9.s1.12.x. [DOI] [PubMed] [Google Scholar]
- 21.Lubon H, Paleyanda RK, Velander WH, Drohan WN. Blood proteins from transgenic animal bioreactors. Transfus Med Rev. 1996;10:131–43. doi: 10.1016/s0887-7963(96)80089-7. [DOI] [PubMed] [Google Scholar]
- 22.Lindsay M, Gil G, Zhang C, Cadiz A, Velander WH, Van Cott KE. Purification of recombinant DNA-derived Factor IX and fractionation of active and inactive subpopulations. J Chromatogr. 2004;1026:149–57. doi: 10.1016/j.chroma.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 23.Paleyanda RK, Velander WH, Lee TK, et al. Transgenic pigs produce functional human factor VIII in milk. Nat Biotechnol. 1997;15:971–5. doi: 10.1038/nbt1097-971. [DOI] [PubMed] [Google Scholar]
- 24.Clark AJ, Bessos H, Bishop JO, et al. Expression of human anti-hemophilic factor IX in the milk of transgenic sheep. Biotechnology. 1989;7:487–92. [Google Scholar]
- 25.Spik G, Coddeville B, Mazurier J, Bourne Y, Cambillaut C, Montreuil J. Primary and three-dimensional structure of lactotransferrin (lactoferrin) glycans. Adv Exp Med Biol. 1994;357:21–32. doi: 10.1007/978-1-4615-2548-6_3. [DOI] [PubMed] [Google Scholar]
- 26.Edmunds T, Van Patten SM, Pollock J, et al. Trans-genically produced human antithrombin: structural and functional comparison to human plasma-derived antithrombin. Blood. 1998;91:4561–71. [PubMed] [Google Scholar]
- 27.Levy JH, Weisinger A, Ziomek CA, Echelard Y. Recombinant antithrombin: production and role in cardiovascular disorder. Semin Thromb Hemost. 2001;27:405–16. doi: 10.1055/s-2001-16893. [DOI] [PubMed] [Google Scholar]
- 28.Ziomek CA. Minimization of viral contamination in human pharmaceuticals produced in the milk of transgenic goats. Dev Biol Stand. 1996;88:265–8. [PubMed] [Google Scholar]
- 29.Giangrande PL, Kessler CM, Jenkins CE, Weatherill PJ, Webb PD. Viral pharmacovigilance study of haemophiliacs receiving porcine factor VIII. Haemophilia. 2002;8:798–801. doi: 10.1046/j.1365-2516.2002.00698.x. [DOI] [PubMed] [Google Scholar]
- 30.Wells GA, Hawkins SA, Austin AR, et al. Studies of the transmissibility of the agent of bovine spongiform encephalopathy to pigs. J General Virol. 2003;84:1021–31. doi: 10.1099/vir.0.18788-0. [DOI] [PubMed] [Google Scholar]
- 31.Russell KE, Read MS, Bellinger DA, et al. Intratracheal administration of recombinant human factor IX (BeneFix) achieves therapeutic levels in hemophilia B dogs. Thromb Haemost. 2001;85:445–9. [PubMed] [Google Scholar]
- 32.Liles D, Landen CN, Monroe DM, et al. Extravascular administration of factor IX: potential for replacement therapy of canine and human hemophilia B. Thromb Haemost. 1997;77:944–8. [PubMed] [Google Scholar]
- 33.McCarthy K, Stewart P, Sigman J, et al. Pharmacokinetics of recombinant factor IX after intravenous and subcutaneous administration in dogs and cynomolgus monkeys. Thromb Haemost. 2002;87:824–30. [PubMed] [Google Scholar]
- 34.Hemker HC, Hermens WT, Muller AD, Zwaal RF. Oral treatment of haemophilia A by gastrointestinal absorption of factor VIII entrapped in liposomes. Lancet. 1980;1:70–1. doi: 10.1016/s0140-6736(80)90495-x. [DOI] [PubMed] [Google Scholar]
- 35.Horikoshi I, Sakuragawa N, Ueno M, Takahashi K. US Patent 4183960. Pharmaceutical Composition for Oral Administration Containing Coagulation Factor VIII or IX. 1982
- 36.National Hemophilia Foundation. Research and development of improved coagulation products that would expedite the transition to total prophylaxis for all persons with coagulation disorders are strongly encouraged. Available at: http://www.hemophilia.org/research/masac/masac151.htm.