

Abstract
Major histocompatibility complex (MHC) molecules play a key roll in cell-mediated immune responses presenting bounded peptides for recognition by the immune system cells. Several in-silico methods have been developed to predict the binding affinity of a given peptide to a specific MHC molecule. One of current in-state-of-art methods for MHC class I is NetMHCpan, which has as a core ingredient the representation of the MHC class I molecule using a pseudo sequence representation of the binding cleft amino acid environment. New and large MHC-peptide binding data sets are constantly being made available and also new structures of MHC class I molecules with bound peptide have been published. In order to test if the NetMHCpan method can be improved by integrating this novel information, we created new pseudo-sequence definitions for the MHC binding cleft environment from sequence and structural analysis of different MHC data sets including human (HLA), non-human primates and other animal alleles (cattle, mouse and swine). From these constructs, we demonstrated that by focusing on MHC sequence positions found to be polymorphic across the MHC molecules used to train the method, the NetMHCpan method achieved a significant increase in predictive performance in particular for non-human MHCs. This study hence demonstrated that an improved performance of MHC binding methods can be achieved not only by accumulation of more MHC-peptide binding data, but also by a refined definition of the MHC binding environment including information from non-human species.
Introduction
Proteins are the essential immune-target structures, which in the MHC class I (MHC-I) pathway are processed to 8-11mer peptides. In this way, peptides that bind to MHC-I molecules are presented and potentially recognized by cytotoxic T cells, which can lead to an immune response. The most selective step in this antigen presentation is the peptide binding to MHC (1).
Each MHC molecule has a potentially unique binding affinity motif (2) and the characterization of this motif for each MHC molecule prevalent in a given population is a central aspect of rational T cell epitope discovery. Due to the immense MHC-I polymorphism (3, 4), an exhaustive characterization of all MHC molecules is a high cost-intensive effort, and as of today in spite of significant advances in high-throughput immune assays only a little more than 100 MHC-I molecules, including 25 non-human molecules have been experimentally characterized at a detail allowing to describe their binding specificity (IEDB date 2012).
To face this problem, several in silico prediction methods have been developed in the last decades (5-12), reviewed in (13). Of these methods, NetMHCpan is the current in state-of the art method (14) for predicting binding affinity of peptides to any MHC-I molecule with a know protein sequence (7). A core ingredient of the NetMHCpan method is the definition of the so-called pseudo sequence defined from the binding cleft amino acid environment of each MHC molecule. In the original NetMHCpan method, this pseudo-sequence was defined from the set of polymorphic residue positions in a set of human MHC crystal structures and sequences available at the time of the study in potential contact with the bound peptide, comprising residues within a distance of 4.0 Å between any pair of atoms from the MHC complex and the bounded peptide. However, since the original NetMHCpan publication, large novel data sets have become available not only for human MHC alleles but also for non-human alleles. Furthermore, the number of crystal structures has increased for human and non-human MHC molecules potentially expanding our definition of which MHC positions can potentially interact with a bound peptide and which positions are polymorphic. It hence seems very likely that the positions defining the pseudo sequence could be altered when investigating the most recent data.
In this study, we investigate how the many novel structural and sequence data available for MHC-I impact the definition of the NetMHCpan pseudo sequence, and how these impacts would alter the predictive performance of the method. We proposed alternative definitions of the pseudo-sequence by analyzing the binding cleft of MHC class I molecule with a bound peptide in 25 different crystallized structures, including eight non-human alleles and compared these constructs with the original pseudo-sequence from the human complex structures used in the original definition of the pseudo-sequence (7). Next, we analyze the impact on the predictive performance of the pan-specific method when novel polymorphic in potential contact positions are incorporated in the pseudo-sequences, and finally evaluate whether this impact has a bias non-human MHC molecules where the difference between the “old” and “new” MHC data is most pronounced.
Materials and Methods
Data sets
The peptide-MHC binding data consisted of 128,935 quantitative nonameric peptide-MHC class I binding data obtained from the IEDB Database (15) and an in-house database. In total, 119 MHC alleles (39 HLA-A, 41 HLA-B, six HLA-C, 11 chimpanzee (Patr), one gorilla (Gogo), 11 Rhesus macaque (Mamu), two cattle (BoLA), six mouse (H-2) and two swine (SLA) alleles) were considered. Binding affinity measurements were obtained as IC50 values and transformed to fall in the range between 0 and 1 using the transformation 1-log(IC50nM)/log(50,000).
HLA sequences were downloaded from the IMGT/HLA Database (3) and animal MHC class I sequences were obtained from the IPD-MHC (4) and the Uniprot (16) databases. The MHC-I sequences used in this study are summarized in table 1.
Table 1.
Number of MHC class I sequences from IMGT/HLA and IPC-MHC Databases.
| MHC class I | Gene | Number of Alleles |
|---|---|---|
| HLA | A | 1388 |
| B | 1909 | |
| C | 1007 | |
| Patr | A | 33 |
| AL | 3 | |
| B | 55 | |
| C | 29 | |
| E | 1 | |
| F | 1 | |
| H | 1 | |
| Gogo | A | 4 |
| B | 9 | |
| C | 7 | |
| E | 1 | |
| H | 1 | |
| Mamu | A | 282 |
| B | 327 | |
| E | 17 | |
| SLA | 1 | 11 |
| 2 | 13 | |
| 3 | 14 | |
| 6 | 9 | |
| BoLA | N | 107 |
| H-2 | Db | 1 |
| Dd | 1 | |
| kb | 1 | |
| kd | 1 | |
| kk | 1 | |
| Ld | 1 |
Available crystal structures of MHC-peptide complexes from different species were downloaded from RCSB PDB. Only alleles containing 9-mer peptides interacting with the MHC were used in this study. In the case of alleles with more than one structure, one representative structure was selected based on the crystal with the most favorable resolution and B-factor. The structure data set is presented in table 2.
Table 2.
Data set consisting of available crystal structures and the PDB ID of the representative structure per allele.
| Allele | Number of available crystal structures |
PDB ID |
|---|---|---|
| HLA-A*01:01 | 1 | 3BO8 |
| HLA-A*02:01 | 48 | 3D25 |
| HLA-A*03:01 | 1 | 2XPG |
| HLA-A*11:01 | 1 | 1X7Q |
| HLA-A*24:02 | 2 | 3I6L |
| HLA-B*14:02 | 1 | 3BXN |
| HLA-B*15:01 | 3 | 1XR9 |
| HLA-B*27:05 | 9 | 1OGT |
| HLA-B*27:09 | 5 | 1K5N |
| HLA-B*35:01 | 2 | 2CIK |
| HLA-B*44:02 | 8 | 3KPM |
| HLA-B*44:03 | 4 | 3KPN |
| HLA-B*44:05 | 3 | 1SYV |
| HLA-B*53:01 | 2 | 1A1M |
| HLA-B*57:01 | 1 | 2RFX |
| HLA-C*03:04 | 1 | 1EFX |
| HLA-C*04:01 | 1 | 1QQD |
| Mamu-A*02 | 1 | 3JTS |
| SLA-1*0401 | 1 | 3QQ3 |
| BoLA-N*01801 | 1 | 3PWU |
| H-2-Db | 18 | 3CC5 |
| H-2-Kb | 6 | 1G7P |
| H-2-Kd | 1 | 2FWO |
| H-2-Kk | 1 | 1ZT7 |
| H-2-Ld | 2 | 1LD9 |
MHC class I pseudo-sequence
The MHC class I molecule was represented by pseudo-sequences consisting of selected amino acids from the α1 and α2 subunits of the molecule. In order to construct pseudo-sequences from the different MHC groups (see Table 3), we first identified polymorphic residues from different multiple alignment data sets: (1) 4304 HLA sequences, (2) 5075 HLA and non-human primates (Mamu, Patr and Gogo alleles) sequences, (3) all 5235 available sequences (including human, non-human primates, BoLA, H-2 and SLA alleles), and (4) 119 sequences for MHC molecules with peptide binding affinity data (hereafter referred to as the training set).
Table 3.
Average PCC and AUC for each pseudo sequence group. The PCC and AUC values are calculated as average over the set of 91 alleles having more than 50 data point and at least 5 binding peptides (affinity < 500 nM). The different pseudo sequence groups are defined as described in the text.
| Group | PCC | AUC |
|---|---|---|
| NetMHCpan | 0.752 | 0.910 |
| HLA | 0.750 | 0.908 |
| Primates | 0.734 | 0.903 |
| Primates expanded | 0.752 | 0.910 |
| Training | 0.750 | 0.910 |
| Training expanded | 0.755 | 0.913 |
MHC-I residues in potential contact with the bound peptide were identified from contact maps generated from set of the representative crystal structures. A contact residue was defined as a residue having a heavy atom within 4.0 Å from a heavy atom of a residue of the bound peptide and with its side chain pointing towards the peptide. Additionally, residues 118 and 158 were added in the pseudo-sequence constructs, even though not found to be in direct contact with the peptide (distance greater than 4.0 Å) as these residues might stabilize the peptide-MHC complex through water mediated interactions (17). In total 39 residues were found to have potential contacts with the bound peptide. Note, that this number remained unchanged when analyzing only human peptide-MHC complexes.
The final MHC class I pseudo-sequence is constructed (figure 1) as the subset of polymorphic residues found in potential contact with the bound peptide. The “HLA group” consists of the polymorphic residues from the HLA alignment that are in potential contact with the binding peptide, this group differs from the NetMHCpan pseudo-sequence definition in excluding those residues that interact via water molecules (positions 118 and 158). The second group of pseudo-sequence constructs, referred as “Primates”, includes contact positions found to be polymorphic in the alignment of HLA and non-human primate sequences. In the “Primate expanded” group, positions 118 and 158 were added to measure their impact in the performance of the method. Finally, the “Training” group is defined from contacts and polymorphisms across the 119 MHC sequences included in the training data. Also here the residues 118 and 158 were added, to from the “Training expanded” group. To quantify the degree of polymorphism at the different pseudo sequence positions we for each position calculate the Shannon information content (18) for the 1) complete set of 5235 MHC sequences, 2) the set of 119 sequences included in the training data set. The results of these calculations are included in the two lower rows in Figure 1. The Shannon information content has a value of 4.32 for fully conserved positions and a value of 0 for positions with equiprobable amino acid distribution. A structural visualization of the NetMHCpan and “Training expanded” pseudo-sequence constructs can be found in Figure 2, where the common and unique residues for each construct are highlighted.
Figure 1.

Pseudo-sequence constructs, length and differences compared to NetMHCpan group. The residues included in the pseudo sequence construct for each group are marked in grey. The two additional residue positions 118 and 158 are marked in dark grey. The last column gives the number of residues included in the given pseudo sequence construct. The Shannon Index for the Primates Expanded and Training Expanded Groups are given in the last two rows.
Figure 2.
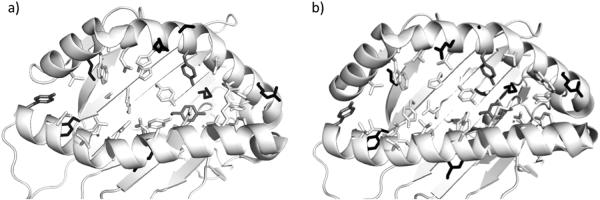
A structural visualization of the NetMHCpan and Training expanded pseudo-sequence definitions in a) Mamu A*02 (PDB ID: 3JTS) and b) HLA A*02:01 (PDB ID: 3D25) alleles. The residues shown in white sticks are found in both constructs, residues exclusive to NetMHCpan are shown in light grey and residues only included in the “Training expanded” set are shown in black.
Artificial neural network training
Artificial Neural Networks (ANN) were trained to quantitatively predict peptide-MHC class I biding affinities as described by Nielsen et al. (7). For each of the pseudo-sequence constructs, we used as input the peptide sequence, the pseudo-sequence of the respective allele and the binding affinity. The input sequences were presented to the ANN as Blosum encoding (where the BLOSUM50 matrix score vector encoded each amino acid as 20 values), as the conventional sparse encoding (where each amino acids is encoded as a vector with 20 elements, one having the value 1 and 19 the value zero) and as mixture of the two (where the peptide was sparse encoded and the pseudo-sequence was Blosum encoded). The final predictions were made as a simple ensemble average of the predictions with the three encoding schemes (7). The neural network architecture used was a feed-forward network with one hidden layer and a single output neuron. A back-propagation procedure was used to update the weights of the network. A traditional fivefold cross-validated training was performed, for each of the pseudo-sequence groups, using the same split of peptides for all the pseudo-sequence definitions in order to let the pseudo-sequence be the only parameter different between each experiment. The Pearson Correlation Coefficient (PCC) for the correlation between the experimental (target) and predicted values, and the area under the ROC curve (AUC) were calculated for each allele that had more than 50 data points with at least five binder peptides for each pseudo-sequence group (see table 3).
Ligands Benchmarking
As an independent benchmark for evaluating the performance of the different predictions methods, we used a data set consisting of 889 known 9-mer ligands with full-resolution of the HLA restriction and 33 9-11-mers non-human primate MHC restricted T cell epitopes together with information about the source protein sequence obtained from the SYFPEITHI Database (19). Also, 65 8-15-mer non-human primate peptides (22 ligands and 43 T cell epitopes) from the IEDB were analyzed in this study. The performance of the different methods was evaluated in terms of AUC values as described in (20). In short, when calculating the AUC value, the source protein of the given ligand was divided into overlapping peptides of the size of the given ligand. All peptides, except the annotated ligands were taken as negative peptides (non-ligands) and the given ligand were taken as positive. A perfect AUC value of 1.0 corresponds to the ligand having the strongest predicted binding value compared to all other possible peptides originating from the source protein. The AUC values were calculated for each ligand-protein pair using the predictor for the different pseudo sequence definitions.
Statistical Analysis
The Pearson correlation coefficient (PCC) and area under the ROC curve (AUC) values from the new methods were compared to the corresponding performance values of NetMHCpan method using a binomial test excluding ties with a 5% significance level.
Results
Five-fold cross validation
The different methods (based on the different definitions of the pseudo sequence) were trained on the set of quantitative peptide-MHC class I binding data as described in Materials and Methods. The cross-validated PCC and AUC performance values averaged over the 93 MHC molecules included in the training data are shown in table 3 (the complete list of performance values for each MHC data set are given in supplementary material table S1). Comparing the NetMHCpan and HLA methods demonstrated that the excluding residues 118 and 158 of the MHC molecule in the pseudo sequence leads to a significantly drop in the predictive performance values (p<0.05 for both PCC and AUC). There was no significant difference in the predictive performance between the Primate and HLA groups (p=0.300 for PCC and p=0.679 for AUC). Only when including the positions 118 and 158 for the Primates expanded group, did the predictive performance become similar to that of NetMHCpan (p=0.300 for PCC and p=0.534 for AUC). Focusing on the pseudo sequence definitions defined from MHC positions that are polymorphic across the training data, we find that the predictive performance of the Training group is similar to the NetMHCpan method, and that finally that the Training expanded group including the two residue positions 118 and 158 achieved the highest performance of all methods, although the performance gain was not statistically significant compared to NetMHCpan (p=0.097 and p=0.213, for the PCC and AUC respectively).
Ligands benchmark
Next we turned to the ligand benchmark. This benchmark was only performed for the NetMHCpan and “Training in contact expanded” groups. For the first benchmark, 889 9-mer HLA ligands were analyzed. Next, 32 9-11-mer non-human primate MHC class I ligands from the same database and, 65 8-15-mers non-human primate MHC class I ligands from the IEDB were tested. Final results are presented in table 4. No significant difference between the two methods was found for the HLA ligand benchmark (p=0.069). However, the predictive performance was significantly increased using the new pseudo sequence construct (Training expanded) when predicting non-human primate ligands for both the SYFPEITHI and IEDB benchmark data sets (p=0.024 and p=0.005 respectively).
Table 4.
Five-fold cross validation values (PCC and AUC) of NetMHCpan, Training Expanded, Inclusion and Exclusion methods and ligands benchmarking results for the SYFPEITHI human and non-human primates datasets and IEDB non-human primates.
| Five-fold cross validation |
SYFPEITHI | ||||
|---|---|---|---|---|---|
|
|
|
||||
| Method | PCC | AUC | Human | Non-human | IEDB |
| NetMHCpan | 0.752 | 0.910 | 0.980 | 0.810 | 0.935 |
| Training Expanded |
0.755 | 0.913 | 0.980 | 0.839* | 0.949* |
| Inclusion | 0.732 | 0.902 | 0.976 | 0.811 | 0.908 |
| Exclusion | 0.753** | 0.909** | 0.978 | 0.848 | 0.927** |
denotes a statistical difference (p<0.05) between the NetMHCpan and Training Expanded methods
denotes a statistical difference between the inclusion and exclusion groups.
Include/Exclude Analysis
A significant gain in the performance was found for the Training Expanded group in the non-human primate ligand benchmark. This construct consisted in the inclusion of positions 5,22,124,155, and 170 and the exclusion of 7,84 and 159 positions compared to the NetMHCpan pseudo sequence (see figure 1). To investigate if the gain in the performance was due to the inclusion or exclusion of these residues, two new pseudo sequence constructs were created: one adding the included five additional positions to the NetMHCpan pseudo-sequence and another removing the three excluded positions. These two new pseudo sequence constructs were analyzed using the five-fold cross validation and the three independent ligands benchmarks. The results of the calculations are summarized in the lower part of table 4. The five-fold cross validation showed that the PCC and AUC values are significantly higher (p<0.01 in both cases) for the exclusion group compared to the inclusion group. When the two groups were compared in the ligands benchmark, no significant difference (p>0.05) was found for the SYFPEITHI human data set. For both non-human primate data sets, did the exclusion group outperform the inclusion group. The difference was only statistically significant for the IEDB data set (p=0.002). No significant difference (p>0.05) was found between the Training Expanded method and these two methods on the SYFPEITHI non-human primates benchmark. However, the Training Expanded method did significantly (p<0.05) outperform both the inclusion and exclusion groups when evaluated on the IEDB non-human primate data set. These results thus strongly suggest that both the inclusion of new polymorphic positions and the exclusion of positions that are non-polymorphic in the training data contribute to the improved predictive performance of the Training Expanded method.
Discussion
Several prediction methods have been developed to predict the affinity of a peptide to the MHC class I (5-12). Among these NetMHCpan is the current considered among the state-of-the-art (14). In the construction of the NetMHCpan method, MHC molecules are represented as so-called pseudo-sequences, consisting of polymorphic residues from the MHC protein sequence in potential contact with bound peptide. The residue positions included in the pseudo sequence of the original NetMHCpan method were derived from an analysis of HLA sequences and structures. Large novel data sets have become available not only for HLA but also for non-human MHC molecules including an extensive number of crystal structures for human and non-human MHC molecules interacting with a bound peptide. Given this large amount of novel information, in this work, we investigated impacts on the definition of the MHC pseudo sequence and subsequent impacts on the predictive performance of the NetMHCpan method.
A key finding was obtained in the structural analysis of the MHC-peptide complexes. Here, we found a common pattern of peptide interaction for both human and non-human molecules, and did not find additional contact positions when including complexes of non-human MHC molecules in the analysis. Hence, confirming that the structure of MHC molecules is highly conserved also between different species.
Using a large extended set of MHC protein sequences, we identified five novel polymorphic positions from the pool of positions found in potential contact with the bound peptide, compared to the positions identified in the original NetMHCpan publication. Residues in positions 118 and 158 of the MHC class I molecule were not found to be in direct contact with the bound peptide. However as they have been suggested to stabilize the peptide-MHC complex via water-mediated interactions (17), we analyzed the impact of including/excluding these positions in the pseudo-sequence construct. Here, we found that the two residues consistently significantly improved the predictive performance for the different pseudo sequence constructs tested, indicating the importance of the long-range interactions for these two positions with the bound peptide.
A novel pseudo-sequence construct (called “Training expanded”) defined from these observations was constructed consisting of residues in potential contact with bound peptides and polymorphic across the MHC alleles included in the training data. We tested this new pseudo-sequence on three different benchmarks of known MHC class I ligands and compared its performance to that of NetMHCpan, and found here a large and highly significant improve predictive performance for non-human primate MHC molecules.
The “Training expanded” pseudo-sequence construct, compared to the original NetMHCpan pseudo-sequence, consists of the exclusion of residues in positions 7,84 and 159, and the inclusion of positions 5,22,124,155, and 170 (see figure 1). To explore if the gain of predictive performance of the “Training expanded” pseudo-sequence was due to the exclusion or the inclusion of these novel residues positions, two new constructs were made: one excluding the three positions from the NetMHCpan pseudo-sequence and the other with the inclusion of the five additional positions. The exclusion group consistently in all benchmark calculation demonstrated a superior performance compared to the inclusion group suggesting that the exclusion of the residues found to be monomorphic across the MHC molecules used in the training was the major factor to explain the gain in performance for the “Training Expanded” method. However, when predicting binding for non-human primate MHC molecule, the “Training expanded group” defined from both the inclusion and exclusion of the residues mentioned above significantly outperformed all other methods, underlining that that both the inclusion of new polymorphic positions and the exclusion of positions that are monomorphic in the training data contribute to the overall improved predictive performance.
In conclusion, the results demonstrate that it is possible to achieve an improved predictive performance for pan-specific MHC peptide binding predictions when more human and, specially, non-human alleles are included not only in the training data as previously stated (21-23), but also in the pseudo sequence representation of the MHC molecules
Supplementary Material
Acknowledgements
This work was supported by the NIH (National Institute of Health) grant (contract number HHSN272200900045C).
References
- 1.Yewdell JW, Bennink JR. Immunodominance in major histocompatibility complex class I-restricted T lymphocyte responses. Annual review of immunology. 1999;17:51–88. doi: 10.1146/annurev.immunol.17.1.51. [DOI] [PubMed] [Google Scholar]
- 2.Falk K, Rotzschke O, Stevanovic S, Jung G, Rammensee HG. Allele-specific motifs revealed by sequencing of self-peptides eluted from MHC molecules. Nature. 1991;351:290–6. doi: 10.1038/351290a0. [DOI] [PubMed] [Google Scholar]
- 3.Robinson J, Mistry K, McWilliam H, Lopez R, Parham P, Marsh SG. The IMGT/HLA database. Nucleic acids research. 2011;39:D1171–6. doi: 10.1093/nar/gkq998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robinson J, Mistry K, McWilliam H, Lopez R, Marsh SG. IPD–the Immuno Polymorphism Database. Nucleic acids research. 2010;38:D863–9. doi: 10.1093/nar/gkp879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jacob L, Vert JP. Efficient peptide-MHC-I binding prediction for alleles with few known binders. Bioinformatics. 2008;24:358–66. doi: 10.1093/bioinformatics/btm611. [DOI] [PubMed] [Google Scholar]
- 6.Jojic N, Reyes-Gomez M, Heckerman D, Kadie C, Schueler-Furman O. Learning MHC I–peptide binding. Bioinformatics. 2006;22:e227–35. doi: 10.1093/bioinformatics/btl255. [DOI] [PubMed] [Google Scholar]
- 7.Nielsen M, Lundegaard C, Blicher T, et al. NetMHCpan, a method for quantitative predictions of peptide binding to any HLA-A and -B locus protein of known sequence. PloS one. 2007;2:e796. doi: 10.1371/journal.pone.0000796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang GL, Khan AM, Srinivasan KN, August JT, Brusic V. MULTIPRED: a computational system for prediction of promiscuous HLA binding peptides. Nucleic acids research. 2005;33:W172–9. doi: 10.1093/nar/gki452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoof I, Peters B, Sidney J, et al. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. 2009;61:1–13. doi: 10.1007/s00251-008-0341-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karosiene E, Lundegaard C, Lund O, Nielsen M. NetMHCcons: a consensus method for the major histocompatibility complex class I predictions. Immunogenetics. 2012;64:177–86. doi: 10.1007/s00251-011-0579-8. [DOI] [PubMed] [Google Scholar]
- 11.Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8-11. Nucleic acids research. 2008;36:W509–12. doi: 10.1093/nar/gkn202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Peters B, Sette A. Generating quantitative models describing the sequence specificity of biological processes with the stabilized matrix method. BMC bioinformatics. 2005;6:132. doi: 10.1186/1471-2105-6-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lundegaard C, Lund O, Buus S, Nielsen M. Major histocompatibility complex class I binding predictions as a tool in epitope discovery. Immunology. 2010;130:309–18. doi: 10.1111/j.1365-2567.2010.03300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang L, Udaka K, Mamitsuka H, Zhu S. Toward more accurate panspecific MHC-peptide binding prediction: a review of current methods and tools. Briefings in bioinformatics. 2012;13:350–64. doi: 10.1093/bib/bbr060. [DOI] [PubMed] [Google Scholar]
- 15.Vita R, Zarebski L, Greenbaum JA, et al. The immune epitope database 2.0. Nucleic acids research. 2010;38:D854–62. doi: 10.1093/nar/gkp1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.UniProt C. Update on activities at the Universal Protein Resource (UniProt) in 2013. Nucleic acids research. 2013;41:D43–7. doi: 10.1093/nar/gks1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ogata K, Wodak SJ. Conserved water molecules in MHC class-I molecules and their putative structural and functional roles. Protein engineering. 2002;15:697–705. doi: 10.1093/protein/15.8.697. [DOI] [PubMed] [Google Scholar]
- 18.Shannon CE. A mathematical theory of communication. The Bell System Technical Journal. 1948;27:379–423. and 623–56. [Google Scholar]
- 19.Rammensee H, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50:213–9. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- 20.Stranzl T, Larsen MV, Lundegaard C, Nielsen M. NetCTLpan: pan-specific MHC class I pathway epitope predictions. Immunogenetics. 2010;62:357–68. doi: 10.1007/s00251-010-0441-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang H, Lund O, Nielsen M. The PickPocket method for predicting binding specificities for receptors based on receptor pocket similarities: application to MHC-peptide binding. Bioinformatics. 2009;25:1293–9. doi: 10.1093/bioinformatics/btp137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang H, Lundegaard C, Nielsen M. Pan-specific MHC class I predictors: a benchmark of HLA class I pan-specific prediction methods. Bioinformatics. 2009;25:83–9. doi: 10.1093/bioinformatics/btn579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yu K, Petrovsky N, Schonbach C, Koh JY, Brusic V. Methods for prediction of peptide binding to MHC molecules: a comparative study. Molecular medicine. 2002;8:137–48. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.