

Abstract
Camptothecins are commonly used chemotherapeutics; in some models, they enhance signaling via the mitogen-activated protein kinase (MAPK) pathway through effects on upstream kinases. To evaluate the impact of camptothecin (CPT) on MAPKs in human colon cancer, we studied HCT116 and CaCo2 colon cancer cells. We found that HCT116 cells highly express mitogen-activated protein kinase phosphatase-1 (MKP1), which selectively inactivates extracellular signal-regulated kinase (ERK), whereas MKP1 levels were undetectable in CaCo2 cells. CPT did not affect ERK activity in CaCo2 cells, but did induce a striking increase in ERK activity in HCT116 cells in association with a corresponding decrease in MKP1. The reduction in MKP1 expression occurred at a posttranscriptional level and was blocked by the proteasome inhibitor MG132, whereas that CPT-induced downregulation of MKP1 was not due to proteasome-mediated degradation. Treatment of HCT116 cells with CPT induced a sustained activation of nuclear ERK, which was required for CPT-induced apoptosis. P38 and JNK activity were unaffected by CPT, suggesting that the effects of CPT are mediated specifically by ERK. These results suggest that targeting dual-specificity MAPK phosphatases in colon cancer cells may be a viable strategy for optimizing camptothecin-based therapeutic protocols.
Keywords: MKP1, ERK, camptothecin, human colon cancer
Introduction
Camptothecin (CPT) is among the most effective and widely used chemotherapeutic agents employed for the treatment of human cancers, including colon cancer. It has been reported that the antitumor activity of CPT is based on its inhibitory effect on topoisomerase activity. CPT stabilizes a transient intermediate of the topoisomerase reaction. In doing so, CPT causes DNA damage, which is generally considered to be the basis for its cytotoxicity.1 Several anti-neoplastic agents that are strong inducers of apoptosis, including cisplatin, etoposide, and CPT, also activate the mitogen-activated protein kinase (MAPK) pathway,2-4 but the specific mechanism by which these agents trigger the apoptotic program remains unclear. In an effort to understand the mechanism by which CPT induces cell death, we studied the role of several molecules that are assumed to be involved in apoptosis.
MAPKs play important roles in diverse cellular processes such as cell proliferation and apoptosis.5 There are three major families of MAPKs: extracellular signal-regulated kinases (ERK), c-Jun N-terminal kinases (JNK), and p38 MAP kinases (p38).6,7 These enzymes are activated through a sequential phosphorylation cascade that amplifies and transduces signals from the cell membrane to the nucleus.8 Whereas p38 and JNK are typically activated by stress-inducing agents, ERK is generally activated by mitogenic agents.9
It has been reported that the ERK pathway, one of the more ubiquitous cellular signaling cascades, is involved in mediating the induction of apoptosis in response to stress stimuli.10-14 The proapoptotic function of the Ras/Raf/ERK pathway in response DNA-damaging agents such as etoposide,12,15 doxorubicin,12,16,17 UV,12 and gamma irradiation18 is well documented. ERK activity has also been implicated in cell death induced by various other antitumor compounds, including resveratrol, quercetin, phenethyl isothiocyanate, betulinic acid, apigenin, oridonin, miltefosine, shikonin, and paclitaxel.8 DNA-damaging agents and antitumor compounds that are associated with ERK activation are often described as inducing the intrinsic pathway of apoptosis. These findings are paradoxical when considered in light of the originally postulated association of the ERK cascade with cellular proliferation, differentiation, and survival.19-21 The regulatory features upstream of the ERK cascade that assign an apoptotic role to ERK in response to DNA damage-associated stress have remained unclear.
Activation of MAPKs requires phosphorylation of both threonine and tyrosine residues in a conserved T-X-Y motif within the activation loop of MAPK.3,22-24 A growing family of MAPK phosphatases (MKPs), also called dual-specificity phosphatases (DUSPs), are able to dephosphorylate both the threonine and tyrosine residues in this motif. Specific MKPs tightly regulate subcellular ERK activity.9 MKP1 (DUSP1), PAC-1 (DUSP2), MKP-2 (DUSP4), and DUSP5 are mainly nuclear, whereas MKP-3 (DUSP6), MKP-X (DUSP7), and MKP-4 (DUSP9) are cytoplasmic.8 Once activated, MAPK family members can be rapidly inactivated through dephosphorylation by MKPs. Among these phosphatases, MKP1, encoded by an immediate early gene, is equally effective in dephosphorylating all three MAPK isoforms.25–27
MKP1 dephosphorylates and inactivates MAPK substrates and has been implicated in neoplasia. The lack of readily available, selective, small-molecule inhibitors of MKP family members has severely limited interrogation of their biological role. However, it has been found that MKP1 protects cells from apoptosis induced by cisplatin, UV irradiation, and proteasome inhibitors. Taken together with the association of MKP1 with human neoplasia, this makes MKP1 an attractive potential therapeutic target.3,28–30
In normal cells, the subcellular localization of ERK is tightly regulated by scaffold proteins and docking phosphatases that allow dephosphorylated ERK to accumulate in the nucleus and terminate signaling. Thus, in addition to sustained ERK activity, aberrant subcellular localization might contribute to the outcome of ERK-mediated cell death. Active ERKs phosphorylate, and thereby regulate, many cytoplasmic and nuclear targets that perform important biological functions. Indeed, the apoptotic response to estradiol, tamoxifen, doxorubicin, resveratrol, and dominant-negative Rac and Cdc42 mutants is correlated with sustained nuclear ERK activity.8,31,32
In this study, we test the hypothesis that CPT activates ERK in colon cancer cells, in part, by suppressing MKP1. Moreover, we propose that this mechanism is a major contributor to CPT-induced apoptosis. We found that CPT promoted nuclear accumulation of active ERK and prolonged ERK activity through inhibition of MKP1, implicating the function of the Ras/Raf/ERK pathway in cell death. Collectively, our results suggest that inhibition of MKP1 activity in tumor cells might improve the therapeutic response to CPT in human colon cancers.
Results
Expression of MAPKs and MKP1 in human colon cancer cell lines
To assess the relative expression of the MAPK kinases, JNK, ERK, and p38, and the phosphatase, MKP1, in colon cancer, we evaluated their expression in four human colon cancer cell lines (CaCo2, WiDr, LoVo, and HCT116). This analysis revealed high basal levels of active (phosphorylated) forms of JNK and ERK, but not p38, suggesting that JNK and ERK are constitutively activated in colon cancer cells. In addition, CaCo2 cells possessed very high basal levels of the active form of ERK and virtually undetectable levels of MKP1 protein; this latter observation might account for the constitutive ERK kinase activity (Fig. 1A). Reverse transcription-polymerase chain reaction (RT-PCR) analyses revealed no differences in MKP1 mRNA levels normalized to those of GAPDH in the four colon cancer cell lines (Fig. 1B). This suggests that differences in MKP1 protein levels among the four colon cancer cells are independent of the transcriptional regulation of MKP1.
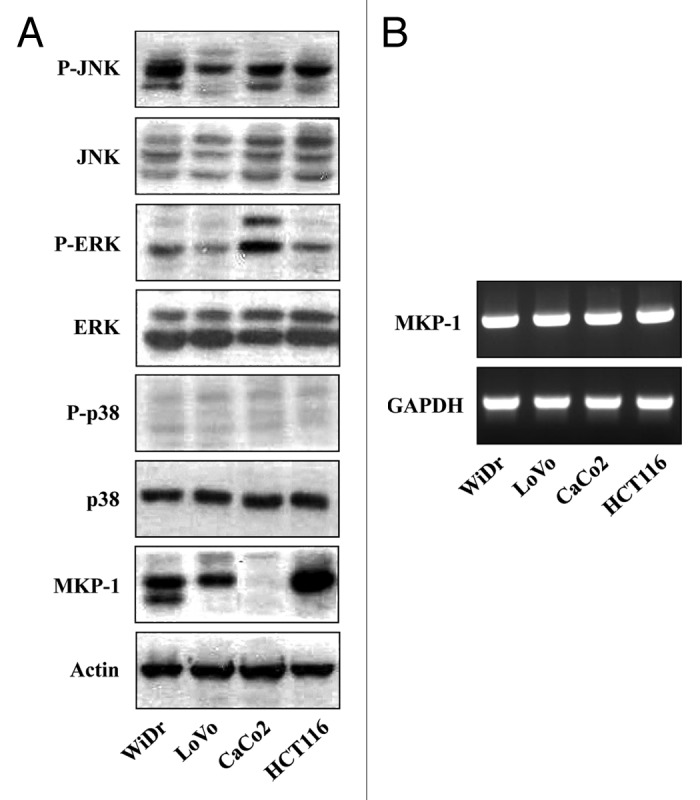
Figure 1. Human colon cancer cell lines exhibit differential activation of MAPKs and expression levels of MKP1. (A) Western blot analysis of total cell extracts from WiDr, LoVo, CaCo2, and HCT116 cells using primary antibodies to phospho-JNKs (P-JNK), JNKs (JNK), phospho-ERKs (P-ERK), ERK1/2 (ERK), phospho-p38 (P-p38), p38 (p38), MKP1 (MKP1), and actin (Actin). (B) RT-PCR analysis of MKP1 mRNA expression normalized to that of GAPDH.
CPT differentially induces growth inhibition in human colon cancer cell lines
CPT is a well-known anticancer drug; however, its mechanism has not been well studied in human colon cancer cell lines. To determine the effects of CPT on cancer cell growth, we screened the four colon cancer cell lines using the MTT (3-[4,5-dimethythiazol-2-yl]-2,5-diphenyl tetrazolium bromide) assay. Table 1 shows growth inhibition assay data after a 24 h exposure to CPT. Data are expressed as half-maximal inhibitory concentration (IC50) values corresponding to the concentration of CPT that produced 50% inhibition of cell growth. The cell lines exhibited inherent differences in sensitivity to CPT. LoVo and HCT116 cells were very CPT-sensitive, with IC50 values of 50 ± 13.21 and 100 ± 12.34 nM, respectively. WiDr cells were resistant, with an IC50 value of 325 ± 12.13 nM. CaCo2 cells were very resistant, with an IC50 value greater than 1000 nM. Taken together with the virtual absence of MKP1 in CaCo2 cells, this suggests that differences in MKP1 protein levels could be important in regulating CPT effects on colon cancer cell growth.
Table 1. Growth inhibition of camptothecin on colon cancer cell in vitro.
| Cell line | IC50 (nM) |
|---|---|
| WiDr | 325 ± 12.13 |
| LoVo | 50 ± 13.21 |
| CaCo2 | >1000 |
| HCT116 | 100 ± 12.34 |
CPT treatment induces a selective increase in ERK signaling
Prompted by the disparity in CPT sensitivity among colon cancer cell lines, we next sought to determine whether the activity of MAPKs or MAPK phosphatase was responsible for CPT-mediated apoptosis using both CaCo2 and HCT116 cells, focusing first on MAPKs. In these experiments, we assessed the activation status of MAPKs in each cell line by western blotting after incubating for various times with or without different concentrations of CPT. In the absence of CPT, the CaCo2 cell line displayed high levels of activated ERK1/2, as evidenced by dually phosphorylated (Thr202/Tyr204) p44 and p42. In contrast, only a low level of basal ERK1/2 phosphorylation was observed in the HCT116 cell line. A western blot analysis using phospho-specific antibodies revealed that CPT effectively increased the activation of ERK1/2 in HCT116 cells (Fig. 2). Neither JNK nor p38 protein levels were affected by CPT, indicating that these MAPK pathways are not involved in CPT-induced cytotoxicity.
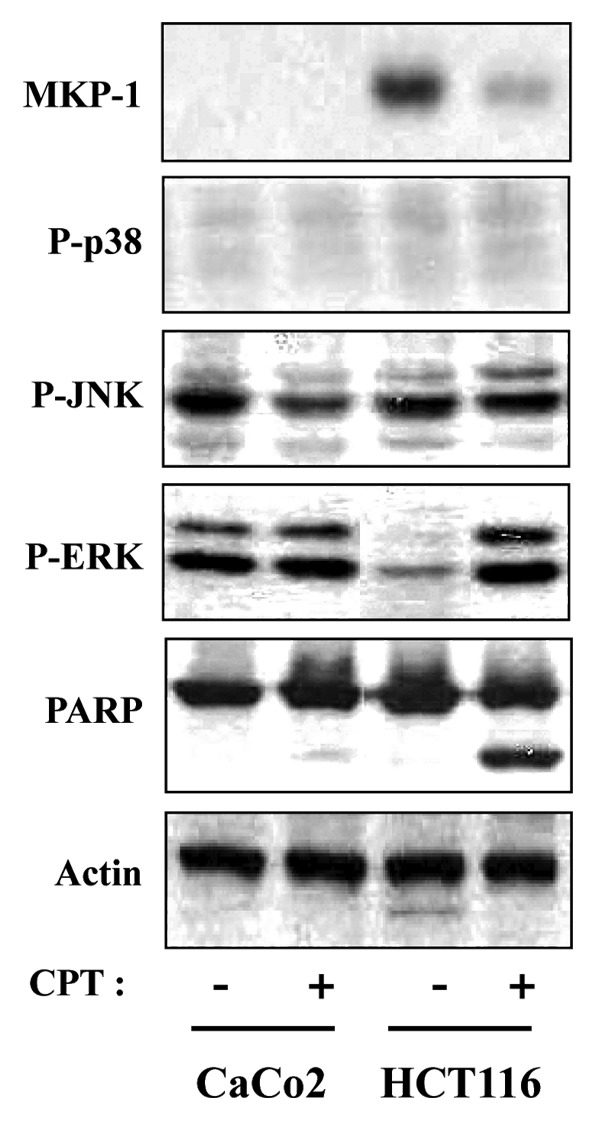
Figure 2. CPT-induced apoptosis in HCT116 cells, but not CaCo2 cells, is associated with decreased expression of MKP1. HCT116 and CaCo2 cells were treated with 500 nM and 1000 nM CPT for 24 h respectively, and total cell extracts were prepared and subjected to western blot analysis using anti-MKP1 (MKP1), anti-phospho-p38 (P-p38), anti-phospho-JNKs (P-JNK), anti-phospho-ERKs (P-ERK), and anti-PARP antibodies. Variations in protein loading among samples were controlled by monitoring actin levels.
MKP1 is downregulated by CPT
Phosphorylation and activation of ERK typically occurs through stimulation of the Ras/Raf/MEK protein kinase cascade. Because the mitogen-activated protein kinase kinase, MEK, is highly specific for phosphorylation and activation of ERK,9,33 we considered the possibility that increased phospho-ERK1/2 levels reflected an increase in dually phosphorylated (Ser217/Ser221), activated MEK. An analysis of HCT116 cells exposed to different concentrations of CPT by western blotting using anti-phospho-MEK antibodies showed no significant increase in MEK activity, indicating that this mechanism does not likely contribute to CPT-induced increases in phospho-ERK1/2 levels (Fig. 3A). CPT increased phospho-ERK1/2 levels in a concentration-dependent fashion, producing a maximal effect in the 500 to 1000 nM range. Time-course experiments revealed that phospho-ERK levels increased as early as 1 h after the addition of CPT, reached their maximum at 3 h, plateaued between 6 and 9 h, and then increased further with prolonged incubation. Total levels of ERK1/2, normalized to those of actin, were unaffected under these conditions, indicating that CPT indeed changed the phosphorylation status of ERK-1/2. Moreover, HCT116 cells express relatively high levels of MKP1, which could account for their low basal levels of phospho-ERK1/2. Changes in MKP1, like those of ERK1/2 phosphorylation, were time dependent, although the loss of MKP1 was somewhat delayed, reaching a nadir 12 h after exposure to CPT (Fig. 3B). To determine whether these changes in MKP1 protein were due to changes at the transcriptional level, we performed a semi-quantitative RT-PCR analysis. After treatment with CPT, MKP1 transcript abundance relative to that of GAPDH was unchanged (Fig. 3C), indicating that CPT-induced changes in MKP1 expression were likely posttranscriptional.

Figure 3. MEK1 does not contribute to CPT-induced downregulation of MKP1. (A) HCT116 cells were treated with the indicated concentrations of CPT and the total cell extracts were prepared. The levels of phosphorylated MEK1/2 (P-MEK1/2), ERKs (P-ERK), total ERKs (ERK), and PARP in total cell extracts were determined by western blot analysis. (B) HCT116 cells were exposed to 500 nM CPT for the indicated times, and analyzed by western blotting using anti-MKP1 (MKP1), anti-phospho-ERKs (P-ERK), anti-ERKs (ERK), anti-p53 (p53), and anti-p21 (p21) antibodies. (C) HCT116 cells were exposed to 500 nM CPT for the indicated times. MKP1, p53, p21 mRNA expression, normalized to that of GAPDH, was analyzed by RT-PCR.
CPT dose not downregulate MKP1 by promoting its proteasome-mediated degradation
Notably, recent work has suggested that the level of phosphorylated ERKs is decreased by proteasome inhibition,33,34 thus, it is possible that the CPT-induced increase in phosphorylated ERKs reflects increased proteasome-mediated degradation of MKP1. To determine the role of the proteasome pathway in CPT-induced ERK signaling in human colon cancer cells, we evaluated activated ERK1/2 and MKP1 in CaCo2 and HCT116 cells by western blotting after treating with CPT and then adding the proteasome inhibitor MG132. Proteasome-mediated degradation of MKP1 in CaCo2 and HCT116 cells were blocked by MG132, whereas co-treatment of CPT-treated cells with MG132 resulted in a decrease in the levels of MKP1 in both cells without a change in actin-normalized, total ERK1/2 protein levels. Exposure of these cells to the proteasome inhibitor MG132 alone also slightly decreased phospho-ERK1/2 levels in both cell lines. Cleaved poly ADP-ribose polymerase (PARP) was detected in CPT-treated HCT116 cells and co-treatment of CPT-treated cells with MG132 in both cells (Fig. 4). Consistent with this interpretation, inhibition of proteasome-mediated degradation of MKP1 by MG132, which decreased phosphorylated ERK levels and attenuate apoptosis in HCT116 cells, sensitized CaCo2 cells to CPT-induced apoptosis.
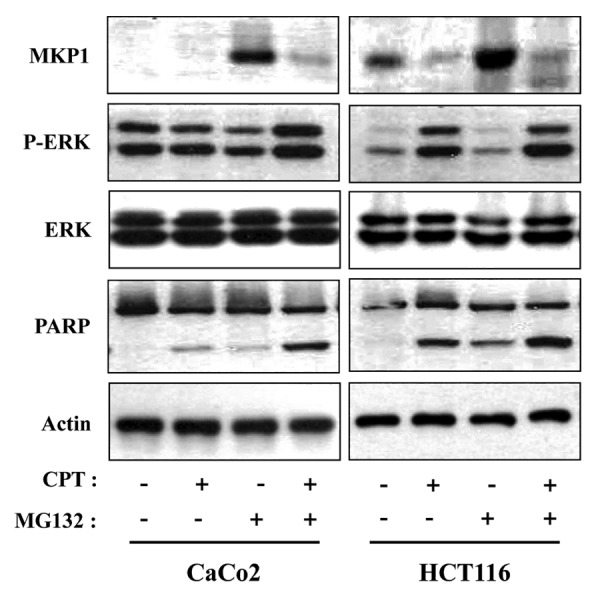
Figure 4. CPT dose not downregulate MKP1 by promoting its proteasome-mediated degradation. Cells were first pretreated with or without the proteasome inhibitor MG132 (10 μM) for 1 h, then co-treated with CPT for 24 h. Total cell extracts were prepared and analyzed by western blotting using anti-MKP1 (MKP1), anti-phospho-ERKs (P-ERK), anti-ERKs (ERK), and anti-PARP antibodies. Variations in protein loading among samples were controlled by monitoring actin levels.
CPT-induced repression of MKP1 alters the nuclear-cytoplasmic location of activated ERK1/2
Using western blot analyses on subcellular fractions, we examined MKP1 and phospho-ERK1/2 expression after 24 h CPT treatment in HCT116 cells. The purity of nuclear fractions was verified by determining the levels of the nuclear protein, U1 snRNP 70 (U1–70) (Fig. 5A). These experiments showed that the abundance of phospho-ERK1/2 in the nucleus was increased by CPT treatment compared with vehicle-treated controls. To visualize CPT-induced nuclear accumulation of active ERK1/2 in intact HCT116 cells, we conducted immunofluorescence assays using confocal microscopy. HCT116 cells were first stained for MKP1 to confirm that CPT inhibited MKP1 protein expression. Under normal growth conditions, MKP1 was located in both the cytoplasm and nucleus (Fig. 5Bb and c). Treatment of cells with 500 nM CPT for 24 h reduced MKP1 levels in both cellular compartments (Fig. 5Bf). Under basal conditions, active ERKs were distributed in both the cytoplasm and nucleus (Fig. 5Cb and c). Consistent with the results of western blotting, after treatment with CPT, phospho-ERK1/2 was mainly localized to the nucleus (Fig. 5Cf). Thus, enhanced nuclear phospho-ERK1/2 levels (Fig. 5C) were associated with correspondingly suppressed MKP1 levels (Fig. 5B). Taken together with the results of proteasome-inhibition experiments, these data imply that CPT-induced MKP1 protein degradation prevents inactivation of phospho-ERK1/2 by nuclear MKP1, allowing ERK1/2 activity to be sustained in the nucleus. Additional support for sustained activity of nuclear ERK1/2 is provided by the observation that the expression of p21, a target of ERK signaling that is known to be induced by CPT treatment, was increased in parallel at both protein (Fig. 3B) and mRNA (Fig. 3C) levels.

Figure 5. Subcellular localization of MKP1 and phosphorylated ERK after CPT treatment. (A) HCT116 cells were incubated for 24 h with CPT (500 nM), then cytoplasmic and nuclear fractions were collected and analyzed by western blotting using anti-MKP1 (MKP1), anti-phospho-ERKs (P-ERK), anti-ERKs (ERK), and anti-U1–70 antibodies. (B) HCT116 cells were incubated in the absence or presence of 500 nM CPT. After 24 h, cells were fixed with 4% (v/v) paraformaldehyde and immunostained for MKP1 using a FITC-conjugated secondary antibody and counterstained with PI. (a and d) Staining for PI (red). (b and e) Localization of MKP1 (green). (c and f) Merged images of MKP1 and PI. (C) Cells were stained as described in (B) except that the antibody against MKP1 was replaced with an antibody against phosphorylated ERKs. (a and d) Staining for PI (red). (b and e) Localization of phosphorylated ERKs (green). (c and f) Merged images of phosphorylated ERKs and PI.
Suppression of ERK1/2 activity by MEK inhibition decreases CPT-induced apoptosis
Although CPT does not increase ERK activity by acting on its upstream kinase (Fig. 3A), if MEK is central to the induction of ERK acitvity, then partially suppressing its basal activation by inhibiting MEK should produce a measurable decrease in CPT-induced apoptosis. To further investigate this possibility, we analyzed HCT116 cells by fluorescence-activated cell sorting (FACS) after treatment with (or without) CPT in the presence or absence of the MEK inhibitors, PD98059 and U012. The effects of CPT and MAPK inhibitors on MEK and ERK1/2 expression and activation were assessed by western blotting. Cells were incubated with or without CPT (500 nM) as indicated, and 1 h later PD98059 (30 µM) or U0126 (10 µM) was added to the media. After an additional 24 h, cells were analyzed by western blotting and FACS. As expected, CPT stimulated an increase in ERK1/2 phosphorylation; it also occasionally induced a modest decrease in phosphorylated MEK (Fig. 6A, top panel). Incubation of HCT116 cells with the specific MEK inhibitors PD98059 and U0126 effectively attenuated both basal and CPT-stimulated ERK1/2 phosphorylation, but the levels of phospho-ERK1/2 in these cells was still greater than those in cells treated with PD98059 or U0126 alone (Fig. 6A, middle panel). To determine the proportion of cells undergoing cell death, we performed a cell-cycle analysis using propidium iodide (PI) and FACS to identify cells with a sub-G1 DNA content (apoptotic cells). HCT116 cells were treated with (or without) CPT in the presence or absence of PD98059 or U0126. At baseline, most cells had at least a diploid DNA content, with only 2% ± 0.5% of cells undergoing apoptosis. CPT increased the proportion of apoptotic cells to 50% ± 3.03%. This increase was blunted by PD98059, which reduced the sub-G1 to 33% ± 2.4%. PD98059 alone induced a small increase in the size of the apoptotic population (6% ± 0.6%). Similar results were obtained for U0126, which reduced the CPT-induced sub-G1 population to 39% ± 2.07%, but modestly increased this compartment (to 6% ± 0.51%) when added alone (Fig. 6A, bottom panel). Notably, a substantial apoptotic population remained following CPT treatment, even with the MAPK pathway inhibited by PD98059 (sub-G1, 33% ± 2.4%) or U0126 (sub-G1, 39% ± 2.07%), consistent with the presence of functionally significant amounts of active ERK1/2 in the nucleus (Fig. 6Bh, i, k, and l).

Figure 6. MEK inhibitors attenuate CPT-induced apoptosis and ERK activation. (A) HCT116 cells were exposed to 500 nM CPT. One hour after treatment, the cells were treated with PD98059 (30 µM) or U0126 (10 µM) in the presence or absence of CPT, as indicated. After an additional 24 h, total cell extracts from HCT116 cells were analyzed by western blotting using primary antibodies to phospho-MEK1/2 (P-MEK1/2), phospho-ERKs (P-ERK), and total ERKs (ERK). Apoptosis levels were determined using PI staining and flow cytometry under the same conditions as above. (B) HCT116 cells were treated with CPT (500 nM), PD989059 (30 µM), or U0126 (10 µM), as indicated, using the same conditions as above. After 24 h, cells were fixed and immunostained for phosphorylated ERKs using a FITC-conjugated secondary antibody and counterstained with PI. (a, d, g, and j) Staining for PI (red). (b, e, h and k) Localization of phosphorylated ERKs (green). (c, f, I, and l) Merged images of phosphorylated ERKs and PI.
Discussion
Among the more ubiquitous cellular signal transduction cascades are the p38, JNK, and ERK1/2 MAPK pathways, which play important roles in cell development, growth, differentiation, and apoptosis.35 Investigations of the role of MAPK cascades in the regulation of apoptosis have led to the general view that activation of the ERK pathway constitutes a survival signal. Inhibition of ERK signaling has indeed been shown to increase the sensitivity of cytotoxic agents in a variety of cellular contexts. Despite this, clear evidence exists that the ERK pathway can mediate apoptosis in response to different stimuli in different tissues.31 In the present study, we provide evidence that activation of ERK is important for mediating CPT-induced apoptosis in human colon cancer cell lines. As reported previously, we found that CPT activates ERK, which is linked with the induction of apoptosis in a variety of cell lines.4 In addition to CPT, many other chemotherapeutic agents also show similar ERK-mediated responses in various cell lines.1 Our observations, taken together with these reports, suggest a pro-apoptotic role of ERK that contrasts with the general view that the ERK pathway confers a survival advantage to cells. The mechanisms by which ERK mediates apoptosis have remained poorly understood and may be multi-faceted. We report here that CPT activates ERK1/2 in HCT116 cells through a novel mechanism that involves inactivation of the ERK1/2 phosphatase, MKP1.
Protein phosphatases play an important role as negative regulators of the Ras/Raf/ERK signaling pathway through actions at different levels.8 Upregulation of MKP1 at both mRNA and protein levels is detected in early stage carcinomas and in various stages of breast and prostate carcinoma. These findings support the idea that upregulation of MKP1 could result in increased resistance to chemotherapy in various human tumors. In cases where MKP1 was specifically suppressed or inactivated, resulting increases in apoptosis were correlated with enhanced phospho-ERK levels.29
Given the ability of MKP1 to inhibit active ERK1/2, we considered the possibility that inhibition of this phosphatase might activate ERK-mediated cell death. However, we first sought to determine whether MAPK signaling was involved in CPT-induced cell death in colon cancer cells. We observed that ERK1/2 was activated by CPT stimulation and this activation was necessary for CPT-induced apoptosis in HCT116 cells. We also found an inverse correlation between the basal activity of ERK1/2 and MKP1 protein expression levels in both CaCo2 and HCT116 cells (Fig. 1), suggesting that changes in ERK1/2 activity might be related to reciprocal changes in MKP1. To investigate this further, we probed CPT effects on MKP1. We found that stimulation of HCT116 cells with CPT decreased MKP1 expression. Importantly, this decrease in MKP1 was accompanied by an increase in the phosphorylated, activated form of ERK1/2 (Fig. 2). Consistent with this, lower levels of MKP1 in CPT-treated cells were associated with higher levels of phospho-ERK1/2, and a western blot analysis of the kinetics of MKP1 and ERK responses to CPT treatment in HCT116 cells showed that MKP1 levels remained suppressed through at least 3 h of CPT treatment, during which phosphorylated ERK1/2 levels remained elevated (Fig. 3B). These results show that MKP1 is a target of CPT action and suggest that the CPT-induced loss of MKP1 contributes to the increase in ERK phosphorylation/activation. CPT-induced downregulation of MKP1 protein also occurred much earlier than the strong activation of ERK. Collectively, these results imply that the upstream signal for CPT-induced activation of ERK is independent of MEK1/2-mediated signaling; instead, the increased ERK1/2 phosphorylation is likely due to a decrease in MKP1 phosphatase.
These results raise the question of how CPT decreases MKP1 expression. An analysis of MKP1 expression by RT-PCR showed no changes in MKP1 mRNA levels in response to CPT, suggesting that CPT-induced downregulation of MKP1 was a posttranscriptional event. Previous studies have shown that MKP1 degradation can be mediated by the proteasome,29 and proteasome inhibition has recently been linked to a decrease in the level of phosphorylated ERKs.33,34 As expected, inhibition of MKP1 proteolysis with the proteasome inhibitor MG132 resulted in diminished activation of ERK in both CaCo2 and HCT116 cells whereas that CPT-induced downregulation of MKP1 was not due to proteasome-mediated degradation (Fig. 4).
The kinetics and duration of ERK activation may play an important role in influencing its effect on cell fate. It has been reported that prolonged ERK activation is necessary for ERK-induced cell death,36 whereas a transient activation of ERK protects cells from death.37 Accordingly, we postulated that, if active ERKs were important for CPT-mediated apoptosis, eliminating the additional activation of ERK due to MEK activity should weaken the programmed cell death response to CPT. This was indeed the case, as evidenced by the fact that the MEK inhibitors PD98059 and U0126 attenuated CPT-induced apoptosis in HCT116 cells (Fig. 6). In each case, MEK inhibition diminished CPT-induced apoptosis by ~20–30% (from 50% ± 3.03% to 33% ± 2.43% and 39% ± 2.07% for PD98059 and U0126, respectively). Thus, these results are consistent with the idea that ERK activity contributes directly to CPT-induced apoptosis in HCT116 cells and support the hypothesis that downregulation of MKP1 by CPT is a significant contributor to preserving sustained levels of nuclear phospho-ERK1/2 (Fig. 5). That CPT induces apoptosis, at least in part, through downregulation of MKP1 is consistent with the identification of MKP1 as a mediator of inducible chemoresistance.29,34
In summary, our data demonstrate that CPT treatment induces concurrent, reciprocal changes in MKP1 expression and ERK activation in HCT116 cells, supporting the view that CPT-induced downregulation of MKP1 results in increased activation of ERK. Moreover, we suggest that CPT-induced ERK activation is necessary for CPT-induced apoptosis, thereby assigning an apoptotic role to ERK instead of its conventional survival function. Further characterization of the factors that link MKP1 to the activation of ERK may contribute to a better understanding of activated ERK-mediated induction of apoptosis in response to chemotherapeutic agents. Our results also suggest that inhibition of endogenous MKP1 phosphatase activity is a good strategy for optimizing CPT therapy. Because prolonged ERK activation has been shown to promote the death of human cancer cell lines from different origins, this pro-apoptotic property of the Ras/Raf/ERK pathway could be used to specifically target cancer cells, because such strategies would only target cancer cells with deregulated ERK activity and not normal cells, in which ERK activation is transient.
Materials and Methods
Reagents and cell culture
RPMI 1640 medium, Dulbecco modified Eagle medium (DMEM), fetal bovine serum (FBS), and a mixed antibiotic solution were purchased from Life Technologies. Antibodies against ERK1/2, phospho-ERK (Thr202/Tyr705), JNK, phospho-JNK (Thr183/Tyr185), p38, phospho-p38 (Thr180/Tyr182), and phospho-MEK1/2 (Ser217/221) were purchased from Cell Signaling Technology. Primary antibodies against p53, p21, U1–70, PARP, and actin, and horseradish peroxidase (HRP)-conjugated goat anti-rabbit and anti-mouse secondary antibodies were purchased from Santa Cruz Biotechnology. Dimethyl sulfoxide (DMSO) and other chemicals used in solutions were obtained from the Sigma-Aldrich Chemical Co. MG132 (carbobenzoxy-l-leucyl-l-leucyl-l-leucinal) was from Calbiochem. Human colon cancer cell lines were purchased from the American Type Culture Collection. CaCo2 and WiDr cells were maintained in DMEM. LoVo and HCT116 cells were grown in RPMI 1640 medium. Cells were grown in a 37 °C incubator with 5% CO2 in culture media supplemented with 10% (v/v) heat-inactivated FBS, penicillin (100 U/mL), and streptomycin (100 μg/mL).
Western immunoblot analyses
Drug-treated cells were washed once with ice-cold 1× PBS (phosphate-buffered saline). Total cell lysates were prepared by incubation in radioimmunoprecipitation assay buffer (RIPA; 50 mM Tris [pH 7.4], 150 mM NaCl, 5 mM EDTA, 1% [v/v] Triton X-100, 1% [w/v] sodium deoxycholic acid, 0.1% [w/v] sodium dodecyl sulfate [SDS], 2 mM phenylmethylsulfonyl fluoride [PMSF], 30 mM Na2HPO4, 50 mM NaF, and 1 mM Na3VO4) containing freshly added protease inhibitor cocktail (Roche Applied Science). Cytoplasmic and nuclear extracts were prepared according to the instructions of the NE-PER Nuclear and Cytoplasmic Extraction Kit (Pierce). Proteins in the supernatants of lysates (30 μg aliquots) were mixed with SDS sample buffer, boiled for 5 min, then separated by SDS-PAGE (SDS-PAGE) on 8% or 10% (w/v) gels and transferred to nitrocellulose membranes (Millipore). Membranes were blocked with 5% (w/v) non-fat dried milk in TBS-T (50 mM Tris-HCl [pH 7.6], 150 mM NaCl and 0.1% [v/v] Tween 20) and incubated with primary antibodies for 3 h. Blots were washed with TBS-T, incubate with appropriate HRP-conjugated secondary antibody for 1 h, and examined using an enhanced chemiluminescence (ECL) detection system (Amersham).
Cytotoxicity assay
The MTT colorimetric assay38 was used to screen for cytotoxic activity. Briefly, cells were seeded at a density of 1 × 104 cells per well in 96-well plates in culture medium containing 10% (v/v) FBS. Following 24 h incubation and attachment, the cells were treated with different concentrations of CPT for 24 h. IC50 values were obtained by assessing cell survival over a CPT concentration range of 25 to 1000 nM. After washing and incubating with MTT solution (20 μL of 5 mg/mL) at 37 °C for 1 h, cells were lysed with DMSO. The absorbance in each well was measured at 540 nm using an ELISA reader (Bio-Rad Laboratories). The results were generated from three independent experiments; each experiment was performed in triplicate.
RT-PCR
Total RNA was isolated using the TRIzol reagent according to the manufacturer’s instructions. The quality of total RNA was tested by formaldehyde-agarose gel electrophoresis. Single-strand cDNA was synthesized from equal amounts of RNA (2 μg) using 0.5 μg of random primers and MMLV reverse transcriptase, following the manufacturer’s instructions. Each cDNA fragment was PCR-amplified using the following specific primer pairs: MKP1, 5′-AAAGGAGGAT ACGAAGCGTT-3′ (sense) and 5′-ATAAGGTAAG CAAGGCAGAT-3′ (antisense); p21, 5′-CTTCGGCCCA GTGGACAGCG-3′ (sense) and 5′-TCTGCCGCCG TTTTCGACCC-3′ (antisense); p53, 5′-TGGGACAGCC AAGTCTGTGA-3′ (sense) and 5′-ACTGGAGTCT TCCAGTGTGA-3′ (antisense); and GAPDH (glyceraldehyde-3-phosphate dehydrogenase), 5′-TGCTGAGTAT GTCGTGGAGT CTA-3′ (sense) and 5′-AGTGGGAGTT GCTGTTGAAG TCG-3′ (antisense).
Apoptosis assay
Apoptosis was evaluated by determining the proportion of cells with sub-G1 DNA content. Cells were trypsinized at specific times after treatment with chemicals and collected by centrifugation at 200 × g for 10 min at room temperature. The supernatant was discarded and the precipitated cells were washed twice with PBS (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4 and 1.4 mM KH2PO4 [pH 7.4]). Pelleted cells were carefully suspended in 500 μL PBS buffer and fixed in 4 mL of ice-cold 70% (v/v) ethanol overnight. Fixed cells were washed twice with PBS, then resuspended in PBS (5 × 105 cells/500 μL) and treated with 100 μg/mL of RNase A at 37 °C for 30 min. PI was next added to a final concentration of 50 μg/mL for DNA staining, and 10 000 fixed cells were analyzed on a FACSCalibur Flow fluorescence-activated cell sorter (Becton Dickinson). Cell-cycle distribution was analyzed using the Modifit program (Becton Dickinson).
Immunofluorescence analysis
Immunofluorescence analyses were done as described previously38 with slight modifications. Briefly, cells were seeded on coverslips and grown for 2 d in culture medium, rinsed with PBS, and immersed in 70% (v/v) ethanol overnight. Coverslips were rinsed with PBS and fixed in 4% (v/v) paraformaldehyde for 15 min at room temperature. This was followed by permeabilization in 0.1% (v/v) Triton X-100 for 10 min and rinsing with PBS. The cells were blocked in sterile PBS with 2% (w/v) bovine serum albumin (BSA) for 1 h. Subsequently, cells were rinsed with PBS/0.1% (w/v) BSA and incubated with primary antibody (1:200) for 3 h at room temperature. Cells were next rinsed five times with PBS/0.1% (w/v) BSA and incubated with FITC-conjugated secondary antibody (Invitrogen) for 1 h at room temperature in the dark. Cells were rinsed three times with PBS and treated with 2 μg/mL PI in PBS for 5 min to stain the chromosomes. Coverslips were rinsed in PBS and mounted on glass slides. Cells were observed using a Zeiss LSM 510 META confocal microscope (Carl Zeiss).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by Basic Science Research Program (2012R1A1A2007317) and the Nuclear Research and Development Program through a National Research Foundation of Korea (NRF) grant funded by the Korea government (Ministry of Education, Science and Technology).
Glossary
Abbreviations:
- MKP1
mitogen-activated protein kinase phosphatase-1
- ERK
extracellular signal-regulated kinase
- CPT
camptothecin
- MTT
3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide
- FBS
fetal bovine serum
- HRP
horseradish peroxidase
- FACS
fluorescence-activated cell sorting
- RT-PCR
reverse transcription-polymerase chain reaction
- PI
propidium iodide
- BSA
bovine serum albumin
- MG132
carbobenzoxy-l-leucyl-l-leucyl-l-leucinal
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/26044
References
- 1.Singh S, Upadhyay AK, Ajay AK, Bhat MK. p53 regulates ERK activation in carboplatin induced apoptosis in cervical carcinoma: a novel target of p53 in apoptosis. FEBS Lett. 2007;581:289–95. doi: 10.1016/j.febslet.2006.12.035. [DOI] [PubMed] [Google Scholar]
- 2.Seimiya H, Mashima T, Toho M, Tsuruo T. c-Jun NH2-terminal kinase-mediated activation of interleukin-1beta converting enzyme/CED-3-like protease during anticancer drug-induced apoptosis. J Biol Chem. 1997;272:4631–6. doi: 10.1074/jbc.272.7.4631. [DOI] [PubMed] [Google Scholar]
- 3.Sánchez-Pérez I, Martínez-Gomariz M, Williams D, Keyse SM, Perona R. CL100/MKP-1 modulates JNK activation and apoptosis in response to cisplatin. Oncogene. 2000;19:5142–52. doi: 10.1038/sj.onc.1203887. [DOI] [PubMed] [Google Scholar]
- 4.Lee S, Lee HS, Baek M, Lee DY, Bang YJ, Cho HN, Lee YS, Ha JH, Kim HY, Jeoung DI. MAPK signaling is involved in camptothecin-induced cell death. Mol Cells. 2002;14:348–54. [PubMed] [Google Scholar]
- 5.Graves LM, Guy HI, Kozlowski P, Huang M, Lazarowski E, Pope RM, Collins MA, Dahlstrand EN, Earp HS, 3rd, Evans DR. Regulation of carbamoyl phosphate synthetase by MAP kinase. Nature. 2000;403:328–32. doi: 10.1038/35002111. [DOI] [PubMed] [Google Scholar]
- 6.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 7.Chen Z, Gibson TB, Robinson F, Silvestro L, Pearson G, Xu B, Wright A, Vanderbilt C, Cobb MH. MAP kinases. Chem Rev. 2001;101:2449–76. doi: 10.1021/cr000241p. [DOI] [PubMed] [Google Scholar]
- 8.Cagnol S, Chambard JC. ERK and cell death: mechanisms of ERK-induced cell death--apoptosis, autophagy and senescence. FEBS J. 2010;277:2–21. doi: 10.1111/j.1742-4658.2009.07366.x. [DOI] [PubMed] [Google Scholar]
- 9.Warmka JK, Mauro LJ, Wattenberg EV. Mitogen-activated protein kinase phosphatase-3 is a tumor promoter target in initiated cells that express oncogenic Ras. J Biol Chem. 2004;279:33085–92. doi: 10.1074/jbc.M403120200. [DOI] [PubMed] [Google Scholar]
- 10.Wang X, Martindale JL, Holbrook NJ. Requirement for ERK activation in cisplatin-induced apoptosis. J Biol Chem. 2000;275:39435–43. doi: 10.1074/jbc.M004583200. [DOI] [PubMed] [Google Scholar]
- 11.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–2. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 12.Tang D, Wu D, Hirao A, Lahti JM, Liu L, Mazza B, Kidd VJ, Mak TW, Ingram AJ. ERK activation mediates cell cycle arrest and apoptosis after DNA damage independently of p53. J Biol Chem. 2002;277:12710–7. doi: 10.1074/jbc.M111598200. [DOI] [PubMed] [Google Scholar]
- 13.Werlen G, Hausmann B, Naeher D, Palmer E. Signaling life and death in the thymus: timing is everything. Science. 2003;299:1859–63. doi: 10.1126/science.1067833. [DOI] [PubMed] [Google Scholar]
- 14.Schweyer S, Soruri A, Meschter O, Heintze A, Zschunke F, Miosge N, Thelen P, Schlott T, Radzun HJ, Fayyazi A. Cisplatin-induced apoptosis in human malignant testicular germ cell lines depends on MEK/ERK activation. Br J Cancer. 2004;91:589–98. doi: 10.1038/sj.bjc.6601919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee ER, Kang YJ, Kim JH, Lee HT, Cho SG. Modulation of apoptosis in HaCaT keratinocytes via differential regulation of ERK signaling pathway by flavonoids. J Biol Chem. 2005;280:31498–507. doi: 10.1074/jbc.M505537200. [DOI] [PubMed] [Google Scholar]
- 16.Lee ER, Kim JY, Kang YJ, Ahn JY, Kim JH, Kim BW, Choi HY, Jeong MY, Cho SG. Interplay between PI3K/Akt and MAPK signaling pathways in DNA-damaging drug-induced apoptosis. Biochim Biophys Acta. 2006;1763:958–68. doi: 10.1016/j.bbamcr.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 17.Liu J, Mao W, Ding B, Liang CS. ERKs/p53 signal transduction pathway is involved in doxorubicin-induced apoptosis in H9c2 cells and cardiomyocytes. Am J Physiol Heart Circ Physiol. 2008;295:H1956–65. doi: 10.1152/ajpheart.00407.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee YJ, Soh JW, Jeoung DI, Cho CK, Jhon GJ, Lee SJ, Lee YS. PKC epsilon -mediated ERK1/2 activation involved in radiation-induced cell death in NIH3T3 cells. Biochim Biophys Acta. 2003;1593:219–29. doi: 10.1016/S0167-4889(02)00392-0. [DOI] [PubMed] [Google Scholar]
- 19.Johnson GL, Vaillancourt RR. Sequential protein kinase reactions controlling cell growth and differentiation. Curr Opin Cell Biol. 1994;6:230–8. doi: 10.1016/0955-0674(94)90141-4. [DOI] [PubMed] [Google Scholar]
- 20.Robinson MJ, Cobb MH. Mitogen-activated protein kinase pathways. Curr Opin Cell Biol. 1997;9:180–6. doi: 10.1016/S0955-0674(97)80061-0. [DOI] [PubMed] [Google Scholar]
- 21.He H, Wang X, Gorospe M, Holbrook NJ, Trush MA. Phorbol ester-induced mononuclear cell differentiation is blocked by the mitogen-activated protein kinase kinase (MEK) inhibitor PD98059. Cell Growth Differ. 1999;10:307–15. [PubMed] [Google Scholar]
- 22.Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–48. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 23.Dérijard B, Hibi M, Wu IH, Barrett T, Su B, Deng T, Karin M, Davis RJ. JNK1: a protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell. 1994;76:1025–37. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 24.Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie EA, Ahmad MF, Avruch J, Woodgett JR. The stress-activated protein kinase subfamily of c-Jun kinases. Nature. 1994;369:156–60. doi: 10.1038/369156a0. [DOI] [PubMed] [Google Scholar]
- 25.Keyse SM. Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Curr Opin Cell Biol. 2000;12:186–92. doi: 10.1016/S0955-0674(99)00075-7. [DOI] [PubMed] [Google Scholar]
- 26.Slack DN, Seternes OM, Gabrielsen M, Keyse SM. Distinct binding determinants for ERK2/p38alpha and JNK map kinases mediate catalytic activation and substrate selectivity of map kinase phosphatase-1. J Biol Chem. 2001;276:16491–500. doi: 10.1074/jbc.M010966200. [DOI] [PubMed] [Google Scholar]
- 27.Liu C, Shi Y, Han Z, Pan Y, Liu N, Han S, Chen Y, Lan M, Qiao T, Fan D. Suppression of the dual-specificity phosphatase MKP-1 enhances HIF-1 trans-activation and increases expression of EPO. Biochem Biophys Res Commun. 2003;312:780–6. doi: 10.1016/j.bbrc.2003.10.186. [DOI] [PubMed] [Google Scholar]
- 28.Franklin CC, Srikanth S, Kraft AS. Conditional expression of mitogen-activated protein kinase phosphatase-1, MKP-1, is cytoprotective against UV-induced apoptosis. Proc Natl Acad Sci U S A. 1998;95:3014–9. doi: 10.1073/pnas.95.6.3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Small GW, Shi YY, Edmund NA, Somasundaram S, Moore DT, Orlowski RZ. Evidence that mitogen-activated protein kinase phosphatase-1 induction by proteasome inhibitors plays an antiapoptotic role. Mol Pharmacol. 2004;66:1478–90. doi: 10.1124/mol.104.003400. [DOI] [PubMed] [Google Scholar]
- 30.Lazo JS, Nunes R, Skoko JJ, Queiroz de Oliveira PE, Vogt A, Wipf P. Novel benzofuran inhibitors of human mitogen-activated protein kinase phosphatase-1. Bioorg Med Chem. 2006;14:5643–50. doi: 10.1016/j.bmc.2006.04.036. [DOI] [PubMed] [Google Scholar]
- 31.Murphy LO, Blenis J. MAPK signal specificity: the right place at the right time. Trends Biochem Sci. 2006;31:268–75. doi: 10.1016/j.tibs.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 32.Ramos JW. The regulation of extracellular signal-regulated kinase (ERK) in mammalian cells. Int J Biochem Cell Biol. 2008;40:2707–19. doi: 10.1016/j.biocel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 33.Orlowski RZ, Small GW, Shi YY. Evidence that inhibition of p44/42 mitogen-activated protein kinase signaling is a factor in proteasome inhibitor-mediated apoptosis. J Biol Chem. 2002;277:27864–71. doi: 10.1074/jbc.M201519200. [DOI] [PubMed] [Google Scholar]
- 34.Huang TH, Chen HC, Chou SM, Yang YC, Fan JR, Li TK. Cellular processing determinants for the activation of damage signals in response to topoisomerase I-linked DNA breakage. Cell Res. 2010;20:1060–75. doi: 10.1038/cr.2010.95. [DOI] [PubMed] [Google Scholar]
- 35.Small GW, Somasundaram S, Moore DT, Shi YY, Orlowski RZ. Repression of mitogen-activated protein kinase (MAPK) phosphatase-1 by anthracyclines contributes to their antiapoptotic activation of p44/42-MAPK. J Pharmacol Exp Ther. 2003;307:861–9. doi: 10.1124/jpet.103.055806. [DOI] [PubMed] [Google Scholar]
- 36.Ho Y, Samarasinghe R, Knoch ME, Lewis M, Aizenman E, DeFranco DB. Selective inhibition of mitogen-activated protein kinase phosphatases by zinc accounts for extracellular signal-regulated kinase 1/2-dependent oxidative neuronal cell death. Mol Pharmacol. 2008;74:1141–51. doi: 10.1124/mol.108.049064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhuang S, Schnellmann RG. A death-promoting role for extracellular signal-regulated kinase. J Pharmacol Exp Ther. 2006;319:991–7. doi: 10.1124/jpet.106.107367. [DOI] [PubMed] [Google Scholar]
- 38.Yun HJ, Kim SY, Kwon YY, Kim CH, Kang CM, Kim EJ. Janus-activated kinases and signal transducer and activator of transcription control tumor growth response to camptothecin in human colon cancer cells. Cancer Biol Ther. 2010;10:354–61. doi: 10.4161/cbt.10.4.12382. [DOI] [PubMed] [Google Scholar]