

Abstract
In the present study we show that histone deacetylase inhibitors (HDACIs) enhance the anti-tumor effects of melanoma differentiation associated gene-7/interleukin 24 (mda-7/IL-24) in human renal carcinoma cells. Similar data were obtained in other GU tumor cells. Combination of these two agents resulted in increased autophagy that was dependent on expression of ceramide synthase 6, with HDACIs enhancing MDA-7/IL-24 toxicity by increasing generation of ROS and Ca2+. Knock down of CD95 protected cells from HDACI and MDA-7/IL-24 lethality. Sorafenib treatment further enhanced (HDACI + MDA-7/IL-24) lethality. Anoikis resistant renal carcinoma cells were more sensitive to MDA-7/IL-24 that correlated with elevated SRC activity and tyrosine phosphorylation of CD95. We employed a recently constructed serotype 5/3 adenovirus, which is more effective than a serotype 5 virus in delivering mda-7/IL-24 to renal carcinoma cells and which conditionally replicates (CR) in tumor cells expressing MDA-7/IL-24 by virtue of placing the adenoviral E1A gene under the control of the cancer-specific promoter progression elevated gene-3 (Ad.5/3-PEG-E1A-mda-7; CRAd.5/3-mda-7, Ad.5/3-CTV), to define efficacy in renal carcinoma cells. Ad.5/3-CTV decreased the growth of renal carcinoma tumors to a significantly greater extent than did a non-replicative virus Ad.5/3-mda-7. In contralateral uninfected renal carcinoma tumors Ad.5/3-CTV also decreased the growth of tumors to a greater extent than did Ad.5/3-mda-7. In summation, our data demonstrates that HDACIs enhance MDA-7/IL-24-mediated toxicity and tumor specific adenoviral delivery and viral replication of mda-7/IL-24 is an effective pre-clinical renal carcinoma therapeutic.
Keywords: MDA-7/IL-24, HDACI, ceramide, apoptosis, bystander, cytokine, ROS, caspase, animal study
Introduction
In the United States, renal cell carcinoma (RCC) is diagnosed in ~51 000 patients per annum. If the disease is localized and has not metastasized, the entire kidney can be removed with the intact tumor, an essential “cure” is achieved.1 However, if the disease has spread beyond the capsule of the kidney the prognosis is dismal and often associated with rapid patient death.1 RCC is highly resistant to well-known cytotoxic chemotherapy treatments.1 Consequently, the need arises to improve therapies against this lethal disease.
Melanoma differentiation associated gene-7 (mda-7) was originally identified through a subtraction hybridization method using human melanoma cells induced to terminally differentiate by treatment with interferon-β (IFN-) and mezerein (MEZ).2,3 An inverse correlation has been shown between MDA-7/IL-24 expression and melanoma progression. Our prior work involving recombinant adenovirus (Ad.5-mda-7) has illuminated numerous mechanisms by which MDA-7/IL-24 regulates cell survival in various tumor types, while sparing non-transformed cells.4-14 These involve Ca2+ elevation, ceramide generation, and reactive oxygen species (ROS) production.15-19 MDA-7/IL-24 is also implicated in the formation of autophagic vesicles, that required ROS induction, and knock down of Beclin 1 or ATG5 protected against MDA-7/IL-24 lethality. Recombinant GST-MDA-7 protein has also been shown to possess anti-proliferative properties as observed in previous work in RCCs.18,19 Even in nanomolar concentrations, the protein facilitates growth arrest attributed to the above mechanisms, and these effects are enhanced further to the point of almost complete growth inhibition and subsequent apoptosis at ~20-fold higher concentrations. The efficacy of this cytokine as an anti-tumor agent is further validated through the successful application of Ad.5-mda-7 (INGN 241) in Phase I clinical trials where its safety with repeated infusions and anti-tumor effects were observed.7
A significant dilemma that exists in the event of using type 5 adenoviral vectors for gene therapy is that the expression of Coxsackie and Adenovirus receptors (CAR) is downregulated in multiple cancer types including kidney cancer, which are required for the virus to infect a cell.20,21 In order to improve infection efficiency, replacing the type 5 virus fiber knobs with the fiber knobs of the type 3 adenovirus has created a novel tropism modified virus. This approach resulted in an elevated infection of tumor cells independent of CAR, which was also evident in our pre-clinical studies in RCC and other tumor cell types providing strong evidence for the enhanced therapeutic efficacy of Ad.5/3-mda-7 vs. Ad.5-mda-7.18,19,22,23 Apart from this, a conditionally replication-competent adenovirus has also been created where expression of the adenoviral early region 1A (E1A) virus gene and conditional virus replication was driven by the cancer-specific promoter progression elevated gene-3 (PEG-3) which has little activity in non-transformed cells (Ad.5-PEG-E1A-mda-7; a cancer terminator virus, Ad.5-CTV).22–26 Athymic nude mice bearing xenografts derived from prostate cancer or melanoma cells injected with Ad.5-CTV encountered complete remission not only in the primary infected tumor but also in the uninfected tumor growing on the opposite flank. This can be attributed to the “bystander” anti-tumor effect of the secreted MDA-7/IL-24 protein, generated from cells infected with Ad.5-mda-7.27,28
In order to further enhance the anti-tumor effects of MDA-7/IL-24, in this study we evaluated the effect of combining it with HDAC inhibitors (HDACIs), which like MDA-7/IL-24 cause increased DNA damage and ceramide and ROS levels, apart from exhibiting selective lethality in tumor cells compared with non-transformed cells.29-34 HDACIs are a chemically varied class of agents, e.g., vorinostat (SAHA; Zolinza) and sodium valproate (Depakote), which block histone deacetylation and neutralization of positively charged lysine residues on histone tails, thereby modifying chromatin structure/condensation and transcription.29,30 HDACIs also cause disruption of transcription factor complexes, increased expression of death receptors and ligands, increased levels of lipid second messengers, and modulate HSP90 activity and acetylation of NFκB.31–34
Thus, the present studies were performed to determine whether MDA-7/IL-24 and HDACIs could be used in combination to kill renal carcinoma cells. We then determined whether a serotype 5/3 adenovirus that conditionally replicates in tumor cells expressing MDA-7/IL-24 (Ad.5/3-PEG-E1A-mda-7; Ad.5/3-CTV), decreased the growth of renal carcinoma tumors to a significantly greater extent than did a non-replicative virus Ad.5/3-mda-7.
Results
Combinatorial treatment with recombinant MDA-7 or Ad.5/3-mda-7 and HDACIs causes growth inhibition in multiple cancer types in vitro
We first determined whether bacterially synthesized GST-MDA-7 and the HDACIs SAHA (vorinostat) and sodium valproate (Depakote) interacted to kill renal carcinoma cells. Concomitant treatment of RCCs with GST-MDA-7 and HDACIs resulted in a greater than additive induction of cell killing (Fig. 1A). Cell killing did not occur in primary human renal epithelial cells. We next determined whether eukaryotic cell synthesized His6-MDA-7/IL-24 also interacted with HDACIs to kill RCCs. HDACIs and His6-MDA-7/IL-24 interacted to kill RCCs (Fig. 1B). HDACIs and Ad.5/3-mda-7 also interacted to kill ovarian and prostate cancer cells in short-term viability assays (Fig. 1C). HDACIs and Ad.5/3-mda-7/GST-MDA-7 interacted to kill GU tumor cells in a greater than additive fashion with combination index (CI) values of less than 1.0 (Table 1; Fig. 1D).

Figure 1. GST-MDA-7 and HDAC inhibitors interact in a greater than additive fashion to kill RCCs but not primary renal epithelial cells. (A) A498 and UOK121LN and primary renal epithelial cells were treated with GST or GST-MDA-7 (100 nM). One hour after GST-MDA-7 treatment cells were treated with vehicle or with the HDACIs (Na Valproate or vorinostat [SAHA]) at the indicated concentrations. Cells were isolated (48 h) later and viability determined by trypan blue (n = 3, ± SEM). *P < 0.05 greater than corresponding GST value; **P < 0.05 greater than corresponding GST-MDA-7 value. (B) UOK121LN cells were treated with His6-MDA-7 synthesized in mammalian cells (200 nM). One hour after MDA-7 treatment cells were treated with vehicle or with the SAHA (500 nM). Cells were isolated (24 h, 48 h) later and viability determined by trypan blue (n = 3, ± SEM). *P < 0.05 greater than corresponding HIS6-MDA-7 value. (C) OVCAR3, DU145, and LNCaP cells were infected with Ad.5/3-cmv or Ad.5/3-mda-7 (10 multiplicity of infection). Twenty-four hours after infection cells were treated with vehicle or with the indicated concentrations of SAHA. Cells were isolated (96 h) after infection and viability determined by trypan blue (n = 3, ± SEM). *P < 0.05 greater than corresponding vehicle value. (D) DU145 cells were plated in sextuplicate as single cells and 12 h later infected with Ad.5/3-cmv or Ad.5/3-mda-7 (10 multiplicity of infection). Twenty-four hours after infection cells were treated with vehicle or with the indicated concentrations of SAHA. Forty-eight hours after infection the media was changed to drug free media and colonies permitted to form for the following 10 d. Colonies of >50 cells were counted (n = 3, ± SEM). #P < 0.05 less than corresponding Ad.5/3-mda-7 value.
Table 1. SAHA and GST-MDA-7 synergize to kill RCCs in colony formation assays.
| GST-MDA-7 (nM) | SAHA (μM) | Fa | CI | GST-MDA-7 (nM) | Na Val. (mM) | Fa | CI |
|---|---|---|---|---|---|---|---|
| 10 | 0.2 | 0.36 | 0.55 | 10 | 0.5 | 0.29 | 0.46 |
| 20 | 0.4 | 0.60 | 0.50 | 20 | 1.0 | 0.40 | 0.46 |
| 30 | 0.6 | 0.81 | 0.41 | 30 | 1.5 | 0.49 | 0.37 |
A498 cells plated in sextuplicate as single cells (250–1500 cells/well) and 12 h after plating treated with GST or GST-MDA-7 (10–30 nM) in parallel with either SAHA (0.2–0.6 μM) or Na Valproate (0.5–1.5 mM). Forty-eight hours later cells are washed free drugs and colonies permitted to form for 20 d. (n = 3, ± SEM). Data were tabulated using the Calcusyn program, which permits the calculation of synergy of drug interaction. Synergy is defined as a combination index (CI) value of less than 1.00 (Fa; fraction affected).
MDA-7/IL-24 plus HDACIs modify multiple signaling pathways in RCCs leading to growth inhibition
SAHA treatment enhanced the induction of LC3-GFP vesicle numbers by GST-MDA-7 that correlated with increased LC3II levels and reduced p62 levels, indicative of autophagosome formation and autophagic flux (Figs. 2A and B). Knock down of Beclin1 expression abolished the induction of LC3-GFP vesicles. Depletion of Beclin 1 levels suppressed the toxicity of GST-MDA-7 and SAHA treatment (Fig. 2C). GST-MDA-7 and SAHA interacted to increase the phosphorylation of PKR-like endoplasmic reticulum kinase (PERK) and its downstream substrate eIF2α and to decrease the expression of the protective BCL-2 family proteins MCL-1 and BCL-XL (Fig. 3A). Expression of a dominant negative kinase dead PERK protein prevented the downregulation of MCL-1 and BCL-XL and suppressed the lethal effects of SAHA and GST-MDA-7 treatment (Figs. 3A and B). Overexpression of MCL-1 or BCL-XL almost abolished GST-MDA-7 lethality but only partially suppressed GST-MDA-7 and SAHA lethality (Fig. 3C). MDA-7 has been shown to kill through generation of reactive oxygen species and increased cytoplasmic Ca2+ levels; expression of the reactive oxygen species quenching protein thioredoxin (TRX) or the calcium quenching protein calbindin (Calb.) suppressed the lethal effects of SAHA and GST-MDA-7 treatment (Fig. 3D). MDA-7 has been shown to kill through generation of ceramide; knockdown of ceramide synthase 6 (LASS6) also suppressed the lethal effects of SAHA and GST-MDA-7 treatment (Fig. 3E). In the presence of SAHA, LASS6 expressed via transfection, was rapidly acetylated (Fig. 3E, upper inset panel). In renal carcinoma cells, as a single agent, GST-MDA-7 lethality relies on ligand independent activation of the death receptor FAS (CD95).18,19 Knock down of CD95 reduced the toxicity of GST-MDA-7 and GST-MDA-7 combined with SAHA (Fig. 3F). In RCCs SAHA treatment increased expression of both CD95 and FAS-ligand (Fig. 3F, upper inset panel).
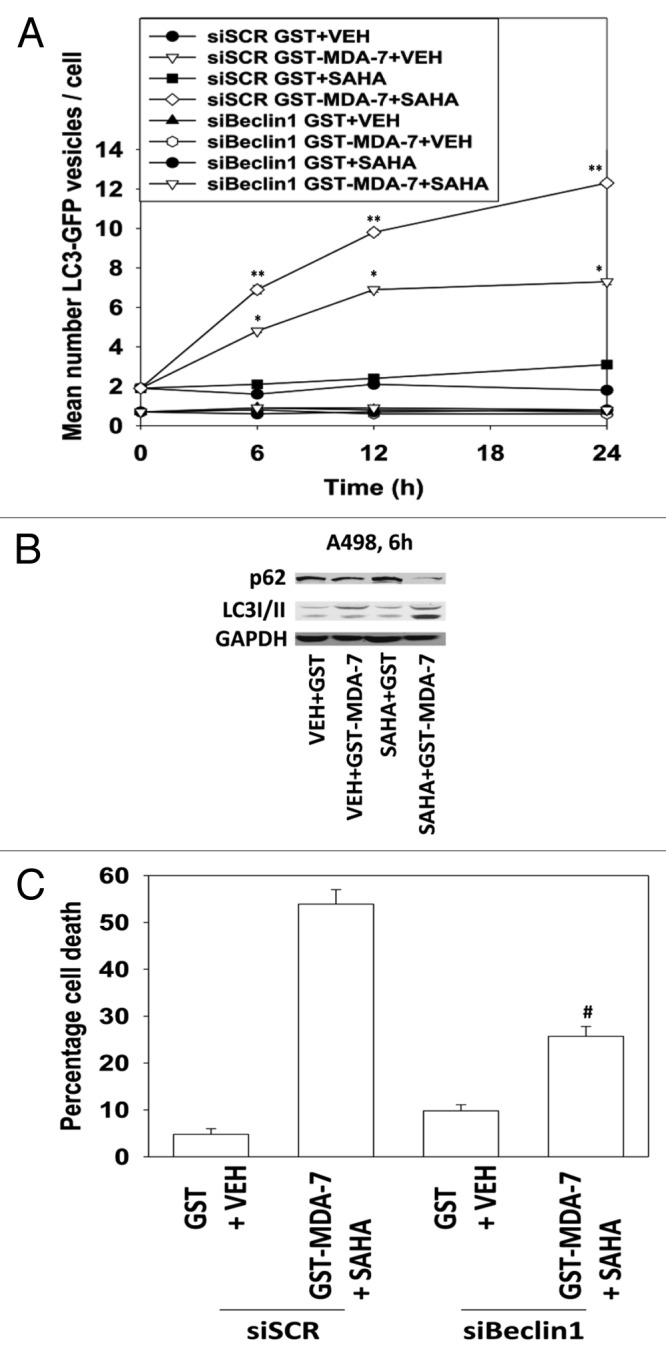
Figure 2. SAHA and GST-MDA-7 interact to promote greater vesicularization of an LC3-GFP construct: knock down of Beclin1 blocks autophagy and enhanced tumor cell killing. (A) A498 cells were transfected with an LC3 (ATG8)–GFP fusion protein and in parallel transfected with a scrambled siRNA (siSCR) or a validated siRNA to knock down Beclin1 (siBeclin1); 24 h after transfection cells were treated with GST-MDA-7 (100 nM), SAHA (0.5 μM) or the agents combined. Cells (a representative of 40) were examined 6 h, 12 h, and 24 h after treatment using an Axiovert microscope (40×) for the formation of punctate vesicles containing LC3-GFP. Data are plotted as the number of LC3-GFP vesicles per cell (n = 2, ± SEM). *P < 0.05 greater than corresponding GST value; **P < 0.05 greater than corresponding GST-MDA-7 value. (B) A498 cells were treated with GST-MDA-7 (100 nM), SAHA (0.5 μM), or the agents combined. Cells were isolated 6 h after exposure and immunoblotting performed to determine expression of p62 and LC3II. (C) Parallel sets of cells to (A) in triplicate were isolated 48 h after GST-MDA-7+SAHA treatment and viability determined via trypan blue exclusion (n = 2, ± SEM). #P < 0.05 less than siSCR (GST-MDA-7 + SAHA) value.

Figure 3. SAHA and GST-MDA-7 interact to promote greater PERK and eIF2α phosphorylation in RCCs. (A) Upper blots: A498 cells were transfected with an empty vector control plasmid or a plasmid to express dominant negative PERK. Twenty-four hours after transfection cells were treated with GST-MDA-7 (50 nM), SAHA (0.5 μM), or the agents combined. Cells were isolated 6 h after exposure and the phosphorylation of PERK and eIF2α; and expression of MCL-1 and BCL-XL determined (representative n = 2). Lower graph: A498 cells were transfected in triplicate with an empty vector control plasmid or a plasmid to express dominant negative PERK. Twenty-four hours after transfection cells were treated with GST-MDA-7 (50 nM), SAHA (0.5 μM), or the agents combined. Cells were isolated 48 h after treatment and viability determined via trypan blue exclusion (n = 2, ± SEM). #P < 0.05 less than siSCR (GST-MDA-7 + SAHA) value. (B) A498 cells were transfected in triplicate with an empty vector control plasmid or plasmids to express BCL-XL or MCL-1. Twenty-four hours after transfection cells were treated with GST-MDA-7 (50 nM), SAHA (0.5 μM), or the agents combined. Cells were isolated 48 h after treatment and viability determined via trypan blue exclusion (n = 3, ± SEM). #P < 0.05 less than corresponding siSCR value; *P < 0.05 greater than GST-MDA-7 + VEH value. (C) A498 cells were transfected in triplicate with an empty vector control plasmid; a plasmid to express TRX to quench ROS; or a plasmid to express Calbindin D28 (Calb.) to quench Ca2+. In parallel cells were infected with empty vector control adenovirus (Ad.5/3-cmv) or virus to express MDA-7/IL-24 (Ad.5/3-mda-7) (10 multiplicity of infection). Twenty-four hours after trans-/infection cells were treated with vehicle (DMSO) or SAHA (0.5 μM). Forty-eight hours after drug exposure, cells were isolated and cell viability determined by Annexin-PI staining and flow cytometry (n = 2, ± SEM). #P < 0.05 less than corresponding CMV value. (D) A498 cells were transfected in triplicate with an empty vector control plasmid or a plasmid to express an siRNA to knock down expression of LASS6 and in parallel cells were infected with empty vector control adenovirus (Ad.5/3-cmv) or virus to express MDA-7/IL-24 (Ad.5/3-mda-7) (10 multiplicity of infection); 24 h after trans-/infection cells were treated with vehicle (DMSO) or SAHA (0.5 μM); 48 h after exposure, cells were isolated and viability determined by Annexin-PI staining and flow cytometry (n = 2, ± SEM). #P < 0.05 less than corresponding Ad.5/3-cmv value. Upper blot: A498 cells were transfected to express FLAG-LASS6 and 24 h later treated with SAHA (0.5 μM). Thirty minutes after SAHA treatment FLAG-LASS6 was immunoprecipitated and the acetylation of the protein determined by immunoblotting against pan-acetyl residues. (E) A498 cells were transfected in triplicate with a control siRNA (siSCR) or an siRNA to knock down expression of CD95 (siCD95). Twenty four hours after transfection cells were treated with GST-MDA-7 (50 nM), SAHA (0.5 μM), or the agents combined. Cells were isolated 48 h after treatment and viability determined via trypan blue exclusion (n = 3, ± SEM). #P < 0.05 less than corresponding siSCR value. Upper blot: A498 cells were treated with SAHA (500 nM). Cells were isolated at the indicated times and immunoblotting performed to determine the expression of CD95 and FAS-L.
Sorafenib is a standard of care therapeutic for RCC patients. We have previously shown that sorafenib and HDACIs synergize to kill RCCs.35 Sorafenib enhanced the lethality of (SAHA + GST-MDA-7) in short-term cell death assays in RCCs (Fig. 4A). In colony formation assays, in a dose-dependent fashion, sorafenib enhanced (SAHA + MDA-7) lethality (Fig. 4B). For tumor cells to spread beyond their primary site and metastasize requires some level of resistance to the toxic effects of tumor cell-to-cell dissociation and from the basal lamina; this is termed anoikis resistance.36 Anoikis resistant (AR) RCCs expressed higher levels of BCL-XL and MCL-1, exhibited SRC activation, as judged by decreased Y527 phosphorylation and increased Y416 phosphorylation, and increased CD95 activity as judged by elevated CD95 tyrosine phosphorylation (Fig. 5A).10,19,37 AR RCCs were more sensitive to GST-MDA-7 than wild type (WT) parental cells (Fig. 5B).
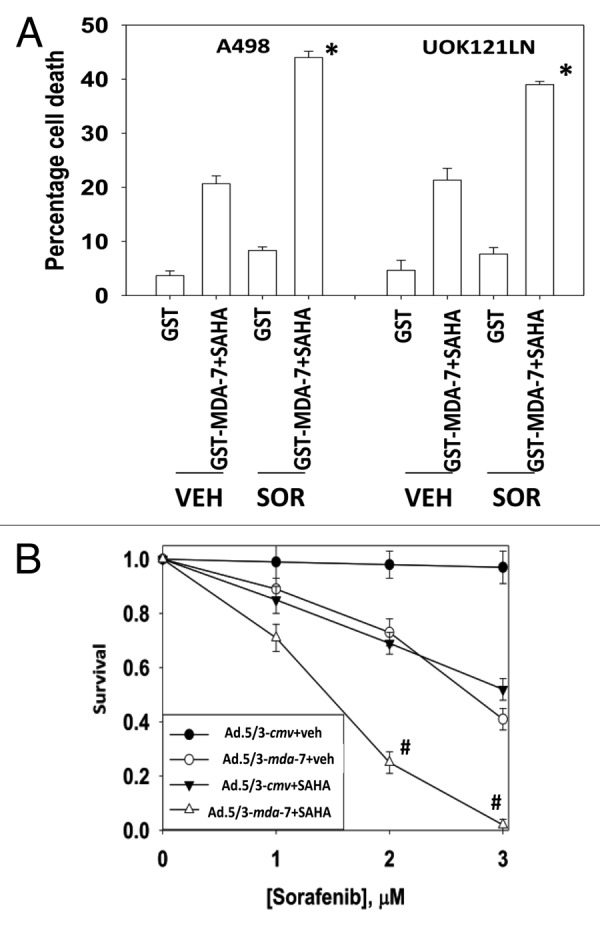
Figure 4. Sorafenib enhances (MDA-7 + SAHA) lethality. (A) A498 and UOK121LN cells were treated with GST-MDA-7 (50 nM), SAHA (0.5 μM), or the agents combined. Thirty minutes after the initial treatment cells were treated with vehicle or sorafenib (Sor, 3 μM) as indicated. Cells were isolated 24 h after treatment and viability determined via trypan blue exclusion (n = 3, ± SEM). *P < 0.05 greater than corresponding (GST-MDA-7 + SAHA) + vehicle value. (B) UOK121LN cells plated as single cells in sextuplicate were infected with limiting amounts of Ad.5/3-cmv or Ad.5/3-mda-7 (1.0 moi); 12 h after infection cells were treated with vehicle (VEH, DMSO) or SAHA (0.5 μM). Twelve hours later cells were treated with sorafenib (1–3 μM). Media was replaced with drug free media 48 h after infection and colonies of > 50 cells permitted to form for the following ~10 d (n = 2, ± SEM). #P < 0.05 less than corresponding (Ad.5/3-mda-7 + SAHA) + vehicle value.
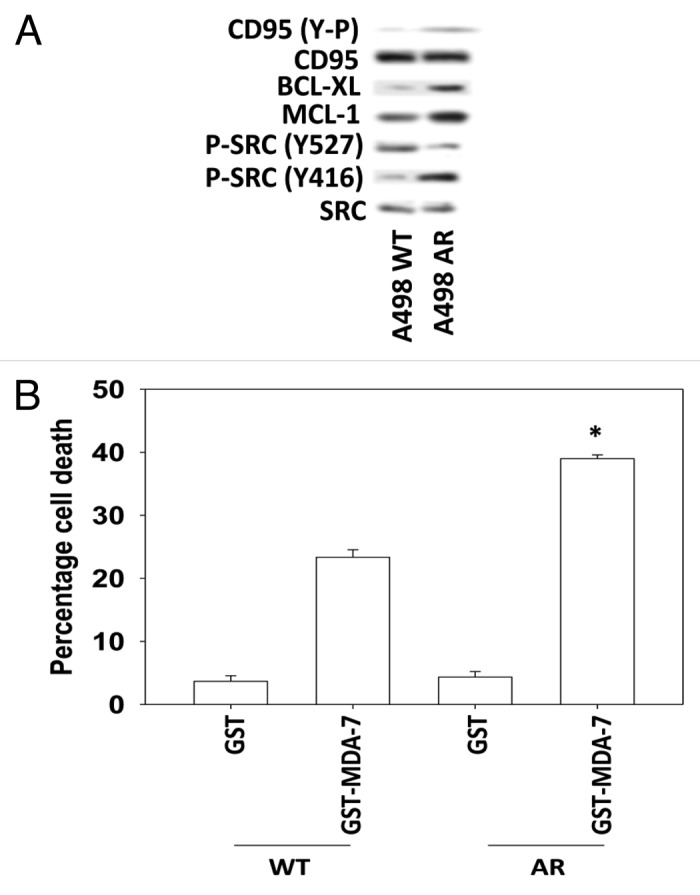
Figure 5. Anoikis resistant RCCs are more sensitive to MDA-7 that correlates with CD95 tyrosine phosphorylation. (A) A498 cells (WT, wild type; AR, anoikis resistant) 24 h after plating were lysed and protein samples subjected to immunoblotting against the indicated phospho-proteins and total protein levels. (B) A498 cells (WT, wild type; AR, anoikis-resistant) 24 h after plating were treated with GST or GST-MDA-7 (50 nM). Forty eight hours after treatment cells were isolated and viability determined via trypan blue exclusion (n = 3, ± SEM). *P < 0.05 greater than corresponding (GST-MDA-7) value in WT cells.
Evaluating the therapeutic effects of Ad.5/3-mda-7 and Ad.5/3-CTV in murine models
Next, we compared and contrasted the abilities of Ad.5/3-mda-7 and the conditionally replicative virus that expresses MDA-7, termed Ad.5/3-CTV (CRAd.5/3-mda-7), to control the growth of RCCs growing in the flanks of athymic mice. Tumors were formed on both flanks and tumors on the left flank infused with virus(es). Infusion of a tumor (on the left side) with CRAd.5/3-cmv, Ad.5/3-mda-7 or CRAd.5/3-mda-7 resulted in growth suppression of the infused tumor below initial tumor volume levels (Fig. 6). The long-term growth suppressive effects of Ad.5/3-mda-7 and CRAd.5/3-mda-7 infusion into the left side tumors were very similar, whereas tumors infused with CRAd.5/3-cmv exhibited less initial growth suppression and eventually began to re-grow. In the contralateral (right side) “bystander” tumor, Ad.5/3-mda-7 and CRAd.5/3-mda-7 exposures both initially suppressed growth to a similar extent however the tumor exposed to Ad.5/3-mda-7 began to re-grow. Our data demonstrate that the conditionally replicative virus that expresses MDA-7, CRAd.5/3-mda-7, is more effective at controlling the growth of infected and bystander tumors than the non-replicative virus Ad.5/3-mda-7.
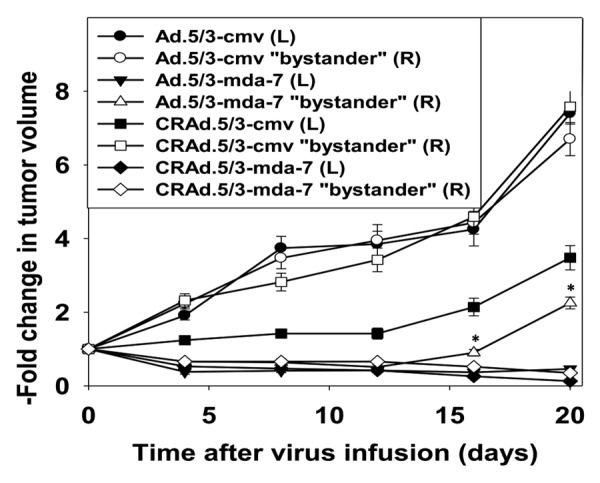
Figure 6. A conditionally replicative virus to express MDA-7 (CRAd.5/3-mda-7) provides a greater tumor control in bystander tumors than a non-replicative virus (Ad.5/3-mda-7). A498 cells (1 × 106) were injected onto each rear flank of an athymic mouse and tumors permitted to form (6 mice total per group; 2 experiments ± SEM; mean tumor size of all tumors prior to start of the study 113 ± 21 mm3). The tumor on the left flank was infused with either Ad.5/3-cmv; Ad.5/3-mda-7; CRAd.5/3-cmv; CRAd.5/3-mda-7 (1 × 109 plaque forming units of virus, each tumor). Tumor volume, both the infused left tumor and the bystander right tumor, was measured every fourth day. *P < 0.05 greater volume than corresponding right side tumor infusion in CRAd.5/3-mda-7 animals.
Ad.5/3-mda-7 in combination with HDACIs is more effective in diminishing tumor growth in vivo.
Finally, we determined whether HDAC inhibitors enhanced MDA-7/IL-24 toxicity in vivo. Using one quarter the number of infective virus particles in Figure 6, we noted that Ad.5/3-mda-7 still significantly reduced the growth of established RCC tumors (Fig. 7). As a single agent SAHA did not alter RCC tumor growth. However, SAHA significantly reduced the growth of Ad.5/3-mda-7 infused tumors, below the initial tumor volume at day 0. Our data demonstrate that Ad.5/3-mda-7 in combination with SAHA is more effective at controlling the growth of infected tumors than Ad.5/3-mda-7 alone.
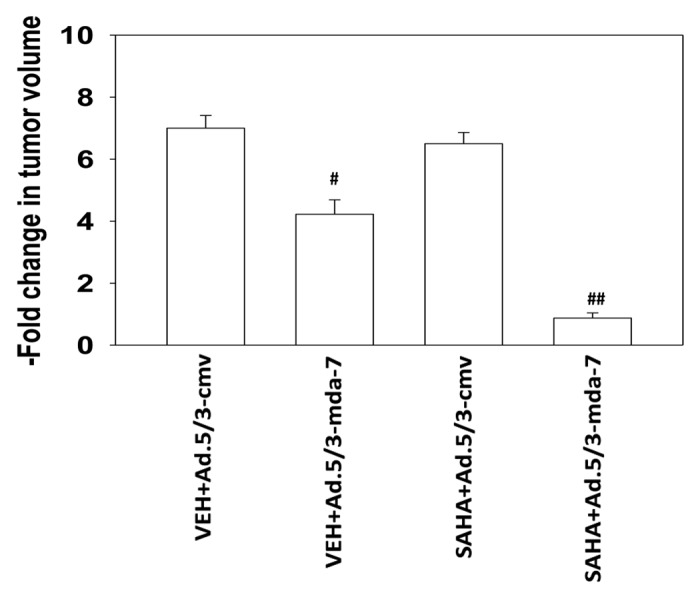
Figure 7. SAHA enhances MDA-7 toxicity in vivo. A498 cells (1 × 106) were injected onto the left rear flank of an athymic mouse and tumors permitted to form (4 mice total per group; 2 experiments ± SEM; mean tumor size of all tumors prior to start of the study 113 ± 21 mm3). The tumor was infused with either Ad.5/3-cmv or Ad.5/3-mda-7 (2 × 108 plaque forming units of virus, each tumor). Twenty four hours after virus infusion animals were treated for three days with either vehicle or SAHA (25 mg/kg, PO, QD). Tumor volume was measured 20 d after virus infusion. #P < 0.05 lower volume than Ad.5/3-cmv infusion; ##P < 0.05 lower volume than Ad.5/3-mda-7 plus vehicle treated tumors.
Discussion
The present studies were designed to ascertain whether the tumor cell selective toxic cytokine MDA-7/IL-24 interacted with HDAC inhibitors to kill renal carcinoma cells, and whether MDA-7/IL-24 interacted with HDAC inhibitors in vivo to kill kidney cancer cells. We discovered that MDA-7/IL-24 interacted in vitro with HDAC inhibitors to kill RCCs and other GU cell types, without apparent toxicity to non-transformed renal epithelial cells, and that this combination caused prolonged growth suppression of RCCs grown as tumors.
We analyzed the mechanisms by which combination cell killing occurred (Fig. 8). Combined exposure of cells to MDA-7/IL-24 and HDAC inhibitors caused an enhanced and prolonged induction of autophagy. Molecular inhibition of autophagy significantly reduced killing by the drug combination. Exposure of RCCs to the combination of agents caused a large increase in the phosphorylation of PERK and its downstream substrate eIF2α, which was directly associated with reduced expression of MCL-1 and BCL-XL.18,19,38 MCL-1 in particular has a short half-life and reduced MCL-1 production i.e., due to eIF2α phosphorylation, will rapidly reduce MCL-1 protein levels (Fig. 8). Although transient overexpression of BCL-XL or MCL-1 suppressed MDA-7/IL-24 toxicity, it only partially protected cells from combined MDA-7/IL-24 and HDAC inhibitor killing. In prior studies ROS and Ca2+ levels were increased by MDA-7 and shown to be involved in killing RCCs by the cytokine.18,19 Quenching of ROS and Ca2+ suppressed the lethal effects of MDA-7/IL-24 and also SAHA plus MDA-7/IL-24. Prior studies had also shown that knock down of ceramide synthase 6 (LASS 6) significantly reduces MDA-7/IL-24 toxicity in multiple tumor cell types, and in RCCs this was associated with a lack of CD95 activation.18,39 Knock down of LASS6 also abolished the ability of SAHA to enhance MDA-7/IL-24 killing. Of perhaps greater interest was the observed change in LASS6 acetylation. Cells were transfected to express a FLAG-tagged form of LASS6 and treated with SAHA for 30 min; the acetylation of immunoprecipitated LASS6 had increased. Whether acetylation of LASS6 alters its enzymatic activity or sub-cellular localization of the enzyme will be the subject of future studies. Knockdown of the death receptor CD95 significantly reduced the toxicity of MDA-7/IL-24 and also SAHA plus MDA-7/IL-2; additionally, it was noted that HDAC inhibitors enhance the expression of both CD95 and FAS-L in RCCs suggestive that pre-treatment of RCCs with HDAC inhibitors may prove even more efficacious in enhancing MDA-7/IL-24 toxicity. Thus our data demonstrate that the agent combination of MDA-7/IL-24 and HDAC inhibitors kills RCCs through a mechanism that requires CD95 signaling and increased levels of dihydro-ceramide, ROS, and Ca2+. The drug combination induces a toxic form of autophagy and endoplasmic reticulum stress, which results in reduced expression of the protective proteins BCL-XL and MCL-1. Collectively, the combination of MDA-7/IL-24 and HDAC inhibitors pre-disposes RCCs to die through multiple overlapping mechanisms.

Figure 8. MDA-7 promotes MDA-7 synthesis in Tumor A that when released into the vasculature can act upon distant tumor cells in Tumor B to induce more MDA-7 that results in tumor cell death. MDA-7 as a single agent acts to suppress MCL-1 levels and increase ROS levels that contributes to cell killing MDA-7 combined with HDAC inhibitors promotes: more ER stress signaling resulting in less MCL-1; more ROS, Ca2+, and ceramide production resulting in more mitochondrial dysfunction and death receptor activation; increased expression of CD95 and FAS-L resulting in more extrinsic pathway death signaling.
The expression of CAR is reduced in many tumor cell types such as RCCs, which is required by serotype 5 adenoviruses to infect cells.21,40 Thus, in order to identify CAR-independent pathways to enhance viral infectivity, we incorporated the viral capsid “knob” from a serotype 3 adenovirus into the adenovirus type 5 “knob”. Subsequently, we demonstrated that a modified serotype 5/3 knob adenovirus was able to infect low-CAR RCCs in vitro and in vivo. Within a tumor the infection of every tumor cell by an infused adenovirus is impossible using a non-replicative adenovirus. Unfortunately not only have many gene therapy studies used serotype 5 viruses, but many studies have also frequently expressed intracellular proteins which means that only those cells which have been virally infected are being subjected to the actions of the therapeutic gene, i.e., infections to express an intracellular protein lack a toxic “bystander” effect that would be generated by a secreted toxic protein on uninfected tumor cells.
In order to overcome the issues of infectivity in localized and loco-regional RCC in situ, we made use of Ad.5/3-PEG-E1A-mda-7 (CRAd.5/3-mda-7; Ad.5/3-CTV), which infects low CAR RCCs and replicates selectively in tumor cells. Infected RCC and stromal cells secrete MDA-7/IL-24 protein into the culture media or surrounding microenvironment, a result which we have recently demonstrated in renal, GBM and prostate cancer cells, where media containing secreted MDA-7/IL-24 induces apoptosis in uninfected tumor cells.18,41,42 In addition to this, MDA-7/IL-24 post-transcriptionally controls its own expression and can induce its own synthesis in tumor cells. This further amplifies the initial effects of viral infection or the initial MDA-7/IL-24 secretion, thus overcoming the problems associated with a lack of a toxic “bystander” effect following adenoviral-mediated gene therapy.
Ad.5/3-CTV and Ad.5/3-mda-7 caused similar levels of prolonged growth suppression in RCC tumors that were directly infused with viruses; however, in uninfected contralateral tumors Ad.5/3-CTV to a greater extent than Ad.5/3-mda-7 suppressed tumor growth. We have performed similar studies with Ad.5/3-CTV using primary human glioblastoma tumors as a model and based on the findings in glioblastoma, as per FDA regulations, we made use of the Syrian hamster to perform our preliminary toxicology testing, which is an approved rodent model for studying human adenovirus replication.43 Infection of a constitutively replicating adenovirus Ad.5/3-cmv-E1A into the hamster brain resulted in significant levels of apoptosis and expression of the viral protein E1A. Conversely, infection of Ad.5/3-CTV into hamster brain did not result in apoptosis and expression of the viral E1A protein was undetectable which emphasizes the fact that viral replication driven by the PEG-3 promoter was tumor cell specific as observed in CNS cell types.
In vitro we demonstrated that MDA-7/IL-24 synergized with HDAC inhibitors to kill RCCs and other GU tumor cell types. HDAC inhibitors have been examined in many tissues in many trials (e.g., refs. 44 and 45). We formed RCC tumors on the flanks of mice and infused low particle levels of Ad.5/3-mda-7 to permit assessment of any combinatorial anti-tumor effects and to isolate the effect of MDA-7/IL-24 expression from any anti-tumor viral replication effects. Expression of MDA-7/IL-24 reduced tumor growth by ~40% whereas treatment with an HDAC inhibitor had no discernible effect on tumor growth. Combined exposure to MDA-7/IL-24 and HDAC inhibitor resulted in tumor volumes that were at or below the day 0 values in uninfected tumors. Thus HDAC inhibitors also enhance the anti-tumor effects of MDA-7/IL-24 against RCC tumors in vivo.
In conclusion, MDA-7/IL-24 toxicity can be enhanced in RCCs by HDAC inhibitors. The combination of these two agents kills through activation of CD95 (and possibly increased CD95 and FAS-L expression), dihydro-ceramide/ROS/Ca2+ generation and ER stress-induced lowering of BCL-XL and MCL-1 levels. Further studies will be required to determine whether Ad.5/3-CTV is a useful tool to treat renal carcinoma and whether MDA-7/IL-24 and HDAC inhibitors interact in patients.
Materials and Methods
Suberohydroxamic acid (SBHA) and vorinostat (SAHA) were supplied by Calbiochem as a powder, dissolved in sterile DMSO, and stored frozen under light-protected conditions at −80 °C. Trypsin-EDTA, DMEM, and RPMI medium, and penicillin–streptomycin were purchased from GIBCOBRL (GIBCOBRL Life Technologies). Cell lines were obtained from the ATCC (A498, CAKI, 786-0) and NCI-Frederick (UOK121LN) and information on the genetic background of such cells has been published. Dr S Spiegel (VCU) supplied the plasmid to express LC3-GFP. Other reagents were of the highest quality commercially available (see refs. 10, 15–19, and 35).
Generation of adenoviruses
Recombinant serotype 5 and serotype 5/3 adenoviruses to express MDA-7/IL-24 and control empty vector were generated as described in references 23 and 41. Ad5/3.PEG-E1.mda-7 (Ad.5/3-CTV) was prepared in collaboration with Drs Igor Dmitriev and David Curiel, Washington University School of Medicine. This recombinant virus was generated in three consecutive steps: (1) Homologous recombination of pAd5/3 genomic plasmid with pShuttlE3 plasmid containing the mda-7/IL-24 expression cassette and kanamycin selection resulted in the pAd5/3.E3-mda-7 genome; (2) pAd5/3.E3-mda-7 was cut with Swa I to excise the kanamycin resistance gene; (3) The resultant pAd5/3.E3-mda-7 plasmid was recombined with pShuttlE1 plasmid containing E1A and E1B genes under control of the PEG-3 promoter resulting in Ad5/3.PEG-E1.mda-7 (Ad5/3-CTV; CRAd.5/3-mda-7) genomic plasmid. This plasmid was digested with Pac I to release viral ITRs and transfected in A549 cells to rescue the CRCA, Ad.5/3-CTV. Similar strategies were used to generate Ad.5/3-cmv-E1A-mda-7 and Ad.5/3-PEG-mda-7. Viruses were expanded and titers determined as previously described.22,23
Cell culture and in vitro exposure of cells to GST-MDA-7, “Ad.mda-7”, and drugs
All RCC lines were cultured at 37 °C (5% (v/v CO2) in vitro using RPMI supplemented with 5% (v/v) fetal calf serum and 10% (v/v) Non-essential amino acids. For short-term cell killing assays and immunoblotting, cells were plated at a density of 3 × 103 per cm2 and 36 h after plating were treated with MDA-7/IL-24 and/or various drugs, as indicated. In vitro small molecule inhibitor treatments were from a 100 mM stock solution of each drug and the maximal concentration of Vehicle (DMSO) in media was 0.02% (v/v). For adenoviral infection, cells were infected 12 h after plating and the expression of the recombinant viral transgene was allowed to occur for at least 12 h prior to any additional experimental procedure. Cells were not cultured in reduced serum media during any study.
Recombinant adenoviral vectors; infection in vitro
Cells were infected with serotype 5/serotype 3 adenoviruses at an approximate m.o.i. of 1–50 based on the experiment (see legends). Cells were incubated for 24 h to ensure adequate expression of transduced gene products prior to drug exposures.
Detection of cell death by trypan blue assays
Cells were harvested by trypsinization with Trypsin/EDTA for ~10 min at 37 °C. As some apoptotic cells detached from the culture substratum into the medium, these cells were also collected by centrifugation of the medium at 1500 rpm for 5 min. The pooled cell pellets were resuspended and mixed with trypan blue dye. Trypan blue stain, in which blue dye incorporating cells were scored as being dead, was performed by counting of cells using a light microscope and a hemacytometer. Five hundred cells from randomly chosen fields were counted and the number of dead cells was counted and expressed as a percentage of the total number of cells counted.
Plasmid transfection
Plasmid DNA (0.5 μg/total plasmid transfected) was diluted into 50 μl of RPMI growth media that lacked supplementation with FBS or with penicillin-streptomycin. Lipofectamine 2000 reagent (1 μl) (Invitrogen) was diluted into 50 μl growth media that lacked supplementation with FBS or with penicillin–streptomycin. The two solutions were then mixed together and incubated at room temperature for 30 min. The total mix was added to each well (4-well glass slide or 12-well plate) containing 200 μl growth media that lacked supplementation with FBS or with penicillin-streptomycin. The cells were incubated for 4 h at 37 °C, after which time the media was replaced with RPMI growth media containing 5% (v/v) FBS and 1× pen-strep.
Microscopy for LC3-GFP expression
Where indicated LC3-GFP transfected cells, were 12 h after transfection infected with either “Ad.cmv” or “Ad.mda-7”, then cultured for 24 h. LC3-GFP transfected cells were visualized at the indicated time points on the Zeiss Axiovert 200 microscope using the FITC filter.
Inoculation of RCCs
Athymic female NCr-nu/nu mice (NCI-Fredrick) weighing ~20 g, were used for this study. Mice were maintained under pathogen-free conditions in facilities approved by the American Association for Accreditation of Laboratory Animal Care and in accordance with current regulations and standards of the US Department of Agriculture, the US Department of Health and Human Services, and the National Institutes of Health. RCCs were cultured in DMEM supplemented with 5% (v/v) fetal calf serum and 100 μg/ml (1% v/v) penicillin-streptomycin. Cells were incubated in a humidified atmosphere of 5% (v/v) CO2 at 37 °C. Flank injection of 1.0 × 106 RCCs in 100 μl of DMEM medium was performed over 10 s. Adenoviral vectors were administered 14 d after tumor cell implantation when tumor volume was ~100 mm3. Mice were anesthetized via i.p. administration of (ketamine, 40 mg/kg; xylazine, 3 mg/kg). Viral vectors suspended in 2 μl of PBS were delivered by slow infusion over a 10 min period (see figure legends).
Data analysis
Comparison of the effects of various treatments was performed using one way analysis of variance and a two tailed Student t test. Differences with a P value of < 0.05 were considered statistically significant. Statistical examination of in vivo animal survival data utilized log rank statistical analyses between the different treatment groups. Experiments shown are the means of multiple individual points from multiple experiments (± SEM).
Disclosure of Potential Conflicts of Interest
PD is the Universal Inc. Professor in Signal Transduction Research. PBF holds the Thelma Newmeyer Corman Chair in Cancer Research at the VCU Massey Cancer Center
Acknowledgments
Support for the present study was provided by PHS grants (P01-CA104177, R01-CA108325, R01-DK52825, R01-CA63753; R01-CA77141, R01-CA150215, R01-CA097318; R01-CA134721); The Jim Valvano “V” foundation, and Department of Defense Award (DAMD17–03–1-0262). We acknowledge Dr Dmitriev and Dr Curiel for assistance in producing Ad.5/3-PEG-E1A-mda-7 (Ad.5/3-CTV).
Glossary
Abbreviations:
- HDACI
histone deacetylase inhibitor
- VEH
vehicle
- dn
dominant negative
- PERK
PKR-like endoplasmic reticulum kinase
- IL
interleukin
- MDA
melanoma differentiation associated
- CTV
cancer terminator virus
- WT
wild type
- SBHA
suberohydroxamic acid
- SAHA
vorinostat (suberoylanilide hydroxamic acid, Zolinza)
- GBM
glioblastoma multiforme
- GFP
green fluorescent protein
- GST
glutathione S-transferase
- si
small interfering RNA
- SCR
scrambled
- Val
sodium valproate
- PEG
progression elevated gene
- LASS
longevity assurance homolog
- ROS
reactive oxygen species
- TRX
thioredoxin
- GX15-070
obatoclax
- E1A
adenoviral early region 1A
- pfu
plaque forming units
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/26110
References
- 1.Gillett MD, Cheville JC, Karnes RJ, Lohse CM, Kwon ED, Leibovich BC, Zincke H, Blute ML. Comparison of presentation and outcome for patients 18 to 40 and 60 to 70 years old with solid renal masses. J Urol. 2005;173:1893–6. doi: 10.1097/01.ju.0000158157.57981.80. [DOI] [PubMed] [Google Scholar]
- 2.Jiang H, Lin JJ, Su ZZ, Goldstein NI, Fisher PB. Subtraction hybridization identifies a novel melanoma differentiation associated gene, mda-7, modulated during human melanoma differentiation, growth and progression. Oncogene. 1995;11:2477–86. [PubMed] [Google Scholar]
- 3.Ekmekcioglu S, Ellerhorst J, Mhashilkar AM, Sahin AA, Read CM, Prieto VG, Chada S, Grimm EA. Down-regulated melanoma differentiation associated gene (mda-7) expression in human melanomas. Int J Cancer. 2001;94:54–9. doi: 10.1002/ijc.1437. [DOI] [PubMed] [Google Scholar]
- 4.Ellerhorst JA, Prieto VG, Ekmekcioglu S, Broemeling L, Yekell S, Chada S, Grimm EA. Loss of MDA-7 expression with progression of melanoma. J Clin Oncol. 2002;20:1069–74. doi: 10.1200/JCO.20.4.1069. [DOI] [PubMed] [Google Scholar]
- 5.Huang EY, Madireddi MT, Gopalkrishnan RV, Leszczyniecka M, Su Z, Lebedeva IV, Kang D, Jiang H, Lin JJ, Alexandre D, et al. Genomic structure, chromosomal localization and expression profile of a novel melanoma differentiation associated (mda-7) gene with cancer specific growth suppressing and apoptosis inducing properties. Oncogene. 2001;20:7051–63. doi: 10.1038/sj.onc.1204897. [DOI] [PubMed] [Google Scholar]
- 6.Emdad L, Lebedeva IV, Su ZZ, Gupta P, Sauane M, Dash R, Grant S, Dent P, Curiel DT, Sarkar D, et al. Historical perspective and recent insights into our understanding of the molecular and biochemical basis of the antitumor properties of mda-7/IL-24. Cancer Biol Ther. 2009;8:391–400. doi: 10.4161/cbt.8.5.7581. [DOI] [PubMed] [Google Scholar]
- 7.Cunningham CC, Chada S, Merritt JA, Tong A, Senzer N, Zhang Y, Mhashilkar A, Parker K, Vukelja S, Richards D, et al. Clinical and local biological effects of an intratumoral injection of mda-7 (IL24; INGN 241) in patients with advanced carcinoma: a phase I study. Mol Ther. 2005;11:149–59. doi: 10.1016/j.ymthe.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 8.Pestka S, Krause CD, Sarkar D, Walter MR, Shi Y, Fisher PB. Interleukin-10 and related cytokines and receptors. Annu Rev Immunol. 2004;22:929–79. doi: 10.1146/annurev.immunol.22.012703.104622. [DOI] [PubMed] [Google Scholar]
- 9.Gupta P, Su ZZ, Lebedeva IV, Sarkar D, Sauane M, Emdad L, Bachelor MA, Grant S, Curiel DT, Dent P, et al. mda-7/IL-24: multifunctional cancer-specific apoptosis-inducing cytokine. Pharmacol Ther. 2006;111:596–628. doi: 10.1016/j.pharmthera.2005.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Park MA, Walker T, Martin AP, Allegood J, Vozhilla N, Emdad L, Sarkar D, Rahmani M, Graf M, Yacoub A, et al. MDA-7/IL-24-induced cell killing in malignant renal carcinoma cells occurs by a ceramide/CD95/PERK-dependent mechanism. Mol Cancer Ther. 2009;8:1280–91. doi: 10.1158/1535-7163.MCT-09-0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lebedeva IV, Emdad L, Su ZZ, Gupta P, Sauane M, Sarkar D, Staudt MR, Liu SJ, Taher MM, Xiao R, et al. mda-7/IL-24, novel anticancer cytokine: focus on bystander antitumor, radiosensitization and antiangiogenic properties and overview of the phase I clinical experience (Review) Int J Oncol. 2007;31:985–1007. [PubMed] [Google Scholar]
- 12.Su Z, Lebedeva IV, Gopalkrishnan RV, Goldstein NI, Stein CA, Reed JC, Dent P, Fisher PB. A combinatorial approach for selectively inducing programmed cell death in human pancreatic cancer cells. Proc Natl Acad Sci U S A. 2001;98:10332–7. doi: 10.1073/pnas.171315198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Su ZZ, Madireddi MT, Lin JJ, Young CS, Kitada S, Reed JC, Goldstein NI, Fisher PB. The cancer growth suppressor gene mda-7 selectively induces apoptosis in human breast cancer cells and inhibits tumor growth in nude mice. Proc Natl Acad Sci U S A. 1998;95:14400–5. doi: 10.1073/pnas.95.24.14400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Su ZZ, Lebedeva IV, Sarkar D, Emdad L, Gupta P, Kitada S, Dent P, Reed JC, Fisher PB. Ionizing radiation enhances therapeutic activity of mda-7/IL-24: overcoming radiation- and mda-7/IL-24-resistance in prostate cancer cells overexpressing the antiapoptotic proteins bcl-xL or bcl-2. Oncogene. 2006;25:2339–48. doi: 10.1038/sj.onc.1209271. [DOI] [PubMed] [Google Scholar]
- 15.Yacoub A, Hamed HA, Allegood J, Mitchell C, Spiegel S, Lesniak MS, Ogretmen B, Dash R, Sarkar D, Broaddus WC, et al. PERK-dependent regulation of ceramide synthase 6 and thioredoxin play a key role in mda-7/IL-24-induced killing of primary human glioblastoma multiforme cells. Cancer Res. 2010;70:1120–9. doi: 10.1158/0008-5472.CAN-09-4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yacoub A, Park MA, Gupta P, Rahmani M, Zhang G, Hamed H, Hanna D, Sarkar D, Lebedeva IV, Emdad L, et al. Caspase-, cathepsin-, and PERK-dependent regulation of MDA-7/IL-24-induced cell killing in primary human glioma cells. Mol Cancer Ther. 2008;7:297–313. doi: 10.1158/1535-7163.MCT-07-2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yacoub A, Liu R, Park MA, Hamed HA, Dash R, Schramm DN, Sarkar D, Dimitriev IP, Bell JK, Grant S, et al. Cisplatin enhances protein kinase R-like endoplasmic reticulum kinase- and CD95-dependent melanoma differentiation-associated gene-7/interleukin-24-induced killing in ovarian carcinoma cells. Mol Pharmacol. 2010;77:298–310. doi: 10.1124/mol.109.061820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park MA, Hamed HA, Mitchell C, Cruickshanks N, Dash R, Allegood J, Dmitriev IP, Tye G, Ogretmen B, Spiegel S, et al. A serotype 5/3 adenovirus expressing MDA-7/IL-24 infects renal carcinoma cells and promotes toxicity of agents that increase ROS and ceramide levels. Mol Pharmacol. 2011;79:368–80. doi: 10.1124/mol.110.069484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eulitt PJ, Park MA, Hossein H, Cruikshanks N, Yang C, Dmitriev IP, Yacoub A, Curiel DT, Fisher PB, Dent P. Enhancing mda-7/IL-24 therapy in renal carcinoma cells by inhibiting multiple protective signaling pathways using sorafenib and by Ad.5/3 gene delivery. Cancer Biol Ther. 2010;10:1290–305. doi: 10.4161/cbt.10.12.13497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Waehler R, Russell SJ, Curiel DT. Engineering targeted viral vectors for gene therapy. Nat Rev Genet. 2007;8:573–87. doi: 10.1038/nrg2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haviv YS, Blackwell JL, Kanerva A, Nagi P, Krasnykh V, Dmitriev I, Wang M, Naito S, Lei X, Hemminki A, et al. Adenoviral gene therapy for renal cancer requires retargeting to alternative cellular receptors. Cancer Res. 2002;62:4273–81. [PubMed] [Google Scholar]
- 22.Azab B, Dash R, Das SK, Bhutia SK, Shen XN, Quinn BA, Sarkar S, Wang XY, Hedvat M, Dmitriev IP, et al. Enhanced delivery of mda-7/IL-24 using a serotype chimeric adenovirus (Ad.5/3) in combination with the Apogossypol derivative BI-97C1 (Sabutoclax) improves therapeutic efficacy in low CAR colorectal cancer cells. J Cell Physiol. 2012;227:2145–53. doi: 10.1002/jcp.22947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dash R, Dmitriev I, Su ZZ, Bhutia SK, Azab B, Vozhilla N, Yacoub A, Dent P, Curiel DT, Sarkar D, et al. Enhanced delivery of mda-7/IL-24 using a serotype chimeric adenovirus (Ad.5/3) improves therapeutic efficacy in low CAR prostate cancer cells. Cancer Gene Ther. 2010;17:447–56. doi: 10.1038/cgt.2009.91. [DOI] [PubMed] [Google Scholar]
- 24.Sarkar D, Lebedeva IV, Su ZZ, Park ES, Chatman L, Vozhilla N, Dent P, Curiel DT, Fisher PB. Eradication of therapy-resistant human prostate tumors using a cancer terminator virus. Cancer Res. 2007;67:5434–42. doi: 10.1158/0008-5472.CAN-07-0195. [DOI] [PubMed] [Google Scholar]
- 25.Sarkar D, Su ZZ, Park ES, Vozhilla N, Dent P, Curiel DT, Fisher PB. A cancer terminator virus eradicates both primary and distant human melanomas. Cancer Gene Ther. 2008;15:293–302. doi: 10.1038/cgt.2008.14. [DOI] [PubMed] [Google Scholar]
- 26.Su ZZ, Sarkar D, Emdad L, Duigou GJ, Young CS, Ware J, Randolph A, Valerie K, Fisher PB. Targeting gene expression selectively in cancer cells by using the progression-elevated gene-3 promoter. Proc Natl Acad Sci U S A. 2005;102:1059–64. doi: 10.1073/pnas.0409141102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sauane M, Su ZZ, Gupta P, Lebedeva IV, Dent P, Sarkar D, Fisher PB. Autocrine regulation of mda-7/IL-24 mediates cancer-specific apoptosis. Proc Natl Acad Sci U S A. 2008;105:9763–8. doi: 10.1073/pnas.0804089105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Emdad L, Lebedeva IV, Su ZZ, Gupta P, Sauane M, Dash R, Grant S, Dent P, Curiel DT, Sarkar D, et al. Historical perspective and recent insights into our understanding of the molecular and biochemical basis of the antitumor properties of mda-7/IL-24. Cancer Biol Ther. 2009;8:391–400. doi: 10.4161/cbt.8.5.7581. [DOI] [PubMed] [Google Scholar]
- 29.Ellis L, Pili R. Histone Deacetylase Inhibitors: Advancing Therapeutic Strategies in Hematological and Solid Malignancies. Pharmaceuticals (Basel) 2010;3:2411–69. doi: 10.3390/ph3082441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frew AJ, Johnstone RW, Bolden JE. Enhancing the apoptotic and therapeutic effects of HDAC inhibitors. Cancer Lett. 2009;280:125–33. doi: 10.1016/j.canlet.2009.02.042. [DOI] [PubMed] [Google Scholar]
- 31.Kahali S, Sarcar B, Fang B, Williams ES, Koomen JM, Tofilon PJ, Chinnaiyan P. Activation of the unfolded protein response contributes toward the antitumor activity of vorinostat. Neoplasia. 2010;12:80–6. doi: 10.1593/neo.91422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spiegel S, Milstien S, Grant S. Endogenous modulators and pharmacological inhibitors of histone deacetylases in cancer therapy. Oncogene. 2012;31:537–51. doi: 10.1038/onc.2011.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruefli AA, Ausserlechner MJ, Bernhard D, Sutton VR, Tainton KM, Kofler R, Smyth MJ, Johnstone RW. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc Natl Acad Sci U S A. 2001;98:10833–8. doi: 10.1073/pnas.191208598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosato RR, Grant S. Histone deacetylase inhibitors in clinical development. Expert Opin Investig Drugs. 2004;13:21–38. doi: 10.1517/13543784.13.1.21. [DOI] [PubMed] [Google Scholar]
- 35.Zhang G, Park MA, Mitchell C, Hamed H, Rahmani M, Martin AP, Curiel DT, Yacoub A, Graf M, Lee R, et al. Vorinostat and sorafenib synergistically kill tumor cells via FLIP suppression and CD95 activation. Clin Cancer Res. 2008;14:5385–99. doi: 10.1158/1078-0432.CCR-08-0469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagaprashantha L, Vartak N, Awasthi S, Awasthi S, Singhal SS. Novel anti-cancer compounds for developing combinatorial therapies to target anoikis-resistant tumors. Pharm Res. 2012;29:621–36. doi: 10.1007/s11095-011-0645-9. [DOI] [PubMed] [Google Scholar]
- 37.Park MA, Reinehr R, Häussinger D, Voelkel-Johnson C, Ogretmen B, Yacoub A, Grant S, Dent P. Sorafenib activates CD95 and promotes autophagy and cell death via Src family kinases in gastrointestinal tumor cells. Mol Cancer Ther. 2010;9:2220–31. doi: 10.1158/1535-7163.MCT-10-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gupta P, Walter MR, Su ZZ, Lebedeva IV, Emdad L, Randolph A, Valerie K, Sarkar D, Fisher PB. BiP/GRP78 is an intracellular target for MDA-7/IL-24 induction of cancer-specific apoptosis. Cancer Res. 2006;66:8182–91. doi: 10.1158/0008-5472.CAN-06-0577. [DOI] [PubMed] [Google Scholar]
- 39.Sauane M, Su ZZ, Dash R, Liu X, Norris JS, Sarkar D, Lee SG, Allegood JC, Dent P, Spiegel S, et al. Ceramide plays a prominent role in MDA-7/IL-24-induced cancer-specific apoptosis. J Cell Physiol. 2010;222:546–55. doi: 10.1002/jcp.21969. [DOI] [PubMed] [Google Scholar]
- 40.Paul CP, Everts M, Glasgow JN, Dent P, Fisher PB, Ulasov IV, Lesniak MS, Stoff-Khalili MA, Roth JC, Preuss MA, et al. Characterization of infectivity of knob-modified adenoviral vectors in glioma. Cancer Biol Ther. 2008;7:786–93. doi: 10.4161/cbt.7.5.5421. [DOI] [PubMed] [Google Scholar]
- 41.Hamed HA, Yacoub A, Park MA, Eulitt PJ, Dash R, Sarkar D, Dmitriev IP, Lesniak MS, Shah K, Grant S, et al. Inhibition of multiple protective signaling pathways and Ad.5/3 delivery enhances mda-7/IL-24 therapy of malignant glioma. Mol Ther. 2010;18:1130–42. doi: 10.1038/mt.2010.29. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 42.Bhutia SK, Das SK, Azab B, Dash R, Su ZZ, Lee SG, Dent P, Curiel DT, Sarkar D, Fisher PB. Autophagy switches to apoptosis in prostate cancer cells infected with melanoma differentiation associated gene-7/interleukin-24 (mda-7/IL-24) Autophagy. 2011;7:1076–7. doi: 10.4161/auto.7.9.16163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dhar D, Toth K, Wold WS. Syrian hamster tumor model to study oncolytic Ad5-based vectors. Methods Mol Biol. 2012;797:53–63. doi: 10.1007/978-1-61779-340-0_4. [DOI] [PubMed] [Google Scholar]
- 44.Emanuele S, Lauricella M, Carlisi D, Vassallo B, D’Anneo A, Di Fazio P, Vento R, Tesoriere G. SAHA induces apoptosis in hepatoma cells and synergistically interacts with the proteasome inhibitor Bortezomib. Apoptosis. 2007;12:1327–38. doi: 10.1007/s10495-007-0063-y. [DOI] [PubMed] [Google Scholar]
- 45.Galanis E, Jaeckle KA, Maurer MJ, Reid JM, Ames MM, Hardwick JS, Reilly JF, Loboda A, Nebozhyn M, Fantin VR, et al. Phase II trial of vorinostat in recurrent glioblastoma multiforme: a north central cancer treatment group study. J Clin Oncol. 2009;27:2052–8. doi: 10.1200/JCO.2008.19.0694. [DOI] [PMC free article] [PubMed] [Google Scholar]