

Abstract
Transforming growth factor β (TGF-β)/Smad signaling is involved in colorectal carcinoma (CRC) development and progression. The frequent loss of SMAD4 is associated with liver metastasis and poor prognosis of CRC, but the underlying mechanism remains elusive. This study aimed to elucidate the role of Smad-independent TGF-β signaling in CRC metastasis. Immunohistochemistry showed that Smad4 level was negatively correlated with TNM stage and phospho-ERK level in human CRCs and liver metastasis samples. Knockdown of Smad4 in CT26 and HCT116 cells activated ERK pathway, altered the expression of MMP2 and COX-2, promoted cell motility, migration, and invasion in vitro, enhanced metastasis, and shortened the survival of metastatic tumor-bearing mice. MEK inhibitor U0126 and GSK1120212 inhibited the motility, migration, and invasion of Smad4 knockdown cells, inhibited metastasis, and prolonged the survival of metastatic tumor-bearing mice. Furthermore, MEK inhibitor could reverse the changes of phospho-ERK, MMP2, and COX-2 levels. In conclusion, our results indicate that ERK pathway plays a key oncogenic role in CRC with SMAD4 inactivation mutations, and implicate ERK as a potential therapeutic target for CRC liver metastasis.
Keywords: ERK pathway, Smad4, TGF-β, colorectal cancer, metastasis
Introduction
Colorectal carcinoma (CRC) is one of the most common causes of cancer-related deaths in the world. Especially, liver metastasis is the predominant cause of CRC-related mortality.1-3 Synchronous disease with liver metastasis may be found in 10–25% of patients at the time of initial diagnosis and the prognosis and survival rate of CRC patients are determined by the presence of metastasis.4,5 Despite great improvements in early diagnostic technique and adjuvant chemotherapy, the 5-year survival rate of CRC patients remains low (21–44%) even in those amenable to surgical resection.4,6 For those patients who have not undergone surgical treatment, the 5-year survival rate following the diagnosis of liver metastasis is below 10%.7 Therefore, there is urgent need to understand the molecular mechanisms underlying colorectal cancer development and metastasis in order to develop novel effective approaches for CRC diagnosis and therapy.
Transforming growth factor β (TGF-β) signaling regulates a wide variety of cellular processes such as proliferation, differentiation, invasion, angiogenesis, and cell survival, and is crucially involved in tumorigenesis.8,9 In response to TGF-β, TGF-β receptor I and receptor II recruit and phosphorylate Smad2 and Smad3, which then associate with Smad4 to translocate into the nucleus to modulate the transcription of target genes. Smad4 is essential for canonical TGF-β pathway. In addition, TGF-β can induce Smad4 independent signaling pathways such as MEK/ERK, JNK, p38MAPK, and PI3K/AKT to regulate the progression of a variety of cancers.10-12
Loss of SMAD4 expression has been shown to be correlated with poor prognosis of CRC patients.13 However, the underlying mechanism remains elusive. Our previous study showed that TGF-β promoted liver metastasis of CRC via both Smad-dependent and Smad-independent pathways, and TGF-β activated extracellular regulated protein kinases (ERK) pathway in CRC cell line CT26.14 Kras is known to be overactivated in approximately 35% of sporadic colorectal cancer, leading to the hyperactivation of mitogen-activated protein kinases (MAPK) cascade, which then cooperates with TGF-β to promote CRC liver metastasis.15,16 Therefore, we speculated that TGF-β may activate Smad-independent ERK pathway to contribute to CRC metastasis.
In this study, by using human CRC samples and CT26 and HCT116 cells, and metastatic mouse model, we investigated the correlation between loss of smad4 and activation of ERK both in vitro and in vivo, and elucidated the important oncogenic role of ERK in mediating TGF-β induced CRC metastasis.
Results
Smad4 is negatively correlated with phosphorylation of ERK in metastatic CRC samples
Fifty-three human CRC metastatic tumors and seven human primary CRC samples were collected, and the expression of SMAD4 and phospho-ERK was determined by immunohistochemistry. SMAD4 staining in primary CRC tumor was significantly stronger than in liver metastasis (Mann–Whitney U-test, P = 0.003), and phospho-ERK staining in primary CRC tumor was significantly lower than in liver metastasis (Mann–Whitney U-test, P < 0.001) (Fig. 1A and B). The Spearman analysis showed that SMAD4 expression was negatively correlated with the TNM stage of CRC primary tumor. In addition, phospho-ERK level was negatively correlated with the level of SMAD4 in CRC samples (Table 1). These data provide evidence that activation of ERK is associated with the loss of SMAD4 in human CRC.
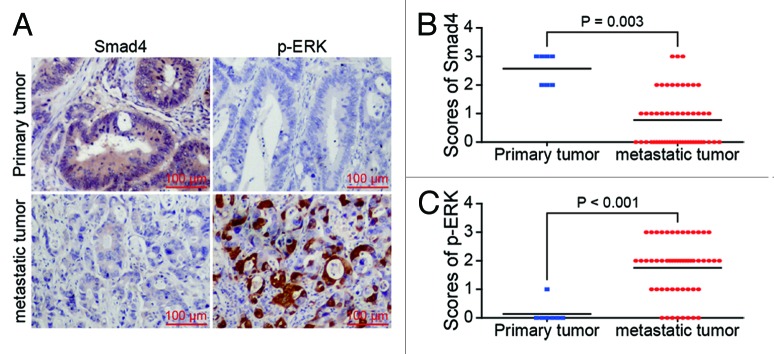
Figure 1. Smad4 is negatively related with phosphorylation of ERK in human metastatic CRCs. (A) Representative images of immunostained Smad4 and phospho-ERK in paired primary tumor and metastasis originated from single patient. (B) The expression of Smad4 in primary tumors was higher than that in liver metastases, (C) phospho-ERK in primary tumors was lower than that in liver metastases (P = 0.003 and P < 0.001, respectively, Mann–Whitney U-test).
Table 1. Correlation analysis of Smad4 level, p-ERK level, and the TNM grade in colorectal cancer samples.
| Smad4 IHC grade | p-ERK IHC grade | TNM stage | ||||
|---|---|---|---|---|---|---|
| low expression | high expression | low expression | high expression | I and II | III | IV |
| 41 | 12 | 18 | 35 | 15a | 16 | 22 |
Smad4 and p-ERK level correlation analysis: Spearman r = -0.374, P = 0.006. Smad4 IHC grade and TNM stage correlation analysis: Spearman r = -0.294, P = 0.033. p-ERK IHC grade and TNM stage correlation analysis: Spearman r = 0.193, P = 0.166. aTwo cases were stratified into TNM stage I, which combined with 15 cases of TNM stage II for the Spearman correlation analysis.
Smad4 knockdown promotes the migration and invasion of CRC cells in vitro
In order to determine the role of Smad4 in colorectal cancer, we generated CT26-Smad4KD cells, HCT116-Smad4KD cells, and their corresponding negative control cells (Fig. 2A). To confirm that Smad-dependent TGF-β pathway is effectively blocked in CT26-Smad4KD cell, we detected the Smad2/3/4 complex by immunoprecipitation. The results showed that TGF-β induced functional complex formation between Smad2/3 and Smad4 in TGF-β treated CT26-NC cells, but only a trace of this functional complex was observed in CT26-Smad4KD cells (Fig. 2B, top panel). We also observed that TGF-β significantly induced Smad2 phosphorylation within 30 min (Fig. 2B, bottom panel). These results indicate that Smad-dependent pathway is inhibited in Smad4KD cells.

Figure 2. Smad4 knockdown promotes the migration and invasion of CT26 and HCT116 cells in vitro. (A) CT26-Smad4KD, HCT116-Smad4KD, and their wild type, negative control cells were subjected to western blot analysis with the indicated antibodies. (B) CT26-NC and CT26-Smad4KD cells were treated with TGF-β1 (10 ng/ml) for the indicated time and the lysates were immunoprecipitated by Smad2/3 antibody followed by western blotting with Smad4 antibody (top panel). Total cell lysates were subjected to western blot analyses with the indicated antibodies (bottom panel). GAPDH served as loading control. (C) CT26-WT, CT26-NC, and Smad4KD cells were treated with or without TGF-β (5 ng/ml) for 72 h. Cells were subjected to CCK-8 assay. (D) CT26-WT, CT26-NC, CT26-Smad4KD, HCT116-WT, HCT116-NC, and HCT116-Smad4KD cells were treated with TGF-β (5 ng/ml), and subjected to wound healing assay. (E and F) CT26-WT, CT26-NC, and Smad4KD cells were treated with or without TGF-β (5 ng/ml), and subjected to transwell migration and invasion assay. The data were presented as the mean ± SD (n = 6). (G and H) HCT116-WT, HCT116-NC, and HCT116-Smad4KD cells were treated with or without TGF-β (5 ng/ml), and subjected to transwell migration and invasion assay. The data were presented as the mean ± SD (n = 6).
To determine the biological consequences after Smad4 knockdown, we performed cell proliferation assay. The result showed that there was no significant difference in cell proliferation among CT26-WT, CT26-NC, and CT26-Smad4KD cells exposed to exogenous TGF-β (Fig. 2C). Wound healing assay showed that CT26-Smad4KD cells and HCT116-Smad4KD cells exhibited better wound healing compared with their negative control cells, respectively (Fig. 2D). Transwell migration assay demonstrated increased migration of CT26-Smad4KD cells compared with CT26-NC cells (in the absence of TGF-β: 45.06 ± 14.80 cells/lpf vs 200.50 ± 16.42 cells/lpf, P < 0.001; in the presence of TGF-β: 58.83 ± 23.73 cells/lpf vs 256.83 ± 23.04 cells/lpf, P < 0.001) (Fig. 2E). Invasion assay demonstrated increased invasion of CT26-Smad4KD cells compared with CT26-NC cells (in the absence of TGF-β: 68.67 ± 27.38 cells/lpf vs 158.38 ± 13.04 cells/lpf, P < 0.001; in the presence of TGF-β: 112.83 ± 35.27 cells/lpf vs 168.00 ± 35.15 cells/lpf, P = 0.022) (Fig. 2F). Additionally, we observed similar results in migration and invasion assay in human CRC cell line HCT116. The number of migrated cells of Smad4KD group was 1.9 or 2.7 times of that in negative control group in the absence or presence of TGF-β (Fig. 2G), while in invasion assay, the invasion rate was 1.8 or 2.2 (Fig. 2H). Taken together, these data indicate that Smad4 inhibits the migration and invasion of CT26 and HCT116 cells in vitro.
Smad4 knockdown enhances CRC liver metastasis in vivo
To investigate the role of Smad4 in CRC metastasis in vivo, we established animal model of metastasis by splenic injection of CT26 cells into Balb/c mice. We found that mice injected with CT26-Smad4KD cells showed significant increase in the growth of metastatic foci (Fig. 3A). The weight of metastatic livers in CT26-Smad4KD group was significantly increased compared with the CT26-NC group (1.50 ± 0.41 g vs 3.22 ± 0.99 g, P = 0.003) (Fig. 3B). Furthermore, the median survival of CT26-Smad4KD injected mice was shortened to 26 d, compared with 44.5 d of CT26-NC injected mice (Fig. 3C; P < 0.001, log-rank test). These results indicate that Smad4 inhibits metastatic potential of CT26 cells in vivo.

Figure 3. Smad4 knockdown enhances CRC liver metastasis in vivo. (A) The morphology of metastatic liver in CT26-WT group, CT26-NC group, and Smad4KD group. Liver metastasis was examined 25 d after injecting CT26-WT, CT26-NC, and Smad4KD cells into the spleens of Balb/c mice. (B) The average weight of the metastatic liver in each group (n = 6). There was significant difference in metastatic liver weight between the CT26-NC group and the Smad4KD group (1.68 ± 0.52 g vs 3.22 ± 0.99 g). (C) The survival curve of mice in CT26-NC group and Smad4KD group (n = 10).
The activation of MEK/ERK pathway contributes to TGF-β induced migration and invasion of CRC cells
MEK/ERK pathway is one of the downstream pathways of TGF-β that is Smad-independent. Thus we first investigated the activation status of MEK/ERK pathway in CT26 and HCT116 cells depleted of Smad4. We found that TGF-β-induced phosphorylation of ERK was significantly higher in Smad4KD cells than in the negative controls (Fig. 4A and B), MMP2 was downregulated and COX-2 was upregulated in Smad4KD cells, respectively (Fig. 4C), suggesting that the knockdown of Smad4 may favor the activation of MEK/ERK pathway in CT26 and HCT116 cells.

Figure 4. MEK inhibitor inhibits TGF-β-induced migration and invasion of CRC cells. (A) CT26-NC and CT26-Smad4KD cells were treated with TGF-β1 (5 ng/ml) for the indicated time and subjected to western blot analysis with the indicated antibodies. GAPDH served as loading control. (B) HCT116-NC and HCT116-Smad4KD cells were treated with TGF-β1 (5 ng/ml) and GSK1120212 as indicated for 24 h, and cell lysates were analyzed by western blotting. (C) Knockdown of Smad4 downregulates MMP2 and upregulates COX-2. (D and E) CT26-Smad4KD and HCT116-Smad4KD cells were treated as indicated, and cell lysates were analyzed by western blotting with antibodies against phospho-ERK, ERK, MMP2, COX-2, and GAPDH. (F) CT26-Smad4KD cells were treated as indicated and subjected to wound healing assay. (G and H) CT26-Smad4KD cells were treated as indicated and subjected to transwell migration and invasion assay. The data were presented as mean ± SD (n = 6). (I) HCT116-Smad4KD cells were treated as indicated and subjected to wound healing assay. (J and K) HCT116-Smad4KD cells were treated as indicated and subjected to transwell invasion assay. The data were presented as mean ± SD (n = 6).
To understand the contribution of MEK/ERK signaling to the biological behaviors of CT26-Smad4KD and HCT116-Smad4KD cells, we treated these cells with U0126 or GSK1120212, two selective inhibitors of MEK, and performed in vitro wound healing, transwell migration and invasion assays. Western blot analysis showed that U0126 and GSK1120212 inhibited the phosphorylation of ERK in CT26-Smad4KD and HCT116-Smad4KD cells, respectively (Fig. 4D and E). Interestingly, the inhibition of ERK signal reversed the changes of MMP2 and COX-2 altered by Smad4 knockdown (Fig. 4D and E). In addition, TGF-β enhanced the motility of Smad4KD cells in wound healing assay, while U0126 and GSK1120212 treatment inhibited TGF-β induced cell motility in CT26-Smad4KD and HCT116-Smad4KD cells, respectively (Fig. 4F and I). In transwell migration assay, we observed that TGF-β enhanced the migration of CT26-Smad4KD cells (21.50 ± 4.47 cells/lpf vs 33.63 ± 5.18 cells/lpf, P < 0.001), but U0126 treatment completely abrogated TGF-β induced migration of CT26-Smad4KD cells (33.63 ± 5.18 cells/lpf vs 10.25 ± 3.15 cells/lpf, P < 0.001) (Fig. 4G). Similarly, we found that U0126 inhibited TGF-β induced invasion of CT26-Smad4KD cells (162.25 ± 28.26 cells/lpf vs 17.00 ± 5.24 cells/lpf, P < 0.001) (Fig. 4H). Additionally, we evaluated the effects of GSK1120212 on migration and invasion of HCT116-Smad4KD cells in vitro. GSK1120212 led to 93% inhibition of cell migration (Fig. 4J), and 91% inhibition of cell invasion (Fig. 4K). Collectively, these results indicate that TGF-β induces the activation of MEK/ERK pathway independently of classical Smad dependent signal pathway, and MEK/ERK activation promotes the migration and invasion of CT26 and HCT116 cells.
U0126 treatment inhibits colorectal cancer liver metastasis and prolongs the survival of metastatic tumor-bearing mice
To further determine the therapeutic potential of U0126 in vivo, we employed liver metastasis mice model. The mice were divided into U0126 group and control group, which were intraperitoneally injected with U0126 and the vehicle, respectively. Mice in U0126 group showed significantly reduced liver metastases (Fig. 5A), and the weight of metastatic liver in U0126 group was significantly decreased compare with vehicle group (2.67 ± 0.37 g vs 1.67 ± 0.64 g, P = 0.008) (Fig. 5B). Furthermore, survival analysis showed that the median survival was prolonged to 45 d for mice in U0126 group compared with 27 d for mice in vehicle group (Fig. 5C; P < 0.001, log-rank test). These results indicate that inhibiting the activation of MEK/ERK pathway reduces colorectal cancer liver metastasis, and prolongs the survival of mice bearing metastasis.

Figure 5. MEK inhibitor inhibits CRC liver metastasis in vivo. (A) The morphology of metastatic liver in vehicle group and U0126 group. (B) The average weight of the metastatic liver in each group (n = 6). There was significant difference in metastatic liver weight between vehicle group and U0126 group. (C) The survival curve of mice in vehicle group and U0126 group (n = 8).
Discussion
It is generally considered that Smad-dependent pathway contributes to the tumor suppression function of TGF-β, while the activation of Smad-independent pathways leads to the loss of tumor suppressor function of TGF-β. In the later stages of CRC, TGF-β is known to play oncogenic role to induce tumor invasion and metastasis. The exact molecular mechanism underlying the functional shift of TGF-β remains elusive. In this study, we demonstrated that the inactivation of Smad-dependent signaling via knockdown of Smad4 in CRC cell line resulted in increased tumor metastasis, and this was associated with the activation of Smad-independent MER/ERK pathway.
Smad4 is one of most important tumor suppressors that plays role in cell proliferation, angiogenesis, migration, invasion, and metastasis.17-19 High SMAD4 level indicates a better prognosis in colon cancers.2,13 Loss of SMAD4 has been reported to occur in 9–67% of CRC and is associated with advanced disease, metastasis and poor prognosis of CRC.13,20-23 It has been proposed that loss of Smad4 may contribute to functional shift of TGF-β from tumor suppressor to tumor promoter.24,25 Notably, overactivation of TGF-β induced MEK/ERK and p38-MAPK pathways are involved in malignancy promotion and drug resistance of colon cancer.26 It is observed that the loss of Smad4 promoted disease progression in the presence of the activated Kras mutation27,28 but the molecular mechanism needs to be further elucidated.
In this study, we found a significant increase of motility, migration and invasion of Smad4KD cells in vitro and in vivo. These data suggest that the inactivation of Smad dependent pathway results in the functional shift of TGF-β, which is in agreement with the view that the downregulation or loss of SMAD4 increases the resistance to TGF-β induced tumor suppression and promotes malignant progression of CRC and pancreatic tumors.29,30 The enhancement of metastasis accounts for the loss of the inhibitory effect of Smad4. On the other hand, to determine whether increased metastatic ability of CT26-Smad4KD and HCT116-Smad4KD cells is caused by the activation of ERK pathway, we performed wound healing, migration and invasion assay in vitro. We observed that the phosphorylation of ERK was increased in CT26-Smad4KD and HCT116-Smad4KD cells compared with the negative control cells, indicating that Smad4 knockdown leads to the overactivation of ERK signal pathway. Furthermore, ERK inhibitor U0126 and GSK1120212 reduced TGF-β induced increase in the motility, migration and invasion of CT26-Smad4KD and HCT116-Smad4KD cells in vitro, respectively. In vivo we found that U0126 decreased the metastatic liver weight and prolonged the survival of metastatic tumor-bearing mice. It is reported that oncogenic Kras mutation is detected in 40–50% CRC and is critical for colorectal carcinogenesis due to its ability to activate MEK/ERK pathway and inhibit the tumor suppressor function of TGF-β.10-12,14 In our study, both CT26 and HCT116 cell lines had activation mutation of Kras gene, indicating that these cells are nice experimental models. Interestingly, Saha et al. reported that Smad4 protein was degraded in the condition of Ras activated mutation, which may provide an Ras/MEK/MAP autostimulatory activation mechanism.31
Notably, our results indicated that the loss of Smad4 alternated the level of MMP2 and COX-2 in CT26 and HCT116 cells, and this could be reversed by U0126 or GSK1120212 treatment. It has been proposed that the expression of MMP2 was associated with an unfavorable prognosis in a variety of tumors,7,32-34 while other studies suggested that MMP2 did not appear to be a significant predictor of the prognosis of CRC.35 However, it was reported recently that the absence of MMP2 expression in liver metastasis was associated with high local recurrence, distant metastasis and death.36,37 On the other hand, COX-2-derived PGE2 has been shown to be involved in the progression of CRC and bone metastasis of breast cancer.38,39 Taken together, these data suggest that TGF-β acts in a Smad-independent manner to activate MEK/ERK pathway, which then regulates the expression of MMP2 and COX-2 to promote cell motility, migration and invasion, leading to metastasis.
In summary, we employed Smad4 knockdown CT26 and HCT116 cells, MEK inhibitor U0126 and GSK1120212, metastatic mouse model, and human CRC samples to provide a series of evidence that loss of smad4 is positively correlated with the activation of ERK pathway in CRC both in vitro and in vivo, and ERK pathway is crucially involved in TGF-β induced CRC metastasis. Our findings suggest that ERK pathway may be exploited for therapeutic intervention to inhibit CRC progression and metastasis.
Materials and Methods
Patients and specimens
Fifty-three tumor patients with CRC liver metastasis were enrolled in this study who were diagnosed at Tongji Hospital (Wuhan, China) from 2004 to 2011. All of metastatic tumors and 7 primary tumor tissues of these patients were collected and frozen immediately. The stages of primary colorectal cancer were stage I, II, III, and IV. Thirty-five were men and 14 were women, with the remaining 4 unknown due to lack of registry information. The median age at diagnosis was 57 y (range: 26 to 77 y). Ethics approval was obtained from the institutional review boards and all patients gave signed consent.
Immunohistochemical analysis
The sections were deparaffinized and rehydrated, and endogenous peroxidase was blocked using 0.3% H2O2. For antigen retrieval, sections were heated in citrate buffer for 15 min. The slides were blocked with 10% BSA and incubated with the following primary antibodies at 4 °C overnight: Smad4 (Epitomics, rabbit monoclonal, diluted 1:100), Phospho-p44/42 MAPK (Erk1/2) (CST, rabbit monoclonal, diluted 1:400), followed by the incubation with horseradish peroxidase (HRP) conjugated secondary antibody for 1 h. The visualization signal was developed with DAB staining, and the slides were counterstained with hematoxylin. For negative controls the primary antibodies were replaced with PBS. Staining scores were calculated as described previously,36 based on the number of tumor cells with positive staining in the cytoplasm as follows: 0 or none (expression <10%), 1+ or weak (10% to 50%), 2+ or strong (50% to 80%), and 3+ or intense (>80%). The scoring was performed by two independent pathologists who were blinded to clinicopathologic data.
Cell culture
Mouse CRC cell line CT26 was an N-nitroso-N-methylurethane-(NNMU)-induced, undifferentiated colon carcinoma cell line (ATCC number: CRL-2638). HCT116 was a human CRC cell line which had a constitutive activation mutation in codon 13 of ras proto-oncogene (ATCC number: CCL-247). These cell lines were purchased from cell bank of Chinese Academy of Sciences where they were characterized by mycoplasma detection, isozyme detection, DNA-fingerprinting, and vitality detection. The cell line was immediately expanded and frozen so that they could be recovered every three months from a frozen vial of the same batch of cells. CT26 cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (Hyclone) at 37 °C and 5% CO2, while HCT116 cells were maintained in the McCoy 5A medium (Hyclone).
CT26 cells infection and selection
For small interfering RNA (siRNA)-mediated Smad4 silencing, the following target siRNA sequences of Smad4 were used: CCAGCTACTT ACCATCATA. pGCSIL-PUR-Smad4 small hairpin RNA (shRNA), a Smad4-RNA interference (RNAi) lentiviral vector was constructed (Shanghai GeneChem Co, Ltd). Double-stranded oligonucleotides corresponding to mouse Smad4-vshRNA (NM_008540; CcggagCCAG CTACTTACCA TCATATTCAA GAGATATGAT GGTAAGTAGC TGGctTTTTT g) were annealed and inserted into the small hairpin RNA (shRNA) expression vector pGCSIL-PUR. pGCSIL-PUR-NC (negative control sequence: TTCTCCGAAC GTGTCACGT) was used as a negative control. The lentivirus was infected into CT26 cells and the infected cells were treated with 7 μg/ml puromycin (ENZO Life Sciences) for 2 weeks. Stable cell line with knockdown of Smad4 was selected and named as CT26-Smad4KD.
Knockdown of Smad4 using small interfering RNA in HCT116 cells
The Smad4 and negative control small interfering RNAs (siRNAs) were purchased from RiboBio. The sequences of Smad4-siRNA and the negative control were described above. Wild type HCT116 cells were seeded of 70% confluent in six-well plates and transfected with 100 nM Smad4 or negative control siRNA oligoduplexes after preincubation for 10 min with Lipofectamine RNAiMAX Reagent (Invitrogen) in serum-free Opti-MEM I Reduced Serum Medium (Invitrogen). The cells were collected 48 h after the transfection, and used for the following assays.
Cell proliferation assay
Cell proliferation assay was performed using the Cell Counting Kit-8 (CCK8) Kit (Beyotime) according to the manufacturer’s instructions. Briefly, cells were seeded at the density of 1200 cells/well in 96-well plates. At the indicated time points, 10% CCK8 was added into each well followed by incubation for 1 h, and then the plates were read using an enzyme-linked immunosorbent assay plate reader at 450 nm.
Wound healing assay
Confluent cells were treated with mitomycin C (1 μg/ml) for 3 h. Then the cell monolayer was scraped in a straight line by using a 200-μl pipette tip. After wounding, cells were treated with TGF-β (5 ng/ml) in the presence or absence of U0126 (10 μM) or GSK1120212 (Trametinib, 10 μM) for 36 h.
Cell invasion and migration assays
For cell invasion assay, transwell chamber (pore size 8 μm) with precoated matrigel (Becton Dickinson) membrane filter was used. 4 × 104 CT26 cells in RPMI-1640 medium or 8 × 104 HCT116 cells in the McCoy 5A medium containing 0.2% BSA were added to the upper chamber and medium containing 7% FBS, TGF-β (5 ng/ml) and DMSO (1/1000 v/v) or U0126 (10 μM) or GSK1120212 (Trametinib, 10 μM) was added to the lower chamber. The chamber was incubated for 28 h at 37 °C. Then, the upper surface of the filters was removed and the invaded cells were fixed with 4% paraformaldehyde solution and stained with 0.1% crystal violet. The number of invaded cells was quantified by counting 6 random fields. For cell migration assay, 2 × 104 CT26 cells or 5 × 104 HCT116 cells were added to the transwell chambers with matrigel uncoated and the procedures was the same as cell invasion assay. Both assays were performed in triplicate.
Experimental liver metastasis model of colorectal cancer
Liver metastases were generated by injecting cells into the spleens of Balb/c mice as described previously.17 Four- to six-week-old male Balb/c mice (Beijing HFK Bioscience) were housed under standardized conditions with free access to a standard chow and water, and were given 1 week to adapt to the laboratory environment prior to manipulation. Mice were divided into 3 groups (n = 6). Briefly, the spleen was exposed by an incision on the left upper abdomen. CT26-wild type (WT) (group 1), CT26-NC (group 2) and CT26-Smad4KD (group 3) cells (1 × 105) in 100 μl serum-free RPMI-1640 were injected into the spleens, respectively. The spleen was removed 5 min after the injection, and the abdominal cavity was closed. The mice were euthanized after 25 d. Livers were removed, weighted and examined for metastases. For survival assay, mice were injected CT26-NC (n = 10) or CT26-Smad4KD (n = 10) cells into spleens, and housed under the above condition. Mice were euthanized when advanced bulky disease was present. The day of sacrifice was considered the day of death for survival evaluation. The mean survival time for each group of mice was determined and the survival was evaluated by log-rank test.
In addition, U0126 was prepared as a 3 mg/ml solution in 30% PEG-400, 20% polypropylene glycol, 15% Cremophor EL (Sigma), and 5% ethanol in sterile saline. Twelve mice were divided into vehicle group and U0126 group (n = 6), which were injected intraperitoneally with vehicle alone or U0126 (15 mg/kg), respectively, 5 d after spleen injection. The mice were euthanized after 25 d and survival assay was conducted as described above.
Western blot and immunoprecipitation assay
Western blotting was performed as described previously.17 Briefly, cells or tumor tissues were lysed in RIPA Lysis Buffer (Beyotime) containing Protease Inhibitor Cocktail, Phosphatase Inhibitor Cocktail (Roche). Protein concentrations of whole-cell lysates were assessed using the Enhanced BCA Protein Assay Kit (Beyotime), and equal amounts (50 μg) were separated on SDS-PAGE and transferred to PVDF membranes (Roche). The membranes were incubated with the following primary antibodies at 4 °C overnight: Phospho-Smad2, Phospho-p44/42 MAPK (Erk1/2), P44/42 MAP Kinase (CST); Smad4, MMP2 (Epitomics); Smad2/3, GAPDH (Santa Cruz); COX-2 (Cayman). Next the membranes were incubated with the second antibody at 37 °C for 1 h. For immunoprecipitation assay, lysate prepared from TGF-β1 (10 ng/ml) treated CT26-NC or CT26-Smad4KD cells were incubated with Smad2/3 antibody at 4 °C overnight. Immune complexes were precipitated and analyzed by western blotting with Smad4 antibody.
Statistical analysis
Data were presented as the mean ± SD from three independent experiments unless otherwise indicated. The Student t test was used for comparison between groups. The Mann–Whitney U-test was used to compare the staining score distributions of primary tumor and metastatic tumor. A Spearman correlation test was used to assess the correlation between Smad4 expression and the TNM stage, phospho-ERK expression and the TNM stage, and the expression of Smad4 and phospho-ERK by the SPSS software package (version 11.5). Cumulative survival time was calculated by the Kaplan–Meier method and analyzed by the log-rank test. P < 0.05 was considered statistically significant.
Disclosure of Potential Conflicts of Interest
The authors have no conflict of interest or financial interest to disclose.
Acknowledgments
This work was supported by the grants from the National Natural Science Foundation of China (30973498 and 81072001), the State Key Project on Infection Disease of China (2012ZX10002016-004 and 2012ZX10002010-001-004), and the Chinese Ministry of Public Health for Key Clinical Project ([2010] 493–51) to XP Chen.
Glossary
Abbreviations:
- CCK8
cell counting kit-8
- CRC
colorectal cancer
- ERK
extracellular regulated protein kinases
- KD
knockdown
- MAPK
mitogen-activated protein kinases
- MEK
mitogen-activated protein kinase kinase
- NC
negative control
- siRNA
small interfering RNA
- WT
wild type
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/26427
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Isaksson-Mettävainio M, Palmqvist R, Dahlin AM, Van Guelpen B, Rutegård J, Oberg A, Henriksson ML. High SMAD4 levels appear in microsatellite instability and hypermethylated colon cancers, and indicate a better prognosis. Int J Cancer. 2012;131:779–88. doi: 10.1002/ijc.26473. [DOI] [PubMed] [Google Scholar]
- 3.de Gramont A, de Gramont A, Chibaudel B, Larsen AK, Tournigand C, André T, GERCOR French Oncology Research Group The evolution of adjuvant therapy in the treatment of early-stage colon cancer. Clin Colorectal Cancer. 2011;10:218–26. doi: 10.1016/j.clcc.2011.10.001. [DOI] [PubMed] [Google Scholar]
- 4.Abbott AM, Parsons HM, Tuttle TM, Jensen EH. Short-term outcomes after combined colon and liver resection for synchronous colon cancer liver metastases: a population study. Ann Surg Oncol. 2013;20:139–47. doi: 10.1245/s10434-012-2515-z. [DOI] [PubMed] [Google Scholar]
- 5.Mina LA, Sledge GW., Jr. Rethinking the metastatic cascade as a therapeutic target. Nat Rev Clin Oncol. 2011;8:325–32. doi: 10.1038/nrclinonc.2011.59. [DOI] [PubMed] [Google Scholar]
- 6.Robertson DJ, Stukel TA, Gottlieb DJ, Sutherland JM, Fisher ES. Survival after hepatic resection of colorectal cancer metastases: a national experience. Cancer. 2009;115:752–9. doi: 10.1002/cncr.24081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ding Q, Chang CJ, Xie X, Xia W, Yang JY, Wang SC, Wang Y, Xia J, Chen L, Cai C, et al. APOBEC3G promotes liver metastasis in an orthotopic mouse model of colorectal cancer and predicts human hepatic metastasis. J Clin Invest. 2011;121:4526–36. doi: 10.1172/JCI45008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klotzsche-von Ameln A, Muschter A, Heimesaat MM, Breier G, Wielockx B. HIF prolyl hydroxylase-2 inhibition diminishes tumor growth through matrix metalloproteinase-induced TGFβ activation. Cancer Biol Ther. 2012;13:216–23. doi: 10.4161/cbt.13.4.18830. [DOI] [PubMed] [Google Scholar]
- 9.Massagué J. TGFbeta in Cancer. Cell. 2008;134:215–30. doi: 10.1016/j.cell.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 11.Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009;19:128–39. doi: 10.1038/cr.2008.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moustakas A, Heldin CH. Non-Smad TGF-beta signals. J Cell Sci. 2005;118:3573–84. doi: 10.1242/jcs.02554. [DOI] [PubMed] [Google Scholar]
- 13.Alazzouzi H, Alhopuro P, Salovaara R, Sammalkorpi H, Järvinen H, Mecklin JP, Hemminki A, Schwartz S, Jr., Aaltonen LA, Arango D. SMAD4 as a prognostic marker in colorectal cancer. Clin Cancer Res. 2005;11:2606–11. doi: 10.1158/1078-0432.CCR-04-1458. [DOI] [PubMed] [Google Scholar]
- 14.Zhang B, Halder SK, Zhang S, Datta PK. Targeting transforming growth factor-beta signaling in liver metastasis of colon cancer. Cancer Lett. 2009;277:114–20. doi: 10.1016/j.canlet.2008.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smakman N, Borel Rinkes IH, Voest EE, Kranenburg O. Control of colorectal metastasis formation by K-Ras. Biochim Biophys Acta. 2005;1756:103–14. doi: 10.1016/j.bbcan.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 16.Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature. 2002;418:934. doi: 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- 17.Zhang B, Halder SK, Kashikar ND, Cho YJ, Datta A, Gorden DL, Datta PK. Antimetastatic role of Smad4 signaling in colorectal cancer. Gastroenterology. 2010;138:969–80, e1-3. doi: 10.1053/j.gastro.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nikolic A, Ristanovic M, Perovic V, Trifunovic J, Perovic M, Radojkovic D. Genetic alterations in SMAD4 and K-ras in Serbian patients with endometrial carcinoma. Int J Gynecol Cancer. 2012;22:442–6. doi: 10.1097/IGC.0b013e31823fabab. [DOI] [PubMed] [Google Scholar]
- 19.Zhong D, Morikawa A, Guo L, Colpaert C, Xiong L, Nassar A, Chen C, Lamb N, Dong JT, Zhou W. Homozygous deletion of SMAD4 in breast cancer cell lines and invasive ductal carcinomas. Cancer Biol Ther. 2006;5:601–7. doi: 10.4161/cbt.5.6.2660. [DOI] [PubMed] [Google Scholar]
- 20.Isaksson-Mettävainio M, Palmqvist R, Forssell J, Stenling R, Oberg A. SMAD4/DPC4 expression and prognosis in human colorectal cancer. Anticancer Res. 2006;26(1B):507–10. [PubMed] [Google Scholar]
- 21.Salovaara R, Roth S, Loukola A, Launonen V, Sistonen P, Avizienyte E, Kristo P, Järvinen H, Souchelnytskyi S, Sarlomo-Rikala M, et al. Frequent loss of SMAD4/DPC4 protein in colorectal cancers. Gut. 2002;51:56–9. doi: 10.1136/gut.51.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Royce SG, Alsop K, Haydon A, Mead L, Smith LD, Tesoriero AA, Giles GG, Jenkins MA, Hopper JL, Southey MC. The role of SMAD4 in early-onset colorectal cancer. Colorectal Dis. 2010;12:213–9. doi: 10.1111/j.1463-1318.2009.01779.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Losi L, Bouzourene H, Benhattar J. Loss of Smad4 expression predicts liver metastasis in human colorectal cancer. Oncol Rep. 2007;17:1095–9. [PubMed] [Google Scholar]
- 24.Inman GJ. Switching TGFβ from a tumor suppressor to a tumor promoter. Curr Opin Genet Dev. 2011;21:93–9. doi: 10.1016/j.gde.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 25.Ikushima H, Miyazono K. TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer. 2010;10:415–24. doi: 10.1038/nrc2853. [DOI] [PubMed] [Google Scholar]
- 26.Papageorgis P, Cheng K, Ozturk S, Gong Y, Lambert AW, Abdolmaleky HM, Zhou JR, Thiagalingam S. Smad4 inactivation promotes malignancy and drug resistance of colon cancer. Cancer Res. 2011;71:998–1008. doi: 10.1158/0008-5472.CAN-09-3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Izeradjene K, Combs C, Best M, Gopinathan A, Wagner A, Grady WM, Deng CX, Hruban RH, Adsay NV, Tuveson DA, et al. Kras(G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell. 2007;11:229–43. doi: 10.1016/j.ccr.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 28.Kojima K, Vickers SM, Adsay NV, Jhala NC, Kim HG, Schoeb TR, Grizzle WE, Klug CA. Inactivation of Smad4 accelerates Kras(G12D)-mediated pancreatic neoplasia. Cancer Res. 2007;67:8121–30. doi: 10.1158/0008-5472.CAN-06-4167. [DOI] [PubMed] [Google Scholar]
- 29.Levy L, Hill CS. Smad4 dependency defines two classes of transforming growth factor beta (TGF-beta) target genes and distinguishes TGF-beta-induced epithelial-mesenchymal transition from its antiproliferative and migratory responses. Mol Cell Biol. 2005;25:8108–25. doi: 10.1128/MCB.25.18.8108-8125.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Diab A, Nikolopoulou-Stamati P, Katostaras T, Safioleas M, Kostakis A, Athanassiadou P, Liossi-Ioakeim A, Marinos G, Konstantopoulos K. Expression of Smad4, E-cadherin and beta-catenin in advanced colorectal cancer: a retrospective study. J BUON. 2012;17:92–6. [PubMed] [Google Scholar]
- 31.Saha D, Datta PK, Beauchamp RD. Oncogenic ras represses transforming growth factor-beta /Smad signaling by degrading tumor suppressor Smad4. J Biol Chem. 2001;276:29531–7. doi: 10.1074/jbc.M100069200. [DOI] [PubMed] [Google Scholar]
- 32.Zeng ZS, Cohen AM, Guillem JG. Loss of basement membrane type IV collagen is associated with increased expression of metalloproteinases 2 and 9 (MMP-2 and MMP-9) during human colorectal tumorigenesis. Carcinogenesis. 1999;20:749–55. doi: 10.1093/carcin/20.5.749. [DOI] [PubMed] [Google Scholar]
- 33.Hilska M, Roberts PJ, Collan YU, Laine VJ, Kössi J, Hirsimäki P, Rahkonen O, Laato M. Prognostic significance of matrix metalloproteinases-1, -2, -7 and -13 and tissue inhibitors of metalloproteinases-1, -2, -3 and -4 in colorectal cancer. Int J Cancer. 2007;121:714–23. doi: 10.1002/ijc.22747. [DOI] [PubMed] [Google Scholar]
- 34.Giannelli G, Falk-Marzillier J, Schiraldi O, Stetler-Stevenson WG, Quaranta V. Induction of cell migration by matrix metalloprotease-2 cleavage of laminin-5. Science. 1997;277:225–8. doi: 10.1126/science.277.5323.225. [DOI] [PubMed] [Google Scholar]
- 35.Schwandner O, Schlamp A, Broll R, Bruch HP. Clinicopathologic and prognostic significance of matrix metalloproteinases in rectal cancer. Int J Colorectal Dis. 2007;22:127–36. doi: 10.1007/s00384-006-0173-y. [DOI] [PubMed] [Google Scholar]
- 36.Wong JC, Chan SK, Schaeffer DF, Sagaert X, Lim HJ, Kennecke H, Owen DA, Suh KW, Kim YB, Tai IT. Absence of MMP2 expression correlates with poor clinical outcomes in rectal cancer, and is distinct from MMP1-related outcomes in colon cancer. Clin Cancer Res. 2011;17:4167–76. doi: 10.1158/1078-0432.CCR-10-1224. [DOI] [PubMed] [Google Scholar]
- 37.Waas ET, Hendriks T, Lomme RM, Wobbes T. Plasma levels of matrix metalloproteinase-2 and tissue inhibitor of metalloproteinase-1 correlate with disease stage and survival in colorectal cancer patients. Dis Colon Rectum. 2005;48:700–10. doi: 10.1007/s10350-004-0854-y. [DOI] [PubMed] [Google Scholar]
- 38.Zhang MZ, Xu J, Yao B, Yin H, Cai Q, Shrubsole MJ, Chen X, Kon V, Zheng W, Pozzi A, et al. Inhibition of 11beta-hydroxysteroid dehydrogenase type II selectively blocks the tumor COX-2 pathway and suppresses colon carcinogenesis in mice and humans. J Clin Invest. 2009;119:876–85. doi: 10.1172/JCI37398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hiraga T, Myoui A, Choi ME, Yoshikawa H, Yoneda T. Stimulation of cyclooxygenase-2 expression by bone-derived transforming growth factor-beta enhances bone metastases in breast cancer. Cancer Res. 2006;66:2067–73. doi: 10.1158/0008-5472.CAN-05-2012. [DOI] [PubMed] [Google Scholar]