

Terminally differentiated somatic cells can be reset to pluripotent capability either by ectopic expression of defined factors to form so-called “induced pluripotent stem cells” (iPSCs)1 or by employing somatic cell nuclear transfer.2 During iPSC induction, the expression of OCT4, SOX2, KLF4, and c-MYC (OSKM) triggers nuclear reprogramming via a mechanism that remains elusive.
Although it is now routine to reprogram somatic cells into iPSCs, the process is extremely inefficient, preventing the immediate translation of this promising technology into clinics. Using lentivirus or retrovirus as a delivery vector, it is possible to observe that > 90% of target fibroblasts are transduced by tracking with a coexpressed EGFP reporter gene. However, only a very small proportion of transduced cells become fully reprogrammed into iPSCs. Even though the majority of the transduced cells express the viral OSKM, the cells are not reprogrammed or are only partially reprogrammed.3 Clearly, it appears that there is a strong epigenetic block that must be overcome before the cells achieve pluripotency.
Tremendous efforts have been made to understand the barriers that block the reprogramming process, with investigators using a variety of small molecules, adding more defined proteins, and altering histone epigenotype and DNA methylation in order to enhance the efficiency of iPSC formation. However, none of these approaches is able to fundamentally improve the methodology. One of the major obstacles to impede iPSC induction is that we know very little about the mechanism by which the 4 defined factors initiate reprogramming in only < 1% of the transduced cells.
To approach this problem, we separated fully reprogrammed iPSCs and the OSKM-expressed unreprogrammed cells (URCs), and tracked them for clues that lead to differential reprogramming in the transduced cells. As the defined factors are all transcription factors, we first focused on the binding of the ectopically expressed factors and the activation of their target gene promoters (OCT4, SOX, NANOG), an essential step in successful reprogramming. Using chromatin immunoprecipitation assay (ChIP), it was surprising to learn that the access of the viral factors to their target genes was not a bottleneck toward cell reprogramming, as no differences were noticed in promoter binding between iPSCs and URCs. However, the binding of OSKM transcription factors to their promoters was not able to activate the pluripotency genes.3 Thus, it appears that even though the virus-derived factors bound to their target promoters, they failed to induce transcription from these genes, suggesting that the activation of endogenous pluripotency genes may represent a critical reprogramming block preventing iPSC induction.
Remodeling of local chromatin structure is an important step required for iPSC induction. DNA looping orchestrated by the cohesin-mediator complex determines the pluripotency of the stem cells.4 Using chromosome conformation capture (3C),5 we found that in iPSCs, the OCT4 promoter DNA interacted frequently with a enhancer DNA region that is located 10 kb downstream of the promoter.3 An intrachromosomal loop brings the downstream enhancer in close proximity to the OCT4 promoter, where it activates the endogenous OCT4 as an essential step in iPSC induction (Fig. 1). These chromatin interactions were very rarely detected in URCs or in fibroblast control cells, where endogenous OCT4 cannot be activated, since the promoter and the downstream enhancer are no longer juxtaposed. Similarly, these promoter/enhancer intrachromosomal interactions were also present in the SOX2 and NANOG genes in the iPSCs but not in the URCs. Together, the formation of intrachromosomal looping in these pluripotency-associated genes may constitute a critical epigenetic barrier that must be overcome for cell reprogramming to occur.
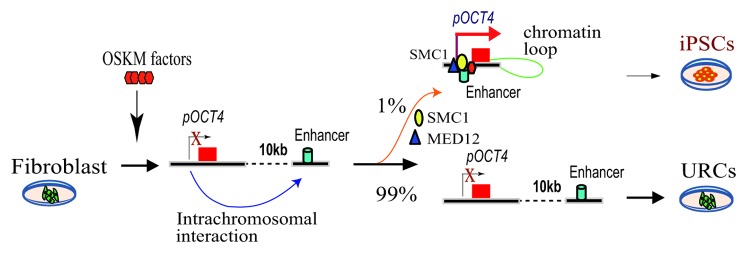
Figure 1. Intrachromosomal loop is required for iPSC induction. After transduction by OSKM factors, only <1% cells form a chromatin loop between the pluripotency gene promoter and the enhancer, leading to full reprogramming into iPSCs. However, majority of the transduced cells that lack the loop are unreprogrammed cells (URCs).
Chromatin looping is coordinated through chromatin factors like cohesion, MED12, and CTCF. Using co-immunoprecipitation, we identified SMC1, a component of the cohesin complex, as a key factor required to maintain the intrachromosomal interaction to activate endogenous OCT4. Expression of SMC1 correlated with the status of cell pluripotency. Knockdown of SMC1 induced differentiation in iPSCs and completely abolished pluripotency induction in fibroblasts.
By assessing chromatin interactions genome-wide, several groups have also demonstrated the importance of the chromatin interaction network in iPSC induction and the maintenance of cell pluripotency.6-8 Enhancer–promoter co-associations were unique to pluripotent cells or intermediates being successfully reprogrammed to iPSCs. These studies highlighted gene loop formation, as necessary, for transcriptional activation, but programmed gene loops may be equally important in order to mask differentiation-specific genes in ESCs or during reprogramming to iPSCs, an issue that remains open for future studies.
Taken together, it is clear that the requirement for chromatin loops between the enhancers and promoters of certain pluripotency genes is a critical epigenetic barrier that must be overcome for a cell to be transformed to pluripotency. The spontaneous formation of such loops may be a rare (< 1% prevalence) and stochastic event, thus explaining the fact that only a very small subset of virally transduced fibroblasts develop into iPS cells (Fig. 1). It will be important to identify endogenous or exogenous factors that organize chromatin loops in order to promote iPSC induction.
Zhang H, et al. Cell Stem Cell. 2013;13:30–5. doi: 10.1016/j.stem.2013.05.012.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/27017
References
- 1.Takahashi K, et al. Cell. 2006;126:663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 2.Tachibana M, et al. Cell. 2013;153:1228–38. doi: 10.1016/j.cell.2013.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang H, et al. Cell Stem Cell. 2013;13:30–5. doi: 10.1016/j.stem.2013.05.012. [DOI] [PubMed] [Google Scholar]
- 4.Kagey MH, et al. Nature. 2010;467:430–5. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dekker J, et al. Science. 2002;295:1306–11. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 6.Apostolou E, et al. Cell Stem Cell. 2013;12:699–712. doi: 10.1016/j.stem.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phillips-Cremins JE, et al. Cell. 2013;153:1281–95. doi: 10.1016/j.cell.2013.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wei Z, et al. Cell Stem Cell. 2013;13:36–47. doi: 10.1016/j.stem.2013.05.010. [DOI] [PubMed] [Google Scholar]