

Abstract
Histone ubiquitination plays a vital role in DNA damage response (DDR), which is important for maintaining genomic integrity in eukaryotic cells. In DDR, ubiquitination of histone H2A and γH2AX by the concerted action of ubiquitin (Ub) ligases, RNF168 and RNF8, generates a cascade of ubiquitination signaling. However, little is known about deubiquitinating enzymes (DUBs) that may catalyze the removal of Ub from these histones. This study demonstrated that USP3, an apparent DUB for mono-ubiquitinated H2A, is indeed the enzyme for deubiquitinating Ub conjugates of γH2AX and H2A from lysine sites, where the ubiquitination is initiated by RNF168. Here, we showed that ectopic expression of USP3 led to the deubiquitination of both H2A and γH2AX in response to UV-induced DNA damage. Moreover, ectopic USP3 expression abrogated FK2 antibody-reactive Ub-conjugate foci, which co-localize with damage-induced γH2AX foci. In addition, USP3 overexpression impaired the accumulation of downstream repair factors BRCA1 and 53BP1 at the damage sites in response to both UV and γ-irradiation. We further identified that the USP3 removes Ub at lysine 13 and 15 of H2A and γH2AX, as well as lysine 118 and 119 of H2AX in response to DNA damage. Taken together, the results suggested that USP3 is a negative regulator of ubiquitination signaling, counteracting RNF168- and RNF8-mediated ubiquitination.
Keywords: 53BP1, BRCA1, DNA repair, RNF168, USP3, deubiquitinating enzyme, histone modification, ubiquitin ligase, γH2AX
Introduction
Genome of living organisms is incessantly challenged by physical and chemical DNA damaging agents, of both physiological and environmental origins. To preserve the integrity of genome, eukaryotic cells evoke a sophisticated DNA damage response (DDR), which is responsible for transiently arresting the cell cycle and allowing faithful lesion repair. As such, the genomic integrity is maintained by the functional interplay between DNA repair processes and DNA damage checkpoint pathways.
In DDR, DNA damage induces a re-localization of damage sensing, signaling, and repair factors into distinct foci at damage sites. Damage induced post-translational histone modification also occur at these foci, reflecting chromatin rearrangements at damage sites.1-4 One of the early and well-characterized events is the phosphorylation of variant H2A (γH2AX) by checkpoint kinases, including Ataxia telangiectasia mutated (ATM), ATM and Rad3-related (ATR), and DNA-dependent protein kinases (DNA-PK).5,6 γH2AX, in turn, is instrumental for efficient accumulation and retention of mediators and repair factors such as MDC1, BRCA1, and 53BP1 at the chromatin surrounding the lesion.7-9
Ubiquitination of γH2AX and H2A is an important event in DDR.10-12 Following ATM and/or ATR recruitment to DNA damage and H2AX phosphorylation, mediator MDC1 is recruited to double/single-strand DNA breaks (DSB/SSB) and serves as a platform for recruiting 2 RING-type ubiquitin (Ub) ligases, RNF8 and RNF168, to modify γH2AX and H2A.13-15 Therefore, ubiquitination of γH2AX and H2A in DDR is a process dependent on ATM/ATR, H2AX phosphorylation, and mediator MDC1. It has been reported that the Ub chains themselves are not sufficient to signal, but that the H2A and γH2AX ubiquitination is the crucial signal that drives the DSB signaling.16 Perhaps, H2A and γH2AX ubiquitination allows a transition of the DSB flanking chromatin to a state permissive for accumulation of BRCA1 and 53BP1.16 These 2 repair factors determine the choice between homologous recombination (HR) and non-homologous end joining (NHEJ) repair pathways.17-19 Therefore, the ubiquitination activities of RNF168 and RNF8 are important for proper DSB repair.15,20,21
Ubiquitination of H2A and γH2AX is a result of the concerted action of RNF168 and RNF8, which generates not only monoubiquinated H2A and γH2AX, but also polyubiquitin conjugates with lysine 63 (K63)-linked Ub chains.14,15,20,22 While RNF168 was thought to be involved in extension of the K63-linked Ub chains, a more recent work showed that lysine 13 and 15 (K13–15) in H2A/H2AX are ubiquitinated by RNF168, and that the K63-Ub chains are extended by RNF8 during DDR.16 The K63-Ub chains formed on K13–15 sites distinguishes modification from Polycomb-mediated monoubiquitination,23 which occurs at K119, generating uH2A. This prevalent modification can constitute up to 10% of cellular H2A in chromatin as a result of ubiquitination by the RING1B E3 ligase present in Polycomb repressive complex 1 (PRC1). It is known that PRC1 plays a role in transcription silencing. Nevertheless, recent studies suggested that PRC1-mediated monoubiquitination is a part of DDR.23 The PRC1 component BMI1 is found to be recruited to damage sites and contributes to K119 ubiquitination of H2A and DNA break repair.23,24
Little is known about H2A and γH2AX deubiquitination, a reverse process which removes Ub moiety from target protein. Ub-specific peptidase 16 (USP16) is shown to deubiquitinate uH2A in regulating Hox gene expression25 and in reversing ATM-dependent transcription silencing in cis to DSB.26 The deubiquitination of uH2A by USP3, however, is described to be involved in S-phase cell cycle progression.27 Interestingly, regulation of damage-induced ubiquitination signaling by USP3 is implicated by several recent studies.13,28 Here, we provided evidence that USP3 targets not only ubiquitinated H2A, but also Ub-conjugates of γH2AX (Ub-γH2AX) for deubiquitination. We found that USP3 can remove Ub from K13–15 on H2A and γH2AX in addition to K118–119 sites. We argue that USP3 negatively regulates DDR via counteracting RNF168 and RNF8-mediated ubiquitination.
Results
USP3 deubiquitinates Ub-γH2AX in response to DNA damage
USP3 was initially identified as a DUB for uH2A.27 However, it is not clear whether USP3 deubiquitinates ubiquitin-conjugated γH2AX, where the ubiquitination occurs at specific lysine sites. In DDR, γH2AX is an ATM/ATR downstream signal transducer, and γH2AX is found to be ubiquitinated by the concerted action of E3 Ub ligases RNF8 and RNF168. To elucidate that Ub-γH2AX is a substrate of USP3, we first knocked down USP3 in HeLa cells using siRNA and analyzed the status of both uH2A and Ub-γH2AX with and without UV-induced DNA damage. Efficiency of USP3 knockdown was confirmed by western blotting (Fig. 1A). As expected, UV irradiation resulted in an increase in γH2AX and Ub-γH2AX level (Fig. 1B; Fig. S1). USP3 siRNA further elevated Ub-γH2AX level. Depletion of USP3 also led to an increase in the levels of uH2A, which were otherwise not significantly affected by UV irradiation (Fig. 1B; Fig. S1). To further probe the role of USP3 at DNA damage sites, we depleted USP3 using siRNA and tested whether USP3 affects DNA damage-induced formation of Ub conjugate foci (also called FK2 foci, as the Ub conjugates are recognized by FK2 monoclonal Ub antibody) at DNA damage spots, which are generated by a localized UV delivery through micropore irradiation. We observed that USP3 knockdown notably increased the formation of FK2 foci (Fig. 1C and E) and, to some extent, increased the uH2A foci (Fig. 1D and E). On the contrary, USP3 knockdown did not affect the formation of γH2AX foci, which colocalize with both FK2 and uH2A foci (Fig. 1C and D). These results indicated that USP3 knockdown only affects the γH2AX and H2A ubiquitination status but had no effect on H2AX phosphorylation, an upstream event of γH2AX ubiquitination.
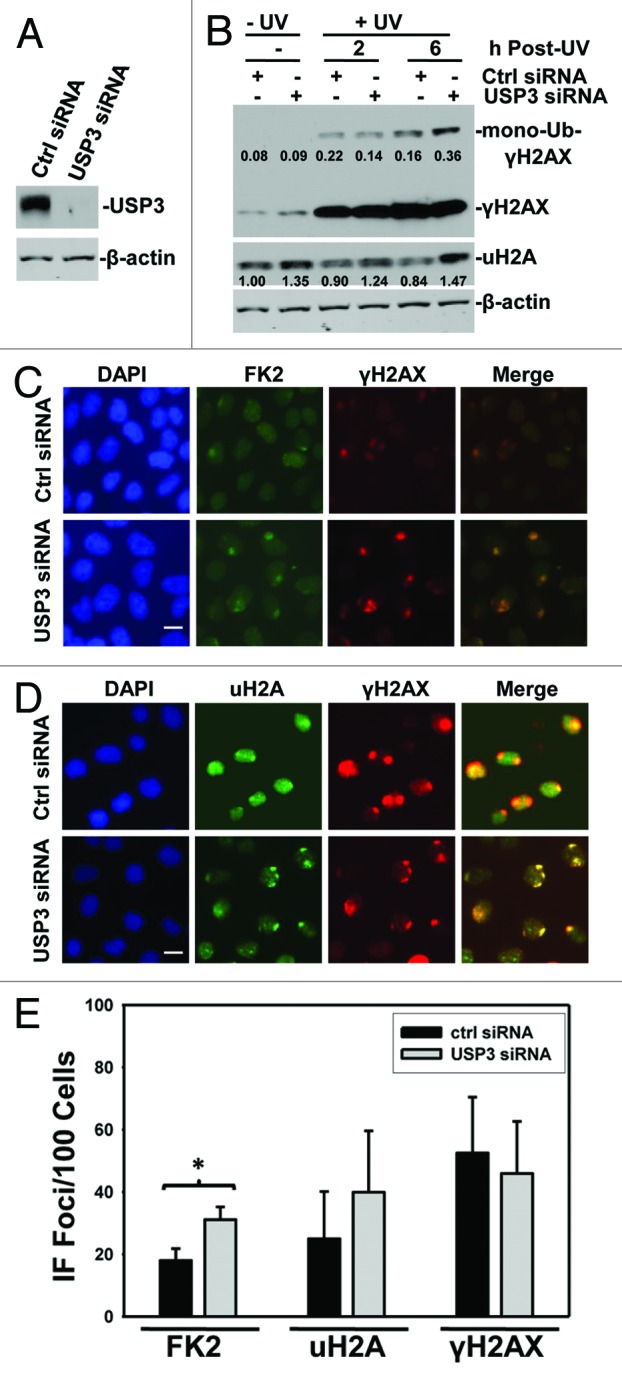
Figure 1. USP3 deubiquitinates γH2AX upon UV irradiation. (A) HeLa cells were transfected with USP3 or control siRNA at a final concentration of 100 nM. The cells were harvested 48 h after transfection. Knockdown efficiency of USP3 siRNA was analyzed by immunoblotting with USP3 antibody. (B) HeLa cells were transfected with siRNA as in (A) and UV-irradiated at 20 J/m2. Cell lysates were prepared at indicated time points and the levels of Ub-conjugates of γH2AX and uH2A were determined by immunoblotting using γH2AX and uH2A antibodies. The protein images in blots were quantitated by ImageJ software, and the number represents relative amount of mono-Ub-γH2AX to γH2AX, and uH2A to that of Ctrl siRNA without UV. (C) HeLa cells were treated as in (A) and UV-irradiated at 100 J/m2 through a 5-μm micropore filter placed on cell monolayers. Six hours after UV irradiation, cells were processed for immunofluorescent staining with FK2 and γH2AX antibodies. The images are representative of multiple experiments. Calibration bar is 10 μm. (D) Cells were treated as described in (C), and the immunofluorescent staining was performed using uH2A and γH2AX antibodies. Calibration bar is 10 μm. (E) Duplicate experiments were performed as described in (C), and the cell numbers positive for FK2, uH2A, and γH2AX were quantified from at least 3 different microscopic fields. The graph depicts percentage of cells positive for the indicated foci, and the error bars show the standard deviation. *Indicates P value 0.05 on Student t test.
Next, we examined the effect of USP3 overexpression on H2A and γH2AX ubiquitination in response to UV-induced DNA damage. Myc-tagged USP3, either wild-type (WT) or catalytically inactive mutant (C1685), was ectopically expressed in HeLa cells, and the ubiquitination status of H2A and γH2AX was then examined. The ectopic USP3 expression was verified by western blotting with anti-USP3 and anti-Myc antibodies (Fig. 2A). As expected, UV irradiation increased γH2AX levels (Fig. 2B), which were not further affected by either WT or mutant USP3. Nevertheless, ectopic expression of WT USP3 led to a marked decrease in mono- and di-ubiquitination of γH2AX (Fig. 2B and C; Fig. S3). The uH2A level was not appreciably changed by UV irradiation alone, but it decreased by WT USP3 expression. In contrast, mutant USP3 did not affect Ub-γH2AX or uH2A levels. We further analyzed whether ectopic expression of USP3 has an effect on foci formation of Ub conjugates. Consistent with the notion that USP3 promotes deubiquitination of γH2AX, we observed that overexpression of WT but not mutant USP3 caused a decrease in FK2 foci formation, while the same WT USP3 did not significantly affect the γH2AX foci formation (Fig. 2D and E). Taken together, these data suggested that USP3 deubiquitinates Ub-γH2AX in addition to uH2A during DDR.
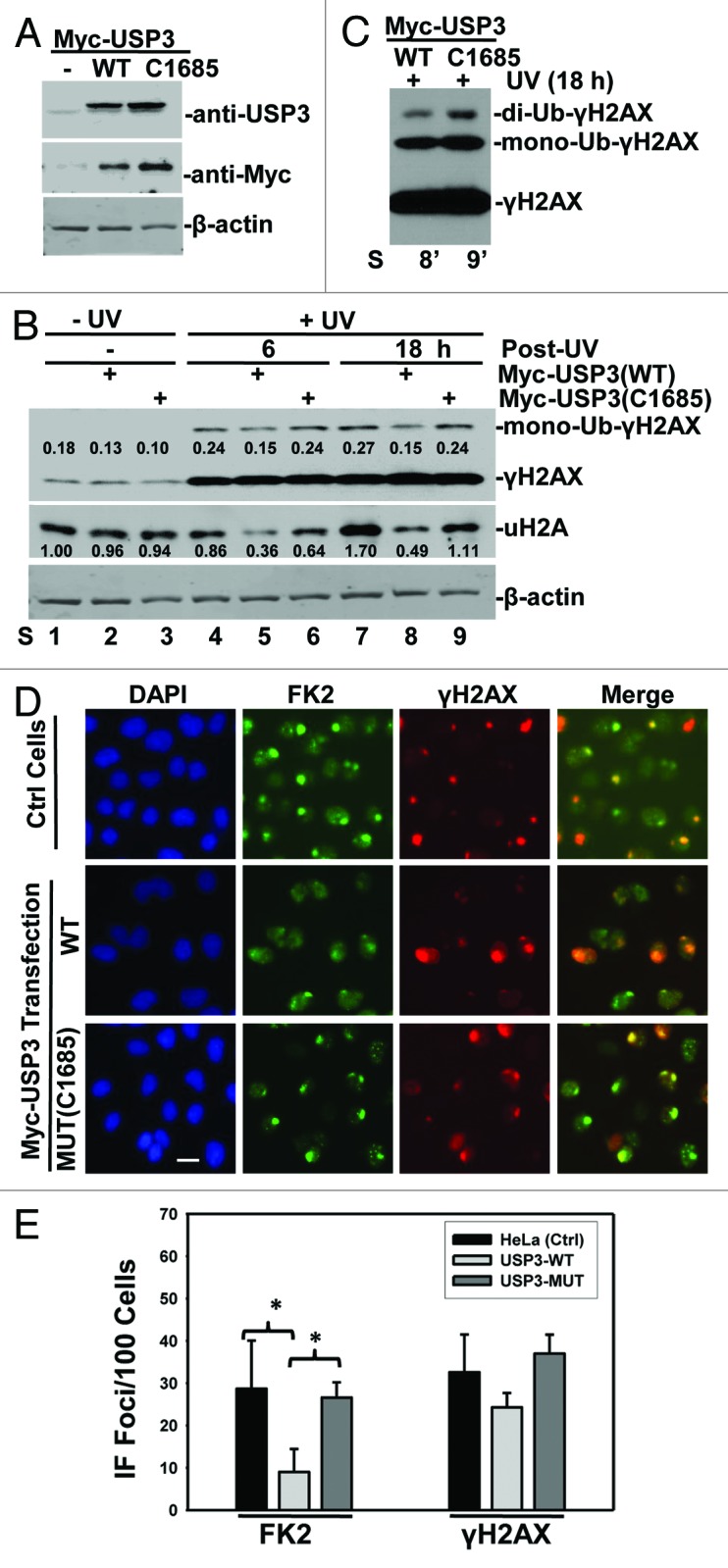
Figure 2. Overexpression of USP3 negatively regulates ubiquitination of uH2A and γH2AX. (A) HeLa cells were transfected with indicated Myc-tagged USP3 constructs. Cell lysates were prepared 48 h post-transfection. The cell lysates were analyzed by western blotting for USP3 or Myc-tagged USP3. The β-actin blot serves as a loading control. (B) HeLa cells were treated as described above in (A). The cells were UV-irradiated at 20 J/m2, and the cell lysates were prepared at the indicated time points. The levels of Ub-conjugates of γH2AX and uH2A were determined by western blotting. The blotting images were quantitated by ImageJ software, and the number represents relative amount of mono-Ub-γH2AX to γH2AX, or uH2A to that of Ctrl siRNA without UV. (C) Heavy exposure image of lane 8 and 9 from panel (B), showing di-ubiquitinated γH2AX. (D) HeLa cells were transfected, UV irradiated, and were processed for immunofluorescent staining as described in Figure 1C. Calibration bar is 10 μm. (E) Experiments as described in (D) were examined for the number of cells positive for FK2 and γH2AX. The nuclear foci counted from at least three microscopic fields were used for the quantification. The graph depicts percentage of cells positive for the indicated foci from 2 independent experiments. The error bars show the standard deviation. *Indicates P value 0.05 on Student t test.
USP3 overexpression leads to an impaired recruitment of DNA damage repair factors
Recent studies suggest that histone ubiquitination by E3 ligases such as RNF8 and RNF168 triggers and facilitates the accumulation of DNA repair factors BRCA1 and 53BP1 at DNA damage sites.13-15,29,30 The assembly of these and other proteins at the DSB-flanking chromatin occurs in a highly ordered and rapid manner. One can envision that deubiquitination of Ub-γH2AX and uH2A by USP3 may eliminate docking sites for these repair factors, thereby regulating their recruitment. Therefore, we assessed whether USP3 affects the accumulation of BRCA1 and 53BP1 at the DNA damage sites. In these experiments, GFP-USP3 fusion protein or GFP-expressing vectors were used such that the GFP-expressing cells can be directly visualized. The results showed USP3 to be a nuclear protein (Fig. S2). More importantly, the GFP-USP3-expressing green cells exhibited significantly less accumulation of BRCA1 as compared with the non-green cells of the same transfection group and green cells of GFP control group (Fig. 3A and B). Similarly, GFP-USP3 expression decreased the accumulation of 53BP1 (Fig. 3C and D). We also analyzed the protein levels of phosphorylated BRCA1 and 53BP1 under the influence of USP3 overexpression (Fig S4). The phosphorylated BRCA1 level was dramatically elevated upon UV irradiation and sustained up to 18 h in control cells. However, phosphorylated BRCA1 level diminished during the 6–18-h time period when USP3 was overexpressed. For 53BP1, there was a decrease in its level in USP3 transfected group. These results indicated that USP3 not only regulates the accumulation of BRCA1 and 53BP1 at the damage sites, but also regulates the duration of phosphorylated forms of these proteins in DDR.
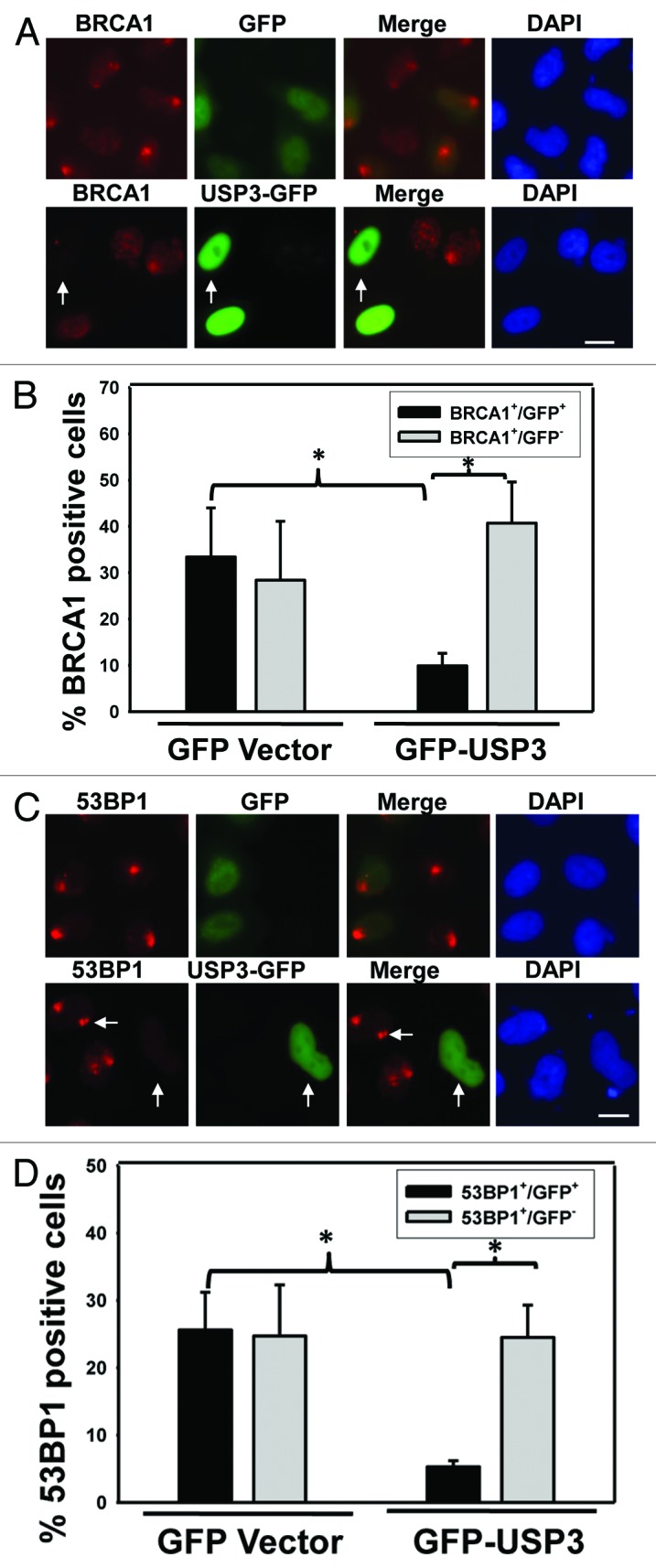
Figure 3. Overexpression of USP3 abrogates the recruitment of repair factors BRCA1 and 53BP1. (A) HeLa cells were transiently transfected with either GFP-USP3 fusion or GFP control construct for 48 h, exposed to 10 J/m2 UV, and fixed 2 h later. Cells were then immunostained with BRCA1 antibody. Arrow marks a cell in which GFP-USP3 impairs the BRCA1 foci formation. Calibration bar is 10 μm. (B) The graph depicts a series of representative experiments in which at least 100 cells from three microscopic fields were scored. The error bars indicate the standard deviation. *Indicates P value 0.05 on Student t test. (C) HeLa cells treated as described in Figure 3A but immunostained with 53BP1 antibody. Arrow marks a cell in which GFP-USP3 impairs the 53BP1 foci formation. Calibration bar is 10 μm. (D) The graph depicts quantitative analysis of data in Figure 3C. *Indicates P value 0.05 on Student t test.
It has been known that BRCA1 and 53BP1 are involved in repair of DNA breaks, which are often caused by ionizing radiation (IR). Thus, we next determined if USP3 regulates the recruitment of repair factors at sites of DSB upon IR. The cells, overexpressing GFP-USP3 or GFP control, were subjected to IR, and formation of ionizing radiation induced foci (IRIF) of BRCA1 and 53BP1 was examined. IR led to the formation of distinctive BRCA1 and 53BP1 foci in control cells expressing GFP. However, the expression of GFP-USP3 attenuated IRIF of both BRCA1 and 53BP1 (Fig. 4A and B), while expression of GFP control had no effect on IRIF of either factor. Despite extensive attempts, no distinguishable IRIF for USP3 were observed. These results confirmed the previous reports in which USP3 has been shown to reverse the RNF168 facilitated IRIF of 53BP1.13,28 The action of USP3 must be swift, as it does not accumulate at IRIF. Altogether, our results suggested that USP3 negatively regulates the accumulation of BRCA1 and 53BP1 at DNA breaks by virtue of deubiquitinating Ub-γH2AX and uH2A.
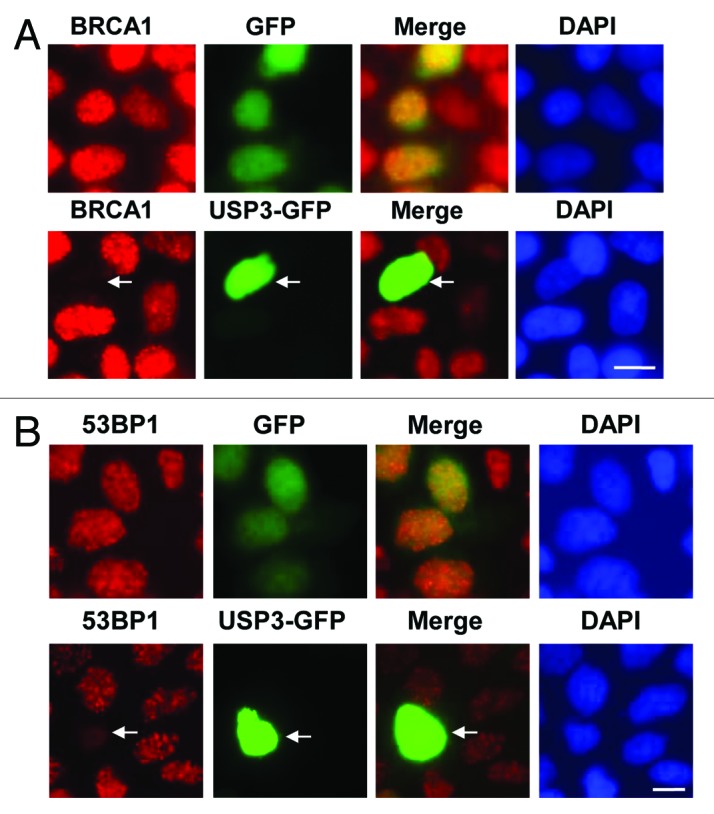
Figure 4. USP3 is involved in regulating DDR to IR. (A) HeLa cells were transiently transfected with expressing constructs of either GFP-USP3 fusion or GFP control for 48 h, subjected to IR at a dose of 4 Gy, and fixed 2 h later. The cells were immunostained with BRCA1 antibody. Arrow marks a cell in which GFP-USP3 impairs the BRCA1 foci formation. (B) HeLa cells were transfected, exposed to IR and processed for immunofluorescent staining with 53BP1 antibody similarly to that described for Figure 4A.
USP3 catalyzes deubiquitination from K13–15 on H2A and H2AX
It has been recently shown that RNF168 catalyzes monoubiquitination of histones at K13–15 on H2A and γH2AX during DDR to IR.16 Thus, we further investigated whether these lysine sites are targeted for deubiquitination by USP3. We overexpressed FLAG-tagged WT H2A or mutant H2A harboring lysine to arginine (R) mutation at amino acid 118 to 119 (K118–119R), or 13 to 15 (K13–15R), alongside with USP3. As shown in Fig. 5A, when FLAG-tagged WT H2A was expressed, a decrease in gel-retained and anti-FLAG-tagged mono-ubiquitinated H2A in presence of USP3 was detected by both anti-FLAG and uH2A antibodies.
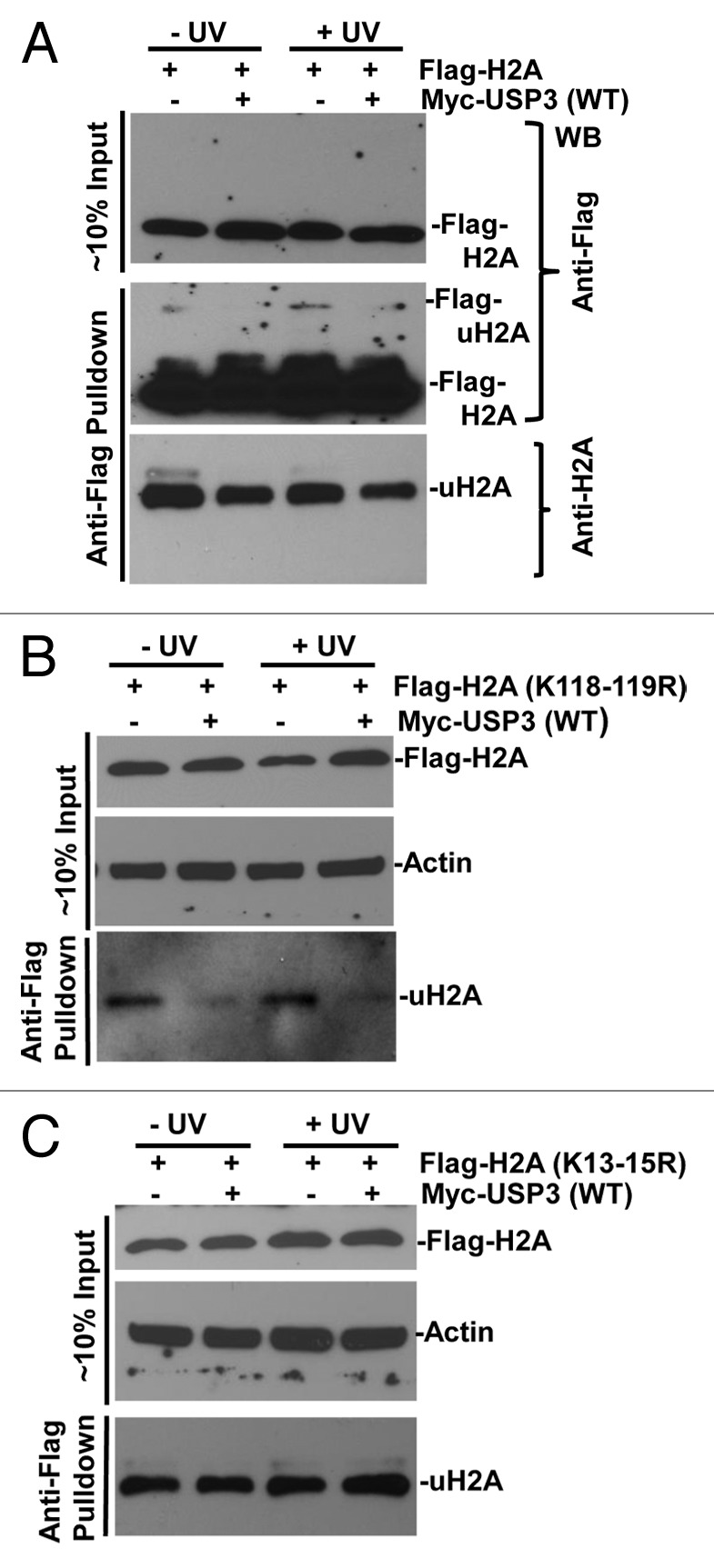
Figure 5. USP3 deubiquitinates at K13–15 on H2A in response to DNA damage. HeLa cells transfected with indicated FLAG-tagged WT or mutant H2A constructs in combination with either Myc-USP3 or control vector and, were UV irradiated. The lysates from transfected cells were prepared and directly used for western blotting shown as input. Alternatively, the lysates were processed for anti-FLAG immunoprecipitates as described in the “Materials and Methods” section. The eluates of FLAG gel retained immunoprecipitates were analyzed by immunoblotting with anti-FLAG or uH2A antibodies. (A) HeLa cells transfected with constructs for FLAG-tagged WT H2A. (B) HeLa cells transfected with constructs for FLAG-tagged H2A with K118 to 119R. (C) HeLa cells transfected with constructs for FLAG-tagged H2A with K13 to 15R.
Upon expression of FLAG-tagged K118–119R H2A, a mono-ubiquitinated H2A signal was detected in anti-FLAG gel-retained fractions by uH2A antibody, indicating that the antibody recognizes not only uH2A, but also mono-ubiquitinated H2A at lysine sites other than K118–119. More importantly, USP3 expression led to a decrease in mono-ubiquitinated H2A with K118–119R regardless of DNA damage (Fig. 5B). In contrast, there was no difference in mono-ubiquitinated H2A with K13–15R (Fig. 5C). Taken together, these results demonstrated that the deubiquitination by USP3 specifically occurs at K13–15.
We further determined if similar deubiquitination occurs at the same lysine sites on γH2AX. We overexpressed FLAG-tagged WT H2AX, mutant H2AX harboring K118–119R, or K13–15R along with USP3, and then assessed the ubiquitination status of γH2AX with or without DNA damage. When WT H2AX was expressed, a damage-dependent increase in mono-ubiquitinated γH2AX in gel-retained FLAG-tagged γH2AX was seen. As expected, co-expression of USP3 decreased such mono-ubiquitinated FLAG-tagged γH2AX (Fig. 6A). For H2AX K118–119R mutant, mono-ubiquitinated FLAG-tagged γH2AX was also elevated by UV but diminished in presence of overexpressed USP3 (Fig. 6B). With H2AX K13–15R mutant, mono-ubiquitinated and FLAG-tagged γH2AX was also increased upon DNA damage, indicating that K118–119 of γH2AX is ubiquitinated in DDR. Surprisingly, the level of such mono-ubiquitinated K13–15R was also considerably lower in presence of overexpressed USP3 (Fig. 6C). Taken together, these results suggested that USP3 targets K13–15 in addition to K118–119 of γH2AX for deubiquitination.
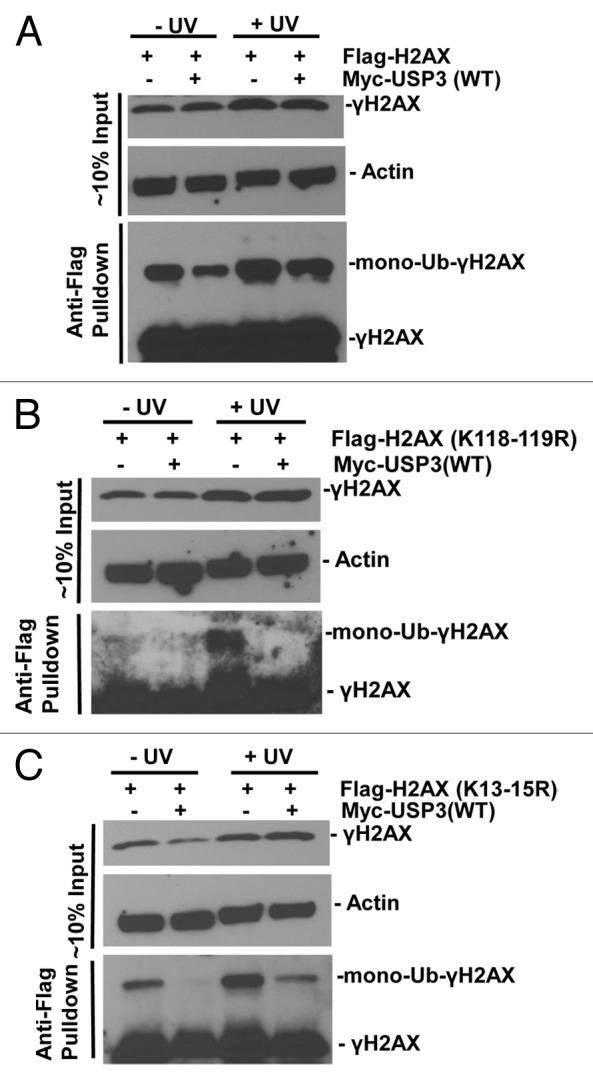
Figure 6. USP3 deubiquitinates at K13–15 and K118–119 on γH2AX. HeLa cells were transfected with the indicated FLAG-H2AX constructs in combination with either Myc-USP3 or empty control vector, and processed similar to that described in Figure 5. The eluates of FLAG gel retained immunoprecipitates were immunoblotting analyzed with anti-FLAG and γH2AX antibodies. (A) HeLa cells transfected with constructs for FLAG-tagged WT H2AX. (B) HeLa cells transfected with constructs for FLAG-tagged H2AX with K118–119R. (C) HeLa cells transfected with constructs for FLAG-tagged H2AX with K13–15R. The images were heavily exposed necessarily to show mono-Ub-γH2AX species.
Discussion
Accumulating evidence in literature supports the role of ubiquitination of histones in DNA damage signaling and repair. However, the importance of DUBs in this process has become evident only recently. Yet, the enzyme–substrate relationship has been established for only a few DUBs. In the present study, we demonstrated that USP3 removes Ub moiety conferred by RNF168/RNF8 on K13–15 of H2A and γH2AX upon DNA damage, and that the deubiquitination by USP3 regulates recruitment of downstream DNA repair factors such as BRCA1 and 53BP1. The data argued that Ub-γH2AX rather than uH2A is indeed the target substrate through which USP3 regulates DDR.
The lack of information on deubiquitination during DDR prompted us to examine the role of USP3. To demonstrate that Ub-γH2AX is a relevant target of deubiquitination by USP3 during DDR, we provided several lines of evidence. First, there was a reduction in protein levels of uH2A and Ub-γH2AX when WT USP3 was overexpressed. Importantly, the deubiquitination activity of USP3 toward Ub-γH2AX and uH2A was attributed to its catalytic activity, as we did not observe the deubiquitination with inactive USP3. In accordance, USP3 knockdown by siRNA increased the levels of Ub-γH2AX and uH2A. Nevertheless, USP3 siRNA only moderately affected the levels of Ub-γH2AX and uH2A. As discussed later, this may be due to functional redundancy among DUBs. Second, we observed an impaired formation of Ub-conjugate foci at damage sites, indicating a reduced level of Ub-γH2AX upon USP3 overexpression. Similarly, a decrease in uH2A foci formation was also seen upon USP3 overexpression. Third, following ectopic expression of GFP-tagged USP3, majority of green cells were impaired in recruitment of repair factors, suggesting that deubiquitination by USP3 abrogates the BRCA1 and 53BP1 docking sites on γH2AX. Fourth, we provided further evidence that USP3 deubiquitinates Ub-γH2AX from K13–15 in addition to canonical K118–119 sites.
Deubiquitination by USP3 from K13–15 is particularly interesting, as it suggests a critical role of USP3 in DDR, especially in DNA break repair. Recently, RNF168 has been shown to ubiquitinate K13–15 of γH2AX and uH2A rather than the canonical K118–119 sites.16 Concurrent with this, the present study highlights the importance of deubiquitination at these sites in counteracting RNF168-dependent ubiquitination in regulating downstream BRCA1 and 53BP1 recruitment events. The ubiquitination of K119 on H2A by RNF8 and RNF168, however, has been shown to play a role in ATM-mediated transcriptional silencing of gene in cis to DSB. The USP16 was shown to be required for reversal of silencing via removal of Ub marks from uH2A in DSB-bearing chromatin. Thus, in contrast with USP3, USP16-mediated removal of Ub from K119 of H2A, at least partially, is responsible for rescue of transcription repression. It should be mentioned that another DUB, USP44, was recently found to target uH2A for deubiquitination.28 Parallel to our USP3 siRNA experiments, depletion of USP44 also moderately enhanced accumulation of Ub conjugates and downstream 53BP1. In the same study, overexpressed USP16, however, did not abrogate 53BP1 foci. Thus, it is possible that Ub-γH2AX rather than uH2A is the target of deubiquitination, through which regulation of 53BP1 recruitment is achieved. We anticipate that USP44 also targets Ub-γH2AX, and that there is considerable functional redundancy among USP3, USP44, and, perhaps, other DUBs.
Our result also showed that K118–119 of Ub-γH2AX can be deubiquitinated by USP3 in response to DNA damage. Ubiquitination at these sites on H2A is considered to be mediated by PRC1 during DDR,23 and it was shown that BMI1 and RING1B components of PRC1 are required for efficient DNA break repair. The ubiquitination at K119 at γH2AX also requires BMI1 and RING1B.31 Thus, USP3 may also counteract PRC1-mediated ubiquitination in DDR, providing an additional regulation of ubiquitination signaling.
In summary, our findings support a model in which USP3 is integral to fine-tuning of DDR by trimming or removing Ub from K13–15 of Ub-γH2AX and uH2A (Fig. 7). Since the ubiquitination at these lysine sites are catalyzed in a RNF168-dependent manner, we further suggest that USP3 counteracts RNF168-dependent ubiquitination signaling. The ubiquitination and deubiquitination occur in tandem and both are important for the cellular determination of DNA break repair pathways and the successful repair. The involvement of USP3 in regulation of DDR warrants further investigation of its functional aspects in DNA break repair, cell cycle and survival, and carcinogenesis. It would be also be interesting to learn whether USP3 plays a role in transcription recovery after DNA damage.
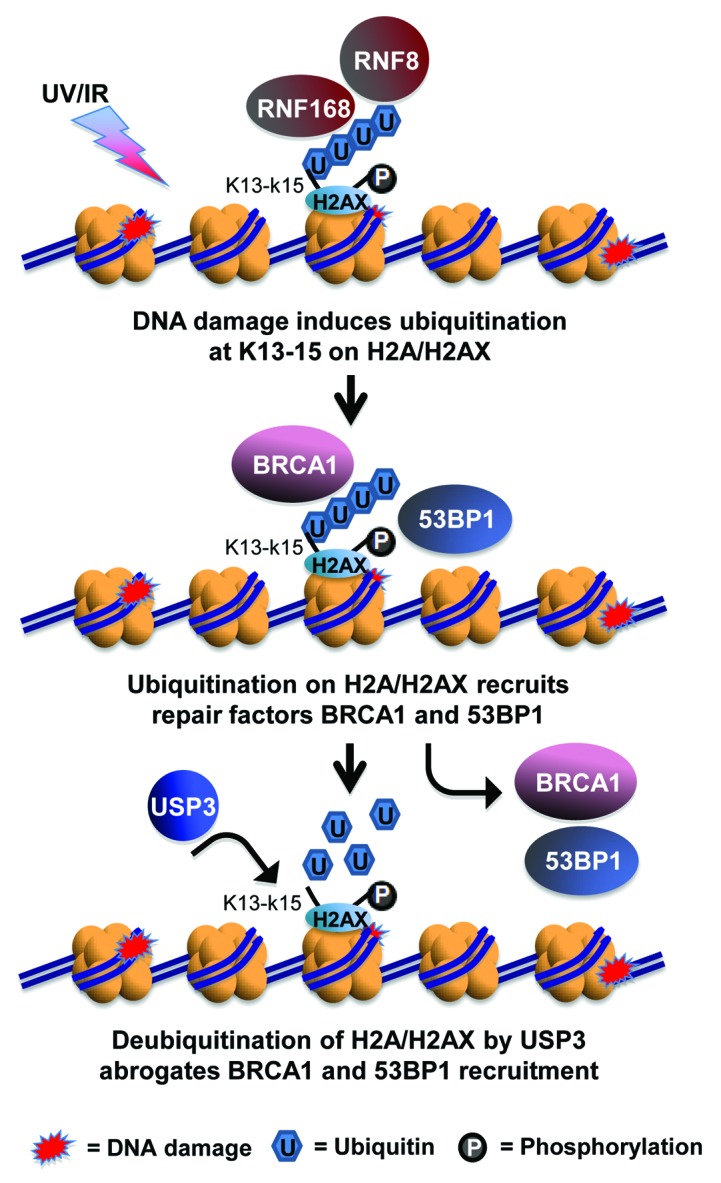
Figure 7. Model for USP3-dependent deubiquitination of γH2AX in regulation of DDR. In response of DNA damage such as UV- or IR-induced DNA breaks, K13–15 of γH2AX is ubiquitinated by the concerted action of RNF168 and RNF8. The ubiquitinated γH2AX, in turn, facilitates the accumulation of DNA break repair factor BRCA1 and 53BP1, leading to the choice of HR or NHEJ repair pathways. USP3 negatively regulates the accumulation of BRCA1 and 53BP1 at DNA breaks by virtue of deubiquitinating γH2AX.
Materials and Methods
UV irradiation, protein isolation, and western blotting
Cells were washed with phosphate-buffered saline (PBS) and irradiated with desired doses of UV-C of 254 nm wavelength, using a germicidal lamp at a dose rate of 1.0 J m−2/s as measured with UVX radiometer (UVP, LLC). The cells were supplied with fresh media, returned to 37 °C incubator, and then harvested at the indicated post-UV times. Whole-cell lysates were extracted by boiling for 5 min in sodium dodecyl sulfate (SDS) lysis buffer, containing 62 mM Tris–HCl [pH 6.8], 2% SDS, 10% glycerol, and freshly added protease and phosphatase inhibitors. Protein content of lysates was estimated through Bio-Rad DCTM protein assay kit. The proteins were resolved with SDS-PAGE (PAGE) using Novex Tris-Glycine gels (Invitrogen), followed by western blotting to detect specific proteins.
Antibodies
γH2AX (phospho-H2AX) (#9718) and phospho BRCA1 (#9009) specific antibodies were purchased from Cell Signaling. Anti-FLAG M2 beads (A2220) and anti-FLAG M2 antibody (F1804) were purchased from Sigma-Aldrich. Anti-53BP1 (SC 22760) and anti-Myc (SC-40) were obtained from Santa Cruz Biotechnology. FK2 monoclonal ubiquitin antibody (#04–268), uH2A antibody, clone E6C5 (#05–678), and anti-USP3 antibody (ab-101473) were acquired from EMB Millipore and Abcam (http://www.abcam.com/), respectively.
RNA interference
USP3 siRNA (individual siGENOME duplex D-006078-01) and control siRNA (5′-UUCUCCGAAC GUGUCACGUd T-3′) were synthesized by Thermo Scientific. The siRNA was transfected at final concentration of 100 nM into cells with Lipofectamine 2000 transfection reagent from Invitrogen, according to manufacturer’s instructions. In brief, HeLa cells were seeded at a density of 0.3 × 106 per 60-mm culture dishes and grown overnight. For cell transfection, Lipofectamine2000 reagent was diluted with proper amount of Opti-MEM medium. The gene-specific or control siRNA was added to the diluted reagent, incubated at room temperature (RT) for 20 min, and then the Lipofectamine–small interfering RNA (siRNA) complex mix was added to the cell cultures for 48 h.
Plasmids and transfection
USP3 expression constructs, both wild type and catalytically inactive mutant, were provided by Dr. Elisabetta Citterio (Division of Molecular Genetics, Netherlands Cancer Institute). Plasmid constructs of FLAG-tagged H2A and H2AX were gifts from Dr Titia K Sixma (Division of Biochemistry, Netherlands Cancer Institute). The plasmid DNAs were transfected into HeLa or other cell lines using Fugene 6 transfection reagents (Promega Corporation), according to the manufacturer’s instructions.
Immunoprecipitation
FLAG-tagged H2A and H2AX immunoprecipitation was conducted according to description by Mattiroli et al. with some modifications.16 Briefly, HeLa cells were transfected, UV-irradiated, and harvested. After a PBS wash, cells were lysed and sonicated in E1A buffer (50 mM HEPES [pH 7.5] 150 mM NaCl, 0.1% Tween-20) in the presence of protease and phosphatase inhibitors. The cells lysates were incubated with the anti-FLAG-M2 beads at 4 °C overnight. The protein bound beads were washed 3 times and eluted with FLAG peptide as described in the manufacturer’s protocol.
Micropore UV irradiation and immunofluorescence analysis
The cells were seeded at an appropriate density and grown on glass coverslips. Micropore UV irradiation was conducted according to the method established in our laboratory.32-34 Briefly, after the cells were washed once with PBS, a 5-μm isopore polycarbonate filter (EMB Millipore) was placed on top of the cell monolayer, followed by UV irradiation at a desired dose. The UV-irradiated cells were maintained in a suitable medium for the indicated time period and processed thereafter. Immunofluorescent staining was conducted according to the method established in our laboratory. In brief, the cells were washed twice with cold PBS and fixed with 2% paraformaldehyde in 0.5% Triton X-100 at 4 °C for 30 min. The fixed cells were rinsed with PBS and blocked with 20% normal goat serum in PBS, stained with an appropriate primary antibody and Fluorescein Isothiocyanate or Texas Red-conjugated secondary antibodies. The coverslips were mounted in Vectashield mounting medium with DAPI. The fluorescence images were obtained at RT with a Nikon fluorescence microscope E80i. The digital images were captured with a cooled SPOT RTKE charge-coupled-device camera (Diagnostic Instruments) and processed with SPOT analysis software.
Supplementary Material
Acknowledgments
The authors thank Dr Elisabetta Citterio at Division of Molecular Genetics, and Dr Titia K Sixma at Division of Biochemistry, Netherlands Cancer Institute for providing USP3-expressing constructs and FLAG-tagged H2A/H2AX constructs, respectively. This work was supported by Public Health service Grants (ES2388, ES12991) to AAW from National Institute of Health.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/26814
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/26814
References
- 1.Bekker-Jensen S, Lukas C, Kitagawa R, Melander F, Kastan MB, Bartek J, Lukas J. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J Cell Biol. 2006;173:195–206. doi: 10.1083/jcb.200510130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stiff T, O’Driscoll M, Rief N, Iwabuchi K, Löbrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004;64:2390–6. doi: 10.1158/0008-5472.CAN-03-3207. [DOI] [PubMed] [Google Scholar]
- 3.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–7. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 4.Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. 2001;276:47759–62. doi: 10.1074/jbc.C100569200. [DOI] [PubMed] [Google Scholar]
- 5.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–8. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell. 2007;28:739–45. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 7.Celeste A, Fernandez-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A. Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol. 2003;5:675–9. doi: 10.1038/ncb1004. [DOI] [PubMed] [Google Scholar]
- 8.Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123:1213–26. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 9.Lukas C, Falck J, Bartkova J, Bartek J, Lukas J. Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat Cell Biol. 2003;5:255–60. doi: 10.1038/ncb945. [DOI] [PubMed] [Google Scholar]
- 10.Bergink S, Jentsch S. Principles of ubiquitin and SUMO modifications in DNA repair. Nature. 2009;458:461–7. doi: 10.1038/nature07963. [DOI] [PubMed] [Google Scholar]
- 11.Lukas J, Bartek J. DNA repair: New tales of an old tail. Nature. 2009;458:581–3. doi: 10.1038/458581a. [DOI] [PubMed] [Google Scholar]
- 12.Messick TE, Greenberg RA. The ubiquitin landscape at DNA double-strand breaks. J Cell Biol. 2009;187:319–26. doi: 10.1083/jcb.200908074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, Ellenberg J, Panier S, Durocher D, Bartek J, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell. 2009;136:435–46. doi: 10.1016/j.cell.2008.12.041. [DOI] [PubMed] [Google Scholar]
- 14.Huen MS, Grant R, Manke I, Minn K, Yu X, Yaffe MB, Chen J. RNF8 transduces the DNA-damage signal via histone ubiquitylation and checkpoint protein assembly. Cell. 2007;131:901–14. doi: 10.1016/j.cell.2007.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mailand N, Bekker-Jensen S, Faustrup H, Melander F, Bartek J, Lukas C, Lukas J. RNF8 ubiquitylates histones at DNA double-strand breaks and promotes assembly of repair proteins. Cell. 2007;131:887–900. doi: 10.1016/j.cell.2007.09.040. [DOI] [PubMed] [Google Scholar]
- 16.Mattiroli F, Vissers JH, van Dijk WJ, Ikpa P, Citterio E, Vermeulen W, Marteijn JA, Sixma TK. RNF168 ubiquitinates K13-15 on H2A/H2AX to drive DNA damage signaling. Cell. 2012;150:1182–95. doi: 10.1016/j.cell.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 17.Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–54. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Callen E, Di Virgilio M, Kruhlak MJ, Nieto-Soler M, Wong N, Chen HT, Faryabi RB, Polato F, Santos M, Starnes LM, et al. 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell. 2013;153:1266–80. doi: 10.1016/j.cell.2013.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao L, Xu X, Bunting SF, Liu J, Wang RH, Cao LL, Wu JJ, Peng TN, Chen J, Nussenzweig A, et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol Cell. 2009;35:534–41. doi: 10.1016/j.molcel.2009.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang B, Elledge SJ. Ubc13/Rnf8 ubiquitin ligases control foci formation of the Rap80/Abraxas/Brca1/Brcc36 complex in response to DNA damage. Proc Natl Acad Sci U S A. 2007;104:20759–63. doi: 10.1073/pnas.0710061104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, Livingston DM, Greenberg RA. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science. 2007;316:1198–202. doi: 10.1126/science.1139516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stewart GS, Panier S, Townsend K, Al-Hakim AK, Kolas NK, Miller ES, Nakada S, Ylanko J, Olivarius S, Mendez M, et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell. 2009;136:420–34. doi: 10.1016/j.cell.2008.12.042. [DOI] [PubMed] [Google Scholar]
- 23.Ginjala V, Nacerddine K, Kulkarni A, Oza J, Hill SJ, Yao M, Citterio E, van Lohuizen M, Ganesan S. BMI1 is recruited to DNA breaks and contributes to DNA damage-induced H2A ubiquitination and repair. Mol Cell Biol. 2011;31:1972–82. doi: 10.1128/MCB.00981-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nacerddine K, Beaudry JB, Ginjala V, Westerman B, Mattiroli F, Song JY, van der Poel H, Ponz OB, Pritchard C, Cornelissen-Steijger P, et al. Akt-mediated phosphorylation of Bmi1 modulates its oncogenic potential, E3 ligase activity, and DNA damage repair activity in mouse prostate cancer. J Clin Invest. 2012;122:1920–32. doi: 10.1172/JCI57477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, Zhang Y. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 2004;431:873–8. doi: 10.1038/nature02985. [DOI] [PubMed] [Google Scholar]
- 26.Shanbhag NM, Rafalska-Metcalf IU, Balane-Bolivar C, Janicki SM, Greenberg RA. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell. 2010;141:970–81. doi: 10.1016/j.cell.2010.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nicassio F, Corrado N, Vissers JH, Areces LB, Bergink S, Marteijn JA, Geverts B, Houtsmuller AB, Vermeulen W, Di Fiore PP, et al. Human USP3 is a chromatin modifier required for S phase progression and genome stability. Curr Biol. 2007;17:1972–7. doi: 10.1016/j.cub.2007.10.034. [DOI] [PubMed] [Google Scholar]
- 28.Mosbech A, Lukas C, Bekker-Jensen S, Mailand N. The deubiquitylating enzyme USP44 counteracts the DNA double-strand break response mediated by the RNF8 and RNF168 ubiquitin ligases. J Biol Chem. 2013;288:16579–87. doi: 10.1074/jbc.M113.459917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huyen Y, Zgheib O, Ditullio RA, Jr., Gorgoulis VG, Zacharatos P, Petty TJ, Sheston EA, Mellert HS, Stavridi ES, Halazonetis TD. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature. 2004;432:406–11. doi: 10.1038/nature03114. [DOI] [PubMed] [Google Scholar]
- 30.Kim H, Chen J, Yu X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science. 2007;316:1202–5. doi: 10.1126/science.1139621. [DOI] [PubMed] [Google Scholar]
- 31.Pan MR, Peng G, Hung WC, Lin SY. Monoubiquitination of H2AX protein regulates DNA damage response signaling. J Biol Chem. 2011;286:28599–607. doi: 10.1074/jbc.M111.256297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arab HH, Wani G, Ray A, Shah ZI, Zhu Q, Wani AA. Dissociation of CAK from core TFIIH reveals a functional link between XP-G/CS and the TFIIH disassembly state. PLoS One. 2010;5:e11007. doi: 10.1371/journal.pone.0011007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu Q, Wani G, Arab HH, El-Mahdy MA, Ray A, Wani AA. Chromatin restoration following nucleotide excision repair involves the incorporation of ubiquitinated H2A at damaged genomic sites. DNA Repair (Amst) 2009;8:262–73. doi: 10.1016/j.dnarep.2008.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu Q, Wani G, Sharma N, Wani A. Lack of CAK complex accumulation at DNA damage sites in XP-B and XP-B/CS fibroblasts reveals differential regulation of CAK anchoring to core TFIIH by XPB and XPD helicases during nucleotide excision repair. DNA Repair (Amst) 2012;11:942–50. doi: 10.1016/j.dnarep.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.