

Abstract
We identified a form of cell death called “liponecrosis.” It can be elicited by an exposure of the yeast Saccharomyces cerevisiae to exogenous palmitoleic acid (POA). Our data imply that liponecrosis is: (1) a programmed, regulated form of cell death rather than an accidental, unregulated cellular process and (2) an age-related form of cell death. Cells committed to liponecrotic death: (1) do not exhibit features characteristic of apoptotic cell death; (2) do not display plasma membrane rupture, a hallmark of programmed necrotic cell death; (3) akin to cells committed to necrotic cell death, exhibit an increased permeability of the plasma membrane for propidium iodide; (4) do not display excessive cytoplasmic vacuolization, a hallmark of autophagic cell death; (5) akin to cells committed to autophagic death, exhibit a non-selective en masse degradation of cellular organelles and require the cytosolic serine/threonine protein kinase Atg1p for executing the death program; and (6) display a hallmark feature that has not been reported for any of the currently known cell death modalities—namely, an excessive accumulation of lipid droplets where non-esterified fatty acids (including POA) are deposited in the form of neutral lipids. We therefore concluded that liponecrotic cell death subroutine differs from the currently known subroutines of programmed cell death. Our data suggest a hypothesis that liponecrosis is a cell death module dynamically integrated into a so-called programmed cell death network, which also includes the apoptotic, necrotic, and autophagic modules of programmed cell death. Based on our findings, we propose a mechanism underlying liponecrosis.
Keywords: mechanisms of programmed cell death, yeast, autophagy, autophagic cell death, mitophagy, apoptosis, necrosis, fatty acids, peroxisomes, lipid droplets
Introduction
In multicellular organisms, the homeostasis of cell number in various tissues and in the entire organism is sustained via a regulated balance between the fundamental biological processes of cell division, cell differentiation, and programmed cell death (PCD).1 The term “PCD” refers to a genetically programmed, regulated form of cell death—as opposed to an accidental, unregulated cellular process.2 As a genetically programmed, regulated cellular process, a PCD subroutine should be intensified or attenuated by genetic manipulations that alter the abundancies and/or activities of only certain proteins.2,3 Furthermore, a PCD subroutine should be a cellular process comprised of a cascade of consecutive cellular events initiated in response to a certain stimulus, following each other in a certain order and orchestrated by a certain signaling pathway or network.2-4 Moreover, a PCD subroutine should be a cellular process that provides a certain benefit for development, survival, and/or stress resistance of a cell population or an organism.2-4 Several PCD subroutines are presently known; these subroutines have different underlying mechanisms, exhibit different morphological and biochemical traits, and are orchestrated by different (although partially overlapping) signaling pathways.3 The initial classification of various types of PCD into apoptosis, autophagic cell death, and necrosis was based mostly on morphological differences between cells committed to these different PCD subroutines.2,4 The most recent classification of PCD subroutines is based on a combination of numerous morphological and biochemical traits typical of each of them; the well-characterized PCD subroutines include extrinsic apoptosis, caspase-dependent or -independent intrinsic apoptosis, regulated necrosis, autophagic cell death, and mitotic catastrophe.3 Some of the currently known PCD subroutines are characterized in much less detail with respect to their morphological traits and biochemical features; these other PCD subroutines include anoikis, excitotoxicity, Wallerian degeneration, paraptosis, pyroptosis, pyronecrosis, entosis, parthanatos, netosis, and cornification.2-4 Molecular mechanisms underlying these PCD subroutines and integrating them into a PCD network remain to be established.
The budding yeast Saccharomyces cerevisiae is a unicellular eukaryote amenable to comprehensive biochemical, genetic, cell biological, chemical biological, and system biological analyses.5 The use of yeast as an advantageous model organism in cell death research has already greatly contributed to the current understanding of the molecular and cellular mechanisms underlying various PCD subroutines.6-13 We recently demonstrated that a short-term exposure of yeast cells to exogenously added palmitoleic fatty acid (POA) causes their death.14,15 In this study, we provide evidence that POA-induced cell death in yeast is an age-related subroutine of genetically programmed, regulated cell death rather than an accidental, unregulated cellular process. We concluded that POA-induced cell death is a PCD subroutine, because: (1) it is intensified or attenuated by genetic manipulations that eliminate only certain proteins involved in maintaining functional mitochondria, metabolizing lipids, or macroautophagically degrading cellular constituents; and (2) it represents a cascade of consecutive cellular events that are initiated in response to POA and follow each other in a certain order. We call this previously unknown PCD subroutine “liponecrosis”. Based on our findings, we propose a model for molecular mechanisms underlying liponecrosis. Our data suggest that liponecrosis represents a cell death module dynamically integrated into a so-called PCD network; this network also includes the apoptotic, necrotic, and autophagic modules of PCD.
Results
Macromitophagy protects yeast from a mode of cell death triggered by exogenous palmitoleic fatty acid (POA)
A short-term (for 2 h) exposure of wild-type (WT) yeast cells to exogenous POA has been shown to cause their death, thereby significantly reducing clonogenic survival of these cells in a POA concentration-dependent manner.14,15 Noteworthy, the pex5Δ mutation, previously known for its ability to impair peroxisomal fatty acid oxidation,16 has been recently demonstrated not only to greatly reduce mitochondrial functionality, but also to enhance the susceptibility of yeast to a form of cell death elicited by a brief exposure to exogenous POA.14 This finding suggested that the maintenance of a healthy population of functional mitochondria may protect yeast from POA-induced cell death modality. Macromitophagy, a selective macroautophagic degradation of dysfunctional mitochondria, is known to sustain such healthy population of functional mitochondria in chronologically aging yeast cells.17 We therefore sought to investigate the importance of macromitophagy in protecting yeast from a mode of cell death triggered by exogenous POA. To attain this objective, we used the single-gene-deletion mutant strain atg32Δ. Because this mutant strain lacks a mitochondrial receptor for macromitophagy, it is known to be impaired only in the mitophagic pathway of selective macroautophagy but not in other pathways of selective or non-selective macroautophagy.18,19 We found that the atg32Δ mutation significantly reduces clonogenic survival of yeast cells briefly exposed to various concentrations of POA (Fig. 1A–C). Thus, by removing dysfunctional mitochondria, macromitophagy protects yeast from a form of cell death triggered by this monounsaturated fatty acid. Of note, the susceptibility of both WT and atg32Δ cells to a death mode elicited by exogenous POA increased with the chronological age of these cells (Fig. 1A–C). Because the degree to which this death mode reduces cell viability appears to progress with the chronological age of a yeast cell, we concluded that it is an age-related modality of cell death.
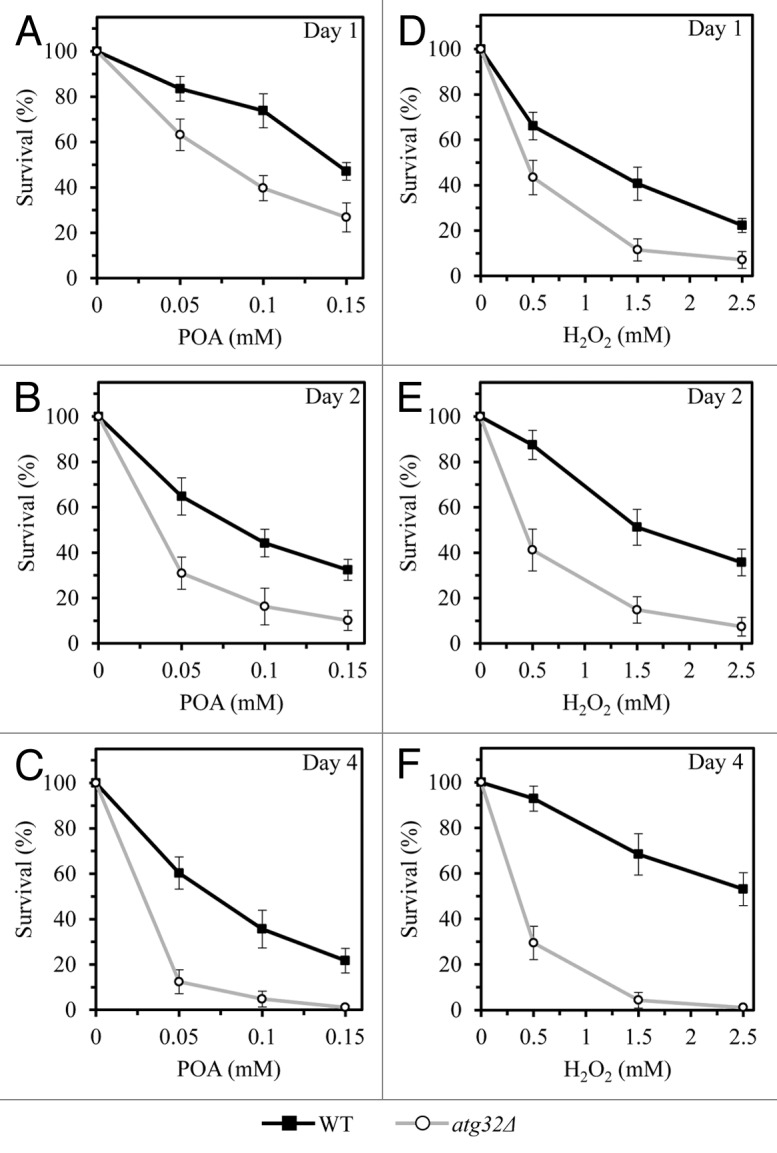
Figure 1. By removing dysfunctional mitochondria, macromitophagy protects yeast from age-related forms of cell death elicited by a short-term exposure to exogenous POA or hydrogen peroxide. WT and atg32Δ cells were recovered at days 1, 2, and 4 of culturing in a nutrient-rich YP medium initially containing 0.2% glucose as carbon source. Cell survival was assessed by measuring the clonogenicity of WT and atg32Δ cells after 2 h of treatment with various concentrations of exogenous POA (A–C) or hydrogen peroxide (D–F).
Notably, we found that the atg32Δ mutation also significantly reduces clonogenic survival of yeast cells following their short-term (2-h) exposure to various concentrations of exogenous hydrogen peroxide (Fig. 1D–F). The susceptibility of WT cells to a death mode elicited by exogenous hydrogen peroxide decreased with the chronological age of these cells, whereas the opposite trend was seen for chronologically aging atg32Δ cells (Fig. 1D–F). Hence, a selective macroautophagic degradation of dysfunctional mitochondria protects yeast from an age-related form of cell death triggered by exogenous hydrogen peroxide.
POA induces “liponecrosis,” a mode of cell death that differs from apoptotic, regulated necrotic, and autophagic cell death subroutines
To provide a mechanistic insight into the demonstrated essential role of macromitophagy in protecting yeast from a POA-induced mode of cell death, we used electron and fluorescence microscopies (EM and FM, respectively) to examine WT and atg32Δ cells exposed to various concentration of this monounsaturated fatty acid. Our objectives were: (1) to define morphological traits characteristic of the cell death subroutine triggered by exogenous POA; and (2) to compare these traits to the well-established3,6,7,10,20 morphological features of the currently known apoptotic, regulated necrotic, and autophagic cell death modalities.
We found that only minor fractions of WT and atg32Δ cells treated with various concentrations of POA exhibit such characteristic traits of a mitochondria-controlled mode of apoptotic cell death as nuclear fragmentation and phosphatidylserine (PS) translocation from the inner to the outer leaflet of the plasma membrane (Figs. 2A and 3A; Fig. S1A and B). Thus, a mode of cell death elicited by an exposure of both WT and atg32Δ cells to exogenous POA is not an apoptotic cell death subroutine.

Figure 2. Morphological traits characteristic of the cell death subroutines triggered by a short-term exposure to exogenous POA or hydrogen peroxide. WT and atg32Δ cells were recovered at day 1 of culturing in a nutrient-rich YP medium initially containing 0.2% glucose as carbon source. Transmission electron micrographs of WT and atg32Δ cells after 2 h of treatment with various concentrations of exogenous POA (A) or hydrogen peroxide (B) are presented. Bar, 1 µm. Abbreviations: ER, endoplasmic reticulum; LD, lipid droplet; N, nucleus; V, vacuole.

Figure 3. Percentage of WT and atg32Δ cells that display nuclear fragmentation (A), plasma membrane rupture (B), LD accumulation (C) or lack of all cellular organelles (D). WT and atg32Δ cells were recovered at days 1, 2, and 4 of culturing in a nutrient-rich YP medium initially containing 0.2% glucose as carbon source. Transmission electron micrographs of WT and atg32Δ cells after 2 h of treatment with various concentrations of exogenous POA or hydrogen peroxide were used for morphometric analysis; at least 100 cells of each strain were used for morphometric analysis at each time-point. Data are presented as means ± SEM (n = 3; *P < 0.01).
Of note, significant portions of WT and atg32Δ cells exposed to exogenous hydrogen peroxide displayed fragmented nucleus and externalized PS (Figs. 2B and 3A; Fig. S1A and B). Although the percentages of both WT and atg32Δ cells exhibiting these hallmarks of the apoptotic cell death subroutine following treatment with hydrogen peroxide were proportional to its concentration, the atg32Δ mutation significantly increased the fraction of such cells (Figs. 2B and 3A; Fig. S1A and B). In sum, these microscopical analyses imply that: (1) a short-term exposure of both WT and atg32Δ cells to exogenous hydrogen peroxide added at the concentrations ranging from 0.5 mM to 2.5 mM triggers an apoptotic form of cell death, and (2) macromitophagy protects yeast from this form of cell death.
Furthermore, our EM analysis revealed that WT and atg32Δ cells treated with various concentrations of POA do not exhibit rupture of the plasma membrane (Figs. 2A and 3B), a hallmark trait of a necrotic mode of programmed cell death.6,21 However, our FM analysis demonstrated that significant portions of WT14 and atg32Δ cells exposed to exogenous POA display propidium iodide (PI)-positive staining (Fig. S1C). This staining pattern is characteristic of a significantly increased permeability of the plasma membrane for PI and other small molecules, a hallmark trait of a necrotic mode of programmed cell death.6,21 Altogether, these EM and FM analyses demonstrate that a form of cell death triggered by an exposure of both WT and atg32Δ cells to exogenous POA is not a typical necrotic cell death subroutine. Indeed, (1) unlike conventional necrotic cell death, this POA-induced form of cell death does not lead to plasma membrane rupture; but (2) akin to conventional necrotic cell death, this POA-induced form of cell death significantly increases plasma membrane permeability for PI and other small molecules.
Importantly, our EM and FM analyses uncovered that WT cells treated with various concentrations of POA exhibit an excessive accumulation of lipid droplets (LD) (Figs. 2A and 3C; Fig. S1D); LD are known to serve as a deposition site for stockpiling non-esterified (“free”) fatty acids and sterols in the forms of triacylglycerols (TAG) and ergosteryl esters (EE), the 2 major neutral lipids synthesized in the endoplasmic reticulum.22,23 This hallmark morphological feature of a POA-induced form of cell death has not been reported for any of the currently known apoptotic, regulated necrotic, and autophagic cell death modalities.3,6-8,20,24 We therefore concluded that POA triggers a previously unknown form of cell death in yeast. We call this novel cell death modality “liponecrosis”.
Macromitophagy, functional mitochondria, and neutral lipids synthesis protect yeast from liponecrotic cell death elicited by POA
It should be stressed that the atg32Δ-dependent mutational block of macromitophagy abolished the accumulation of LD in yeast cells committed to liponecrotic death triggered by cell exposure to POA (Figs. 2A and 3C; Fig. S1D). Moreover, the atg32Δ mutation also significantly reduced clonogenic survival of yeast cells exposed to this monounsaturated fatty acid (Fig. 1A–C). Based on these findings, we hypothesized that: (1) the excessive accumulation of LD, a deposition site for stockpiling non-esterified fatty acids (including POA), protects yeast from a liponecrotic mode of cell death; (2) functional mitochondria are required for such accumulation of LD, likely because these organelles provide energy needed for a pro-survival process of depositing non-esterified fatty acids (including POA) within LD; and (3) macromitophagy protects yeast cells from liponecrotic death by sustaining a healthy population of functional mitochondria capable of providing energy for depositing non-esterified fatty acids (including POA) within LD. In support of this hypothesis, we found that the single-gene-deletion mutations dga1Δ and are2Δ reduce clonogenic survival of yeast cells committed to POA-induced liponecrotic death (Fig. S2A and B); these 2 mutations are known to attenuate LD formation by eliminating redundant enzymes involved in the synthesis of TAG and EE (respectively), the 2 major neutral lipids synthesized in the ER.23 Furthermore, this hypothesis is also supported by our observation that the single-gene-deletion mutation cyc3Δ reduces clonogenic survival of yeast cells committed to liponecrotic death triggered by POA (Fig. S2A and B); this mutation is known to abolish mitochondrial respiration by eliminating cytochrome c heme lyase and thereby impairing cytochrome c functionality.25
In sum, these findings validate our hypothesis that macromitophagy protects yeast cells from liponecrosis by maintaining a healthy population of functional mitochondria capable of providing energy that is needed for a pro-survival process of depositing non-esterified fatty acids (including POA) within LD.
Peroxisomal fatty acid oxidation protects yeast from liponecrotic cell death triggered by POA
We previously demonstrated that the single-gene-deletion mutation pex5Δ decreases clonogenic survival of yeast cells exposed to POA;14 this mutation is known to impair peroxisomal import of the first 2 enzymes of the fatty acid β-oxidation pathway,16 thereby decelerating a conversion of immature (dysfunctional) peroxisomal precursors to mature (functional) peroxisomes.26 We therefore hypothesized that β-oxidation of non-esterified fatty acids (including POA) within functional peroxisomes may protect yeast from liponecrotic cell death by operating as a pro-survival process of reducing the cellular level of POA. In support of this hypothesis, we found that the single-gene-deletion mutation fox1Δ reduces clonogenic survival of yeast committed to POA-induced liponecrotic death (Fig. S2D); this mutation is known to eliminate the first enzyme of the fatty acid β-oxidation pathway, thereby abolishing β-oxidation of non-esterified fatty acids (including POA) within functional peroxisomes.16
Non-selective macroautophagy executes liponecrotic cell death elicited by POA
Our EM analysis revealed that an exposure of WT cells to various concentrations of POA caused a POA concentration-dependent rise in the fraction of cells lacking all cellular organelles (Figs. 2 and 3D). Importantly, we found that the atg32Δ mutation significantly increases the portion of such organelle-less cells that are committed to POA-induced liponecrotic death (Figs. 2 and 3D). Of note, the percentages of WT and atg32Δ cells lacking all cellular organelles following an exposure to POA increased with the chronological age of these cells (Figs. 2 and 3D). Based on these findings, we hypothesized that a non-selective en masse degradation of cellular organelles executes a liponecrotic form of cell death. This hypothesis is supported by our observation that the single-gene-deletion mutation atg1Δ increases clonogenic survival of yeast cells committed to liponecrotic death triggered by POA (Fig. 4A–C). This mutation is known to abolish all pathways of non-selective and selective autophagic degradation of cellular organelles and macromolecules in yeast cells by eliminating a cytosolic serine/threonine protein kinase that governs these pathways.27 We therefore concluded that liponecrosis is an age-related form of programmed cell death that is executed by a non-selective en masse autophagic degradation of cellular organelles and macromolecules in a process orchestrated by the cytosolic serine/threonine protein kinase Atg1p.
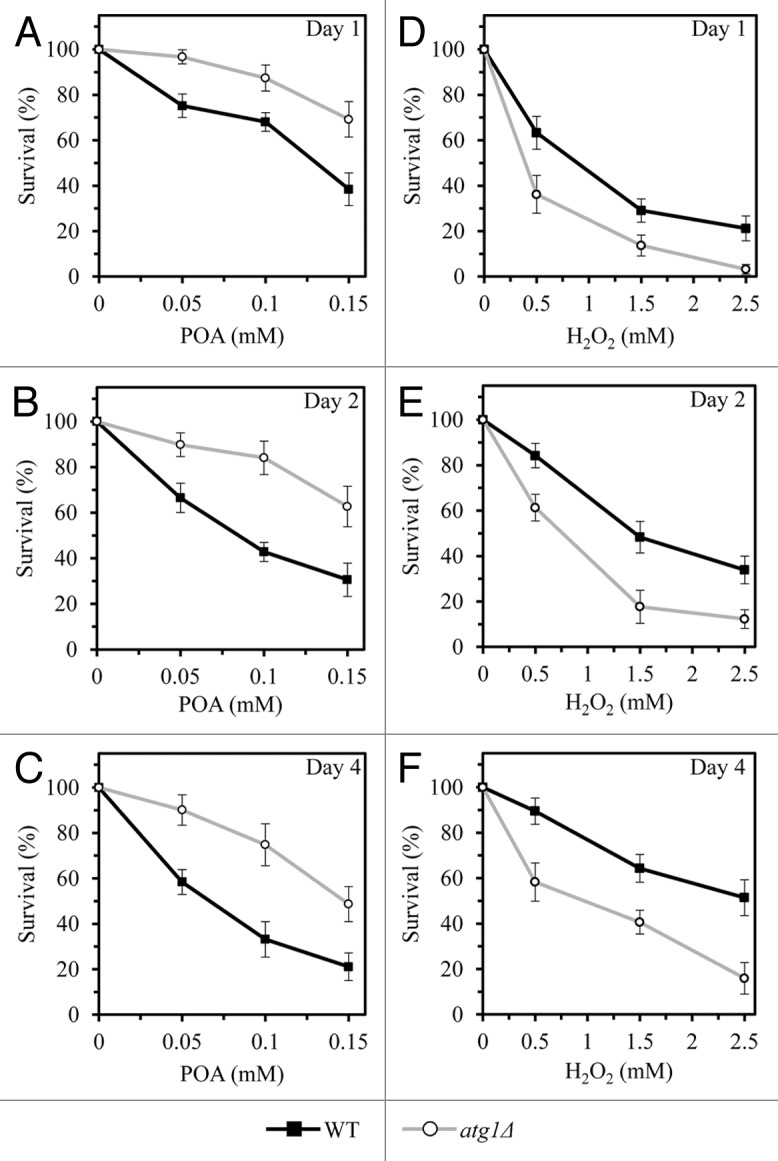
Figure 4. A non-selective en masse autophagic degradation of cellular organelles and macromolecules executes an orchestrated by Atg1p form of POA-induced liponecrotic cell death, whereas such degradation operates as a pro-survival process in yeast committed to apoptotic death that is triggered by cell exposure to exogenous hydrogen peroxide. WT and atg1Δ cells were recovered at days 1, 2, and 4 of culturing in a nutrient-rich YP medium initially containing 0.2% glucose as carbon source. Cell survival was assessed by measuring the clonogenicity of WT and atg1Δ cells after 2 h of treatment with various concentrations of exogenous POA (A–C) or hydrogen peroxide (D–F).
Of note, unlike the effect of atg1Δ on POA-elicited liponecrotic cell death, this mutation significantly reduced clonogenic survival of yeast cells following an exposure to various concentrations of exogenous hydrogen peroxide (Fig. 4D–F). This study demonstrated that a short-term exposure of yeast to exogenous hydrogen peroxide added at the concentrations ranging from 0.5 mM to 2.5 mM triggers an apoptotic form of cell death (see above). Thus, although a non-selective en masse autophagic degradation of cellular organelles and macromolecules executes an orchestrated by Atg1p form of POA-induced liponecrotic cell death, such degradation operates as a pro-survival process in yeast committed to apoptotic death that is triggered by cell exposure to exogenous hydrogen peroxide.
Discussion
This study revealed a previously unknown form of cell death, which we call “liponecrosis”. Liponecrosis is initiated in response to a short-term exposure of yeast to exogenous POA, a monounsaturated fatty acid. We demonstrate that: (1) mutations eliminating certain proteins involved in maintaining functional mitochondria, metabolizing lipids, and macroautophagically degrading cellular constituents either enhance or attenuate liponecrosis, and (2) liponecrosis is a cascade of consecutive cellular events that are initiated in response to POA and follow each other in a certain order. Thus, akin to apoptotic, regulated necrotic and autophagic cell death modalities,3,6-8,21,24 liponecrosis is a programmed, regulated form of cell death rather than an accidental, unregulated cellular process. Moreover, liponecrosis is an age-related form of cell death. Indeed, we found that the degree to which it reduces cell viability progresses with the chronological age of a yeast cell.
Our conclusion that the liponecrotic cell death modality differs from the currently known apoptotic, regulated necrotic and autophagic cell death subroutines is based on the following findings. First, yeast cells committed to POA-induced liponecrotic death do not exhibit nuclear fragmentation and PS externalization; these 2 morphological features are known to be hallmarks of the apoptotic cell death subroutine.8 Second, yeast cells committed to POA-induced liponecrotic death do not display plasma membrane rupture, a morphological trait known to be characteristic of a necrotic cell death subroutine.6,21 However, it should be stressed that liponecrosis and regulated necrosis have at least one common feature, specifically a significantly increased permeability of the plasma membrane for PI.6,21 Our unpublished data suggest that the abnormally high permeability of the plasma membrane for PI and other small molecules in yeast cells committed to POA-induced liponecrotic death is likely due to: (1) a specific remodeling of the plasma membrane lipidome and (2) an excessive internalization of phosphatidylethanolamine caused by its translocation from the outer to the inner leaflet of the plasma membrane (Richard et al., in preparation). Third, yeast cells committed to POA-induced liponecrotic death do not exhibit excessive cytoplasmic vacuolization caused by the accumulation of autophagosomes; this morphological trait is known to be a hallmark of the autophagic cell death subroutine.3,7,20,24 However, it needs to be emphasized that liponecrosis and autophagic cell death share at least 2 characteristic features, including: (1) a non-selective en masse degradation of cellular organelles and (2) a requirement for Atg1p, a cytosolic serine/threonine protein kinase that orchestrates both these subroutines of programmed cell death.3,7,20,24 Fourth, yeast cells committed to POA-induced liponecrotic death exhibit an excessive accumulation of LD, a deposition site for stockpiling non-esterified fatty acids (including POA) in the form of neutral lipids. This characteristic morphological trait of the liponecrotic cell death subroutine has not been reported for any of the currently known apoptotic, regulated necrotic and autophagic cell death modalities.3,6-8,20,24
A body of recent evidence supports the view that the individual pathways known to orchestrate the apoptotic, regulated necrotic and autophagic subroutines of cell death constitute modules that are dynamically integrated into a so-called programmed cell death (PCD) network.13,28-34 The concept of a PCD network provides a suitable explanation for the reported here and in several recent studies observations that: (1) in response to some death triggers, in certain cell types and/or under specific circumstances a cell committed to a programmed death subroutine can exhibit a mix of morphological and biochemical traits that are characteristic of different currently known cell death modalities, and (2) several proteins can be actively involved in orchestrating more than one programmed death subroutine.13,28-34 We therefore hypothesize that liponecrosis is a previously unknown module dynamically integrated into the PCD network. In our hypothesis, the liponecrotic cell death module partially overlaps with 3 other modules comprising the network, namely the apoptotic, necrotic, and autophagic modules of programmed cell death.
Based on our findings, we propose the following model for molecular mechanisms underlying liponecrosis (Fig. 5). This programmed cell death subroutine is initiated in response to an excessive cellular stress that is created due to an incorporation of bulk quantities of POA into POA-containing phospholipids and the subsequent buildup of these POA-containing phospholipids in various cellular membranes. Our unpublished data support this hypothesis; as we found, excessive quantities of POA-containing phospholipids accumulate in both membranes of mitochondria as well as in the endoplasmic reticulum and the plasma membrane (Richard et al., in preparation). A non-selective en masse autophagic degradation of cellular organelles and macromolecules executes the liponecrotic subroutine of programmed cell death that is triggered by the extreme cellular stress caused by the excessive accumulation of POA-containing phospholipids in various cellular membranes (Fig. 5). This autophagic execution of the liponecrotic cell death program is orchestrated by the cytosolic serine/threonine protein kinase Atg1p. Our model posits that several cellular processes play a pro-survival role in yeast exposed to POA by reducing the flow of POA into phospholipid synthesis pathways. These pro-survival processes protect yeast from liponecrosis by alleviating the excessive cellular stress caused by the buildup of POA-containing phospholipids in various cellular membranes. One of these pro-survival processes is an incorporation of POA into neutral lipids that are then deposited in LD (Fig. 5). Indeed, we found that liponecrosis can be enhanced by genetic manipulations that impair neutral lipids synthesis and, thus, attenuate LD formation. Our model envisions that mitochondria provide energy driving the pro-survival process of depositing non-esterified fatty acids (including POA) within LD in the form of neutral lipids (Fig. 5). This assumption is based on our findings that liponecrosis can be enhanced by genetic manipulations that abolish mitochondrial respiration by impairing cytochrome c functionality, or impede a selective macroautophagic degradation of dysfunctional mitochondria. In our model, β-oxidation of non-esterified (“free”) fatty acids (including POA) within functional peroxisomes also plays a pro-survival role in yeast exposed to POA (Fig. 5). By reducing the flow of POA into phospholipid synthesis pathways, this pro-survival process contributes to the alleviation of the excessive cellular stress that is elicited by the buildup of POA-containing phospholipids in various cellular membranes. Indeed, we found that liponecrosis can be enhanced by genetic manipulations that impair peroxisomal import of the first 2 enzymes of the fatty acid β-oxidation pathway or eliminate the first enzyme of this pathway normally confined to mature, functional peroxisomes.

Figure 5. A model for molecular mechanisms underlying programmed liponecrotic cell death elicited by POA. An incorporation of POA into POA-containing phospholipids (PL) and their consequent accumulation in various cellular membranes may operate as pro-death processes that create excessive cellular stress, thereby triggering liponecrosis. This subroutine of programmed cell death is executed by a non-selective en masse autophagic degradation of cellular organelles and macromolecules in a process orchestrated by the cytosolic serine/threonine protein kinase Atg1p. Alternatively, in a pro-survival process POA can be incorporated into neutral lipids (NL) that are then deposited in lipid droplets (LD). Macromitophagy protects yeast cells from liponecrosis by maintaining a healthy population of functional mitochondria capable of providing energy that is needed for a pro-survival process of depositing non-esterified fatty acids (including POA) within LD. Moreover, in a pro-survival process POA can be oxidized in peroxisomes. β-oxidation of non-esterified (“free”) fatty acids (FFA; including POA) within functional peroxisomes protects yeast from liponecrotic cell death by operating as a pro-survival process of reducing the cellular level of POA. See text for details.
It needs to be emphasized that, based on a unique combination of morphological traits characteristic of liponecrosis, this PCD modality differs from: (1) a mitochondria-dependent necrotic cell death subroutine initiated in response to an exposure of the yeast S. cerevisiae to exogenous polyunsaturated fatty acids;9 and (2) a mitochondria-dependent, metacaspase-independent cell death subroutine exhibiting morphological features of both apoptosis and necrosis following an exposure of the yeast S. cerevisiae to exogenous membrane-permeable C2-ceramide.35 Moreover, unlike cells of the yeast S. cerevisiae committed to liponecrosis, cells of the fission yeast Schizosaccharomyces pombe mutant strain deficient in TAG synthesis undergo a lipoapoptotic form of death upon entry into stationary phase; these cells display hallmarks of an apoptotic cell death subroutine, including chromatin condensation, nuclear DNA fragmentation, PS externalization, and reactive oxygen species accumulation.36,37
In the future, it would be important to identify other cellular processes that play essential pro-death or pro-survival roles in the liponecrotic cell death subroutine. These cellular processes may include: (1) phospholipid synthesis in the endoplasmic reticulum and mitochondrial membranes and a bidirectional phospholipid exchange between them through membrane contact sites; (2) a transfer of phospholipids from the endoplasmic reticulum to the plasma membrane via membrane contact sites; (3) maintenance of the non-random distribution of several phospholipid species within a bilayer of the plasma membrane; (4) cellular signal transduction modulated by alterations in plasma membrane phospholipid asymmetry; and (5) cellular protein homeostasis. Another challenge for the future will be to explore mechanisms underlying the predicted here integration of the liponecrotic cell death module into the PCD network known to include the apoptotic, necrotic, and autophagic modules of programmed cell death.
Materials and Methods
Yeast strains, media, and growth conditions
The wild-type strain Saccharomyces cerevisiae BY4742 (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0) as well as the single-gene-deletion mutant strains atg1Δ (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 atg1Δ::kanMX4), atg32Δ (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 atg32Δ::kanMX4), are2Δ (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 are2Δ::kanMX4), cyc3Δ (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 cyc3Δ::kanMX4), dga1Δ (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 dga1Δ::kanMX4), and fox1Δ (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0 fox1Δ::kanMX4) in the BY4742 genetic background (all from Thermo Scientific/Open Biosystems) were grown in YP medium (1% yeast extract, 2% peptone; both from Fisher Scientific; #BP1422-2 and #BP1420–2, respectively) initially containing 0.2% glucose (#D16–10; Fisher Scientific) as carbon source. Cells were cultured at 30 °C with rotational shaking at 200 rpm in Erlenmeyer flasks at a “flask volume/medium volume” ratio of 5:1.
Cell viability assay for monitoring the susceptibility of yeast to a mode of cell death induced by palmitoleic acid (POA)
A sample of cells was taken from a culture at days 1, 2, and 4 of culturing. A fraction of the sample was diluted in order to determine the total number of cells using a hemacytometer. 8 × 107 cells were harvested by centrifugation for 1 min at 21 000 × g at room temperature and resuspended in 8 ml of YP medium containing 0.2% glucose as carbon source. Each cell suspension was divided into 8 equal aliquots. Three pairs of aliquots were supplemented with POA (#P9417; Sigma) from a 50 mM stock solution (in 10% chloroform, 45% hexane and 45% ethanol; #650498, #248878 and #34852, respectively; all from Sigma). The final concentration of POA was 0.05 mM, 0.1 mM, or 0.15 mM for each pair of aliquots; in all these aliquots, the final concentrations of chloroform, hexane, and ethanol were 0.03%, 0.135%, and 0.135%, respectively. One pair of aliquots was supplemented only with chloroform, hexane, and ethanol added to the final concentrations of 0.03%, 0.135%, and 0.135%, respectively. All aliquots were then incubated for 2 h at 30 °C on a Labquake rotator (#400110; Thermolyne/Barnstead International) set for 360° rotation. Serial dilutions of cells were plated in duplicate onto plates containing YP medium with 2% glucose as carbon source. After 2 d of incubation at 30 °C, the number of colony forming units (CFU) per plate was counted. The number of CFU was defined as the number of viable cells in a sample. For each aliquot of cells exposed to POA, the % of viable cells was calculated as follows: (number of viable cells per ml in the aliquot exposed to POA/number of viable cells per ml in the control aliquot that was not exposed to POA) ×100.
Cell viability assay for monitoring the susceptibility of yeast to a mode of cell death induced by hydrogen peroxide
A sample of cells was taken from a culture at days 1, 2, and 4 of culturing. A fraction of the sample was diluted in order to determine the total number of cells using a hemacytometer. 8 × 107 cells were harvested by centrifugation for 1 min at 21 000 × g at room temperature and resuspended in 8 ml of YP medium containing 0.2% glucose as carbon source. Each cell suspension was divided into 8 equal aliquots. Three pairs of aliquots were supplemented with hydrogen peroxide (#H325–500; Fisher Scientific) to the final concentration of 0.5 mM, 1.5 mM, or 2.5 mM for each pair. One pair of aliquots remained untreated. All aliquots were then incubated for 2 h at 30°C on a Labquake rotator (#400110; Thermolyne/Barnstead International) set for 360° rotation. Serial dilutions of cells were plated in duplicate onto plates containing YP medium with 2% glucose as carbon source. After 2 d of incubation at 30 °C, the number of CFU per plate was counted. The number of CFU was defined as the number of viable cells in a sample. For each aliquot of cells exposed to hydrogen peroxide, the % of viable cells was calculated as follows: (number of viable cells per ml in the aliquot exposed to hydrogen peroxide/number of viable cells per ml in the control aliquot that was not exposed to hydrogen peroxide) × 100.
Miscellaneous procedures
BODIPY 493/503 (4,4-Difluoro-1,3,5,7,8-Pentamethyl-4-Bora-3a,4a-Diaza-s-Indacene; #D-3922; Life Technologies/Molecular Probes) staining for monitoring neutral lipids deposited in lipid droplets,38 DAPI (4′,6-Diamidino-2-phenylindole dihydrochloride; #D-9542; Sigma) staining for visualizing nuclei,39 Annexin V (#630109; Clontech Laboratories, Inc) staining for visualizing externalized phosphatidylserine (PS),39 and propidium iodide (PI; #P3566; Life Technologies/Molecular Probes) staining for visualizing the extent of plasma membrane permeability for small molecules39 were performed according to established procedures. Fluorescence40 and electron41 microscopies followed by morphometric analyses of the resulting images were performed according to established procedures.
Statistical analysis
Statistical analysis was performed using Microsoft Excel’s (2010) Analysis ToolPack-VBA. All data are presented as mean ± SEM. The P values were calculated using an unpaired 2-tailed t test.
Supplementary Material
Acknowledgments
We acknowledge the Centre for Structural and Functional Genomics at Concordia University and Facility for Electron Microscopy Research at McGill University for outstanding services. We thank Jeannie Mui and Sevan Mattie for help with electron microscopy. This study was supported by grants from the NSERC of Canada and Concordia University Chair Fund. AB and VRR were supported by Frederick Banting and Charles Best Doctoral Scholarship Awards from the Canadian Institutes of Health Research. LK was supported by an NSERC Undergraduate Summer Research Award. VIT is a Concordia University Research Chair in Genomics, Cell Biology and Aging.
Glossary
Abbreviations:
- CFU
colony forming units
- EE
ergosteryl esters
- EM
electron microscopy
- FM
fluorescence microscopy
- LD
lipid droplets
- PCD
programmed cell death
- PI
propidium iodide
- POA
palmitoleic fatty acid
- PS
phosphatidylserine
- TAG
triacylglycerols
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/26885
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/26885
References
- 1.Melino G, Knight RA, Ameisen JC. The Siren’s Song: This Death That Makes Life Live. In: Melino G, Vaux D, eds. Cell Death. Chichester, West Sussex, UK: John Wiley & Sons, 2010:1-12. [Google Scholar]
- 2.Kroemer G, El-Deiry WS, Golstein P, Peter ME, Vaux D, Vandenabeele P, Zhivotovsky B, Blagosklonny MV, Malorni W, Knight RA, et al. Nomenclature Committee on Cell Death Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2005;12(Suppl 2):1463–7. doi: 10.1038/sj.cdd.4401724. [DOI] [PubMed] [Google Scholar]
- 3.Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, Dawson TM, Dawson VL, El-Deiry WS, Fulda S, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–20. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, et al. Nomenclature Committee on Cell Death 2009 Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guide to Yeast Genetics: Functional Genomics, Proteomics, and Other Systems Analysis. In: Weissman J, Guthrie C, Fink GR., eds. Methods in Enzymology 2010: 470:1-892. [Google Scholar]
- 6.Eisenberg T, Carmona-Gutierrez D, Büttner S, Tavernarakis N, Madeo F. Necrosis in yeast. Apoptosis. 2010;15:257–68. doi: 10.1007/s10495-009-0453-4. [DOI] [PubMed] [Google Scholar]
- 7.Kourtis N, Tavernarakis N. Autophagy and cell death in model organisms. Cell Death Differ. 2009;16:21–30. doi: 10.1038/cdd.2008.120. [DOI] [PubMed] [Google Scholar]
- 8.Carmona-Gutierrez D, Eisenberg T, Büttner S, Meisinger C, Kroemer G, Madeo F. Apoptosis in yeast: triggers, pathways, subroutines. Cell Death Differ. 2010;17:763–73. doi: 10.1038/cdd.2009.219. [DOI] [PubMed] [Google Scholar]
- 9.Rockenfeller P, Ring J, Muschett V, Beranek A, Buettner S, Carmona-Gutierrez D, Eisenberg T, Khoury C, Rechberger G, Kohlwein SD, et al. Fatty acids trigger mitochondrion-dependent necrosis. Cell Cycle. 2010;9:2836–42. doi: 10.4161/cc.9.14.12346. [DOI] [PubMed] [Google Scholar]
- 10.Carmona-Gutiérrez D, Bauer MA, Ring J, Knauer H, Eisenberg T, Büttner S, Ruckenstuhl C, Reisenbichler A, Magnes C, Rechberger GN, et al. The propeptide of yeast cathepsin D inhibits programmed necrosis. Cell Death Dis. 2011;2:e161. doi: 10.1038/cddis.2011.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dziedzic SA, Caplan AB. Identification of autophagy genes participating in zinc-induced necrotic cell death in Saccharomyces cerevisiae. Autophagy. 2011;7:490–500. doi: 10.4161/auto.7.5.14872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dziedzic SA, Caplan AB. Autophagy proteins play cytoprotective and cytocidal roles in leucine starvation-induced cell death in Saccharomyces cerevisiae. Autophagy. 2012;8:731–8. doi: 10.4161/auto.19314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Munoz AJ, Wanichthanarak K, Meza E, Petranovic D. Systems biology of yeast cell death. FEMS Yeast Res. 2012;12:249–65. doi: 10.1111/j.1567-1364.2011.00781.x. [DOI] [PubMed] [Google Scholar]
- 14.Goldberg AA, Richard VR, Kyryakov P, Bourque SD, Beach A, Burstein MT, Glebov A, Koupaki O, Boukh-Viner T, Gregg C, et al. Chemical genetic screen identifies lithocholic acid as an anti-aging compound that extends yeast chronological life span in a TOR-independent manner, by modulating housekeeping longevity assurance processes. Aging (Albany NY) 2010;2:393–414. doi: 10.18632/aging.100168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burstein MT, Kyryakov P, Beach A, Richard VR, Koupaki O, Gomez-Perez A, Leonov A, Levy S, Noohi F, Titorenko VI. Lithocholic acid extends longevity of chronologically aging yeast only if added at certain critical periods of their lifespan. Cell Cycle. 2012;11:3443–62. doi: 10.4161/cc.21754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Léon S, Goodman JM, Subramani S. Uniqueness of the mechanism of protein import into the peroxisome matrix: transport of folded, co-factor-bound and oligomeric proteins by shuttling receptors. Biochim Biophys Acta. 2006;1763:1552–64. doi: 10.1016/j.bbamcr.2006.08.037. [DOI] [PubMed] [Google Scholar]
- 17.Richard VR, Leonov A, Beach A, Burstein MT, Koupaki O, Gomez-Perez A, Levy S, Pluska L, Mattie S, Rafesh R, et al. Macromitophagy is a longevity assurance process that in chronologically aging yeast limited in calorie supply sustains functional mitochondria and maintains cellular lipid homeostasis. Aging (Albany NY) 2013;5:234–69. doi: 10.18632/aging.100547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell. 2009;17:98–109. doi: 10.1016/j.devcel.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell. 2009;17:87–97. doi: 10.1016/j.devcel.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 20.Shen HM, Codogno P. Autophagic cell death: Loch Ness monster or endangered species? Autophagy. 2011;7:457–65. doi: 10.4161/auto.7.5.14226. [DOI] [PubMed] [Google Scholar]
- 21.Eisenberg T, Knauer H, Schauer A, Büttner S, Ruckenstuhl C, Carmona-Gutierrez D, Ring J, Schroeder S, Magnes C, Antonacci L, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol. 2009;11:1305–14. doi: 10.1038/ncb1975. [DOI] [PubMed] [Google Scholar]
- 22.Goldberg AA, Bourque SD, Kyryakov P, Boukh-Viner T, Gregg C, Beach A, Burstein MT, Machkalyan G, Richard V, Rampersad S, et al. A novel function of lipid droplets in regulating longevity. Biochem Soc Trans. 2009;37:1050–5. doi: 10.1042/BST0371050. [DOI] [PubMed] [Google Scholar]
- 23.Kohlwein SD, Veenhuis M, van der Klei IJ. Lipid droplets and peroxisomes: key players in cellular lipid homeostasis or a matter of fat--store ‘em up or burn ’em down. Genetics. 2013;193:1–50. doi: 10.1534/genetics.112.143362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Denton D, Nicolson S, Kumar S. Cell death by autophagy: facts and apparent artefacts. Cell Death Differ. 2012;19:87–95. doi: 10.1038/cdd.2011.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ocampo A, Liu J, Schroeder EA, Shadel GS, Barrientos A. Mitochondrial respiratory thresholds regulate yeast chronological life span and its extension by caloric restriction. Cell Metab. 2012;16:55–67. doi: 10.1016/j.cmet.2012.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Titorenko VI, Rachubinski RA. Spatiotemporal dynamics of the ER-derived peroxisomal endomembrane system. Int Rev Cell Mol Biol. 2009;272:191–244. doi: 10.1016/S1937-6448(08)01605-5. [DOI] [PubMed] [Google Scholar]
- 27.Reggiori F, Klionsky DJ. Autophagic processes in yeast: mechanism, machinery and regulation. Genetics. 2013;194:341–61. doi: 10.1534/genetics.112.149013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gozuacik D, Bialik S, Raveh T, Mitou G, Shohat G, Sabanay H, Mizushima N, Yoshimori T, Kimchi A. DAP-kinase is a mediator of endoplasmic reticulum stress-induced caspase activation and autophagic cell death. Cell Death Differ. 2008;15:1875–86. doi: 10.1038/cdd.2008.121. [DOI] [PubMed] [Google Scholar]
- 29.Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 2009;16:966–75. doi: 10.1038/cdd.2009.33. [DOI] [PubMed] [Google Scholar]
- 30.Bialik S, Zalckvar E, Ber Y, Rubinstein AD, Kimchi A. Systems biology analysis of programmed cell death. Trends Biochem Sci. 2010;35:556–64. doi: 10.1016/j.tibs.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 31.Zalckvar E, Bialik S, Kimchi A. The road not taken: a systems level strategy for analyzing the cell death network. Autophagy. 2010;6:813–5. doi: 10.4161/auto.6.6.12589. [DOI] [PubMed] [Google Scholar]
- 32.Zalckvar E, Yosef N, Reef S, Ber Y, Rubinstein AD, Mor I, Sharan R, Ruppin E, Kimchi A. A systems level strategy for analyzing the cell death network: implication in exploring the apoptosis/autophagy connection. Cell Death Differ. 2010;17:1244–53. doi: 10.1038/cdd.2010.7. [DOI] [PubMed] [Google Scholar]
- 33.Rubinstein AD, Kimchi A. Life in the balance - a mechanistic view of the crosstalk between autophagy and apoptosis. J Cell Sci. 2012;125:5259–68. doi: 10.1242/jcs.115865. [DOI] [PubMed] [Google Scholar]
- 34.Young MM, Kester M, Wang HG. Sphingolipids: regulators of crosstalk between apoptosis and autophagy. J Lipid Res. 2013;54:5–19. doi: 10.1194/jlr.R031278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carmona-Gutierrez D, Reisenbichler A, Heimbucher P, Bauer MA, Braun RJ, Ruckenstuhl C, Büttner S, Eisenberg T, Rockenfeller P, Fröhlich KU, et al. Ceramide triggers metacaspase-independent mitochondrial cell death in yeast. Cell Cycle. 2011;10:3973–8. doi: 10.4161/cc.10.22.18212. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Q, Chieu HK, Low CP, Zhang S, Heng CK, Yang H. Schizosaccharomyces pombe cells deficient in triacylglycerols synthesis undergo apoptosis upon entry into the stationary phase. J Biol Chem. 2003;278:47145–55. doi: 10.1074/jbc.M306998200. [DOI] [PubMed] [Google Scholar]
- 37.Low CP, Liew LP, Pervaiz S, Yang H. Apoptosis and lipoapoptosis in the fission yeast Schizosaccharomyces pombe. FEMS Yeast Res. 2005;5:1199–206. doi: 10.1016/j.femsyr.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 38.Bascom RA, Chan H, Rachubinski RA. Peroxisome biogenesis occurs in an unsynchronized manner in close association with the endoplasmic reticulum in temperature-sensitive Yarrowia lipolytica Pex3p mutants. Mol Biol Cell. 2003;14:939–57. doi: 10.1091/mbc.E02-10-0633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Madeo F, Fröhlich E, Fröhlich KU. A yeast mutant showing diagnostic markers of early and late apoptosis. J Cell Biol. 1997;139:729–34. doi: 10.1083/jcb.139.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goldberg AA, Bourque SD, Kyryakov P, Gregg C, Boukh-Viner T, Beach A, Burstein MT, Machkalyan G, Richard V, Rampersad S, et al. Effect of calorie restriction on the metabolic history of chronologically aging yeast. Exp Gerontol. 2009;44:555–71. doi: 10.1016/j.exger.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 41.Guo T, Gregg C, Boukh-Viner T, Kyryakov P, Goldberg A, Bourque S, Banu F, Haile S, Milijevic S, San KH, et al. A signal from inside the peroxisome initiates its division by promoting the remodeling of the peroxisomal membrane. J Cell Biol. 2007;177:289–303. doi: 10.1083/jcb.200609072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.