

Abstract
Gamma secretase inhibitors (GSI), cell-permeable small-molecule inhibitors of gamma secretase activity, had been originally developed for the treatment of Alzheimer disease. In recent years, it has been exploited in cancer research to inhibit Notch signaling that is aberrantly activated in various cancers. We previously found that GSI could synergize with anti-microtubule agent, vincristine (VCR) in a Notch-independent manner. Here, we delineate the underlying cell cycle-related mechanism using HeLa cells, which have strong mitotic checkpoints. GSI enhanced VCR-induced cell death, although GSI alone did not affect cell viability at all. GSI augmented VCR-induced mitotic arrest in a dose-dependent manner, which was preceded by apoptotic cell death, as shown by an increase in Annexin V-positive and caspase-positive cell population. Furthermore, GSI amplified multi-polar spindle formation triggered by VCR. Altogether, we show the evidence that GSI enhances VCR-induced apoptosis in HeLa cells via multi-polar mitotic spindle formation, independent of Notch signaling. These data suggest that one or more GS substrates, yet to be identified, in a post-GS processed form, may play a role in maintaining functional centrosomes/mitotic spindles. More significantly, the synergistic effect of GSI in combination with VCR could be exploited in clinical setting to improve the efficacy of VCR.
Keywords: gamma secretase inhibitor, Vincristine, mitotic arrest, apoptosis, spindle assembly checkpoint, multi-polar spindle, centrosome, Notch
Introduction
γ-secretase, an aspartyl protease, belongs to the iCLiPs (intramembrane cleaving proteases) family.1 It is a multiprotein complex that consists of presenilin (PS), anterior pharynx defective-1 (APH1), presenilin enhancer 2 (PSEN2), and nicastrin (NCT).2 It processes a wide range of substrates, mostly type I transmembrane proteins.3 The list of substrates is rapidly growing, with more than 90 substrates indentified so far, including amyloid precursor protein (APP), Notch, ErbB4, neuregulin, and CD44.3
Notch is one of the well-characterized γ-secretase substrates due to its essential roles in many biological processes, including apoptosis, proliferation, and migration during mammalian development.4 Following cleavage by γ-secretase, the intracellular domain of Notch (ICN) is released, allowing ICN to enter the nucleus. ICN interacts with the DNA-binding transcription proteins, modulating the expression of downstream target genes.5
Recently, the roles of Notch signaling in cancers have been investigated with great intensity, because Notch pathway has been found to be dysregulated in a variety of cancers including leukemia, breast cancer, lung cancer, and glioma, among others.6 Furthermore, accumulating evidences have shown the essential role of Notch signaling in maintaining cancer stem cell populations,7-16 which are responsible for resistance of various cancers to conventional therapies. Hence, there has been tremendous effort on targeting Notch signaling in cancers by inhibiting γ-secretase activity using small molecules. Although various γ-secretase inhibitors (GSI) have been shown to inhibit Notch signaling by blocking cleavage of the Notch receptor, their therapeutic efficacies as a single agent were, at most, modest in most cancers. Subsequently, the combination of chemotherapy with GSI has been reported to be more effective than chemotherapy or GSI alone in many cancers and possibly prevents the emergence of chemotherapy-resistant cancer cells.15,17-25 Upon a similar effort, we found that GSI specifically synergized with Vincristine (VCR) in inducing cell death in T-cell acute lymphoblastic leukemia (T-ALL; manuscript in preparation). However, the GSI effect in combination with VCR was not associated with inhibition of Notch signaling. Similar to our observation, Katano et al. reported that GSI synergized with Taxol in killing colon and pancreatic cancers in a Notch-independent manner.26,27 Above reports along with our finding with T-ALL suggest that GSI may synergize with anti-microtubule agents through a mechanism other than inhibiting Notch signaling.
Anti-microtubule agents are among the most effective anti-cancer therapeutics currently used in clinics. They inhibit spindle function by disrupting microtubule dynamics, leading to mitotic arrest followed by cell death.28 Mitotic arrest is mediated by the spindle assembly checkpoint (SAC), which is activated by defects in spindle tension or attachment to chromosome kinetochores.29 The spindle assembly checkpoint thereby prevents mitotic errors and thus maintains genome integrity.
To investigate how GSI enhances VCR-induced cell death in a comprehensive manner, HeLa cells were chosen for this study as a model system. HeLa cells have been documented to have relatively strong mitotic assembly checkpoints as evidenced by (1) induction of a nearly indefinite mitotic arrest in the absence of spindle assembly;30,31 and (2) clear manifestation of discrete spindle-assembly and spindle-tension checkpoints.32
Here, we report that GSI augments VCR-induced mitotic arrest by increasing activated BubR1 expression. Furthermore, GSI amplifies multi-polar spindle formation triggered by VCR, which leads to augmentation in number of cells undergoing apoptosis.
Results
GSI enhances VCR-induced cell death in HeLa cells
We started by determining whether GSI can synergize with VCR in HeLa cells, similar to T-ALL cells we previously observed (data not shown). HeLa cells were treated with 10–20 nM VCR for 48 h, and the cell viability was measured by WST-1 assays. VCR reduced the cell viability in a dose-dependent manner, as shown by a decrease in absorbance (Fig. 1A). In contrast, 10 μM DAPT alone did not affect cell viability at all. Higher concentrations of DAPT, up to 100 μM, did not have any effect on cell viability as well (data not shown). However, when HeLa cells were treated with DAPT in conjunction with VCR, the cell viability decreased significantly compared to the treatment with either VCR or DAPT alone. The cell viability data measured by WST-1 assay was further confirmed by manual cell counting. When HeLa cells were treated with VCR and DAPT together, the viable cell numbers were 5 times less than those treated with VCR alone and were below the cell numbers originally seeded (Fig. 1B). These data show a correlation between cell number and absorbance as measured by manual cell counting and WST-1 assay and further suggest that DAPT enhances VCR-induced cell death, rather than merely inhibiting cell proliferation.
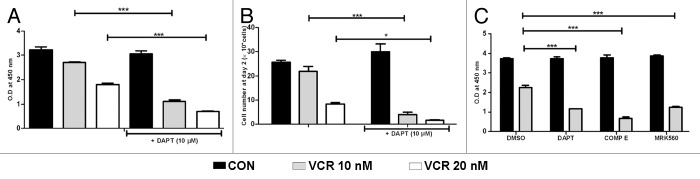
Figure 1. GSI enhances VCR-induced cell death in HeLa cells. HeLa cells were treated with VCR (10–20 nM) and/or GSI (10 μM) for 48 h. Control cells were treated with DMSO. (A and C) Cell viability was measured by WST-1 assays. Relative cell viability is presented as O.D at 450 nm. (B) Viable cell number was counted by trypan blue exclusion assay. Results are presented as mean ± SEM of triplicate assays. The statistical significance of differences was determined by ANOVA test; *P < 0.05; **P < 0.01; ***P < 0.001.
To ensure that the DAPT effect in enhancing VCR-induced cell death is not the result of off-target effect of DAPT, HeLa cells were treated with different GSIs, such as Compound E and MRK560, in combination with VCR. Consistent with DAPT, both Compound E and MRK560 enhanced VCR-induced cell death, although the GSIs alone did not affect HeLa cell viability (Fig. 1C). Since DAPT is as effective as other potent GSIs, we used DAPT for the following experiments. This data corroborates our unpublished data with T-ALL (manuscript in preparation) that GSI synergizes with VCR in inducing cell death.
GSI augments mitotic arrest induced by VCR
Since GSI enhances VCR- induced cell death, we investigated possible mechanisms for the synergism between VCR and GSI. VCR acts by destabilizing microtubules, and thus results in mitotic arrest that plays an important role in VCR-induced cell death.33 We, hence, examined the effect of the combination of VCR and DAPT on cell cycle progression (Fig. 2A). As expected, VCR increased the G2/M population compared with the control (media) in a dose-dependent manner (Fig. 2A, 6.9% for CON; 15.4% for 10 nM VCR; 39.0% for 20 nM VCR). When DAPT was added in increasing doses to the culture along with VCR, the G2/M population was further increased in a DAPT dose-dependent manner (Fig. 2A, 15.4% for 10 nM VCR vs. 33.2% for 10 nM VCR + 5 μM DAPT vs. 42.6% for 10 nM VCR +10 μM DAPT; 39.0% for 20 nM VCR vs. 54.1% for 20 nM VCR + 5 μM DAPT). The combination of 10 nM VCR and 10 μM DAPT was more effective than 20 nM VCR alone in inducing G2/M population, although 10 μM DAPT did not have any effect on cell cycle distribution (Fig. 2A). DAPT by itself (tested up to 25 μM) did not affect the cell cycle progression (Fig. 2A). We further distinguished the cells in M phase from those in G2 phase by staining the cells against MPM-2, mitosis marker (Fig. 2B). Two variable flow cytometry analyses revealed that DAPT increased the cell population in M phase more significantly than that of G2 phase when combined with VCR (Fig. 2B). Five- and 10-micromolar concentrations of DAPT increased M-phase cells by 313% and 377%, respectively, compared with that of 10 nM VCR alone, whereas the same concentrations of DAPT increased G2 phase cells by 114% and 139%, respectively. These data suggest that GSI augments mitotic arrest induced by VCR.

Figure 2. GSI augments VCR-induced mitotic arrest in a dose dependent manner. HeLa cells were treated with increasing concentrations of DAPT (5, 10, 25 μM) and/or VCR (10, 20 nM) for 24 h. (A) Cell cycle progression was analyzed after PI staining. The percentage of cells in each cell cycle phase is presented. (B) Cell population in mitotic phase was measured by double staining with PI and MPM-2. The percentage of cells in M phase (upper box) and G2 phase (lower box) are presented. (C) Cell lysates were analyzed for BubR1 and Mad2 by western blot. Beta-actin served as a loading control.
Since mitotic block induced by antimitotic drugs including VCR occurs through the spindle assembly checkpoint activation,28 we examined the status of spindle checkpoint proteins BubR1 and Mad2. After treating the cells with DAPT or VCR alone or both together, cell lysates were evaluated for BubR1 and Mad2 by western blotting (Fig. 2C). As reported previously,34 VCR treatment increased activated BubR1 expression compared to untreated control HeLa cells. DAPT treatment alone also increased BubR1 protein expression, albeit to a lesser degree. However, when combined with VCR, DAPT substantially increased the expression levels of activated BubR1. In contrast, addition of DAPT to VCR did not change the expression levels of Mad2. An increase in activated BubR1 expression by DAPT indicates that DAPT facilitates the spindle assembly checkpoint induced by VCR. As reported,35 both BubR1 and Mad2 proteins were localized at kinetochores of metaphase (data not shown).
GSI enhances VCR-induced apoptosis
The addition of GSI to VCR seems to increase apoptosis following mitotic arrest, as shown by a concomitant emergence of a sub-G1 population in flow cytometry analyses (Fig. 2A). Hence, we determined whether GSI increases the cell death induced by VCR via apoptosis. To detect early apoptotic process, cells treated with different drugs were stained with FITC-labeled Annexin V and 7AAD and quantified using flow cytometry (Fig. 3A). Similar to control untreated cells, DAPT-treated cells contained very few Annexin V-positive apoptotic cell populations. In contrast, when DAPT was added to the culture in combination with VCR, it significantly increased Annexin V-positive cells. Furthermore, when cells were divided into 2 populations, such as detached cells (mitotic cells) and attached cells (interphase cells), significantly more detached cells were Annexin V-positive compared with attached cells, suggesting that indeed mitotic arrest precedes apoptosis (Fig. 3A, right panel). Since caspases play a central role in apoptotic cell death and are activated in response to a variety of anticancer agents, including anti-microtubule drugs,36 we also measured intracellular active caspases induced by the treatment. To detect active caspases, fluorochrome inhibitor of poly-caspases (SR-VAD-FMK) reagent that binds to active caspases was added to the cells at the time of harvest following treatment with different drugs, and active caspase-positive cells were quantified using FACS analysis (Fig. 3B). Consistent with Annexin V staining data, adding GSI to VCR resulted in a significant increase in the caspase-positive population. Active caspase-positive cells were increased in a DAPT dose-dependent manner. DAPT alone did not increase the proportion of caspase-positive cells. The induction of apoptosis was further confirmed by monitoring the cleavage of PARP that is executed by activated caspase 3. PARP cleavage detected after treating with VCR was significantly increased by addition of DAPT. These data suggest that GSI enhances VCR-induced cell death through activation of apoptotic pathways.

Figure 3. GSI enhances VCR-induced apoptosis. HeLa cells were treated with varying concentrations of DAPT and/or VCR as indicated for 24 h. (A) Annexin V-FITC and 7AAD binding were measured by flow cytometry. Cell debris was excluded from analysis and apoptotic Annexin V-positive cells are shown as percentage. To compare apoptosis between detached and attached populations, cells treated with VCR (10 nM) plus DAPT (10 μM) were harvested separately and staining was performed. One representative image is shown. (B) Poly-caspase activity was measured using polycaspase FLICA SR-VAD-FMK reagent. Caspase-positive cells were quantified by flow cytometry and are presented as percentage. Cell debris was excluded from analysis. (C) Cell lysates were analyzed for PARP cleavage by western blot. Beta-actin served as a loading control. All results are presented as mean ± SEM of triplicate assays. The statistical significance of differences was determined by ANOVA test; *P < 0.05; **P < 0.01; ***P < 0.001.
GSI amplifies VCR-triggered multi-polar mitotic spindle formation
Since DAPT augments VCR-induced mitotic arrest by facilitating spindle assembly checkpoint (Fig. 2C), we sought to look for mitotic abnormalities in HeLa cells when treated with VCR plus DAPT, compared to VCR treatment alone. To address this question, HeLa cells were treated with VCR, DAPT, or VCR plus DAPT, or left untreated for 24 h, after which the mitotic cells were shaken loose and collected for fluorescent immunocytochemistry. Consistent with cell cycle progression data, treating HeLa cells with DAPT alone did not induce the mitotic cell population. The proportion of mitotic population induced by DAPT was similar to that of untreated control cells as evidenced by rare round-up cells. Cells were stained with α-tubulin antibody and DAPI to detect microtubule spindle and DNA, respectively, and microscopied (Fig. 4A). Untreated cells and DAPT treated cells demonstrated normal bipolar spindle in mitosis. Aberrant mitotic figures were rare in these samples. VCR at the concentration of 20 nM showed some multi-polar spindles (around 24.6%) and some mono-polar spindles (around 32%) were also observed as shown by mitotic spindles usually radiating from a central focus. Although DAPT alone (up to 25 μM) did not induce noticeable number of aberrant spindle formation, when added together with VCR, it significantly increased the appearance of multi-polar spindles in a concentration-dependent manner (Fig. 4A and B, 64.6% for 5 μM DAPT; 84% for 10 μM DAPT; 93.6% for 25 μM DAPT). Most of the multi-polar cells with higher concentrations of DAPT included more than 3 poles (Fig. 4B, 62% out of 84% for 10 μM DAPT; 85.4% out of 93.6% for 25 μM DAPT), and the number of poles increased in a DAPT concentration-dependent manner. To confirm the multi-polar spindle formation by VCR plus DAPT, cells were also stained with γ-tubulin. Immunofluorescence analysis of γ-tubulin demonstrated that VCR and DAPT produced abnormal centrosome numbers in majority of mitotic cells (Fig. 4C). Multiple centrosomes as evidenced by the presence of multiple γ-tubulin positive spindle poles in the cells treated with VCR and DAPT supported the data with multi-polar spindle formation observed with α-tubulin staining. DAPT-treated cells as well as untreated control had 2 centrosomes in mitotic cells as detected by γ- tubulin staining (Fig. 4C).

Figure 4. GSI amplifies VCR-triggered multi-polar mitotic spindle formation. HeLa cells were treated with VCR (20 nM) alone and/or increasing concentrations of DAPT (5, 10, 25 μM) for 24 h. (A) Representative examples of mitotic spindles stained with anti-α tubulin (green) and DAPI (blue) for the indicated treatment. Scale bars represent 10 µm. Inserts show representative image from each sample at higher magnification. (B) Observations demonstrated in (A) were quantified by evaluating at least 100 cells per treatment. Untreated control and DAPT treatment alone were left out from analysis because of lack of abnormality in mitotic spindles. Data from three independent experiments were graphed as a stacked mean percentage ± SEM. The statistical significance of differences was determined by ANOVA test. Statistical P values shown in figure were calculated with comparison to VCR treatment alone (*P < 0.05; **P < 0.01; ***P < 0.001). (C) Representative immunofluorescence images of mitotic spindles stained with anti-α tubulin (green), centrosomes stained with γ-tubulin (red), and DAPI (blue) for the indicated treatment.
Notch signaling is not involved in the GSI effect in combination with VCR
Although it appears that GSI synergizes with VCR in inducing apoptosis of T-ALL in a Notch-independent manner (unpublished data), we wanted to confirm whether this is the case for HeLa cells. First, the expression of Notch receptors in HeLa cells was examined by RT- PCR. HeLa cells expressed Notch1, 2, and 3, while Notch4 was not detected (Fig. S1). Lack of Notch4 transcript in HeLa cells was consistent with the report by Wu et al.37 The expression of activated Notch1, 2, and 3 on the protein level was further examined by western blot (Fig. 5A and Fig. S2). To validate antibody specificity, intracellular domain of each Notch receptor (ICN1, ICN2, and ICN3) was transfected into HeLa cells as positive controls, and shRNAs against each Notch receptor were transduced into HeLa cells as negative controls (Fig. S2). It appeared that HeLa cells express activated forms of Notch1, 2, and 3. Consistent with mRNA data, the activated form of Notch4 was not detected in HeLa cells (Fig. S2). The endogenous, activated forms of Notch1 and Notch3 in HeLa cells were significantly decreased after treatment with 10 μM DAPT (Fig. 5A). However, the activated form of Notch2 was resistant to DAPT treatment (Fig. 5A). Higher concentrations of DAPT, up to 50 μM, did not reduce the expression of the activated form of Notch2 in HeLa cells (data not shown). This concurs with the observations by others that GSIs, including DAPT, have differential capacities in blocking cleavage of different Notch receptors. For instance, Notch1 in mammalian cancer cells was sensitive to DAPT treatment, whereas Notch4 in the same cell lines was insensitive to DAPT treatment.9,38

Figure 5. Notch Signaling is not involved in the GSI effect in combination with VCR. (A) Western blot analysis on expression of activated forms of Notch receptors (ICN1-ICN3) in HeLa cells with or without DAPT treatment. Beta-actin served as a loading control. (B) HeLa cells expressing shRNAs against Notch1, Notch2, Notch3, and scramble (N1, N2, N3, and C) were treated with 10 μM DAPT and/or VCR (10 nM or 20 nM) for 24 h. Cell cycle distribution was determined based on DNA content of PI stained populations. (C) Western blot analysis on expression of ICN1–3, and Hes1 after silencing Notch receptors using shRNAs. Beta-actin served as a loading control. (D) HeLa cells were transduced with a retroviral vector expressing ICN1, ICN2, ICN3, or GFP and treated with 20 nM VCR and/or 10 μM DAPT for 48 h. Cell viability was measured by WST-1 assays. Relative cell viability is presented as O.D at 450 nm. Results are presented as mean ± SEM of triplicate assays. The statistical significance of differences was determined by t test; *P < 0.05; **P < 0.01; ***P < 0.001.
Next, it was determined whether Notch inhibition contributes to the GSI effect in combination with VCR. To address this in a more specific manner, each Notch receptor was knocked-down using shRNAs. Silencing Notch1, Notch2, or Notch3 did not potentiate HeLa cells to the VCR treatment as shown in Figure 5B. Furthermore, DAPT synergized with VCR in inducing mitotic arrest and apoptotic sub-G1 in Notch1-, Notch2-, and Notch3-silenced cells as effectively as that in control cells. This was not attributed to insufficient knockdown of each receptor, as shown in Figure 5C. Silencing Notch1 and Notch3 reduced Notch transcriptional activities as shown by a decrease in Notch target gene HES1 expression (Fig. 5C). However, silencing Notch2 did not affect HES1 expression. This data suggests that the GSI effect in combination with VCR is independent of Notch signaling. To substantiate our conclusion, the construct that encodes ICN1 (or ICN2, ICN3) and GFP, or control vector with only GFP was used to transduce HeLa cells as previously described39 and examined whether ICN could reverse the GSI effect (Fig. 5D). HeLa cells transduced with each ICN or control vector (>95% GFP+ cells) were treated with 20 nM VCR in the presence or absence of 10 μM DAPT for 48 h, and the cell viability was measured by WST-1 assays. DAPT was still potent in enhancing VCR-induced cell death in these ICN-overexpressing cells, showing that ICN cannot rescue the GSI effect in the presence of VCR. Altogether, these data suggest that Notch is not involved in the synergistic GSI effect in combination with VCR.
Discussion
The potential use of GSI in conjunction with chemotherapy is an active area of cancer research, considering that Notch signaling is often hyper-activated in many types of cancer and confers survival advantages to cancer cells, eventually leading to poor patient outcomes.6 However, our recent data using T-ALL cells showed that GSI synergized with VCR in inducing cell death in a Notch-independent manner, indicating that GSI has other functions besides inhibiting Notch signaling.
Here, we showed that the GSI effect in enhancing VCR efficacy was not the result of inhibition of Notch signaling in HeLa cells by both loss-of-function and gain-of-function assays. First, silencing Notch receptors using shRNAs did not sensitize HeLa cells to VCR treatment. Second, overexpressing ICN1–3 in HeLa cells did not reverse the GSI effect. Instead, it appeared that GSI augmented VCR-induced mitotic arrest by amplifying VCR-triggered multi-polar spindle formation in mitosis, which eventually led to apoptotic cell death.
Multi-polar spindle formation is a consequence of centrosome amplification.40 Centrosomes play a fundamental role in organizing the mitotic spindle.41-43 To assure spindle bipolarity, centrosomes are normally duplicated exactly once per cell cycle in parallel with DNA replication in a tightly regulated manner.41-43 Centrosome amplifications can be mediated by 2 different mechanisms;40 one is fragmentation of existing centrosomes, and the other is a true amplification in terms of new formation of additional centrosomes. Actually, centrosome amplification represented by latter is a characteristic of cancer.44 Abnormalities in oncogenes and tumor suppressor genes that are often found in cancer cells cause centrosome amplification, leading to supernumerary centrosomes.45 However, cancer cells manage to coalesce multiple centrosomes into 2 functional spindle poles (centrosome clustering) to prevent lethal multi-polar division.46 It is not clear at this point whether the combination of VCR and GSI induce fragmentation of centrosome or disruption of centrosome clustering. It seems that reducing spindle tension using a depolymerizing drug has opposite effect on inducing multi-polar spindle formation depending on the 2 different mechanisms. It has been reported that Nocodazole promoted multi-polar spindle formation induced by centrosome declustering,47 while it reduced multi-polar spindle formation when induced by centrosome fragmentation.48 These observations by others support centrosome declustering as a mechanism for GSI mediated multi-polar spindle formation in our current study. Furthermore, recent genome-wide RNAi screening in cells with supernumerary centrosomes by 2 different groups revealed that spindle tension is, indeed, the principal means by which multiple centrosomes are clustered into a bipolar spindle, preventing multipolarity.47,49 However, we cannot exclude the possibility that both centrosome declustering and centrosome fragmentation contribute to spindle multipolarity augmented by GSI. For example, Arsenite induced multipolar spindle formation in CGL-2 cells by both mechanisms.50 These 2 mechanisms need to be examined carefully for further analysis of the synergism between VCR and GSI. Irrespective of the underlying mechanisms, multi-polar spindle formation leads to apoptotic cell death.47 Consistent with this notion, treatment of HeLa cells with GSI plus VCR induced apoptotic cell death as shown by Annexin V and caspase assays.
The exact mechanism through which GSI synergizes with VCR in triggering multi-polar spindle formation remains unclear. However, it is worth noting that PS, a subunit of γ-secretase complex that possesses substrate binding and catalytic site,51,52 and to which GSI physically bind to,53,54 has been reported to be localized in centrosomes.55,56 In addition, APP, one of the GS substrates, was also found to be localized at centrosomes.56,57 Furthermore, extranumerary centrosomes were more frequently found in cells from PS-K/O and APP-K/O mice compared to those of wild-type mice.56 In contrast, when PS1 was overexpressed in hTERT-HME1 cell line, abnormalities in mitotic spindle structure are often observed.58 For example, centrosomes do not develop in metaphase, although the DNA is tightly arrayed along the metaphase, and lagging chromosomes remain at the metaphase plate as the rest of the DNA moves to opposite poles of the cells.58 Based on the previous reports and our findings here, it is tempting to speculate that the APP fragments generated by γ-secretase have a role in maintaining functional centrosomes/mitotic spindles directly or through interaction with proteins associated with these structures. Disrupting this function by depleting PS or APP or by blocking cleavage of APP via GSI may produce centrosome/mitotic spindle abnormalities, which exacerbate VCR-induced multi-polar spindle formation.
Although an array of questions remain to be answered for the synergism between VCR and GSI; nevertheless, our findings here give new insights on the functions of GSI in mitosis and further open the possibility of combined use of VCR and GSI for more effective cancer therapy.
Materials and Methods
Antibodies and reagents
The following antibodies were used: α-tubulin (Sigma, T5168 or Cell Signaling, 8058), γ-tubulin (Abcam, Ab27076), FTIC-MPM-2 (Millipore, 16–155), β-actin (Sigma, A5441), PARP (Santa Cruz, sc-7150), Mad2 (BD biosciences, 610678), BubR1 (BD biosciences, 612503), Notch1 (Abcam, ab8925), Notch2 (Cell Signaling, 4530), Notch3 (Cell Signaling, 2889), and HES1 (Millipore, ab5702).
VCR (Tocris) was prepared in culture media as concentrated stock solution and added to the final concentrations as indicated. The γ-secretase inhibitors, DAPT (Millipore), Compound E (Millipore), and MRK 560 (Tocris) were dissolved in DMSO and added to final concentrations as indicated.
Cell culture and cell viability measurement
HeLa cell line was obtained from ATCC and maintained in EMEM supplemented with 10% fetal calf serum and 2 mM L-glutamine. Cell growth was measured by a colorimetric cell proliferation assay (WST-1, Roche) according to the protocol provided by the supplier. In brief, cells were seeded at 3–5 × 103 cells/200 μl/well in 96-well culture plates and incubated overnight with the complete media. The cells were then treated with different doses of VCR and GSI as indicated for 48 h. After 48 h, 20 μl of dye solution was added to each well, and the plate was incubated for 1–4 h at 37 °C, 5% CO2. The absorbance was measure at 450 nm using an ELISA microplate reader. Each assay was performed in triplicates. For manual cell counting, cells were seeded at 4 × 104 cells/1 ml/well in 24-well culture plates and incubated overnight with the complete media. The cells were then treated with different doses of VCR and GSI as indicated for 48 h. The cells were collected after trypsinization and the viable cells were counted after staining with trypan blue.
RNA interference of Notch receptors and Stable expression of ICN1–3
The lentivirus encoding shRNAs against human Notch1, Notch2, Notch3, and scramble were obtained from Santa Cruz and used to transduce HeLa cells to silence Notch1, 2, and 3. The transduced cells were selected with puromycin. Retroviral vectors encoding the intracellular domains of human Notch1–3 (ICN1–3) and control vector MigR1 were kindly provided by Dr Zweidler-McKay (MD Anderson). Retroviral supernatants were produced by transient co-transfection of the retroviral vectors along with pVSVG (Clontech) into GP2–293 cells (Clontech) using TransIT-LT1 (Mirus). Supernatants were collected at 48 h after transfection and used directly for transduction of the cells after 0.45 μM filtration.
Cell cycle analysis and flow cytometry
Cells were treated with the drugs as indicated and harvested at 24 h. After fixing with ice-cold 70% ethanol, the cells were washed twice with PBS and incubated with PI staining solution (PBS containing 0.1% Triton X-100, 10 μg RNase, and 50 μg propidium iodide). The cells were read on flow cytometry for DNA contents and analyzed using ModFit LT software. Mitotic cells were determined by staining the cells with MPM-2 antibody. Following fixation in 70% ethanol and subsequent washing with PBS, the cells were incubated with FITC-conjugated MPM-2 antibody for 1 h, followed by incubation with PI solution, as described above. Samples were read on flow cytometry and analyzed.
Apoptosis assay
Cells were treated with the drugs as indicated and harvested at 24 h for measurement of apoptotic cells. To detect early apoptosis, Annexin V-FITC/7AAD (BD bioscience) staining was performed according to the manufacturer’s instructions. Samples were read on flow cytometry and analyzed with Flowjo software. To detect caspase-related apoptosis, sulforhodamine FLICA apoptosis detection kit (SR-VAD-FMK, Immunochemistry Technologies) was utilized. Manufacturer’s protocol was followed as provided, following which the samples were read on flow cytometry and activated caspase-positive cells were quantified. For all the analyses, cell debris was excluded.
Western blot
Cells were treated with VCR and/or DAPT as indicated for 24 h. Cells were harvested and incubated in RIPA buffer supplemented with protease and phosphatase inhibitors. Following centrifugation, total protein concentrations in the lysates were determined using Biorad DC Assay kit (Biorad). The same amount of total protein lysates per sample was run per lane on an SDS-PAGE gel and transferred to PVDF membrane. After blocking with 5% milk in TBST buffer, membranes were probed with primary antibodies. Detection was done using HRP-conjugated secondary antibodies and ECL chemiluminesence.
Fluorescent immunostaining
Cells were plated and treated with DMSO for control, DAPT (5, 10, 25 μM), VCR (20 nM), and DAPT (5, 10, 25 μM) plus VCR (20 nM) for 24 h. Cells were collected and fixed with methanol followed by staining for α-tubulin and γ-tubulin. Cells were fixed with 1% PFA and cytospun onto glass slides using cytocentrifuge (Thermo Scientific). Coverslip was glued (Invitrogen) and dried overnight in dark. Imaging was done with 40× or 63× oil immersion lens and analyzed on Slidebook 5.0 and Adobe Photoshop 7.0 software. All images were deconvoluted after acquisition to improve image clarity and sharpness.
Statistical analysis
GraphPad Prism was used for all statistical analysis. Results were plotted on graph as mean ± SEM of 3 independent experiments. Statistical P values were calculated using analysis of variance (ANOVA) with the Bonferroni post-hoc test or Tukey multiple comparison tests, as applicable or using Student t test (*P < 0.05; ** P < 0.01; ***P < 0.001).
Supplementary Material
Acknowledgments
We are grateful to Dr Zweidler-McKay for sharing retroviral constructs. We thank the Louisiana Cancer Research Consortium FACS core for flow cytometry analysis (P20GM103518). This work was supported by NIH grants P20GM103501 to SOY and R01CA121039 to YSC.
Glossary
Abbreviations:
- VCR
Vincristine
- GSI
gamma secretase inhibitor
- GS
gamma secretase
- T-ALL
T cell acute lymphoblastic leukemia
- SAC
spindle assembly checkpoint
- ICN
intracellular domain of Notch
- APP
amyloid precursor protein
- PS
presenilin
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/26951
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/26951
References
- 1.Wolfe MS. Intramembrane-cleaving proteases. J Biol Chem. 2009;284:13969–73. doi: 10.1074/jbc.R800039200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Strooper B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron. 2003;38:9–12. doi: 10.1016/S0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- 3.Haapasalo A, Kovacs DM. The many substrates of presenilin/γ-secretase. J Alzheimers Dis. 2011;25:3–28. doi: 10.3233/JAD-2011-101065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. 1999;284:770–6. doi: 10.1126/science.284.5415.770. [DOI] [PubMed] [Google Scholar]
- 5.Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–89. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- 6.Capaccione KM, Pine SR. The Notch signaling pathway as a mediator of tumor survival. Carcinogenesis. 2013;34:1420–30. doi: 10.1093/carcin/bgt127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fan X, Matsui W, Khaki L, Stearns D, Chun J, Li YM, Eberhart CG. Notch pathway inhibition depletes stem-like cells and blocks engraftment in embryonal brain tumors. Cancer Res. 2006;66:7445–52. doi: 10.1158/0008-5472.CAN-06-0858. [DOI] [PubMed] [Google Scholar]
- 8.Fan X, Khaki L, Zhu TS, Soules ME, Talsma CE, Gul N, Koh C, Zhang J, Li YM, Maciaczyk J, et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells. 2010;28:5–16. doi: 10.1002/stem.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Harrison H, Farnie G, Howell SJ, Rock RE, Stylianou S, Brennan KR, Bundred NJ, Clarke RB. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010;70:709–18. doi: 10.1158/0008-5472.CAN-09-1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hovinga KE, Shimizu F, Wang R, Panagiotakos G, Van Der Heijden M, Moayedpardazi H, Correia AS, Soulet D, Major T, Menon J, et al. Inhibition of notch signaling in glioblastoma targets cancer stem cells via an endothelial cell intermediate. Stem Cells. 2010;28:1019–29. doi: 10.1002/stem.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sikandar SS, Pate KT, Anderson S, Dizon D, Edwards RA, Waterman ML, Lipkin SM. NOTCH signaling is required for formation and self-renewal of tumor-initiating cells and for repression of secretory cell differentiation in colon cancer. Cancer Res. 2010;70:1469–78. doi: 10.1158/0008-5472.CAN-09-2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tatarek J, Cullion K, Ashworth T, Gerstein R, Aster JC, Kelliher MA. Notch1 inhibition targets the leukemia-initiating cells in a Tal1/Lmo2 mouse model of T-ALL. Blood. 2011;118:1579–90. doi: 10.1182/blood-2010-08-300343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu TS, Costello MA, Talsma CE, Flack CG, Crowley JG, Hamm LL, He X, Hervey-Jumper SL, Heth JA, Muraszko KM, et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 2011;71:6061–72. doi: 10.1158/0008-5472.CAN-10-4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gürsel DB, Berry N, Boockvar JA. The contribution of Notch signaling to glioblastoma via activation of cancer stem cell self-renewal: the role of the endothelial network. Neurosurgery. 2012;70:N19–21. doi: 10.1227/01.neu.0000410937.38828.6f. [DOI] [PubMed] [Google Scholar]
- 15.Hassan KA, Wang L, Korkaya H, Chen G, Maillard I, Beer DG, Kalemkerian GP, Wicha MS. Notch pathway activity identifies cells with cancer stem cell-like properties and correlates with worse survival in lung adenocarcinoma. Clin Cancer Res. 2013;19:1972–80. doi: 10.1158/1078-0432.CCR-12-0370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lu J, Ye X, Fan F, Xia L, Bhattacharya R, Bellister S, Tozzi F, Sceusi E, Zhou Y, Tachibana I, et al. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of Jagged-1. Cancer Cell. 2013;23:171–85. doi: 10.1016/j.ccr.2012.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dong Y, Li A, Wang J, Weber JD, Michel LS. Synthetic lethality through combined Notch-epidermal growth factor receptor pathway inhibition in basal-like breast cancer. Cancer Res. 2010;70:5465–74. doi: 10.1158/0008-5472.CAN-10-0173. [DOI] [PubMed] [Google Scholar]
- 18.Gilbert CA, Daou MC, Moser RP, Ross AH. Gamma-secretase inhibitors enhance temozolomide treatment of human gliomas by inhibiting neurosphere repopulation and xenograft recurrence. Cancer Res. 2010;70:6870–9. doi: 10.1158/0008-5472.CAN-10-1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cook N, Frese KK, Bapiro TE, Jacobetz MA, Gopinathan A, Miller JL, Rao SS, Demuth T, Howat WJ, Jodrell DI, et al. Gamma secretase inhibition promotes hypoxic necrosis in mouse pancreatic ductal adenocarcinoma. J Exp Med. 2012;209:437–44. doi: 10.1084/jem.20111923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Domingo-Domenech J, Vidal SJ, Rodriguez-Bravo V, Castillo-Martin M, Quinn SA, Rodriguez-Barrueco R, Bonal DM, Charytonowicz E, Gladoun N, de la Iglesia-Vicente J, et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer Cell. 2012;22:373–88. doi: 10.1016/j.ccr.2012.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McAuliffe SM, Morgan SL, Wyant GA, Tran LT, Muto KW, Chen YS, Chin KT, Partridge JC, Poole BB, Cheng KH, et al. Targeting Notch, a key pathway for ovarian cancer stem cells, sensitizes tumors to platinum therapy. Proc Natl Acad Sci U S A. 2012;109:E2939–48. doi: 10.1073/pnas.1206400109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu YP, Yang CJ, Huang MS, Yeh CT, Wu AT, Lee YC, Lai TC, Lee CH, Hsiao YW, Lu J, et al. Cisplatin selects for multidrug-resistant CD133+ cells in lung adenocarcinoma by activating Notch signaling. Cancer Res. 2013;73:406–16. doi: 10.1158/0008-5472.CAN-12-1733. [DOI] [PubMed] [Google Scholar]
- 23.Schott AF, Landis MD, Dontu G, Griffith KA, Layman RM, Krop I, Paskett LA, Wong H, Dobrolecki LE, Lewis MT, et al. Preclinical and clinical studies of gamma secretase inhibitors with docetaxel on human breast tumors. Clin Cancer Res. 2013;19:1512–24. doi: 10.1158/1078-0432.CCR-11-3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yabuuchi S, Pai SG, Campbell NR, de Wilde RF, De Oliveira E, Korangath P, Streppel MM, Rasheed ZA, Hidalgo M, Maitra A, et al. Notch signaling pathway targeted therapy suppresses tumor progression and metastatic spread in pancreatic cancer. Cancer Lett. 2013;335:41–51. doi: 10.1016/j.canlet.2013.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang CC, Yan Z, Zong Q, Fang DD, Painter C, Zhang Q, Chen E, Lira ME, John-Baptiste A, Christensen JG. Synergistic effect of the γ-secretase inhibitor PF-03084014 and docetaxel in breast cancer models. Stem Cells Transl Med. 2013;2:233–42. doi: 10.5966/sctm.2012-0096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Akiyoshi T, Nakamura M, Yanai K, Nagai S, Wada J, Koga K, Nakashima H, Sato N, Tanaka M, Katano M. Gamma-secretase inhibitors enhance taxane-induced mitotic arrest and apoptosis in colon cancer cells. Gastroenterology. 2008;134:131–44. doi: 10.1053/j.gastro.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 27.Tasaka T, Akiyoshi T, Yamaguchi K, Tanaka M, Onishi H, Katano M. Gamma-secretase complexes regulate the responses of human pancreatic ductal adenocarcinoma cells to taxanes. Anticancer Res. 2010;30:4999–5010. [PubMed] [Google Scholar]
- 28.Janssen A, Medema RH. Mitosis as an anti-cancer target. Oncogene. 2011;30:2799–809. doi: 10.1038/onc.2011.30. [DOI] [PubMed] [Google Scholar]
- 29.Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8:379–93. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- 30.Jordan MA, Thrower D, Wilson L. Effects of vinblastine, podophyllotoxin and nocodazole on mitotic spindles. Implications for the role of microtubule dynamics in mitosis. J Cell Sci. 1992;102:401–16. doi: 10.1242/jcs.102.3.401. [DOI] [PubMed] [Google Scholar]
- 31.Andreassen PR, Margolis RL. Microtubule dependency of p34cdc2 inactivation and mitotic exit in mammalian cells. J Cell Biol. 1994;127:789–802. doi: 10.1083/jcb.127.3.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skoufias DA, Andreassen PR, Lacroix FB, Wilson L, Margolis RL. Mammalian mad2 and bub1/bubR1 recognize distinct spindle-attachment and kinetochore-tension checkpoints. Proc Natl Acad Sci U S A. 2001;98:4492–7. doi: 10.1073/pnas.081076898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jordan MA, Thrower D, Wilson L. Mechanism of inhibition of cell proliferation by Vinca alkaloids. Cancer Res. 1991;51:2212–22. [PubMed] [Google Scholar]
- 34.Shin HJ, Baek KH, Jeon AH, Park MT, Lee SJ, Kang CM, Lee HS, Yoo SH, Chung DH, Sung YC, et al. Dual roles of human BubR1, a mitotic checkpoint kinase, in the monitoring of chromosomal instability. Cancer Cell. 2003;4:483–97. doi: 10.1016/S1535-6108(03)00302-7. [DOI] [PubMed] [Google Scholar]
- 35.Andreassen PR, Skoufias DA, Margolis RL. Analysis of the spindle-assembly checkpoint in HeLa cells. Methods Mol Biol. 2004;281:213–25. doi: 10.1385/1-59259-811-0:213. [DOI] [PubMed] [Google Scholar]
- 36.Lowe SW, Lin AW. Apoptosis in cancer. Carcinogenesis. 2000;21:485–95. doi: 10.1093/carcin/21.3.485. [DOI] [PubMed] [Google Scholar]
- 37.Wu J, Iwata F, Grass JA, Osborne CS, Elnitski L, Fraser P, Ohneda O, Yamamoto M, Bresnick EH. Molecular determinants of NOTCH4 transcription in vascular endothelium. Mol Cell Biol. 2005;25:1458–74. doi: 10.1128/MCB.25.4.1458-1474.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yun J, Pannuti A, Espinoza I, Zhu H, Hicks C, Zhu X, Caskey M, Rizzo P, D’Souza G, Backus K, et al. Crosstalk between PKCα and Notch-4 in endocrine-resistant breast cancer cells. Oncogenesis. 2013;2:e60. doi: 10.1038/oncsis.2013.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kannan S, Sutphin RM, Hall MG, Golfman LS, Fang W, Nolo RM, Akers LJ, Hammitt RA, McMurray JS, Kornblau SM, et al. Notch activation inhibits AML growth and survival: a potential therapeutic approach. J Exp Med. 2013;210:321–37. doi: 10.1084/jem.20121527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saunders W. Centrosomal amplification and spindle multipolarity in cancer cells. Semin Cancer Biol. 2005;15:25–32. doi: 10.1016/j.semcancer.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 41.Doxsey S, Zimmerman W, Mikule K. Centrosome control of the cell cycle. Trends Cell Biol. 2005;15:303–11. doi: 10.1016/j.tcb.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 42.Nigg EA, Raff JW. Centrioles, centrosomes, and cilia in health and disease. Cell. 2009;139:663–78. doi: 10.1016/j.cell.2009.10.036. [DOI] [PubMed] [Google Scholar]
- 43.Zyss D, Gergely F. Centrosome function in cancer: guilty or innocent? Trends Cell Biol. 2009;19:334–46. doi: 10.1016/j.tcb.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 44.Nigg EA. Origins and consequences of centrosome aberrations in human cancers. Int J Cancer. 2006;119:2717–23. doi: 10.1002/ijc.22245. [DOI] [PubMed] [Google Scholar]
- 45.Fukasawa K. Oncogenes and tumour suppressors take on centrosomes. Nat Rev Cancer. 2007;7:911–24. doi: 10.1038/nrc2249. [DOI] [PubMed] [Google Scholar]
- 46.Krämer A, Maier B, Bartek J. Centrosome clustering and chromosomal (in)stability: a matter of life and death. Mol Oncol. 2011;5:324–35. doi: 10.1016/j.molonc.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leber B, Maier B, Fuchs F, Chi J, Riffel P, Anderhub S, Wagner L, Ho AD, Salisbury JL, Boutros M, et al. Proteins required for centrosome clustering in cancer cells. Sci Transl Med. 2010;2:33ra38. doi: 10.1126/scitranslmed.3000915. [DOI] [PubMed] [Google Scholar]
- 48.Lawo S, Bashkurov M, Mullin M, Ferreria MG, Kittler R, Habermann B, Tagliaferro A, Poser I, Hutchins JR, Hegemann B, et al. HAUS, the 8-subunit human Augmin complex, regulates centrosome and spindle integrity. Curr Biol. 2009;19:816–26. doi: 10.1016/j.cub.2009.04.033. [DOI] [PubMed] [Google Scholar]
- 49.Kwon M, Godinho SA, Chandhok NS, Ganem NJ, Azioune A, Thery M, Pellman D. Mechanisms to suppress multipolar divisions in cancer cells with extra centrosomes. Genes Dev. 2008;22:2189–203. doi: 10.1101/gad.1700908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yih LH, Tseng YY, Wu YC, Lee TC. Induction of centrosome amplification during arsenite-induced mitotic arrest in CGL-2 cells. Cancer Res. 2006;66:2098–106. doi: 10.1158/0008-5472.CAN-05-2308. [DOI] [PubMed] [Google Scholar]
- 51.De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–90. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- 52.Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 1999;398:513–7. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- 53.Esler WP, Kimberly WT, Ostaszewski BL, Diehl TS, Moore CL, Tsai JY, Rahmati T, Xia W, Selkoe DJ, Wolfe MS. Transition-state analogue inhibitors of gamma-secretase bind directly to presenilin-1. Nat Cell Biol. 2000;2:428–34. doi: 10.1038/35017062. [DOI] [PubMed] [Google Scholar]
- 54.Li YM, Xu M, Lai MT, Huang Q, Castro JL, DiMuzio-Mower J, Harrison T, Lellis C, Nadin A, Neduvelil JG, et al. Photoactivated gamma-secretase inhibitors directed to the active site covalently label presenilin 1. Nature. 2000;405:689–94. doi: 10.1038/35015085. [DOI] [PubMed] [Google Scholar]
- 55.Li J, Xu M, Zhou H, Ma J, Potter H. Alzheimer presenilins in the nuclear membrane, interphase kinetochores, and centrosomes suggest a role in chromosome segregation. Cell. 1997;90:917–27. doi: 10.1016/S0092-8674(00)80356-6. [DOI] [PubMed] [Google Scholar]
- 56.Nizzari M, Venezia V, Repetto E, Caorsi V, Magrassi R, Gagliani MC, Carlo P, Florio T, Schettini G, Tacchetti C, et al. Amyloid precursor protein and Presenilin1 interact with the adaptor GRB2 and modulate ERK 1,2 signaling. J Biol Chem. 2007;282:13833–44. doi: 10.1074/jbc.M610146200. [DOI] [PubMed] [Google Scholar]
- 57.Judge M, Hornbeck L, Potter H, Padmanabhan J. Mitosis-specific phosphorylation of amyloid precursor protein at threonine 668 leads to its altered processing and association with centrosomes. Mol Neurodegener. 2011;6:80. doi: 10.1186/1750-1326-6-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boeras DI, Granic A, Padmanabhan J, Crespo NC, Rojiani AM, Potter H. Alzheimer’s presenilin 1 causes chromosome missegregation and aneuploidy. Neurobiol Aging. 2008;29:319–28. doi: 10.1016/j.neurobiolaging.2006.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.