

Abstract
During vessel sprouting, a migratory endothelial tip cell guides the sprout, while proliferating stalk cells elongate the branch. Tip and stalk cell phenotypes are not genetically predetermined fates, but are dynamically interchangeable to ensure that the fittest endothelial cell (EC) leads the vessel sprout. ECs increase glycolysis when forming new blood vessels. Genetic deficiency of the glycolytic activator PFKFB3 in ECs reduces vascular sprouting by impairing migration of tip cells and proliferation of stalk cells. PFKFB3-driven glycolysis promotes the tip cell phenotype during vessel sprouting, since PFKFB3 overexpression overrules the pro-stalk activity of Notch signaling. Furthermore, PFKFB3-deficient ECs cannot compete with wild-type neighbors to form new blood vessels in chimeric mosaic mice. In addition, pharmacological PFKFB3 blockade reduces pathological angiogenesis with modest systemic effects, likely because it decreases glycolysis only partially and transiently.
Keywords: endothelial cell, angiogenesis, glycolysis, metabolism, vessel sprouting
Results and Discussion
The vasculature is of crucial importance for delivering oxygen and nutrients to tissues. When this supply is limiting, new blood vessels are formed. This process (termed angiogenesis) is not only critical during embryonic development, but also promotes pathological conditions such as cancer and inflammatory disorders.1 During vessel sprouting, pro-angiogenic factors such as VEGF promote the induction of a migratory endothelial tip cell, while proliferating endothelial stalk cells elongate the sprout.1 Most anti-angiogenic therapies currently approved for clinical use or in (pre)-clinical development block molecules like VEGF. However, anti-cancer therapies targeting VEGF signaling suffer from resistance and toxicity, necessitating the development of novel anti-angiogenic strategies.2,3 Research over the past decades has identified several genetic signals that regulate angiogenesis, but the role of endothelial cell (EC) metabolism in angiogenesis has not been intensely studied.
ECs remain quiescent for years, but can rapidly start to proliferate and migrate when stimulated by angiogenic signals. These processes require energy and molecular building blocks for biomass duplication. Since angiogenic ECs have different metabolic needs than quiescent ECs, it has been postulated that a metabolic switch accompanies the angiogenic switch.4,5 However, little is known about EC metabolism in relation to vessel branching and on which metabolic pathways ECs rely to form new vessels.
ECs line blood vessels and, thus, have immediate access to oxygen in the blood. Yet, they do not take advantage of this availability of oxygen to rely on oxidative metabolism, but instead are highly glycolytic and generate up to 85% of their ATP from glycolysis.4,6 To evaluate the importance of glycolysis in vessel sprouting, we used pharmacological blockade or genetic silencing of phosphofructokinase-fructose-2,6-bisphosphatase-3 (PFKFB3), an enzyme producing fructose-2,6-biphosphate, which is a potent allosteric activator of the rate-limiting glycolytic enzyme phosphofructokinase-1. PFKFB3 inhibition in ECs reduced glycolysis, but by no more than 40%, and did not cause general energy hypo-metabolism.4 Nonetheless, even this partial inhibition of glycolysis sufficed to reduce EC proliferation and migration, and vessel sprouting from EC spheroids in vitro.4,5 Furthermore, zebrafish treated with the pharmacological PFKFB3 blocker 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (3PO) and mice lacking PFKFB3 in ECs displayed vascular sprouting and branching defects.4,5 3PO was not cytotoxic for ECs, and its anti-angiogenic effects were reversible.4,5
Consistent with reports that PFKFB3-driven glycolysis promotes cellular proliferation,7,8 endothelial loss of PFKFB3 reduced EC proliferation and rendered stalk cells hypo-proliferative.4 However, blockade or loss of PFKFB3 in ECs also impaired various tip cell functions, such as directional migration and filopodia and lamellipodia formation, processes that require actin cytoskeletal remodeling.4 Blockade of PFKFB3-driven glycolysis also affected additional processes of EC dynamics, involving cytoskeleton remodeling. Indeed, for a cell to establish contact with another cell, it must alter its shape. Since a sphere has a low surface tension, a cell preferably adopts a spheroid shape, and any deviation from this shape, such as when a cell is spreading or establishes cell–cell contacts, requires “adhesion energy”.9 Time-lapse imaging of sparsely plated GFP+ ECs showed that control cells established long-lasting cell–cell contacts with each other (Fig. 1A). In contrast, PFKFB3-silenced cells (using published methods4) only made transient “kiss and run” contacts, and after brief touching, they disconnected without maintaining stable contact (Fig. 1B). This was due to the fact that PFKFB3-silenced ECs formed only small, irregular, and transient lamellipodial projections (Fig. 1B), as quantified earlier.4
Figure 1:
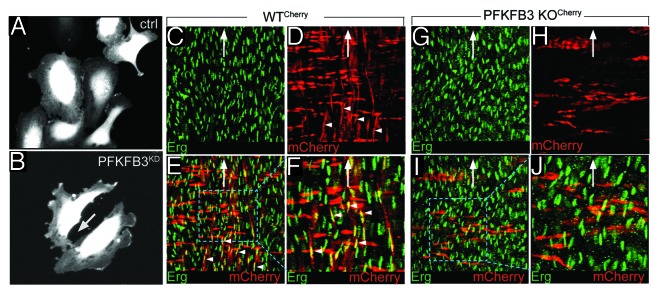
PFKFB3 blokade impairs cell–cell contact and vessel formation. (A and B) Representative snapshot images of time-lapse imaging, showing that control cells make long lasting cell–cell contact (A), while PFKFB3-silenced cells make only transient contact (B). (C–J) Representative images of the postnatal aorta (open book preparation) of chimeric PFKFB3WT/Cherry (C–F) and PFKFB3KO/Cherry pups (G–J), stained for the endothelial nuclear marker Erg (green, C, E–G, I, and J) or expressing mCherry (red, labeling transgenic cells in D–F and H–J). (F and J) show a high magnification of the insets in (E and I). (C–F) A fraction of the ECs are PFKFB3WT/Cherry (arrowheads, red cells oriented in the vertical axis); likewise, a fraction of the smooth muscle cells is also PFKFB3WT/Cherry (red cells oriented in the horizontal axis). (G–J) All ECs are wild-type (host-derived), while PFKFB3KO/Cherry ECs are not detected (evidenced by the absence of vertically oriented red cells), even though other PFKFB3KO/Cherry cell types are present (smooth muscle cells in red, oriented in the horizontal axis). These findings indicate that PFKFB3KO/Cherry ECs are out-competed by wild-type ECs originating from the wild-type host blastocyst. The white arrow indicates the direction of blood flow. Open book preparation: the ECs are facing upwards (the adventitia is facing downwards).
Tip and stalk cell phenotypes are not fixed predetermined cell fates, but instead are interchangeable phenotypes. Hence, a stalk cell can plastically differentiate to a tip cell over a period of 6 h.10 ECs must thus be competitive to reach the tip position. We recently reported that PFKFB3 deficiency impairs tip cell competitiveness during embryonic vascular development in the hindbrain.4 The process of tip and stalk cell differentiation is under tight control of genetic signaling by VEGF (a pro-tip signal) and Notch (a pro-stalk signal), among other angio-modulatory signals.1 Use of mosaic EC spheroids and transgenic zebrafish showed that overexpression of PFKFB3 provided ECs a competitive advantage for the tip position, not only in baseline conditions, but even when Notch signaling was increased in transgenic ECs.4 The finding that PFKFB3 overexpression was capable of overruling the pro-stalk activity of Notch and promoted stalk cells to become tip cells is remarkable, as no other signal has been documented to overcome the strong pro-stalk activity of Notch. These data thus show that glycolysis provides a competitive advantage for ECs to reach the tip and establish that metabolism (glycolysis) co-determines vascular branching, in parallel to genetic signals like VEGF and Notch.4 Importantly, the role of PFKFB3 in vascular branching did not rely on a change in the genetic angiogenic signature, as the expression levels of pro-tip signals (such as VEGF) and pro-stalk signals (such as Notch) were unaffected when PFKFB3 levels were altered.4 Conversely, VEGF or Notch signaling regulated PFKFB3 and glycolysis levels, indicating that glycolysis is downstream and required for angiogenic signaling.4
The tip cell responses that were affected by PFKFB3 blockade relied on remodeling of the actin cytoskeleton,4 a process that requires rapid production of large amounts of ATP by glycolysis in ECs. Indeed, bulky mitochondria were excluded from the thin lamellipodia and narrow filopodia, and altering mitochondrial respiration did not affect vessel sprouting.4 In contrast, PFKFB3 and other glycolytic enzymes were present in the perinuclear cytosol in quiescent ECs, but were relocated to lamellipodia in migrating ECs.4 ATP biosensor imaging revealed ATP hotspots in motile lamellipodia, which were reduced by silencing of PFKFB3 but not by mitochondrial poisons.4 At the molecular level, PFKFB3 interacted with F-actin at increased levels in the leading membrane ruffles of lamellipodia of motile ECs.4 Since binding of various glycolytic enzymes to F-actin promotes the formation of their active tetramerized state and favors the formation of an efficient assembly line, whereby the product of one glycolytic enzyme becomes the substrate of the next glycolytic enzyme,11 compartimentalization of glycolytic enzymes with F-actin provides a mechanistic explanation of how glycolysis fuels EC tip functions.4
The impaired tip and stalk cell activities of ECs lacking PFKFB34 prompted us to investigate whether PFKFB3-deficient (PFKFB3KO) ECs would be able to compete with wild-type (WT) ECs for the formation and maintenance of blood vessels after birth. We therefore used PFKFB3-deficient (PFKFB3KO) ES cells, stably expressing a CherryRed marker, to visualize transgenic cells (PFKFB3KO/Cherry).4 Wild-type (PFKFB3WT) ES cells transfected with CherryRed were used as a control (PFKFB3WT/Cherry).4 PFKFB3KO/Cherry or PFKFB3WT/Cherry ES cells were then injected into WT blastocysts to generate highly (>90–95%) mosaic chimeras. We then assessed the contribution of PFKFB3KO/Cherry or PFKFB3WT/Cherry ECs to the formation and maintenance of the dorsal aorta at postnatal day 5 by staining for the EC-specific transcription factor Erg. Surprisingly, no PFKFB3KO/Cherry ECs were detectable in the aorta; even though PFKFB3WT/Cherry ECs were abundantly present (Fig. 1C–J). This was remarkable, since PFKFB3KO/Cherry ES cells differentiated to smooth muscle cells, which were readily identified by their orientation, perpendicular to the alignment of ECs in the direction of blood flow (Fig. 1C–J). These findings indicate that ECs, lacking PFKFB3, were outcompeted during embryonic development by their WT neighbors originating from the host blastocyst cells, likely because of their impaired tip and stalk cell functions, overall illustrating the importance of PFKFB3-driven glycolysis for ECs for vessel formation.
Overall, these findings show that not only genetic signals, but also metabolism regulates vessel branching. This insight raised the question whether PFKFB3-driven glycolysis might be a target for inhibiting angiogenesis in pathological conditions. Indeed, pharmacological PFKFB3 blockade, using the compound 3PO, reduces vessel sprouting in vitro and pathological angiogenesis in models of choroidal neovascularization, retinopathy of prematurity and inflammatory skin and bowel diseases.5 Remarkably, however, treatment of adult mice (in which most vessels are quiescent) with 3PO for 15 d did not affect the healthy established vasculature in the retina (Fig. 2A and D); neither did it change the vascular area in other organs such as the kidney (Fig. 2B and E) and the heart (Fig. 2C and F). 3PO also did not affect the perfusion of retinal vessels (Fig. 2G–L) or cause vessel wall leakage of various organs in adult healthy mice (Fig. 2M and N). These findings thus illustrate that proliferating and migrating ECs during pathological angiogenesis are more susceptible to PFKFB3 blockade than quiescent ECs in established vessels, in accordance with reports that sprouting ECs have higher levels of glycolysis than quiescent ECs, and that 3PO inhibits the hypermetabolism associated with sprouting ECs.5

Figure 2. PFKFB3 blockade does not target the quiescent vasculature. (A–F) Vascular densities in the retina (A and D, isolectin B4), kidney (B and E, CD31) and heart (C and F, endoglin) were not affected by 15 d treatment of adult healthy mice with 3PO. (A–C) control, (D–F) 3PO. Scale bars: 200 µm (A and D), 50 µm (B and E), and 100 µm (C and F). (G–L) Confocal images of dextran-FITC injected (G and J) and isolectin-B4 (H and K) stained retina of healthy mice that were treated with 3PO (J–L) or vehicle (G–I) for 15 d showing that 3PO does not induce vessel leakiness in the quiescent vessels of the retina. (I and L) show the merged images. (M) Quantification of the Evans blue dye leakage in heart, lung, liver, and kidney, showing no effect of 3PO treatment (50 mg/kg/day for 4 d) on vessel leakiness (mean ± SEM, n = 5, P = NS). (N) Quantification of FITC-dextran+ area in the kidney after injection of FITC-dextran (MW 40 000 Da) followed by saline perfusion 10 min later, showing no effect of 3PO treatment on vessel leakiness (mean ± SEM, n = 5, P = NS).
We also explored whether 3PO treatment induced systemic effects, given that glycolysis is part of the central metabolism in multiple other cell types. We therefore treated adult healthy mice for 15 d with the same dose of 3PO that decreased angiogenesis. The following effects were noticed. First, 3PO caused a <10% body weight loss and reduced red blood cell parameters by <10% (Table 1). Intestinal crypt stem cell proliferation was reduced by 18%, but crypts and villi were normal in morphology and size (Table 1), suggesting that 3PO reduced but did not deplete the intestinal stem cell pool. Other parameters were not affected. For instance, 3PO did not induce striking differences in biochemical blood parameters (Table 1), nor did it induce histological abnormalities in liver, muscle, colon, and kidney (Fig. 3A–H) or of the intestines, brain and heart (not shown). No overt signs of liver inflammation were detected (Fig. 3I), and no signs of eye toxicity were observed as shown by staining for the glial cell marker GFAP (Fig. 3J) and quantification of the thickness of the retinal nuclear layers (Fig. 3K–M). Plasma creatine kinase (CK) levels were also not elevated, suggesting normal cardiomyocyte viability (Fig. 3N). We also tested if 3PO affected the recovery from myelosuppression by doxorubicin, cisplatin, capecitabine, and sorafenib, administered at doses inhibiting tumor growth. Treatment with 3PO slightly aggravated myelosuppression by cisplatin and sorafenib (at a single time point after 14 or 7 d, respectively), but white blood cell counts fully recovered in all 3PO-treated mice (Fig. 3O).
Table 1. Systemic effects of PFKFB3 blockade.
| DMSO control | 3PO | |
|---|---|---|
| Vascular parameters | ||
| •Heart (endoglin+ area, % of total) •Kidney medulla (CD31+ area, % of total) •Kidney cortex (CD31+ area, % of total) |
17.4 ± 0.7 15.8 ± 0.3 55.4 ± 2.8 |
15.8 ± 0.9 14.4 ± 0.6 54.2 ± 1.3 |
| Liver parameters | ||
| •ALT plasma level (U/L) •AlcP plasma levels (U/L) •γ-GT plasma levels (U/L) •Bilirubine plasma levels (mg/dL) |
12 ± 0.5 33 ± 3 <3 <0.18 |
16 ± 1.4 33 ± 11 <3 <0.18 |
| Renal parameters | ||
| •Ureum (mg/dL) •Creatinine (mg/dL) |
53 ± 6.5 <0.06 |
55 ± 6.2 <0.06 |
| Small intestine parameters | ||
| •Crypt cell proliferation (BrdU+ cells, % of total) •Villus height (μm) •Crypt depth (μm) |
28 ± 0.8 379 ± 8 70 ± 2 |
23 ± 0.8* 378 ± 17 69 ± 2.3 |
| Hematological parameters | ||
| •WBCs (x103/μL) •Platelets (x103/μL) •Hematocrit (%) •RBCs (x106/μL) •Hemoglobin (g/dL) |
6.1 ± 1.1 564 ± 63 42 ± 0.3 8.9 ± 0.1 15.8 ± 0.2 |
5.4 ± 0.9 690 ± 64 39 ± 0.9 8.3 ± 0.2* 14.4 ± 0.3* |
Values are mean ± SEM, n = 5. ALT, alanine aminotransferase; AlcP, alkaline phosphatase; γ-GT, γ-glutamyl transpeptidase; WBCs, white blood cell counts; RBCs, red blood cells; *P < 0.05 vs DMSO.

Figure 3. PFKFB3 blockade does not affect organ morphology and heart function. (A–H) No histological abnormalities were detected by H&E staining of the colon (A and E), liver (B and F), muscle (C and G), and kidney (D and H) of adult healthy mice treated for 15 d with 3PO. (A–D) control, (E–H) 3PO. (I) Quantification of the CD45+ area in the liver, showing comparable numbers of inflammatory cells upon 3PO treatment (50 mg/kg/day for 15 d) (mean ± SEM, n = 5, P = NS). (J) Quantification of the glial fibrillary acidic protein (GFAP) positive area in the retina, showing similar number of glial cells and thus absence of gliosis upon 3PO treatment (50 mg/kg/day for 15 d) (mean ± SEM, n = 5, P = NS). (K–M) Quantification of the thickness of the outer nuclear layer (ONL), outer plexiform layer (OPL), and inner nuclear layer (INL), showing no thinning of these retinal layers (a sign of neuroxocity) by 3PO (mean ± SEM, n = 5, P = NS). (N) Plasma creatine kinase levels showing similar levels after 3PO treatment (50 mg/kg/day for 15 d), suggesting normal cardiomyocyte viability (mean ± SEM, n = 10, P = NS). (O) White blood cell counts (WBCs) after treatment with the chemotherapeutics, cisplatin, capecitabine, doxorubicin, and the anti-angiogenic agent sorafenib at doses inhibiting tumor growth and angiogenesis, showing that 3PO aggravated the myelosuppressive toxicity only for cisplatin and sorafenib at a single time point, but WBCs in all 3PO-treated mice fully recovered (mean ± SEM; n = 5; *P < 0.05 vs. ctrl; &P < 0.05 vs. day 0).
We explored the underlying reasons of why 3PO did not induce more severe adverse effects than perhaps initially anticipated. First, 3PO had a short half-life (30 min)12 and reduced glycolysis only partially (by 35–40%) and transiently (during <6 h) in vivo, though 3PO was again effective in reducing glycolysis upon re-administration.5 Nonetheless, this transient partial decrease in glycolysis was sufficient to block pathological angiogenesis,5 thus providing a new paradigm that anti-glycolytic therapy does not need to block glycolysis completely and permanently in ECs to impair angiogenesis. Second, given that cells need glycolysis to proliferate, we also hypothesized that quiescent cells or cells not relying critically on glycolysis would be less sensitive to PFKFB3-blockade. Indeed, 3PO did not affect glycolysis or survival of neurons and resident macrophages, known to have negligible PFKFB3 levels (Table 2).13,14 In other cells, such as hepatocytes and adipocytes, 3PO reduced glycolysis, but their survival was not compromised, indicating that these cells did not critically rely on PFKFB3-driven glycolysis as their predominant metabolic pathway and likely were able to compensatorily switch to another type of metabolism (Table 2). By contrast, rapidly proliferating cells, such as ECs or tumor cells, have high levels of glycolysis, and their glycolytic flux and proliferation were reduced by 3PO (Table 2). Taken together, PFKFB3-blockade primarily targets rapidly proliferating/migrating cell types that critically rely on glycolysis.
Table 2. Effects of 3PO on glycolysis, survival and proliferation of cells.
| Cell type | Glycolysis | Effects of 3PO on | ||
|---|---|---|---|---|
| Glycolysis | Survival (%) | |||
| Quiescent | neurons | 651 ± 50 | 658 ± 65 | 109 ± 11 |
| macrophages | 367 ± 34 | 428 ± 16 | 121 ± 7 | |
| adipocytes | 199 ± 13 | 120 ± 7* | 107 ± 2 | |
| hepatocytes | 219 ± 24 | 146 ± 11* | 110 ± 15 | |
| Glycolysis | Proliferation (%) | |||
| Proliferating | HUVECs | 1800 ± 37 | 1280 ± 57* | 79 ± 2* |
| hemangioma (MS1) | 3300 ± 60 | 2100 ± 43* | 40 ± 3* | |
| angiosarcoma (SVR) | 6350 ± 288 | 3780 ± 23* | 64 ± 1* | |
| glioblastoma (CT2A) | 4780 ± 110 | 3530 ± 67* | 66 ± 3* | |
Data show the effect of 3PO treatment (20–40 µM) on glycolysis (nmol glucose/hr/mg protein) and survival (LDH release in medium; % of control) or proliferation (3H-thymidine incorporation; % of control) in different quiescent and proliferating cells. Values are mean ± SEM, n = 5 for adipocytes and hepatocytes, n = 4 for neurons and n = 3 for all other cell types. *P < 0.05 vs. glycolysis in control conditions.
Overall, the partial, transient blockade of glycolysis sufficed to reduce sprouting, yet was less toxic than perhaps intuitively expected based on the widely held belief that many cell types require glycolysis. However, PFKFB3 inhibition does not indiscriminately affect any type of healthy cells, but primarily targets those cells that rely on glycolysis as a critical metabolic pathway, such as proliferating and migrating cells. Noteworthy in this respect, the vast majority (>90%) of cells in a healthy adult are quiescent. The finding that ECs rely on glycolysis more than other cell types explains why ECs are more vulnerable to PFKFB3 blockade. In addition, it is important to stress that 3PO treatment decreased glycolysis only partially and transiently. A more sustained and more complete inhibition of glucose metabolism by 80%, as obtained when treating ECs with the non-metabolizable glucose analog 2-deoxy-D-glucose, is indeed more toxic, inducing cellular distress and killing ECs, ultimately, in particular in sprouting conditions.5
In this respect, it is also noteworthy that only <2.5% of the total ATP in the body is produced by glycolysis,15 thus implying that most other cells rely on non-glycolytic mechanisms for energy homeostasis and that blocking glycolysis (certainly when only transiently and partially) should not necessarily be expected to cause widespread cellular demise. In contrast, proliferating ECs, tumor cells16,17 and activated T cells18 critically rely on high glycolysis levels. Hence, 3PO inhibited their proliferation, thereby reducing pathological neovascularization, as well as tumor growth and inflammatory diseases.18,19 Our findings are also consistent with reports that human hereditary mutations in glycolytic genes are well tolerated as long as they do not deplete cellular ATP levels.20 Careful assessment of potential toxicity and identification of possible contra-indications will nevertheless be critical to design anti-angiogenic therapy based on partial, transient glycolysis inhibition.
Experimental Methods
Cell culture
Primary cells
Human umbilical vein endothelial cells (HUVECs) were freshly isolated from different donors and cultured as described.4 Murine hepatocytes were isolated using a 2-step collagenase perfusion technique and fixed after short-term culture for 6 h, as described.21 Murine macrophages were collected from the peritoneal cavity and cultured in RPMI (Invitrogen, Life Technologies) containing 10% FBS. Murine adipocytes were isolated from epididymal/gonadal fat pads using 0.1% collagenase II, separated from the stromal vascular fraction via centrifugation, and were cultured in DMEM/F12 supplemented with 10% FBS.22 Neuronal cultures were prepared from the cortices of embryonic day 14 (E14) mice and maintained in serum-free NeuroBasal medium (Gibco) containing 25 mM glucose.23 After 4 d in culture, 8 µM cytosine arabinoside was added to prevent non-neuronal proliferation.
Tumor cell lines
CT2A murine glioblastoma cells were cultured in DMEM containing 10% FBS and 2 mM glutamine (Invitrogen, Life Technologies) supplemented with conditioned medium obtained from CT2A cells. Murine endothelial cells, infected with an ecotropic retrovirus encoding SV40 large T 58–3 allele (MS1 hemangioma) and subsequently infected with a second retrovirus encoding activated H-ras (SVR angiosarcoma) were obtained from Dr Arbiser and cultured in DMEM with glutamine and 10% FBS.24 All cells were routinely maintained in 5% CO2 and 95% air at 37 °C.
Knockdown strategies
Transduction of lentiviral vectors encoding a PFKFB3 or a scrambled control shRNA were performed as described.4
In vitro assays
Live cell imaging
GFP+ control and PFKFB3-silenced ECs were plated on a glass bottom 24-well plate and images were acquired continuously overnight at 37 °C using a Zeiss LSM 510 Meta NLO confocal microscope (LD Plan-Neofluor 20×/0.4 Korr M27 objective).
Proliferation and survival
Proliferation was quantified using [3H]-thymidine incorporation4 and survival was measured by determining the LDH release in the medium using the Cytotoxicity Detection Kit (Roche applied sciences).
Glycolysis
Flux through glycolysis was determined as described.4
In vivo assays
Housing of mice and experimental procedures were approved by the Animal Care and Use Committee of the University of Leuven.
Mouse mosaics
Chimeric pups were generated by injecting 15–20 wild-type or PFKFB3KO ES cells, stably transfected with mCherry, into CD-1 or FVB host blastocysts. Injected blastocysts were transplanted into 2.5 d pseudopregnant CD-1 foster mothers and allowed to develop to term. Aortas were dissected at postnatal day 5, fixed in 4% PFA, mounted as an open book preparation (by making a longitudinal section to spread open the inner surface of the aorta), and stained for anti-Erg (Santa Cruz Biotechnology) and isolectin GS-IB4-Alexa 488 (Molecular Probes).4 Images were captured with a Zeiss laser scanning LSM 780 microscope (Carl Zeiss).
Analysis of systemic effects
General
C57BL/6 mice (Charles River) were used in all experiments. Adult (8-wk-old) mice were treated intraperitoneally with 3PO for 15 d (50 mg/kg/day; dissolved in DMSO). Organs (heart, liver, intestines, brains, lungs, kidneys, muscle, eyes) were dissected, embedded in paraffin, and stained with H&E and for CD105 (endoglin), CD31, CD45, or GFAP as described.25,26 Crypt stem cell proliferation was assessed after BrdU injection and expressed as BrdU+ crypt cells relative to the total number of crypt cells. General morphology was examined on H&E stained sections and quantification of the vascular area, retinal layer thickness,26 GFAP+, CD31+, CD105+, and CD45+ area was done using NIH Image J and AxioVision (Carl Zeiss) software packages. Daily i.p. injections of 3PO resulted in the formation of precipitates, which caused local peritoneal inflammation beyond 2 wk.
Hematological profiling
Hematological profiling was performed using Cell Dyn 3700 equipment (Abbott Diagnostics). Plasma measurements of renal, liver and heart parameters were performed in the clinical laboratory of the university hospital of Leuven.
Cytotoxicity agent
Mice were treated with single doses of cisplatin or doxorubicin via i.p. injection (7 mg/kg). Capecitabine (250 mg/kg) and sorafenib (30 mg/kg) were provided daily via gavage. 3PO treatment was started the first day after initiation of cytotoxic therapy. Blood samples were obtained for hematological profiling at day 0, 3, 7, 10, 14 and 17 d.
Vessel leakiness
Mice were injected daily with 3PO (50 mg/kg/day) for 4 d. For analysis of vessel leakiness, Evans blue (16 h) or FITC dextran (10 min; MW 40 000) was injected into the tail vein after which mice were euthanized and organs dissected. Evans blue leakiness in organs was quantified spectrophotometrically after formamide based extraction of the dye. FITC dextran leakiness outside the vessels was quantified on cryosections counterstained for the blood vessel marker CD105.
Vessel perfusion
Vessel perfusion in the retinal vasculature was determined after retrobulbar FITC dextran injection (3PO treatment: 50 mg/kg/day for 15 d). All analyses were done with NIH ImageJ and AxioVision (Carl Zeiss, Munich, Germany) software packages.
Disclosure of Potential Conflicts of Interest
PC declares being named as inventor on patent applications claiming subject matter related to the results described in this paper.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/27519
References
- 1.Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873–87. doi: 10.1016/j.cell.2011.08.039. [DOI] [PubMed] [Google Scholar]
- 2.Ebos JM, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol. 2011;8:210–21. doi: 10.1038/nrclinonc.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singh M, Ferrara N. Modeling and predicting clinical efficacy for drugs targeting the tumor milieu. Nat Biotechnol. 2012;30:648–57. doi: 10.1038/nbt.2286. [DOI] [PubMed] [Google Scholar]
- 4.De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquière B, Cauwenberghs S, Eelen G, et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013;154:651–63. doi: 10.1016/j.cell.2013.06.037. [DOI] [PubMed] [Google Scholar]
- 5.Schoors S, De Bock K, Cantelmo AR, Georgiadou M, Ghesquière B, Cauwenberghs S, Kuchnio A, Wong BW, Quaegebeur A, Goveia J, et al. Partial and transient reduction of glycolysis by PFKFB3-blockade reduces pathological angiogenesis. Cell Metab. 2013 doi: 10.1016/j.cmet.2013.11.008. accepted. [DOI] [PubMed] [Google Scholar]
- 6.De Bock K, Georgiadou M, Carmeliet P. Role of endothelial cell metabolism in vessel sprouting. Cell Metab. 2013;18:634–47. doi: 10.1016/j.cmet.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 7.Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–64. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 8.Almeida A, Bolaños JP, Moncada S. E3 ubiquitin ligase APC/C-Cdh1 accounts for the Warburg effect by linking glycolysis to cell proliferation. Proc Natl Acad Sci U S A. 2010;107:738–41. doi: 10.1073/pnas.0913668107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maître JL, Heisenberg CP. The role of adhesion energy in controlling cell-cell contacts. Curr Opin Cell Biol. 2011;23:508–14. doi: 10.1016/j.ceb.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jakobsson L, Franco CA, Bentley K, Collins RT, Ponsioen B, Aspalter IM, Rosewell I, Busse M, Thurston G, Medvinsky A, et al. Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat Cell Biol. 2010;12:943–53. doi: 10.1038/ncb2103. [DOI] [PubMed] [Google Scholar]
- 11.Sola-Penna M, Da Silva D, Coelho WS, Marinho-Carvalho MM, Zancan P. Regulation of mammalian muscle type 6-phosphofructo-1-kinase and its implication for the control of the metabolism. IUBMB Life. 2010;62:791–6. doi: 10.1002/iub.393. [DOI] [PubMed] [Google Scholar]
- 12.Clem BF, O’Neal J, Tapolsky G, Clem AL, Imbert-Fernandez Y, Kerr DA, 2nd, Klarer AC, Redman R, Miller DM, Trent JO, et al. Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Mol Cancer Ther. 2013;12:1461–70. doi: 10.1158/1535-7163.MCT-13-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodríguez-Prados JC, Través PG, Cuenca J, Rico D, Aragonés J, Martín-Sanz P, Cascante M, Boscá L. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol. 2010;185:605–14. doi: 10.4049/jimmunol.0901698. [DOI] [PubMed] [Google Scholar]
- 14.Herrero-Mendez A, Almeida A, Fernández E, Maestre C, Moncada S, Bolaños JP. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat Cell Biol. 2009;11:747–52. doi: 10.1038/ncb1881. [DOI] [PubMed] [Google Scholar]
- 15.Rolfe DF, Brown GC. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol Rev. 1997;77:731–58. doi: 10.1152/physrev.1997.77.3.731. [DOI] [PubMed] [Google Scholar]
- 16.Kilburn DG, Lilly MD, Webb FC. The energetics of mammalian cell growth. J Cell Sci. 1969;4:645–54. doi: 10.1242/jcs.4.3.645. [DOI] [PubMed] [Google Scholar]
- 17.Locasale JW, Cantley LC. Altered metabolism in cancer. BMC Biol. 2010;8:88. doi: 10.1186/1741-7007-8-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Telang S, Clem BF, Klarer AC, Clem AL, Trent JO, Bucala R, Chesney J. Small molecule inhibition of 6-phosphofructo-2-kinase suppresses t cell activation. J Transl Med. 2012;10:95. doi: 10.1186/1479-5876-10-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clem B, Telang S, Clem A, Yalcin A, Meier J, Simmons A, Rasku MA, Arumugam S, Dean WL, Eaton J, et al. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Mol Cancer Ther. 2008;7:110–20. doi: 10.1158/1535-7163.MCT-07-0482. [DOI] [PubMed] [Google Scholar]
- 20.Climent F, Roset F, Repiso A, Pérez de la Ossa P. Red cell glycolytic enzyme disorders caused by mutations: an update. Cardiovasc Hematol Disord Drug Targets. 2009;9:95–106. doi: 10.2174/187152909788488636. [DOI] [PubMed] [Google Scholar]
- 21.Dirkx R, Meyhi E, Asselberghs S, Reddy J, Baes M, Van Veldhoven PP. Beta-oxidation in hepatocyte cultures from mice with peroxisomal gene knockouts. Biochem Biophys Res Commun. 2007;357:718–23. doi: 10.1016/j.bbrc.2007.03.198. [DOI] [PubMed] [Google Scholar]
- 22.Pospisilik JA, Schramek D, Schnidar H, Cronin SJ, Nehme NT, Zhang X, Knauf C, Cani PD, Aumayr K, Todoric J, et al. Drosophila genome-wide obesity screen reveals hedgehog as a determinant of brown versus white adipose cell fate. Cell. 2010;140:148–60. doi: 10.1016/j.cell.2009.12.027. [DOI] [PubMed] [Google Scholar]
- 23.Thathiah A, Spittaels K, Hoffmann M, Staes M, Cohen A, Horré K, Vanbrabant M, Coun F, Baekelandt V, Delacourte A, et al. The orphan G protein-coupled receptor 3 modulates amyloid-beta peptide generation in neurons. Science. 2009;323:946–51. doi: 10.1126/science.1160649. [DOI] [PubMed] [Google Scholar]
- 24.Arbiser JL, Moses MA, Fernandez CA, Ghiso N, Cao Y, Klauber N, Frank D, Brownlee M, Flynn E, Parangi S, et al. Oncogenic H-ras stimulates tumor angiogenesis by two distinct pathways. Proc Natl Acad Sci U S A. 1997;94:861–6. doi: 10.1073/pnas.94.3.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mazzone M, Dettori D, Leite de Oliveira R, Loges S, Schmidt T, Jonckx B, Tian YM, Lanahan AA, Pollard P, Ruiz de Almodovar C, et al. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell. 2009;136:839–51. doi: 10.1016/j.cell.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zacchigna S, Oh H, Wilsch-Bräuninger M, Missol-Kolka E, Jászai J, Jansen S, Tanimoto N, Tonagel F, Seeliger M, Huttner WB, et al. Loss of the cholesterol-binding protein prominin-1/CD133 causes disk dysmorphogenesis and photoreceptor degeneration. J Neurosci. 2009;29:2297–308. doi: 10.1523/JNEUROSCI.2034-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]