

Abstract
The shelterin proteins are required for telomere integrity. Shelterin dysfunction can lead to initiation of unwarranted DNA damage and repair pathways at chromosomal termini. Interestingly, many shelterin accessory proteins are involved in DNA damage signaling and repair. We demonstrate here that in normal human fibroblasts, telomeric ends are protected by phosphorylation of CGG triplet repeat-binding protein 1 (CGGBP1) at serine 164 (S164). We show that serine 164 is a major phosphorylation site on CGGBP1 with important functions. We provide evidence that one of the kinases that can phosphorylate S164 CGGBP1 is ATR. Overexpression of S164A phospho-deficient CGGBP1 exerted a dominant-negative effect, causing telomeric dysfunction, accelerated telomere shortening, enhanced fusion of telomeres, and crisis. However, overexpression of wild-type or phospho-mimicking S164E CGGBP1 did not cause these effects. This telomere damage was associated with reduced binding of the shelterin protein POT1 to telomeric DNA. Our results suggest that CGGBP1 phosphorylation at S164 is a novel telomere protection signal, which can affect telomere-protective function of the shelterin complex.
Keywords: CGGBP1, DNA damage, POT1, telomere fusions, telomeres
Introduction
Telomeres, the ends of eukaryotic linear chromosomes, consist of tandem repeats of the highly conserved unit sequence 5′-TTAGGG-3′, and terminate asymmetrically with a 3′G-rich overhang. This asymmetry arises through an incomplete terminal synthesis of the lagging strand during semi-conservative replication and a proposed exonuclease digestion of the C-rich strand.1 The chromosomal end protection by telomeres depends on their association with the shelterin-complex proteins (TRF1, TRF2, TIN2, RAP1, TPP1, and POT1) and shelterin accessory factors.2-6 Replicative telomere shortening leads to TP53-dependent senescence. Dysfunction of telomeres, however, results in their resection, and chromosome fusions resulting from telomeric DNA repair and telomere length crisis even in cells with normal TP53 activity.3,6 Functional insufficiency of different proteins, including the shelterin complex, can cause telomere dysfunction and fusions.6 The TRF proteins bind to the double-stranded telomeric repeats, block ATM activity,7-10 and prevent chromatid-type fusions at the telomeres synthesized by leading strand.11,12 ATR activation at the single-stranded G-rich overhang is inhibited by POT1,13-16 which together with TRF2 maintains it in the t-loop.17,18 POT1 insufficiency is associated with double minute chromosomes, G-rich overhang shortening, senescence-like phenotype, and chromatid-type fusions, which at least partially involve repair initiated by aberrant ATR activation at telomeres.19-21
CGGBP1 was first described as a CGG repeat-binding transcription repressor.22,23 Being intrigued by the scarcity of information on the cellular functions of CGGBP1 since then, we have addressed questions related to its role in normal and neoplastic cells and unraveled hitherto unknown functions in cell regulation. We had reported that it also binds to small CGG repeats as well as CGG repeat-devoid regions,24 suggesting more widespread functions. Our work showed that CGGBP1 localizes to the mitotic spindles and midbodies,25 is required for abscission in normal human fibroblasts,25 and localizes to the kinetochore ring and chromosomal termini during metaphase.22,25 The function of the terminal localization of CGGBP1 is not clear and also whether that localization included the telomeres. We therefore investigated if CGGBP1 plays a role in telomere function in normal human fibroblasts (1064Sk cells). We found that phosphorylation of CGGBP1 at S164 is required for telomere protection, and its abrogation resulted in widespread telomere damage associated with reduced telomere-POT1 binding, telomere fusions, abscission defects, elicitation of a DNA damage response, and accelerated senescence. We thus report a novel mechanism of telomere protection through CGGBP1.
Results and Discussion
CGGBP1 suppresses DNA damage at telomeres
Instability of repetitious DNA, which are potential CGGBP1 binding sites,26 has been described. We thus wanted to know if CGGBP1 plays a role in DNA damage response. CGGBP1 depletion by CGGBP1 siRNA resulted in DNA damage as demonstrated by an increase in the number of γH2AX foci as compared with control siRNA-treated 1064Sk cells (Fig. 1A and B; Fig S1). Nuclei or micronuclei with ubiquitous γH2AX signal, not resolvable as discrete foci (scored as having 100 foci), were also increased in the CGGBP1-depleted samples. Compared with control siRNA, CGGBP1 depletion by UTR siRNA in otherwise untreated cells or cells stably expressing empty vector also showed an increase in the number of γH2AX foci per nucleus, which was rescued by overexpression of WT CGGBP1 (Fig. 1C and D; Fig S1). The γH2AX foci showed clear co-localization with telomere FISH foci. This co-localization was strongly and significantly increased in CGGBP1-depleted cells as compared with the control siRNA-treated cells (Fig. 1E; Fig S4). The level of CGGBP1 knockdown or overexpression is shown in Figure S3.

Figure 1. CGGBP1 inhibits DNA damage partially occurring at the telomeres in 1064Sk cells. (A) A frequency distribution plot of nuclei with a specific number of γH2AX foci. Cells treated with CGGBP1 siRNA (red bars), control siRNA-treated cells (black bars). Two-tailed heteroscedastic t test for significance of difference (n = 145 nuclei) gives P = 0.000881 (n = 145 nuclei for each sample). The number of cells with more than 5 or 10 γH2AX spots per nucleus was significantly increased from 52% to 75% (P = 2.0779 × 10−10) and from 17% to 31% (P = 3.8 × 10−9), respectively, in CGGBP1 siRNA-treated cells compared with control siRNA-treated cells. (B) Representative micrographs of γH2AX immuno-detection in samples depicted in (A). (C) A frequency plot of the number of γH2AX foci per nucleus in WT CGGBP1 (green bars) or empty vector (red bars) or no vector (blue bars) expressing cells treated with UTR siRNA. Two-tailed heteroscedastic t test for significance of difference (n = 145 nuclei) gives P = 0.773449 for UTR siRNA vs. empty vector, P = 0.049634 for UTR siRNA vs. WT CGGBP1+UTRsiRNA and P = 0.045411 for empty vector+UTRsiRNA vs. WT CGGBP1+UTRsiRNA. (D) Representative micrographs of γH2AX immuno-detection in the samples depicted in (C). For all calculations of mean, standard deviation and statistical significance, the nuclei densely positive for γH2AX (scored as having 100 spots per nucleus) were excluded. (E) Micrographs showing partial co-localization between γH2AX foci (green) and telomeres (red) in control or CGGBP1 siRNA-treated cells. The histograms show the quantification of results shown in the micrographs (the black regions in the bars represent the percentage of foci at which telomeric signal and γH2AX signal overlap; the white regions of the bars represent the remaining percentages of the non-overlapping spots). F test P value for upper panel = 2.87 × 10−13 and for the lower panel = 4.13 × 10−114 show highly significant enrichment of co-localization in CGGBP1-depleted cells. (F) Western blot analysis of DNA damage response markers ATR-pS728, CHK1-pS496, ATM, pS1981, and CHK2-pT68 in total lysates of cells treated with CGGBP1 or UTR siRNA. ACTB shows loading control and CGGBP1 levels show the efficacy of siRNA against CGGBP1 levels. (G) Western blot analysis of ATR-pS728 levels in cells overexpressing WT CGGBP1 (N-terminal HA tag), treated with CGGBP1 siRNA (targets both endogenous and transgenic CGGBP1) or UTR siRNA (targets only endogenous CGGBP1). The HA blot shows overexpression of CGGBP1 and specificity of UTR siRNA. ACTB blot shows loading control.
To know if telomere dysfunction caused by CGGBP1 depletion was accompanied by a DNA damage response (DDR), we measured the activation of ATR, ATM, and downstream checkpoint kinases. ATR, CHK1, as well as ATM and CHK2 were moderately activated upon CGGBP1 depletion by CGGBP1 or UTR siRNA (Fig. 1F). In WT CGGBP1-overexpressing cells only the CGGBP1 siRNA and not the UTR siRNA elicited increased ATR activation as compared with control siRNA-treated cells, thereby establishing that CGGBP1 specifically quenches endogenous DDR (Fig. 1G). Together these data demonstrated that CGGBP1 depletion elicits a DNA damage response, which, in part, occurs at the telomeres.
ATR phosphorylates CGGBP1
To understand how CGGBP1 is involved in preventing endogenous DDR, we focused on the previously described PI3 kinase-like kinase family substrate target site (an SQ motif) at Serine164 on CGGBP1. Noticeably, ATR and ATM can phosphorylate CGGBP1 at S164 upon induced DNA damage.27
To verify the possibility that the PI3 kinase-like kinases phosphorylate CGGBP1, we tested the ability of activated ATR to phosphorylate CGGBP1. Using a pan-phosphoserine antibody (generating CGGBP1 phospho-S164-specific antibodies was not successful, perhaps due to poor immunogenicity), we analyzed the total phosphoserine levels on recombinant human CGGBP1 after incubation with immunoprecipitated activated ATR in an in vitro kinase assay. The phosphoserine levels on CGGBP1 were considerably higher when precipitates from WT mouse embryonic fibroblasts (MEFs) were used, as compared with immunoprecipitates from ATR-deficient MEFs or mock immunoprecipitates from WT MEFs. This confirmed the previous findings27 that ATR can phosphorylate CGGBP1 (Fig S4A). Moreover, the serine phosphorylation on endogenous WT CGGBP1 in 1064Sk cells was reduced by ATR knockdown by siRNA. On overexpressed CGGBP1 in 1064Sk cells, the serine 164 to alanine (S164A) mutation also caused a reduction in serine phosphorylation levels (Fig S4B). Although these results do not demonstrate that S164 is specifically phosphorylated by ATR, they separately show that ATR is capable of phosphorylating CGGBP1, and S164 is a major phosphorylation site on CGGBP1, thus corroborating a previously reported finding that ATR and PI3 kinase-like kinase family members do phosphorylate CGGBP1 at S164.27 We then asked what was the significance of S164 phosphorylation of CGGBP1, and if it was important for the role of CGGBP1 in endogenous DDR.
S164A CGGBP1 overexpression reduces cell proliferation, impairs mitotic chromatin segregation, and elicits DNA damage response
To determine the function of S164 phosphorylation on CGGBP1, we analyzed the phenotypes of 1064Sk cells, which stably expressed 1 of the 3 different forms of HA-tagged CGGBP1 (WT, phospho-site mutant S164A, and phospho-mimicking mutant S164E). The 3 stable cells lines are hereafter referred to as “S164S cells”, “S164A cells”, and “S164E cells”, respectively. The 3 forms of CGGBP1 were expressed at similar levels in stably transfected cells (Fig. S5). Proliferation assays starting at 3 wk after first passage post-transfection and stable selection showed strongly reduced cell proliferation in S164A cells (Fig. 2A). The slow dividing S164A cells acquired the morphology of senescent fibroblasts and a very strong increase in β-galactosidase positivity (used to detect cells permanently exiting cell cycle due to senescence) (Fig. 2B). Cell cycle analysis showed an accumulation of cells in S- and G2/M-phases in S164A cells, suggesting a block or slow passage through these phases of cell cycle (Fig. 2C). Consistent with the pattern of the cell cycle block, the duration from anaphase onset till abscission was also increased in S164A cells compared with S164S cells or S164E cells (Fig. 2D). Lagging chromatin in anaphase cells and unresolved chromatin bridges in telophase and post-telophase cells were increased in S164A cells. The percentage of chromatin-containing midbodies increased from 0.4% in S164S- to 11.7% in S164A-overexpressing cells (n = 100 midbodies each). Micronuclei and chromatin bridges broken off from the daughter nuclei in interphase were frequently seen in S164A cells only (Fig. 3A–F). ATR and CHK1 activation, similar to that seen in CGGBP1-depleted cells was observed in S164A cells as compared with S164S cells or S164E cells, suggesting that unlike S164E CGGBP1, S164A CGGBP1 exhibited a dominant-negative effect over the endogenous WT CGGBP1 (Fig. 3G). These results suggested that the cell cycle defects in S164A cells were affected by the endogenous DNA damage. Enhanced co-localization between γH2AX and telomeres detected by FISH was also seen in the S164A cells compared with S164S cells or S164E cells (Fig. 3H and I; Fig S6). These findings suggested that the DNA damage response activation seen in S164A cells partially occurred at the telomeres and resulted in lagging chromatin and aberrant mitotic chromatin segregation.

Figure 2. In 1064Sk cells S164A CGGBP1 overexpression retards cell cycle and accelerates senescence/crisis and adversely affects cytokinetic abscission. (A) Cell proliferation curves (starting at 3 wk post-transfection) of WT, S164A, and S164E CGGBP1-overexpressing cells. Data are from 3 different cell counting done in parallel. At the fourth time point, for S164A compared with WT or S164E, the difference is significant (t test; P < 0.001). (B) Scatter plot showing the effect of S164A overexpression on the occurrence of endogenous β-galactosidase activity (a marker of cellular senescence/crisis). The y-axis shows percentage of total number of cells per field of view, which were positive for β-gal activity (n = 50 fields of view in each case). S164A value is significantly different from WT or S164E (P < 0.001). The estimated population doublings of different samples are 30 (WT), 20 (S164A), and 40 (S164E) post-transfection. (C) Propidium iodide staining-based flow cytometric cell cycle distribution analysis of WT CGGBP1-overexpressing cells, the S164A CGGBP1-overexpressing cells. Larger S-phase and G2/M-phase fractions in S164A cells are clearly visible (data from three technical replicates of pooled biological duplicates). (D) Scatter plot showing the duration of abscission (anaphase onset to completion of abscission) by time-lapse imaging. S164A CGGBP1-overexpressing cells took significantly longer to finish cytokinetic abscission (n = 73 cells for WT, 33 for S164A and 25 for S164E; P < 0.01). There was no significant difference between WT and S164E CGGBP1-overexpressing cells, however. Some intercellular connections between abscising S164A CGGBP1-overexpressing cells showed Hoechst 33342-positive chromatin (not shown).

Figure 3. Overexpression of S164A CGGBP1 causes chromatin segregation defects during mitosis, DNA damage response activation, and enhanced γH2AX localization at telomeres in 1064Sk cells. (A–F) Abnormal metaphase plate (A), lagging chromatin in anaphase (B and C), and unresolved chromatin between telophase nuclei (D and E) are clearly seen in S164A CGGBP1-overexpressing cells. Micronuclei accompanying nuclei with unresolved chromatin can be seen. (F) Breakage of chromatin bridges between interphase nuclei shows that these bridges could not be resolved. (G) Western blot analysis of DNA damage response markers ATR-pS728, CHK1-pS496 and CHK2-pT68 in total lysates of cells expressing empty vector (lanes 1 and 3), WT CGGBP1 (lanes 4 and 6), or S164A CGGBP1 (lanes 5 and 7) transfected with no siRNA (lane 1), control siRNA (lanes 2, 4, and 5), CGGBP1 siRNA (lane 3), or UTR siRNA (lanes 6 and 7). Ponceau stain of the membrane shows protein levels in each lane. (H) Micrographs showing partial co-localization of γH2AX foci with telomere-FISH foci. (I) Histograms showing the quantification of results shown in (H) (the black regions in the bars represent the percentage of foci at which telomeric signal and γH2AX signal overlap. The white regions of the bars represent the remaining percentages of the non-overlapping spots). The F test P values for the left graph were 3.99 × 10−26, 2.39 × 10−7, and 9.00 × 10−35 for comparisons of empty vector, WT, and S164E samples with S164A sample respectively. The F test P values for the right graph were 5.35 × 10−168, 7.01 × 10−31, and 2.91 × 10−164 for comparisons of empty vector, WT, and S164E samples with S164A sample, respectively.
S164A CGGBP1 overexpression causes accelerated telomere shortening, telomere dysfunction, and telomeric fusions
To specifically determine the effect of observed telomeric DNA damage, we started by studying the effect of CGGBP1 S164 phosphorylation on telomere integrity. We first measured the alterations in telomere lengths in S164S, S164A, and S164E cells by telomere qPCR,28 immediately after the selection of stably transfected cells (at 2 and 4 wk post-transfection). The S164A cells showed a catastrophic and highly significant (all P values <0.0167) reduction in telomere repeat content as compared with S164S cells or S164E cells (P = 1.555 × 10−5 for S and 5.871 × 10−5 for E compared with A) (Fig. 4A). The telomere shortening was confirmed by single telomere length analysis (STELA) for XpYp and 17p telomeres. STELA showed significantly shorter telomeres in S164A cells as early as 5 PDs after stable transfection as compared with S164S and S164E cells (P values for comparisons of A with S and E <0.02) (Fig. 4B). Using same samples as those used in STELA, a fusion analysis also detected significantly increased frequency of telomere fusions in S164A cells (Fig. 4C), suggesting that the shortened telomeres had become dysfunctional and fused in the S164A cells, which were seen as chromatin bridges between dividing cells.

Figure 4A and B. S164 phosphorylation of CGGBP1 is required to prevent telomere shortening and fusions. (A) A real-time PCR-based telomere content analysis at different time points in genomic DNA of cells stably overexpressing WT CGGBP1 (S), S164A CGGBP1 (A), or S164E CGGBP1 (E) at different time points post-transfection (n = 9 technical replicates from 3 runs of PCRs, each in triplicates, on a pool of 2 DNA samples derived from 2 parallel cultures); PDs estimated to be between 1 for S164A and 5 for S164E. At all the time points, the reductions observed in the sample A was statistically significant as compared with S and E (all P values < 0.0167). (B) Measurement of telomere length by STELA in WT, S164A, and S164E cells. STELA (at 5 PD for S164A, 10 PD for WT, and over 20 PD for S164E cells) at the XpYp and 17p telomeres showed significantly shorter telomeres length in S164A cells compared with S164S and S164E cells. Mean and SD are detailed below. Pairwise t tests for 17p: S164S vs. S164A P < 0.0001; S164A vs. S164E P = 0.0055, and for XpYp telomere: S164S vs. S164A P = 0.0062; S164A vs. S164E P = 0.0148. (C) Using single-molecule telomere fusion analysis, fusions were detected in S164A cells with enhanced frequency as compared with WT or S164E cells. Each sample was analyzed with 18 separate reactions, each containing 56 ng of DNA. The Southern blots were hybridized with the XpYp and 17p telomere-adjacent probes as indicated on the right. Fusion frequencies are indicated below. We calculated the fusion frequencies based on the number of input diploid genomes and the number of events we observed. For S164S it was 1 event in 168 000 genomes frequency = 5.95 × 10−6 (95% confidence interval 0 to 3.81 × 10−5) and this was the same for S164E. For S164A we detected 8 events in 168 000 genomes, frequency = 4.76 × 10−5 (95% confidence interval, 2.26 × 10−5, to 9.64 × 10−5). For the HEK293T control cells we detected 11 events in 168000 frequency = 6.55 × 10−5 (95% confidence interval, 3.53 × 10−5, to 1.19 × 10−4). The differences between the fusion frequencies are significant. Comparing the data S164S and S164E combined with the S164A gives a Chi of 7.813 (df = 1), P = 0.0052.
CGGBP1 S164 phosphorylation regulates the binding of POT1 to single-stranded telomeric DNA
Shelterin complex proteins are required for telomere protection. To understand how CGGBP1 participates in telomere protection, we first tried to find out a shelterin component, which interacts with and is possibly regulated by CGGBP1 in a phospho-S164-dependent manner. The shelterin proteins TRF1, TRF2, and POT1 were studied in this regard.
In 1064Sk cells, endogenous CGGBP1 could not be immunoprecipitated with endogenous TRF1 and TRF2 (not shown). However, endogenous CGGBP1 and POT1 did co-immunoprecipitate with each other (Fig S7A). POT1 co-immunoprecipitated with CGGBP1 was seen as an expected 70-KDa band, and a larger than expected 90 KDa band (Fig S7A). A recent work demonstrates that the multiple forms of POT1 detected by us are different post-translationally modified forms of POT1.29 Co-immunoprecipitations of POT1 with overexpressed WT or S164A and S164E mutant CGGBP1 showed no difference in interactions between POT1 and CGGBP1 (not shown). This interaction between CGGBP1 and POT1 suggested that POT1 dysfunction could underlie the telomeric damage in S164A cells, as the protection of telomeric ends from DNA damage surveillance and inappropriate DNA repair requires binding of POT1 to the single-stranded telomeric DNA. We performed ChIP for POT1 and specifically assessed the quantity of single-stranded telomeric DNA pulled down with it by combining telomere qPCR with DSN digestion (employed to quantitatively detect single-stranded telomeric DNA30). In S164A cells, the binding of POT1 to single-stranded telomeric DNA was reduced as compared with the S164S or S164E cells (Fig S7B). In vitro DNA–protein interaction assays showed that the single-stranded telomeric TTAGGCx6 oligonucleotide pulled down by the POT1 antibody was, indeed, reduced in S164A cells. Reciprocally, POT1 pulled down with the TTAGGCx6 oligonucleotide (only the 70 KDa), was reduced in the S164A cells (Fig S7C). Interestingly, we also found that CGGBP1 itself is recruited to telomeres (Fig S7D) and its binding to single-stranded telomeric DNA is reduced upon S164A mutation (Fig S7E). These results suggested that POT1 and p-S164 CGGBP1 could bind to telomeric DNA as a complex, while S164 phosphodeficiency reduces the ability of CGGBP1-POT1 complex to bind to and protect the single-stranded telomeric DNA.
Phospho-mimicking CGGBP1 rescues telomere fusion-like chromatin segregation defects caused by ATR depletion
As our results show, CGGBP1 phosphorylation at S164 is important to prevent telomere dysfunction and fusions. Interestingly, it has been previously shown that ATR also regulates telomeric integrity and prevents telomeric fusions. We then asked if the telomere-protective functions of ATR are routed through CGGBP1 S164 phosphorylation. If phosphorylation at S164 on CGGBP1 is, indeed, required for the telomere-protective functions of ATR, then the S164-phosphomimicking CGGBP1 could rescue telomere dysfunction and fusions in ATR-depleted cells. We hence studied the effects of CGGBP1 S164A and S164E overexpression on telomeric damage (by identifying telomere dysfunction foci by γH2AX staining of telomere foci) and fusions (by detecting presence of chromatin at midbodies between cells undergoing cytokinetic abscission) in cells with depleted levels of ATR. As the levels of γH2AX in ATR-depleted cells were very high, we could not establish clear difference in the number of telomere dysfunction-induced foci was seen upon S164S, S164A, or S164E overexpression in ATR-depleted (using siRNA) 1064Sk cells, immortalized conditional ATR-null31 MEFs and immortalized as well as primary ATR-hypomorph MEFs (not shown). However, ATR siRNA increased the occurrence of chromatin-positive midbodies in S164S cells to the levels seen in S164A-overexpressing cells treated with control siRNA, but failed to increase the already high levels of chromatin-positive midbodies further in S164A cells, suggesting that ATR depletion and CGGBP1 S164 phospho-deficiency employ common mechanisms to manifest chromatin segregation defects (Fig. 5). ATR siRNA-induced increase in chromatin-associated midbodies was lower in S164S- and the weakest in S164E-overexpressing cells (Fig. 5). These results suggested that the mechanisms of telomere fusions and chromatin segregation defects caused by ATR deficiency are routed through a lack of S164-phosphorylation on CGGBP1.
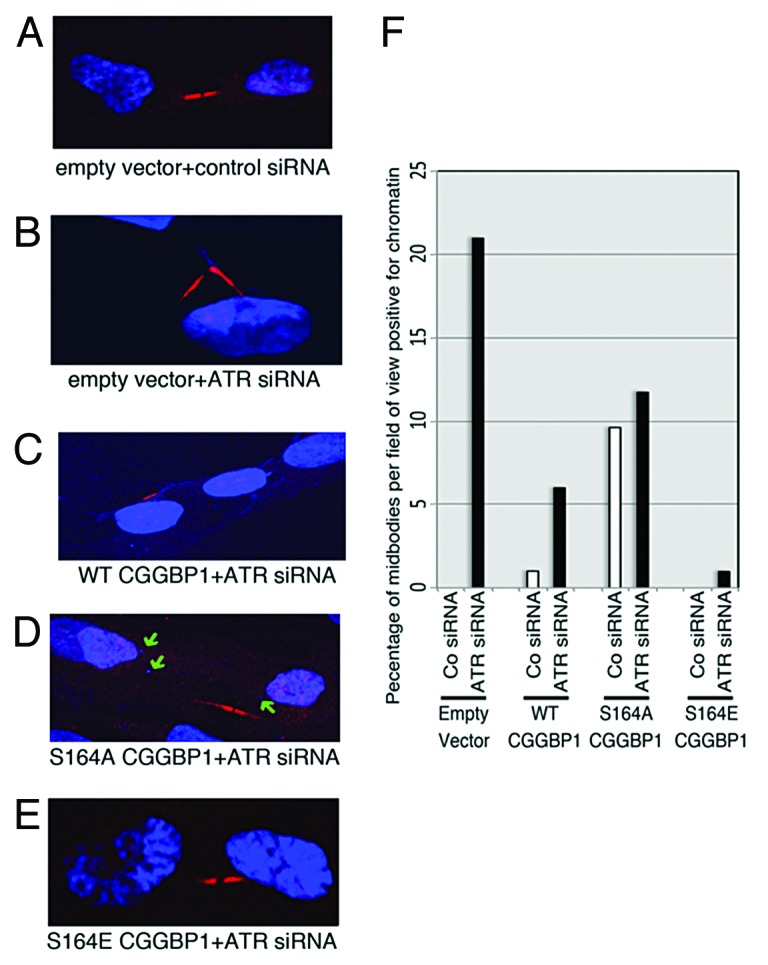
Figure 5. Phospho-mimicking mutation of CGGBP1 on S164 rescues the occurrence of lagging chromatin in abscising 1064Sk cells upon ATR depletion. (A–E) Representative micrographs of midbodies (detected by CGGBP1 staining) in control siRNA-treated empty vector cells (A) and ATR siRNA-treated cells overexpressing the following: empty vector (B), S164A CGGBP1 (C), WT CGGBP1 (D), and S164E CGGBP1 (E). Chromatin in the midbody region of ATR siRNA-treated empty vector and S164A can be clearly seen (B and C). ATR depletion in WT cells caused micronuclei (green arrows) but not fibrous chromatin in the midbody region (D). (F) Quantitation of micrographs (A–E). The values plotted on the y-axis represent the percentage of total midbodies observed per field of view, which were positive for chromatin (n = 100 in each case except 58 for S164A+control siRNA and 27 for S164A+ATR siRNA). The P value for the difference between S164A+control siRNA and S164E+control siRNA = 0.0015, and for the difference between S164A+ATR siRNA and S164E+ATR siRNA = 0.015.
Our results demonstrate that CGGBP1 is involved in telomere protection. This involves its shelterin-accessory properties determined by phosphorylation at S164 as CGGBP1 localized to telomeres and interacted with the shelterin protein POT1. Further, the effect of CGGBP1-S164 phosphorylation on telomere protection also correlates with POT1 binding to single-stranded telomeric DNA. POT1 and CGGBP1 associate with each other and S164A CGGBP1 exhibited reduced binding to telomeres. Thus, S164A CGGBP1 could sequester POT1 into a non-DNA binding pool. It is possible that these mechanisms could all act together to achieve and maintain a dynamic balance between DNA-bound and unbound POT1.
The serine at SQ[T] motifs, such as S164 on CGGBP1, is a target site for PI3 kinase-like kinase family members, which includes ATR and ATM.27 The rapid telomere erosion caused by S164A overexpression exceeds the rate of replicative telomere loss32 and instead indicates catastrophic telomere resection. Also, the DNA damage, beginning as shortening and consequential fusions of telomeres, culminates in chromatin segregation defects, intercellular chromatin bridges between post-telophase nuclei, slowed down S phase, and increased tetraploid-like cell population and formation of micronuclei. Such a widespread and multi-dimensional DNA damage is expected to activate DNA damage-sensing and repair pathways well beyond ATR. However, the effect of overexpression of the S164A mutation was cumulative, and the relative abundance of the cells with such widespread DNA damage would decline due to crisis, cell cycle arrest, premature senescence, and cell death. This could be the reason why ATR, ATM, and downstream kinases exhibited low levels of activation. The interplay between CGGBP1 phosphorylation, cell cycle traverse, and POT1-DNA binding is interesting. Since S164A CGGBP1 overexpression caused an increase in the S- and G2 fractions only, it seems that the effects of S164-phosphodeficiency are manifested from replication onwards. Our data suggest that CGGBP1 S164A phospho-deficiency would compromise the post-replication reloading of POT1 to telomeres (due to reduced POT1-DNA binding), thereby eliciting a telomere-centered DNA damage and S/G2 phase cell cycle arrest.
These findings collectively show the important role of CGGBP1 in telomeric integrity in normal human cells, manifested through a concert between kinases of the ATR family, POT1 and CGGBP1. Considering that CGGBP1 evolved relatively recently, these findings show how the shelterin accessory functions of this protein constitute minute yet indispensable taxa-specific differences in the otherwise well-conserved mechanisms of telomere protection.
Materials and Methods
Cell culture
1064Sk normal human fibroblasts, passage (8–12), were cultured in Eagle minimum essential medium (SIGMA). At passage (14–15), the cells were transfected with either WT or S164G CGGBP1-expressing constructs Four days post-transfection, transfected cells were selected in 800 µg/ml G418 in the medium for 1 wk and 300 µg/ml G418 in the medium later. To avoid the effects of variations between the clones, colonies were pooled and cultured. The pooling of colonies defined the first passage post-transfection. The numbers of passages post-transfection in which different experiments were performed are as follows: 5–7 for the phospho-serine levels measurement and ATR-CGGBP1 association, 5 for the start of the proliferation assay, 8 for cell cycle analysis by FACS, 10–15 for the measurement of the abscission time, 15–20 for β-galactosidase activity assay, 10–15 for detection of chromatin bridges in dividing cells and endogenous DNA damage response activation, 15–20 for in vitro telomerase activity assay and telomere length measurement. Experiments on G-strand shortening and POT1-GCGBP1 interactions were done on transiently transfected cells 4 d post-transfection. Equal transfection efficiencies were established by no differences in the expression of the overexpressed CGGBP1 levels.
Expression constructs and transfections
Full-length cDNA of CGGBP1 was amplified from RNA of 1064Sk human fibroblasts using primers TATCCTTACG ATGTACCAGA CTATGCTGAG CGATTTGTAG TAACAGC (with HA tag) at the 5′ end and TATAGCGGCC GCTCAACAAT CTTGTGAGTT GAG (with stop codon) at the 3′ end. A re-PCR using the same reverse primer but TGTAGGTACC GCCACCATGG GATATCCTTA CGATGTACCA GACTATGCT as forward primer (with Kozak sequence and start codon upstream of HA tag), thus adding HA-tag and Kozak sequence into the CGGBP1 cDNA. Eluted PCR product was restricted with KpnI and XhoI (New England Biolabs) endonucleases and the purified product was ligated into pcDNA3.1+ (Invitrogen), also restricted in the same way, using quick ligation reagents from New England Biolabs. The ligation mix was transformed into competent E. coli (Promega), selected on LB agar–ampicillin plates, and plasmids were extracted and sequenced. Fugene (Roche) was used to transfect the plasmids and generate stably transfected cells, which were selected as a pool to avoid clonal variations using G418 in the culture media.
siRNA and transfections
CGGBP1 and ATR siRNA (Dharmacon) were separate pools of 4 different siRNA duplexes directed against CGGBP1 ORF and ATR ORF, respectively (SmartPool, Dharmacon). The CGGBP1 UTR siRNA (Dharmacon) duplexes were custom made with Dharmacon modifications (the target sequences were CCATTGTGAT CAAGATAAA in the 3′UTR and ACGGAAAGTG CCAGGATTT in the 5′UTR of CGGBP1 mRNA having the RefSeq identification NM_001008390.1). Equimolar mixture of the two UTR siRNA was used as the UTR siRNA pool. Dharmafect 2 was used as the transfection reagent and about 50% confluent cultures were transfected after about 30 h of plating. The final siRNA concentration was 100 nM. Western blots or qRTPCRs were routinely performed to ascertain the efficacy of siRNA knockdown and has been described elsewhere in details.33
Cell cycle analysis by flow cytometry
The method used for flow cytometry (BD Biosciences) measures the DNA content per cell. Briefly, the cells were washed twice in ice-cold 1 × PBS trypsined, resuspended in culture medium with serum and filtered through a 40 micron strainer, and stained with Hoechst 33 342 at 500 µg/ml. ModFit was used to analyze the results with automated settings for removal of multi-nuclear aggregates as well as fragmented nuclei from the dead cells.
Immunofluorescence assays
Immunofluorescence was performed by a previously described method.25
Co-immunoprecipitation assays and western blotting
Phosphate-buffered RIPA, supplemented with protease and phosphatase inhibitor cocktails (SIGMA), was used to lyse cells on ice for 30 min, and the lysates were centrifuged to clear them of insoluble fraction. Cleared lysates were incubated with with protein G sepharose beads for 1 h for clearing. Then 2–4 μg of antibody was added to the beads-free lysates and samples were gently mixed overnight at 4 °C. Protein G sepharose beads were used to pull down the antibody–protein complex conjugates by incubating the antibody–lysate mixture with beads for 2 h at 4 °C. Samples were centrifuged to precipitate the beads, which were then washed with ice-cold phosphate buffered RIPA, mixed with the denaturing agent (Invitrogen), and heated at 85 °C for 10 min for reducing conditions. All samples were simultaneously run on NuPage 4–12% Bis-Tris or 3–8% TA gels, and samples were maintained on ice unless mentioned otherwise.
γH2AX stainings and telomere FISH foci
γH2AX stainings were performed according to the standard immunofluorescence protocol as mentioned above. Following the last wash in PBST after incubation with secondary antibody, the coverslides (cells attached on them) were rinsed in 1 × TBS and incubated for 2 min in 4% formaldehyde in 1 × TBS. Further rinsing in 1 × TBS was followed by denaturing of DNA using a pre-treatment solution (DAKO 5326) and rinsing again in 1 × TBS. The slides were then dehydrated in ascending grades of ethanol maintained at −20 °C. Telomeric-PNA probe was laid on dry slides, covered with coverslides, and enclosed in slide hybridization chambers (Cornig) with 10 μl of water spots at both ends. Closed chambers were incubated in waterbath at 80 °C for 5 min and room temperature in water for 16 h. The coverslides were washed in rinse and wash buffers as directed (DAKO 5326), dehydrated, and mounted with DAPI-containing mounting medium and sealed with nailpaint. Slides were stored at 4 °C and micrographs were obtained using a Leica camera attached to Leica fluorescence microscope using Adobe Photoshop-import function.
DSN digestion and for single-stranded telomeric DNA qPCR
DSN (Evrogen) digestion was performed as standerd manufacturer’s instructions. Ten units (excess) of the enzyme was used, and duration of digestion was 1 h. DSN digestion was done at 65 °C. ChIP DNA obtained from each sample was not digested or digested with 10 units of DSN. Subsequently, the samples were purified using Qiagen columns for gel extraction and eluted in equal volumes of water. Samples were then denatured at 95 °C for 5 min and snap chilled on ice for 10 min. Equal volumes of samples were then used as templates in qPCR.
Additional details of materials and methods used in this study are included in the supplementary information.
Supplementary Material
Acknowledgments
The authors thank Dr Eric J Brown for the ATR conditional knockout MEFs, Dr Oscar Fernandez-Capetillo and Matilde Murga for ATR-seckel hypomorph immortalized and primary MEFs, Dr Ola Söderberg and Karin Grannas for hTERT-transformed fibroblasts, and Geron Corporation and Dr Sergei Gryaznov at Geron Corporation for Imetelstat. The work presented here was funded by grants from the Swedish Cancer Society, the Swedish Research Council and the Swedish Childhood Cancer Foundation to BW, and from Cancer Research UK (C17199/A13490) to DMB. US was funded by the Swedish Cancer Society.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here:
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/26813
References
- 1.Makarov VL, Hirose Y, Langmore JP. Long G tails at both ends of human chromosomes suggest a C strand degradation mechanism for telomere shortening. Cell. 1997;88:657–66. doi: 10.1016/S0092-8674(00)81908-X. [DOI] [PubMed] [Google Scholar]
- 2.Capper R, Britt-Compton B, Tankimanova M, Rowson J, Letsolo B, Man S, Haughton M, Baird DM. The nature of telomere fusion and a definition of the critical telomere length in human cells. Genes Dev. 2007;21:2495–508. doi: 10.1101/gad.439107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–10. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- 4.Farr C, Fantes J, Goodfellow P, Cooke H. Functional reintroduction of human telomeres into mammalian cells. Proc Natl Acad Sci U S A. 1991;88:7006–10. doi: 10.1073/pnas.88.16.7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanish JP, Yanowitz JL, de Lange T. Stringent sequence requirements for the formation of human telomeres. Proc Natl Acad Sci U S A. 1994;91:8861–5. doi: 10.1073/pnas.91.19.8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–34. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- 7.Bianchi A, Stansel RM, Fairall L, Griffith JD, Rhodes D, de Lange T. TRF1 binds a bipartite telomeric site with extreme spatial flexibility. EMBO J. 1999;18:5735–44. doi: 10.1093/emboj/18.20.5735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Griffith J, Bianchi A, de Lange T. TRF1 promotes parallel pairing of telomeric tracts in vitro. J Mol Biol. 1998;278:79–88. doi: 10.1006/jmbi.1998.1686. [DOI] [PubMed] [Google Scholar]
- 9.Nishikawa T, Okamura H, Nagadoi A, König P, Rhodes D, Nishimura Y. Solution structure of a telomeric DNA complex of human TRF1. Structure. 2001;9:1237–51. doi: 10.1016/S0969-2126(01)00688-8. [DOI] [PubMed] [Google Scholar]
- 10.Hanaoka S, Nagadoi A, Nishimura Y. Comparison between TRF2 and TRF1 of their telomeric DNA-bound structures and DNA-binding activities. Protein Sci. 2005;14:119–30. doi: 10.1110/ps.04983705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bailey SM, Cornforth MN, Kurimasa A, Chen DJ, Goodwin EH. Strand-specific postreplicative processing of mammalian telomeres. Science. 2001;293:2462–5. doi: 10.1126/science.1062560. [DOI] [PubMed] [Google Scholar]
- 12.Martínez P, Thanasoula M, Muñoz P, Liao C, Tejera A, McNees C, Flores JM, Fernández-Capetillo O, Tarsounas M, Blasco MA. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. 2009;23:2060–75. doi: 10.1101/gad.543509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baumann P, Cech TR. Pot1, the putative telomere end-binding protein in fission yeast and humans. Science. 2001;292:1171–5. doi: 10.1126/science.1060036. [DOI] [PubMed] [Google Scholar]
- 14.Lei M, Baumann P, Cech TR. Cooperative binding of single-stranded telomeric DNA by the Pot1 protein of Schizosaccharomyces pombe. Biochemistry. 2002;41:14560–8. doi: 10.1021/bi026674z. [DOI] [PubMed] [Google Scholar]
- 15.Lei M, Podell ER, Baumann P, Cech TR. DNA self-recognition in the structure of Pot1 bound to telomeric single-stranded DNA. Nature. 2003;426:198–203. doi: 10.1038/nature02092. [DOI] [PubMed] [Google Scholar]
- 16.Denchi EL, de Lange T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature. 2007;448:1068–71. doi: 10.1038/nature06065. [DOI] [PubMed] [Google Scholar]
- 17.Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H, de Lange T. Mammalian telomeres end in a large duplex loop. Cell. 1999;97:503–14. doi: 10.1016/S0092-8674(00)80760-6. [DOI] [PubMed] [Google Scholar]
- 18.Stansel RM, de Lange T, Griffith JD. T-loop assembly in vitro involves binding of TRF2 near the 3′ telomeric overhang. EMBO J. 2001;20:5532–40. doi: 10.1093/emboj/20.19.5532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Veldman T, Etheridge KT, Counter CM. Loss of hPot1 function leads to telomere instability and a cut-like phenotype. Curr Biol. 2004;14:2264–70. doi: 10.1016/j.cub.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 20.Wu L, Multani AS, He H, Cosme-Blanco W, Deng Y, Deng JM, Bachilo O, Pathak S, Tahara H, Bailey SM, et al. Pot1 deficiency initiates DNA damage checkpoint activation and aberrant homologous recombination at telomeres. Cell. 2006;126:49–62. doi: 10.1016/j.cell.2006.05.037. [DOI] [PubMed] [Google Scholar]
- 21.Yang Q, Zheng YL, Harris CC. POT1 and TRF2 cooperate to maintain telomeric integrity. Mol Cell Biol. 2005;25:1070–80. doi: 10.1128/MCB.25.3.1070-1080.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Müller-Hartmann H, Deissler H, Naumann F, Schmitz B, Schröer J, Doerfler W. The human 20-kDa 5′-(CGG)(n)-3′-binding protein is targeted to the nucleus and affects the activity of the FMR1 promoter. J Biol Chem. 2000;275:6447–52. doi: 10.1074/jbc.275.9.6447. [DOI] [PubMed] [Google Scholar]
- 23.Naumann F, Remus R, Schmitz B, Doerfler W. Gene structure and expression of the 5′-(CGG)(n)-3′-binding protein (CGGBP1) Genomics. 2004;83:106–18. doi: 10.1016/S0888-7543(03)00212-X. [DOI] [PubMed] [Google Scholar]
- 24.Singh U, Roswall P, Uhrbom L, Westermark B. CGGBP1 regulates cell cycle in cancer cells. BMC Mol Biol. 2011;12:28. doi: 10.1186/1471-2199-12-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh U, Westermark B. CGGBP1 is a nuclear and midbody protein regulating abscission. Exp Cell Res. 2011;317:143–50. doi: 10.1016/j.yexcr.2010.08.019. [DOI] [PubMed] [Google Scholar]
- 26.Voineagu I, Surka CF, Shishkin AA, Krasilnikova MM, Mirkin SM. Replisome stalling and stabilization at CGG repeats, which are responsible for chromosomal fragility. Nat Struct Mol Biol. 2009;16:226–8. doi: 10.1038/nsmb.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–6. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 28.Cawthon RM. Telomere measurement by quantitative PCR. Nucleic Acids Res. 2002;30:e47. doi: 10.1093/nar/30.10.e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Singh U, Maturi V, Westermark B. Evidence for multiple forms and modifications of human POT1. DNA Repair (Amst) 2013 doi: 10.1016/j.dnarep.2013.08.014. In press. [DOI] [PubMed] [Google Scholar]
- 30.Zhao Y, Hoshiyama H, Shay JW, Wright WE. Quantitative telomeric overhang determination using a double-strand specific nuclease. Nucleic Acids Res. 2008;36:e14. doi: 10.1093/nar/gkm1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown EJ, Baltimore D. Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev. 2003;17:615–28. doi: 10.1101/gad.1067403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baird DM, Rowson J, Wynford-Thomas D, Kipling D. Extensive allelic variation and ultrashort telomeres in senescent human cells. Nat Genet. 2003;33:203–7. doi: 10.1038/ng1084. [DOI] [PubMed] [Google Scholar]
- 33.Singh U, Bongcam-Rudloff E, Westermark B. A DNA sequence directed mutual transcription regulation of HSF1 and NFIX involves novel heat sensitive protein interactions. PLoS One. 2009;4:e5050. doi: 10.1371/journal.pone.0005050. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.