

Abstract
Objectives
T-helper (Th)-17 lymphocytes play a crucial role in maintenance and regulation of gut immunity. Our laboratory has demonstrated that acute ethanol (EtOH) exposure before burn injury results in intestinal T cell suppression and enhanced bacterial translocation.
Background
To extend these studies, we examined the effects of EtOH exposure and burn injury on Th17 responses within intestinal lymphoid Peyer’s patches (PP). We further investigated whether restitution of interleukin (IL)-23 enhances PP cell IL-17 and IL-22 after EtOH and burn injury.
Methods
Male mice, approximately 25 g, were gavaged with EtOH (2.9 mg/kg) before receiving an approximately 12.5% total body surface area full thickness burn. One day postinjury, PP mixed cells were cultured in the presence of plate-bound anti-CD3/soluble anti-CD28 in the presence or absence of IL-23 for 48 hours. Supernatants were harvested for IL-17 and IL-22 levels.
Results
When combined with EtOH intoxication, burn injury significantly decreased IL-17 and IL-22, as compared with sham injury. IL-23 treatment successfully increased levels of IL-22 but not IL-17. This restoration was prevented when PP cells were treated with CH-223191, an aryl hydrocarbon receptor inhibitor. To further delineate the mechanism of differential IL-17 and IL-22 suppression, PP cells were treated with phorbol 12-myristate 13-acetate (PMA) and ionomycin, which signal via protein kinase C (PKC) and calcium flux. Treatment with PMA and ionomycin significantly prevented the decrease in IL-17 but not IL-22 after EtOH exposure and burn injury.
Conclusions
These findings suggest that IL-23-mediated restoration of IL-22 is aryl hydrocarbon receptor dependent, whereas IL-17 requires activation of protein kinase C and intracellular calcium signaling.
Keywords: alcohol, burn, immunity, Peyer’s patch, T cells, Th17, trauma
More than 1 million burn injuries are reported yearly within the United States.1 These injuries translate into an approximate 500,000 emergency department visits and 40,000 hospitalizations annually.1 Interestingly, nearly one-half of these injuries occur under the influence of alcohol/ethanol (EtOH) intoxication.2–5 Furthermore, burn victims who sustain injury post-EtOH exposure are more susceptible to infection, exhibit higher morbidity, and are more likely to die than patients without EtOH exposure at the time of injury.2–6 Previous data suggest that gut pathogens and their products may play a pivotal role in the development of sepsis and multiple organ failure reported in burn and trauma patients.7–9 In line with these studies, our laboratory has demonstrated increased intestinal tissue damage, leakiness, and bacterial translocation and T-helper (Th) cell suppression after EtOH intoxication and burn injury.7,10–13 Namely, we observed that EtOH combined with burn injury results in decreased Th1 cytokine interferon (IFN)-γ production.10,13 Similarly, alterations in T cell effector functions were reported after major trauma, including burn injury, in the absence of prior EtOH exposure.14–18 Furthermore, these latter studies suggest that a suppression of Th1 responses after burn and other traumatic injuries are often accompanied with a decrease in host resistance and increased susceptibility to infection.14–18
T cell activation is primarily induced via stimulation of the T cell receptor (TCR); however, differentiation of T cells into Th1, Th2, or Th17 cells is dependent on the presence of costimulatory molecules and the surrounding cytokine milieu.19 The stimulation of TCR induces a series of intracellular signaling cascade that includes the activation of protein kinases and the release of intracellular calcium ions.20,21 We have shown that the decrease in T cell IFN-γ may result from alterations in T cell intracellular signaling cascade including alterations in mitogen activated protein kinases.10,13,22
Recent findings suggest that Th17 lymphocytes maintain intestinal immune homeostasis and barrier function.19,23–26 Importantly, interleukin (IL)-23, a heterodimeric cytokine and member of the IL-12 family, has been shown to play a critical role in the development, expansion, and survival of Th17 lymphocytes.19,24,25 Binding of IL-23 to its receptor complex on differentiating Th lymphocytes activates signal transducer and activator of transcription (STAT)-3 to maintain upregulation of transcription factor retinoic acid–related orphan receptor (ROR)-γt, which has been reported to be indispensable in the production and secretion of Th17 effector cytokines IL-17 and IL-22.19,24,25 In addition, aryl hydrocarbon receptor (AhR), a ubiquitous transcription factor found in the cytoplasm of vertebrate cells has been recently implicated in the regulation of IL-22,27–29 but whether IL-23 utilizes AhR to modulate IL-17/IL-22 remains poorly described. IL-17 has been shown to protect against gut pathogens, including Bacteroides fragilis and Klebsiella pneumoniae,19,23 and to enhance tight junction formation.30 Conversely, IL-22 acts on epithelial cells to induce release of antimicrobial peptides31,32 and stimulate epithelial cell regeneration and proliferation.19,33 Furthermore, dysregulation of Th17 lymphocytes has been implicated in both forms of inflammatory bowel disease.24,25,34 More recently, suppression of the generation of Th17 cells was reported in patients with severe burn injuries.19,35 Similar studies in mice have implicated Th17 cells in immune suppression after burn injury.36 Because Th17 cells are implicated in defense against gut pathogens as well in the maintenance of gut barrier function, our study examined whether Th17 effector cytokines IL-17 and IL-22 are affected by combined EtOH exposure and burn injury in gut-associated secondary lymphoid organs, Peyer’s patches (PPs). PPs are important intestinal secondary lymphoid organs that play a critical role in T cell immunity and containment of gut bacterial translocation.37,38 We further explored the role of intracellular signaling in altered Th17 effector functions. Our results demonstrate that EtOH exposure and burn injury results in suppressed CD3-dependent Th17 cytokines IL-17 and IL-22. Furthermore, we observed that IL-23-mediated restoration of IL-22 is AhR dependent, whereas IL-17 requires activation of protein kinase C (PKC) and intracellular calcium signaling.
MATERIALS AND METHODS
Animals and Reagents
Male C57BL/6 mice, 6 to 7 weeks old, were obtained from Harlan Laboratories (Indianapolis, IN). Hamster antimouse CD3ε and antimouse CD28 were obtained from BD Biosciences (San Diego, CA). Rat affinity purified antimouse CD16/32, hamster PE-conjugated antimouse CD11c, rat FITC-conjugated antimouse MHC II, rat APC-conjugated antimouse F4/80, hamster PE-Cy7-conjugated antimouse CD3ε, and recombinant IL-12 and IL-23 were obtained from eBioscience (San Diego, CA). Goat APC-conjugated antimouse IL-23R and IL-17 and IL-22 enzyme-linked immunosorbent assay (ELISA) kits were obtained from R&D Systems (Minneapolis, MN). Collagenase D was obtained from Roche Applied Science (Indianapolis, IN). Concanavalin A (ConA), ionomycin calcium salt, phorbol 12-myristate 13-acetate (PMA), and AhR inhibitor CH-223191 were obtained from Sigma-Aldrich (St. Louis, MO).
Mouse Model of Acute EtOH Intoxication and Burn Injury
As previously described,39 adult C57BL/6 male mice were randomly divided to receive sham or burn injury and either EtOH or vehicle (water) to yield 4 experimental groups: sham vehicle, sham EtOH, burn vehicle, and burn EtOH. Mice were gavaged with either 0.4 mL of 25% EtOH in water (~2.9 g/kg) or water. Four hours after gavage, mice were anesthetized by intraperitoneal injection of ketamine hydrochloride/xylazine cocktail (~80 mg/kg and 1.2 mg/kg, respectively). Dorsal surfaces were shaved, and animals were transferred into a template fabricated to expose approximately 12.5% of the total body surface area. Total body surface area was calculated by using Meeh’s formula as described by Walker and Mason.40 Burn-injured mice were immersed into a water bath maintained at 85 to 87°C for 7 seconds, which results in a full-thickness scald injury. Sham-injured mice were subjected to identical anesthesia and treatment but immersed into isothermic water (37°C) for 7 seconds. Immediately after burn or sham procedure, animals were dried and resuscitated with 1.0-mL physiological saline by intraperitoneal injection. Animals were allowed food and water ad libitum. This model produces less than 10% mortality, 1-day postburn injury. All experiments were conducted in accordance with the guidelines set forth in the Animal Welfare Act and were approved by the Institutional Animal Care and Use Committee at the Loyola University Health Sciences Division.
Cell Isolation From PPs
One day after injury, mice were anesthetized, and the abdominal cavity exposed via midline incision. PPs were collected aseptically and placed into a collagenase D bath for 15 minutes at 37°C, as described previously.41 Collagenase D (0.5 mg/mL) was prepared in Hank’s Buffered Saline Solution (HBSS, Fisher Scientific) containing Ca2+ and Mg2+ and supplemented with 10 mM HEPES, 50 μg/mL gentamicin, 100 U/mL penicillin with 100 μg/mL streptomycin, and 5% fetal calf serum. After collagenase D treatment, PPs were crushed to prepare single-cell suspensions, filtered through a 70-μm nylon filter, and washed and resuspended at a concentration of 5 × 106 cells/mL in complete media (RPMI 1640 supplemented with 2 mM L-glutamine, 10 mM HEPES, 50 μg/mL gentamicin, 100 U/mL penicillin with 100 μg/mL streptomycin, and 10% fetal calf serum).
Flow Cytometry Analysis
For the measurement of PP T cell, macrophage and dendritic cell populations, PP mixed cells were resuspended in fluorescence-activated cell sorting (FACS) buffer (phosphate buffered saline with 5% fetal calf serum) at a concentration of 2 × 106 cells/mL. Cell suspensions were blocked with purified antimouse CD16/32 for 20 minutes at 4°C and stained with PE-Cy7-conjugated antimouse CD3ε, PE-conjugated antimouse CD11c, APC-conjugated anti-F4/80, and FITC-conjugated antimouse MHC II for 30 minutes in the dark at 4°C, as previously described.10 For the measurement of IL-23 receptor expression, PP-mixed cells (2 × 106 cells/mL) were cultured in 96-well plates in the presence of plate-bound anti-CD3 (5 μg/mL) and soluble anti-CD28 (1 μg/mL) for 4 hours. Cells were collected, blocked with purified antimouse CD16/32 for 20 minutes at 4°C, and stained with PE-Cy7 conjugated anti-CD3ε and PE-conjugated anti-IL-23R for 30 minutes in the dark at 4°C. The cells were washed twice and resuspended in 0.5-mL FACS buffer. All samples were analyzed at the Loyola University Health Sciences Division FACS Core Facility using a 6-color flow cytometer (BD FACSCanto) and FlowJo Software (Treestar). CD3ε+ cells were identified as T cells, CD11c+ MHC II+ cells were considered dendritic cells, and F4/80+ cells were considered macrophages.
Measurement of IL-23
For the measurement of IL-23 PP mixed cells (2 × 106 cells/mL) were cultured in 96-well plates in complete media in the presence of lipopolysaccharide (LPS; 1 μg/mL) for 24 hours. After culture, cell supernatants were collected for IL-12/23p40 or IL-23p19 protein measurement by ELISA. Cells were lysed for intracellular IL-23p19 protein levels by ELISA and normalized to protein levels.
Measurement of IFN-γ, IL-2, IL-17, and IL-22
To determine the effects of EtOH exposure and burn injury on Th effector cytokines IFN-γ, IL-2, IL-17, and IL-22, mixed cells (2 × 106 cells/mL) were cultured in the presence of ConA (5 μg/mL) or T cell specific stimuli, plate-bound anti-CD3 (5 μg/mL) and soluble anti-CD28 (1 μg/mL), in 96-well plates at 37°C and 5% CO2 for 48 hours.10,42 After culture, supernatants were harvested and tested for IFN-γ, IL-2, IL-17, and IL-22 levels, using respective ELISA kits (R & D Systems).
To assess whether rIL-23 restores IL-17 and IL-22 after combined injury, mixed cells (2 × 106 cells/mL) were cultured in the presence of plate-bound anti-CD3 (5 μg/mL) + soluble anti-CD28 (1μg/mL) ± rIL-23 (10 ng/mL) ± rIL-12 (10 ng/mL) in 96-well plates at 37°C and 5% CO2 for 48 hours.10,42 Supernatants were harvested and IFN-γ, IL-17, and IL-22 measured.
To investigate the role of AhR transcription factor in IL-23-dependent restoration of IL-22, mixed cells (2 × 106 cells/mL) were pretreated with AhR inhibitor CH-223191 (10 μM) for 2 hours, as previously described.43–45 CH-2233191 has been shown to have maximal effects at 10 μM with no additional effects at 25 μM.44,45 Cells were then cultured in the presence of plate-bound anti-CD3 (5 μg/mL) + soluble anti-CD28 (1 μg/mL) ± rIL-23 (10 ng/mL) in 96-well plates at 37°C and 5% CO2 for 48 hours. IL-17 and IL-22 were measured in culture supernatants by ELISA.
To explore the role of PKC activation and intracellular calcium signaling on Th17 effector cytokines IL-17 and IL-22 after EtOH exposure and burn injury, mixed cells (2 × 106 cells/mL) were cultured in the presence of plate-bound anti-CD3 (5 μg/mL) + soluble anti-CD28 (1 μg/mL) ± PMA (10 ng/mL) and ionomycin (50 ng/mL) in 96-well plates at 37°C and 5% CO2 for 48 hours. IL-17 and IL-22 were measured in culture supernatants.
Statistical Analysis
The data, wherever applicable, are presented as means + SEM and were analyzed using analysis of variance with Tukey post hoc test or Student t test (GraphPad InStat). P < 0.05 was considered statistically significant.
RESULTS
PP Immune Cells After EtOH Exposure and Burn Injury
We determined the effect of EtOH exposure and burn injury on PP T cells (CD3ε+), dendritic cells (CD11c+ MHC II+) and macrophages (F4/80+) by flow cytometry. As summarized in Table 1, the percentage of PP immune cells remained unaffected after EtOH and/or burn injury.
TABLE 1.
Percentage of T Cells, Dendritic Cells, and Macrophages in PPs After EtOH Intoxication and Burn Injury
| T Cells (%)(CD3ε+) | Dendritic Cells (%) (CD11c+ MHC II+) | Macrophages (%) (F4/80+ MHC II+) | |
|---|---|---|---|
| Sham vehicle | 19.57 ± 1.74 | 3.23 ± 0.21 | 0.26 ± 0.04 |
| Sham EtOH | 18.97 ± 1.84 | 3.03 ± 0.20 | 0.26 ± 0.06 |
| Burn vehicle | 24.46 ± 2.25 | 3.09 ± 0.20 | 0.24 ± 0.02 |
| Burn EtOH | 22.58 ± 1.82 | 3.21 ± 0.18 | 0.25 ± 0.02 |
PPs were harvested 1 day postinjury, processed for single-cell suspension, and analyzed by flow cytometry. CD3ε+ cells were identified as T cells, CD11c+ MHC II+ cells were considered dendritic cells, and F4/80+MHC II+ cells were considered macrophages. Data represent frequency of T cells, dendritic cells, and macrophages per live events. Data are represented as mean ± SEM, n = 8 to 12 animals per group, combined from 2 independent experiments.
EtOH Exposure and Burn Injury Suppresses PP Th17 Effector Cytokines IL-17 and IL-22
Our laboratory has previously demonstrated that EtOH intoxication and burn injury suppress gut associated T cell IFN-γ and IL-2 in a rat model.10,11,13 To further elucidate the effects of EtOH intoxication and burn injury on Th responses, we examined whether combined insult affects Th17 effector responses in PPs. To test this, PP mixed cells were cultured with ConA (5 μg/mL, data not shown) or T cell specific stimuli, plate-bound anti-CD3 (5 μg/mL), and soluble anti-CD28 (1 μg/mL) for 48 hours. After culture, cell supernatants were harvested and IL-17 (Fig. 1A) and IL-22 (Fig. 1B) were measured. As demonstrated in Figure 1, we found no significant change in PP IL-17 and IL-22 in mice gavaged with EtOH alone compared with sham vehicle group. After burn injury, IL-17 and IL-22 were both decreased as compared with sham vehicle. This burn-induced suppression of IL-17 and IL-22 was significantly exacerbated in the presence of EtOH intoxication.
FIGURE 1.

EtOH exposure and burn injury suppresses PP Th17 effector cytokines IL-17 and IL-22. PP mixed cells (2 × 106 cells/mL) were cultured in 96-well plates in the presence of plate-bound anti-CD3 (5 μg/mL) and soluble anti-CD28 (1 μg/mL) for 48 hours. Supernatants were harvested for the measurement of IL-17 (panel A) and IL-22 (panel B). Values are means + SEM, n = 5 to 11 animals per group, combined from 3 independent experiments. *P < 0.001 and †P < 0.01 as compared with sham vehicle. ‡P < 0.01 as compared with sham EtOH by analysis of variance with Tukey post hoc test. §P < 0.05 as compared with burn vehicle by Student t test.
In our preliminary studies, we used ConA as a T cell stimulant (data not shown) and found similar results to T cell–specific CD3/CD28. Thus, to explicitly study the effects of EtOH exposure and burn injury on CD3-/CD28-mediated Th17 effector responses, further experiments utilized anti-CD3 and anti-CD28 as T cell stimuli. Moreover, the greatest suppression of Th17 effector cytokines was found in animals subjected to combined EtOH exposure and burn injury; thus, the remaining studies were carried out using only the sham vehicle and burn EtOH groups.
EtOH Exposure and Burn Injury Suppresses PP Th1 Effector Cytokines
Our laboratory has previously demonstrated that EtOH intoxication and burn injury suppress gut-associated T cell, including PP, IFN-γ, and IL-2 in a rat model.10,11,13 To determine whether decreased Th1 effector cytokines, IFN-γ, and IL-2, after EtOH exposure and burn injury were recapitulated in our mouse model, PP mixed cells were cultured with ConA for 48 hours. After culture, cell supernatants were harvested for the measurement of IFN-γ and IL-2. As shown in Figure 2, combined insult suppressed Th1 effector cytokines IFN-γ (Fig. 2A) and IL-2 (Fig. 2B), as compared with sham injury.
FIGURE 2.

PP IFN-γ and IL-2 are decreased after EtOH exposure and burn injury. PP mixed cells (2 × 106 cells/mL) were cultured in 96-well plates in the presence of ConA (5 μg/mL) for 48 hours. Supernatants were harvested for the measurement of IFN-γ (panel A) and IL-2 (panel B). Values are means + SEM, n = 4 to 6 animals per group. *P < 0.05 and †P < 0.005 as compared with sham vehicle group by Student t test.
PP IL-23 and IL-23 Receptor Expression
IL-23 is synthesized by a variety of cells, including monocytes, macrophages, dendritic cells, T cells, B cells, and endothelial cells.19,24,25 Given its central role in mediating Th17 effector responses, we tested whether EtOH exposure and burn injury perturbs IL-23 levels. Mixed cells were cultured in the presence of LPS (1 μg/mL) for 24 hours; cell supernatants were collected for extra-cellular IL-23 protein levels and cells were lysed for intracellular IL-23 protein levels. EtOH exposure and burn injury suppressed IL-12/23p40, a subunit shared between IL-12 and IL-23, in cell culture supernatants, as compared with sham injury (Fig. 3A). We next measured IL-23p19, the subunit unique to IL-23, in cell culture supernatants and found that the levels of IL-23p19 were below the level of detection in all treatment groups (data not shown). To circumvent this, we tested IL-23p19 in cell lysates. Levels of IL-23p19 were detectable only in samples from sham vehicle animals (Fig. 3B), suggesting that combined EtOH exposure and burn injury suppresses IL-23p19.
FIGURE 3.

PP IL-23. PP mixed cells (2 × 106 cells/mL) were cultured in 96-well plates in the presence of LPS (1 μg/mL) for 24 hours. Supernatants were harvested for the measurement of IL-12/23p40 (panel A). Cells were collected and lysed for the measurement of intracellular IL-23p19 protein levels (panel B) by ELISA. Values are means + SEM, n = 3 to 8 animals per group. *P < 0.05 as compared with sham vehicle by Student t test.
We further determined whether EtOH exposure and burn injury perturb expression of the IL-23 receptor (IL-23R). To accomplish this, PP cells were stimulated with plate-bound anti-CD3 (5 μg/mL) and soluble anti-CD28 (1 μg/mL) for 4 hours. After incubation, cells were harvested and stained with PE-Cy7-conjugated antimouse CD3ε and PE-conjugated antimouse IL-23R, CD3ε and IL-23R coexpression was determined by flow cytometry. We found that only 2% to 3% of PP cells coexpress CD3ε and IL-23R after 4 hours of culture. However, EtOH intoxication and burn injury did not significantly affect the frequency (percent) of CD3ε+ IL-23R+ events (data not shown), nor the per cell expression of IL-23R within CD3ε+ cells (Fig. 4).
FIGURE 4.

EtOH exposure and burn injury does not influence IL-23 receptor expression. PP mixed cells (2 × 106 cells/mL) were cultured in 96-well plates in the presence of plate-bound anti-CD3 (5μg/mL) and soluble anti-CD28 (1 μg/mL) for 4 hours. Cells were collected and stained with PE-Cy7-conjugated anti-CD3ε and PE-conjugated anti-IL-23R. Expression of CD3ε and IL-23R were determined by flow cytometry. Panel A, representative FACS plots demonstrate the mean fluorescence intensity (MFI) of IL-23R antibody conjugate: gray histogram, fluorescence minus 1 control; solid black line, respective treatment. Panel B, cumulative IL-23R MFI expression data; values are means + SEM from 4 animals per group.
IL-23 Specifically Restores IL-22, But Not IL-17, After Combined Injury
Differentiation of Th17 lymphocytes depends on IL-1β, IL-6, and transforming growth factor β; however, IL-23 is crucial to Th17 effector functions. As mentioned previously, IL-23 and IL-12 share the p40 subunit; where p40 joins with the p19 subunit to form IL-23, p40 can also join with the p35 subunit to form IL-12.19,25 Because IL-23 shares 1 subunit with IL-12 and we have previously shown decreased IL-12 and IFN-γ after EtOH exposure and burn injury,10 we tested whether IL-23 and IL-12 influence IFN-γ, IL-17, and IL-22. PP mixed cells were cultured with plate-bound anti-CD3 (5 μg/mL) and soluble anti-CD28 (1 μg/mL) in the presence or absence of rIL-12 (10 ng/mL) or rIL-23 (10 ng/mL) for 48 hours. After culture, cell supernatants were harvested, and IFN-γ (Fig. 5A), IL-17 (Fig. 5B), and IL-22 (Fig. 5C) were measured by ELISA. In line with our previous findings,10 we showed that IL-12 reestablishes IFN-γ to sham vehicle levels after EtOH intoxication and burn injury in PP cells (Fig. 5A). Restoration of post-EtOH exposure and burn injury–induced IFN-γ suppression is specific to IL-12, as treatment with IL-23 does not significantly affect IFN-γ. In regard to Th17 effector cytokines, our results demonstrate a differential role of IL-23 in modulation of IL-17 and IL-22. Treatment with IL-12 or IL-23 did not modulate PP IL-17 levels (Fig. 5B). Conversely, IL-23 treatment significantly increased IL-22 in both sham and burn EtOH samples, suggesting that the IL-23 restores IL-22 after EtOH intoxication and burn injury (Fig. 5C). Moreover, IL-12 did not affect IL-22 in either sham or burn EtOH, confirming that restoration of IL-22 is truly IL-23 dependent. Together, these results suggest that IL-23 reestablishes IL-22 but not IL-17 after EtOH exposure and burn injury.
FIGURE 5.

IL-23 specifically restores PP IL-22, but not IL-17, after EtOH intoxication and burn injury. PP mixed cells (2 × 106 cells/mL) were cultured in 96-well plates in the presence of plate-bound anti-CD3 (5μg/mL) and soluble anti-CD28 (1 μg/mL) in the presence or absence of rIL-23 (10 ng/mL) or rIL-12 (10 ng/mL) for 48 hours. Supernatants were harvested for the measurement of IFN-γ (panel A), IL-17 (panel B), and IL-22 (panel C). Values are means + SEM, n = 6 to 14 animals per group, combined from 2 independent experiments. *P < 0.001 as compared with all groups. †P < 0.05 as compared with respective sham vehicle. ‡P < 0.001 as compared with respective anti-CD3/28 and anti-CD3/28 + rIL-12 groups by analysis of variance with Tukey post hoc test. §P < 0.05 as compared with respective sham vehicle by Student t test.
AhR Modulates IL-23-Dependent Restoration of IL-22 After EtOH Exposure and Burn Injury
Given the differential response of IL-17 and IL-22 after IL-23 treatment, we chose to examine the role of AhR, a ubiquitous transcription factor found in the cytoplasm of vertebrate cells, in IL-23-dependent restoration of IL-22. Although AhR has been recently implicated in the regulation of IL-22,27–29 and less so IL-17, the mechanisms by which IL-23 and AhR modulate IL-22 remain poorly described. To examine whether AhR plays a role in IL-23-induced IL-22 after EtOH exposure and burn injury, mixed PP cells were pretreated with AhR inhibitor CH-223191 (10 μM) for 2 hours before being cultured with plate-bound anti-CD3 (5 μg/mL) and soluble anti-CD28 (1 μg/mL) in the presence or absence of rIL-12 or rIL-23. CH-223191 has been used in previous studies and is shown to maximally inhibit AhR activity at 10 μM, without having agonist activity at high doses.43–45 After 48 hours of culture, cell supernatants were collected and tested for IL-17 (Fig. 6A) and IL-22 (Fig. 6B). PP cells cultured with an AhR inhibitor, before IL-23 treatment, produced significantly less IL-22 (Fig. 6B). However, inhibition of AhR did not affect IL-17 (Fig. 6A). These results suggest that AhR may play a predominant role in mediating IL-23-dependent restoration of IL-22 and further support divergent roles of IL-23 in Th17 effector functions after EtOH intoxication and burn injury.
FIGURE 6.
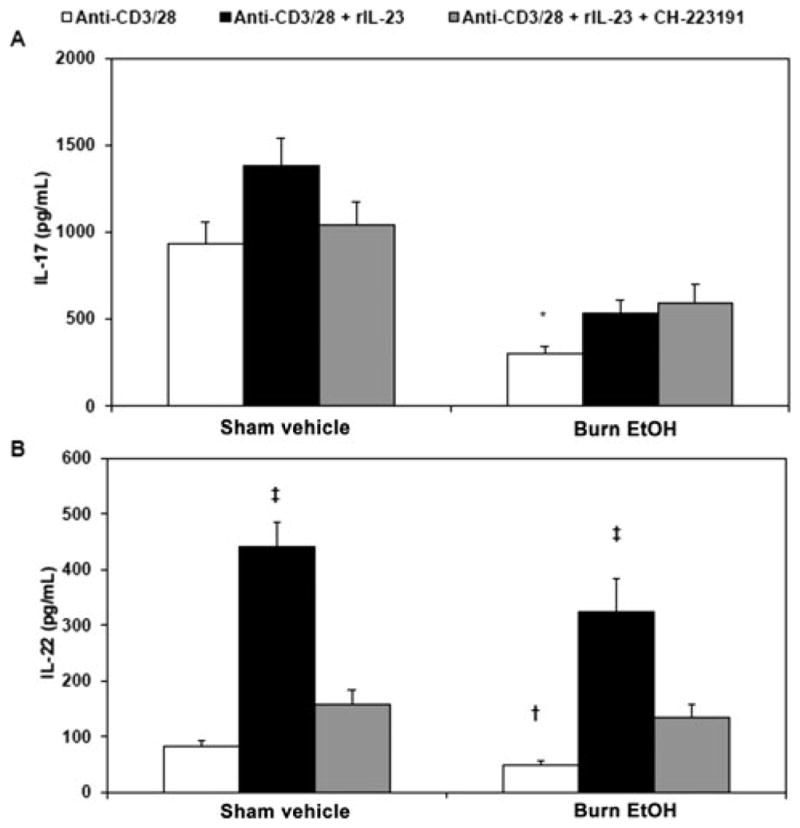
AhR modulates IL-23-dependent restoration of IL-22 after combined injury. PP mixed cells (2 × 106 cells/mL) were treated with AhR inhibitor CH-223191 (10 μM) for 2 hours before being cultured in 96-well plates in the presence of plate-bound anti-CD3 (5 μg/mL) and soluble anti-CD28 (1 μg/mL) in the presence or absence of rIL-23 (10 ng/mL) for 48 hours. Supernatants were harvested for the measurement of IL-17 (panel A) and IL-22 (panel B). Values are means + SEM, n = 9 to 17 animals per group, combined from 3 independent experiments. *P < 0.01 as compared with respective sham vehicle. †P < 0.05 as compared with respective sham vehicle by Student t test. ‡P < 0.01 as compared with respective anti-CD3/28 and anti-CD3/28 + CH-223191 groups by analysis of variance with Tukey post hoc test.
IL-17 Requires Intracellular Calcium Signaling After EtOH Exposure and Burn Injury
To further decipher the differential effects of EtOH exposure combined with burn injury on IL-17 and IL-22, PP cells were treated with PMA and ionomycin, which directly activates PKC and intracellular calcium signaling. Mixed PP cells were cultured with plate-bound anti-CD3 (5 μg/mL)/soluble anti-CD28 (1 μg/mL) alone or with PMA (10 ng/mL) and ionomycin (50 ng/mL) for 48 hours. After cell culture, cell supernatants were collected and tested for IL-17 (Fig. 7A) and IL-22 (Fig. 7B). We found that treatment of cells with PMA and ionomycin restores PP IL-17 levels to that of sham vehicle levels. However, PMA and ionomycin treatment does not affect IL-22. Together, these data highlight the importance of PKC and intracellular calcium in the induction of IL-17, but not IL-22, which supports recent data indicating differential regulation of IL-17 and IL-22.
FIGURE 7.

IL-17 requires intracellular calcium signaling after EtOH exposure and burn injury. PP mixed cells (2 × 106 cells/mL) were cultured in 96-well plates in the presence of plate-bound anti-CD3 (5 μg/mL), soluble anti-CD28 (1 μg/mL), PMA (10 ng/mL), and ionomycin (50 ng/mL) for 48 hours. Supernatants were harvested for the measurement of IL-17 (panel A) and IL-22 (panel B). Values are means + SEM, n = 6 to 16 animals per group, combined from 3 independent experiments. *P < 0.05 as compared with respective sham vehicle (t test for IL-22). †P < 0.01 as compared with respective anti-CD3/28 group by analysis of variance with Tukey post hoc test.
DISCUSSION
The data presented in this article clearly demonstrate that acute EtOH exposure before burn injury results in suppressed CD3/28-dependent expression of Th17 effector cytokines IL-17 and IL-22 in PP cells. Our findings suggest that restitution of IL-23 effectively increases IL-22 but does not modulate IL-17. We further found that IL-23-mediated restoration of IL-22 is dependent on AhR. IL-17, on the contrary, requires activation of PKC and intracellular calcium signaling, as demonstrated by increased levels of IL-17 after treatment with PMA and ionomycin. Altogether, our results provide novel evidence suggesting that IL-23 differentially regulates Th17 effector cytokines IL-17 and IL-22 after EtOH exposure and burn injury and further indicate a cross talk between IL-23 and AhR.
T-helper cells, including Th17 cells, are of critical importance to mucosal immunity, barrier function, and the containment of pathogenic gut bacteria.23,26,30,32 Gut bacterial pathogens are often implicated in EtOH and/or burn injury–associated morbidity and mortality. For these studies, we focused on gut-associated lymphoid organs PPs, because they are of central importance to gut bacteria translocation and their containment.38 Specifically, PPs are secondary lymphoid organs along the small intestine, which are lined by specialized epithelial cells, “M” cells. Similar to antigen-presenting cells such as dendritic cells, M cells come in direct contact with luminal content. Under normal conditions, pathogens that cross the epithelial barrier are limited from invading the host by the immune cells within PPs, including Th cells.37,38 Specifically, (nu/nu) mice, which lack mature T cells, have increased bacterial translocation to mesenteric lymph nodes (MLN), spleen, and liver.46 Together, these findings define an interdependent relationship between PPs, T cells, and containment of gut bacteria.
In the context of EtOH exposure, chronic EtOH feeding has been linked with decreased total, T and B cells in PPs as compared with control animals.37 As demonstrated in Table 1, we did not find any change in the frequency of T cells, macrophages, or dendritic cells. Thus, the lack of compositional changes in PPs after EtOH exposure and burn injury indicate that EtOH exposure and burn injury may compromise the ability of T cells to respond to antigenic stimuli and/or impair TCR signaling.47 This hypothesis is supported by reduced levels of Th17 effector cytokines IL-17 and IL-22 in EtOH and burn-injured animals. Our laboratory has previously demonstrated decreased T cell proliferation and Th1 immune functions in T cells from PPs and MLN and increased bacterial translocation to MLN after EtOH exposure and burn injury.7,10,12,13 In addition, our laboratory found that depletion of CD3+ T cells from normal animals increases bacterial accumulation in MLN. We further showed that depletion of T cells in animals receiving EtOH exposure and burn injury results in systemic invasion of bacteria, as evidenced by bacterial contamination of the spleen and systemic circulation.11 The results presented in this article are the first to elucidate the effects of EtOH exposure and burn injury on Th17 immune responses within PPs. Because Th17 cells have been implicated in defense against gut pathogens,19,23,26,32 suppression of Th17 effector functions may further contribute to the increased bacterial translocation and gut barrier dysfunction after combined insult of EtOH intoxication and burn injury.
As stated previously, IL-23 is a known modulator of Th17 differentiation and cytokine effector secretion.19,24,25 In this study, we found decreased IL-12/23p40 in cells from EtOH and burn-injured mice. Moreover, we were able to measure only IL-23 in cell lysates from the sham vehicle group, as levels of IL-23 were undetectable in cell lysates from the burn EtOH group. Because we observed a decrease in IL-23 levels in PP after EtOH exposure and burn injury, we focused on determining the role of IL-23 in post-EtOH and burn-induced IL-17 and IL-22 suppression. Our laboratory recently demonstrated decreased IL-12 after EtOH exposure and burn injury and successful restoration of Th1 cytokines IFN-γ and IL-2 after restitution of IL-12.10 IL-12 and IL-23 share a common subunit, the p40 subunit, and have been suggested to have common roles in driving Th1 and Th17 adaptive immune responses, although this remains controversial. Therefore, we tested the effects of IL-12 and IL-23 on PP cell IFN-γ, IL-17, and IL-22 in our murine model of EtOH exposure and burn injury. Our results demonstrate effective restoration of IFN-γ after IL-12 treatment, with no significant change in IFN-γ after IL-23 treatment. Although we expected restoration of IL-17 and IL-22 after restitution of IL-23, the addition of IL-23 only induced IL-22, not IL-17. Levels of IL-17 and IL-22 were not changed in response to IL-12. These effects were not due to changes in IL-23 receptor expression on CD3+ T cells. Although not examined in these studies, it is possible that restoring levels of IL-17 may require combined stimulation with other Th17-inducing cytokines, including IL-6 and transforming growth factor β as suggested in other studies.19 New data investigating the molecular pathways that control IL-17 and IL-22 expression increasingly demonstrate differential regulation of these Th17 effector cytokines.27–29 Among the multiple regulators of IL-17 and IL-22 are various transcription factors, including STAT3, ROR-γt, and, most recently, AhR. Previously, the accepted dogma suggested that IL-17 and IL-22 expression was via ligation of the IL-23R, STAT3 activation, and induction of ROR-γt, the homolog to RORC (RAR-related orphan receptor C) in humans. However, recently Duhen et al29 reported low expression of RORC in IL-22 producing cells, as compared with IL-17 producing cells, and concluded that RORC is nonessential to expression of IL-22. Thus, EtOH exposure and burn injury may inhibit IL-23-dependent induction of ROR-γt and prevent IL-23 induction of IL-17. Further studies will examine the effects of EtOH exposure and burn injury on the IL-23 signaling pathway, including activation of STAT3 and expression of ROR-γt.
Although it is generally accepted that both IL-6 and IL-23 activate STAT3 to in turn induce upregulation of Th17 hallmark transcription factor ROR-γt, how AhR regulates IL-22 remains unclear. AhR is a cytosolic ligand–activated transcription factor. Upon ligand binding, AhR translocates to the nucleus, where it dimerizes with AhR nuclear translocator and activates gene transcription.48 Using an inhibitor that binds AhR and prevents its nuclear translocation,44 we demonstrate that IL-23 induction of IL-22 is, in part, AhR dependent. We recognize that although this finding is based on single pharmacological agent, this AhR inhibitor (CH-223191) has been used in many previous studies and found to be highly specific at 10 μM.43–45 AhR null mice are available; however, these mice do not have fully developed cryptopatches (intestine-associated lymphoid organs), which may confound immune responses in PPs.49 Therefore, although our data indicate cross talk between IL-23 and AhR, further studies need to be conducted to elucidate how these 2 pathways converge. Previous studies have used coimmunoprecipitation to demonstrate interaction of AhR and various STAT proteins.50 Thus, it is possible that IL-23 activates AhR/STAT interactions. Moreover, determining the role of ROR-γt in IL-22 immunity also warrants further investigation, because controversy regarding the importance of ROR-γt in IL-22 expression still exists.27,29 Duhen et al29 found minimal expression of RORC in the IL-22-producing cells and concluded that RORC is nonessential to IL-22. Conversely, Trifari et al27 used siRNA to demonstrate that silencing of RORC or AhR diminished IL-22 production from memory CD4+ T cells.
Stimulation of T cells with anti-CD3/CD28 results in the activation of a number signaling molecules (eg, Zap-70, P59fyn, P56lck, and PLC-γ) before inducing calcium mobilization and activating PKC; therefore, any change in these upstream signaling molecules may influence the activation of calcium and PKC. To further decipher the intracellular mechanism of suppressed Th17 effector cytokines after EtOH intoxication and burn injury, we determined whether direct activation of intracellular calcium and activation of PKC, with PMA and ionomycin, prevents the suppression of Th17 effector function. More specifically, several lines of evidence suggest that the activation of PKC and the sustained elevation of intracellular calcium are required for T cell activation.20 These studies suggest that signals emanating from TCR result in the activation of protein tyrosine kinases and phospholipase C-γ (PLC-γ).21 PLC-γ hydrolyzes phosphatidylinositol 4,5-bisphosphate into inositol 1,4,5-trisphosphate (IP3) and 1,2-diacylglycerol (DAG). IP3 causes a calcium release from intracellular stores, followed by calcium influx through the plasma membrane leading to a sustained elevation in intracellular calcium concentration, DAG activates PKC.21 In line with these findings, burn injury alone has been correlated with suppressed intracellular calcium signaling.22 We further found that EtOH combined with burn injury suppresses p38/ERK activation in T cells.10,51 In addition, unpublished data from our laboratory suggest a decrease in Zap70 phosphorylation. So although we did not directly measure PKC and calcium mobilization in this study, our previous and current unpublished findings clearly suggest alterations in signaling molecules both up-(Zap-70) and downstream (p38/ERK) to calcium and PKC activation. In this study, using the combination of anti-CD3/CD28 with PMA/ionomycin allowed us to determine whether the changes in calcium/PKC up- or downstream molecules are critical to the changes in T cell effector function after EtOH exposure and burn injury. We used PMA and ionomycin to activate calcium and PKC to determine whether restoration of calcium- and PKC-dependent pathways maintains Th17 effector functions after EtOH exposure and burn injury. The results from these experiments suggest that PP cells cultured with plate-bound anti-CD3 (5μg/mL), soluble anti-CD28 (1 μg/mL) in the presence of PMA (10 ng/mL), and ionomycin (50 ng/mL) restored IL-17 levels to that of sham vehicle levels. On the contrary, PMA and ionomycin treatment did not affect IL-22. Thus, these findings suggest that alterations in signaling molecules upstream to PKC and calcium are more critical to the decrease in IL-17 and not IL-22 release. Together, these data suggest the importance of effective intracellular calcium influx and PKC activation in the induction of IL-17. Furthermore, our findings support recent data indicating differential regulation of IL-17 and IL-22. However, the mechanism underlying this divergent regulation of PP T cell IL-17 and IL-22 remains to be explored.
Concern may be raised for not seeing an effect of burn injury alone on T cell effector responses in our study. This is in contrast to many previous studies, suggesting that burn influences Th1 effector functions.14,15,35 However, many of these previous studies were performed using a relatively large burn area. The severity of the post-burn pathogenesis is directly proportional to the burn size.52 We used an approximately 12.5% total body surface area burn injury, which is relatively moderate burn insult and may not be severe enough to produce any adverse effects on T cell responses 1 day after injury. In addition, age, sex, and other preclinical manifestation can also negatively affect the outcome of burn patients, specially the patients with small burn injury.53 Therefore, although a relatively small burn by itself may not have any deleterious effect on host defense, when combined with existing conditions, such as EtOH intoxication, it may become detrimental.
CONCLUSIONS
In summary, our findings suggest that EtOH combined with burn injury differentially regulates the expression of PP T cell IL-17 and IL-22. Although IL-22 is successfully reestablished by restitution of IL-23, IL-17 restoration requires reestablishment of PKC and intracellular calcium signaling. The divergent transcriptional regulation of IL-17 and IL-22 may offer a target for immunomodulation in the treatment of patients who sustain burn injury under the influence of EtOH intoxication. Although IL-23 did not increase IL-17 in our in vitro studies, it is possible that in vivo modulation of IL-23 might increase IL-17 and contribute to an exacerbated immune response by promoting inflammation and neutrophil recruitment. Our previous studies demonstrate heightened neutrophil activation54,55; IL-23-dependent induction of IL-17 would intensify gut damage after EtOH and burn. Thus, solely targeting IL-22 may promote gut epithelial and immune and barrier function maintenance, improving outcomes after trauma. Moreover, IL-22’s transcription regulation by AhR offers a unique opportunity for targeted therapy. Two recent studies independently identified plant-derived phytochemicals as ligands of AhR.56,57 Specifically, various vegetables, including broccoli, cabbage, and brussel sprouts, are enriched with glucosinolate glucobrassicin, which is broken down into indole-3-carbino and in the acidic gut environment is further converted to diinodelylmethane and indolo[3,2-b]carbazole, high affinity AhR ligands. After injury, including EtOH and burn, these compounds may be incorporated into diet regimens to induce IL-22 and promote epithelial cell regeneration, proliferation, and boost innate immunity through induced expression of antimicrobial peptides, without inducing pathogenic levels of IL-17. Thus, future studies will explore the translational potential of modulating IL-22 after EtOH exposure and burn injury.
Footnotes
Disclosure: Supported by NIH grants R01AA015731 (MAC) and R01AA015731-04S1 (MAC). Juan L. Rendon is supported by NIH grants F30AA020167 (JLR), T32AA013527 (EJK), the Loyola University Chicago Stritch School of Medicine Combined MD/PhD Program, and the Dr Ralph and Marian C. Falk Medical Research Trust. The authors declare that they have no competing interests.
References
- 1.American Burn Association. [Accessed April 7, 2010];Burn incidence and treatment in the US: 2007 fact sheet. Available at: http://www.ameriburn.org/resourcesfactsheet.php. Updated 2007.
- 2.Maier RV. Ethanol abuse and the trauma patient. Surg Infect (Larchmt) 2001;2:133–141. doi: 10.1089/109629601750469456. discussion 141–144. [DOI] [PubMed] [Google Scholar]
- 3.McGill V, Kowal-Vern A, Fisher SG, et al. The impact of substance use on mortality and morbidity from thermal injury. J Trauma. 1995;38:931–934. doi: 10.1097/00005373-199506000-00019. [DOI] [PubMed] [Google Scholar]
- 4.Kelley D, Lynch JB. Burns in alcohol and drug users result in longer treatment times with more complications. J Burn Care Rehabil. 1992;13:218–220. doi: 10.1097/00004630-199203000-00008. [DOI] [PubMed] [Google Scholar]
- 5.Albright JM, Kovacs EJ, Gamelli RL, et al. Implications of formal alcohol screening in burn patients. J Burn Care Res. 2009;30:62–69. doi: 10.1097/BCR.0b013e3181921f31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Silver GM, Albright JM, Schermer CR, et al. Adverse clinical outcomes associated with elevated blood alcohol levels at the time of burn injury. J Burn Care Res. 2008;29:784–789. doi: 10.1097/BCR.0b013e31818481bc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choudhry MA, Rana SN, Kavanaugh MJ, et al. Impaired intestinal immunity and barrier function: a cause for enhanced bacterial translocation in alcohol intoxication and burn injury. Alcohol. 2004;33:199–208. doi: 10.1016/j.alcohol.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 8.Keshavarzian A, Holmes EW, Patel M, et al. Leaky gut in alcoholic cirrhosis: a possible mechanism for alcohol-induced liver damage. Am J Gastroenterol. 1999;94:200–207. doi: 10.1111/j.1572-0241.1999.00797.x. [DOI] [PubMed] [Google Scholar]
- 9.Deitch EA, Xu D, Kaise VL. Role of the gut in the development of injury- and shock induced SIRS and MODS: the gut-lymph hypothesis, a review. Front Biosci. 2006;11:520–528. doi: 10.2741/1816. [DOI] [PubMed] [Google Scholar]
- 10.Li X, Chaudry IH, Choudhry MA. ERK and not p38 pathway is required for IL-12 restoration of T cell IL-2 and IFN-gamma in a rodent model of alcohol intoxication and burn injury. J Immunol. 2009;183:3955–3962. doi: 10.4049/jimmunol.0804103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choudhry MA, Fazal N, Goto M, et al. Gut-associated lymphoid T cell suppression enhances bacterial translocation in alcohol and burn injury. Am J Physiol Gastrointest Liver Physiol. 2002;282:G937–947. doi: 10.1152/ajpgi.00235.2001. [DOI] [PubMed] [Google Scholar]
- 12.Li X, Rana SN, Schwacha MG, et al. A novel role for IL-18 in corticosterone-mediated intestinal damage in a two-hit rodent model of alcohol intoxication and injury. J Leukoc Biol. 2006;80:367–375. doi: 10.1189/jlb.1205745. [DOI] [PubMed] [Google Scholar]
- 13.Li X, Schwacha MG, Chaudry IH, et al. A role of PP1/PP2A in mesenteric lymph node T cell suppression in a two-hit rodent model of alcohol intoxication and injury. J Leukoc Biol. 2006;79:453–462. doi: 10.1189/jlb.0705369. [DOI] [PubMed] [Google Scholar]
- 14.Cairns BA, Yamamoto H, Smith D, et al. Dehydroepiandrosterone fails to improve immunoglobulin synthesis and lymphocyte mitogenic response after burn injury. J Burn Care Rehabil. 1994;15:509–514. doi: 10.1097/00004630-199411000-00008. [DOI] [PubMed] [Google Scholar]
- 15.MacConmara MP, Tajima G, O’Leary F, et al. Regulatory T cells suppress antigen-driven CD4 T cell reactivity following injury. J Leukoc Biol. 2011;89:137–147. doi: 10.1189/jlb.0210082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Furukawa K, Kobayashi M, Herndon DN, et al. Appearance of monocyte chemoattractant protein 1 (MCP-1) early after thermal injury: role in the subsequent development of burn-associated type 2 T-cell responses. Ann Surg. 2002;236:112–119. doi: 10.1097/00000658-200207000-00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jeschke MG, Chinkes DL, Finnerty CC, et al. Pathophysiologic response to severe burn injury. Ann Surg. 2008;248:387–401. doi: 10.1097/SLA.0b013e3181856241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murphy T, Paterson H, Rogers S, et al. Use of intracellular cytokine staining and bacterial superantigen to document suppression of the adaptive immune system in injured patients. Ann Surg. 2003;238:401–410. doi: 10.1097/01.sla.0000086661.45300.14. discussion 410–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rendon JL, Choudhry MA. Th17 cells: critical mediators of host responses to burn injury and sepsis. J Leukoc Biol. 2012;92:529–538. doi: 10.1189/jlb.0212083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choudhry MA, Sayeed MM. Calcium signaling restitution prevents T-cell proliferative suppression by prostaglandin E2. Shock. 1996;6:101–105. doi: 10.1097/00024382-199608000-00004. [DOI] [PubMed] [Google Scholar]
- 21.Cardenas ME, Heitman J. Role of calcium in T-lymphocyte activation. Adv Second Messenger Phosphoprotein Res. 1995;30:281–298. doi: 10.1016/s1040-7952(05)80011-4. [DOI] [PubMed] [Google Scholar]
- 22.Fazal N, Choudhry MA, Sayeed MM. Inhibition of T cell MAPKs (Erk 1/2, p38) with thermal injury is related to down-regulation of Ca2+ signaling. Biochim Biophys Acta. 2005;1741:113–119. doi: 10.1016/j.bbadis.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 23.Chung DR, Kasper DL, Panzo RJ, et al. CD4+ T cells mediate abscess formation in intra-abdominal sepsis by an IL-17-dependent mechanism. J Immunol. 2003;170:1958–1963. doi: 10.4049/jimmunol.170.4.1958. [DOI] [PubMed] [Google Scholar]
- 24.Kobayashi T, Okamoto S, Hisamatsu T, et al. IL23 differentially regulates the Th1/Th17 balance in ulcerative colitis and Crohn’s disease. Gut. 2008;57:1682–1689. doi: 10.1136/gut.2007.135053. [DOI] [PubMed] [Google Scholar]
- 25.Sarra M, Pallone F, Macdonald TT, et al. IL-23/IL-17 axis in IBD. Inflamm Bowel Dis. 2010;16:1808–1813. doi: 10.1002/ibd.21248. [DOI] [PubMed] [Google Scholar]
- 26.Blaschitz C, Raffatellu M. Th17 cytokines and the gut mucosal barrier. J Clin Immunol. 2010;30:196–203. doi: 10.1007/s10875-010-9368-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trifari S, Kaplan CD, Tran EH, et al. Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from T(H)-17, T(H)1 and T(H)2 cells. Nat Immunol. 2009;10:864–871. doi: 10.1038/ni.1770. [DOI] [PubMed] [Google Scholar]
- 28.Ramirez JM, Brembilla NC, Sorg O, et al. Activation of the aryl hydrocarbon receptor reveals distinct requirements for IL-22 and IL-17 production by human T helper cells. Eur J Immunol. 2010;40:2450–2459. doi: 10.1002/eji.201040461. [DOI] [PubMed] [Google Scholar]
- 29.Duhen T, Geiger R, Jarrossay D, et al. Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells. Nat Immunol. 2009;10:857–863. doi: 10.1038/ni.1767. [DOI] [PubMed] [Google Scholar]
- 30.Kinugasa T, Sakaguchi T, Gu X, et al. Claudins regulate the intestinal barrier in response to immune mediators. Gastroenterology. 2000;118:1001–1011. doi: 10.1016/s0016-5085(00)70351-9. [DOI] [PubMed] [Google Scholar]
- 31.Zheng Y, Valdez PA, Danilenko DM, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 32.Kolls JK, McCray PB, Jr, Chan YR. Cytokine-mediated regulation of antimicrobial proteins. Nat Rev Immunol. 2008;8:829–835. doi: 10.1038/nri2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ki SH, Park O, Zheng M, et al. Interleukin-22 treatment ameliorates alcoholic liver injury in a murine model of chronic-binge ethanol feeding: role of signal transducer and activator of transcription 3. Hepatology. 2010;52:1291–1300. doi: 10.1002/hep.23837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fujino S, Andoh A, Bamba S, et al. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Inatsu A, Kogiso M, Jeschke MG, et al. Lack of Th17 cell generation in patients with severe burn injuries. J Immunol. 2011;187:2155–2161. doi: 10.4049/jimmunol.1003235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neely CJ, Maile R, Wang MJ, et al. Th17 (IFN gamma- IL17+) CD4+ T cells generated after burn injury may be a novel cellular mechanism for postburn immunosuppression. J Trauma. 2011;70:681–690. doi: 10.1097/TA.0b013e31820d18a6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lopez MC, Watzl B, Colombo LL, et al. Alterations in mouse Peyer’s patch lymphocyte phenotype after ethanol consumption. Alcohol. 1997;14:107–110. doi: 10.1016/s0741-8329(96)00104-8. [DOI] [PubMed] [Google Scholar]
- 38.Neutra MR, Frey A, Kraehenbuhl JP. Epithelial M cells: gateways for mucosal infection and immunization. Cell. 1996;86:345–348. doi: 10.1016/s0092-8674(00)80106-3. [DOI] [PubMed] [Google Scholar]
- 39.Li X, Akhtar S, Kovacs EJ, et al. Inflammatory response in multiple organs in a mouse model of acute alcohol intoxication and burn injury. J Burn Care Res. 2011;32:489–497. doi: 10.1097/BCR.0b013e3182223c9e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walker HL, Mason AD., Jr A standard animal burn. J Trauma. 1968;8:1049–1051. doi: 10.1097/00005373-196811000-00006. [DOI] [PubMed] [Google Scholar]
- 41.Kadaoui KA, Corthesy B. Secretory IgA mediates bacterial translocation to dendritic cells in mouse Peyer’s patches with restriction to mucosal compartment. J Immunol. 2007;179:7751–7757. doi: 10.4049/jimmunol.179.11.7751. [DOI] [PubMed] [Google Scholar]
- 42.Harrington LE, Hatton RD, Mangan PR, et al. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 43.Veldhoen M, Hirota K, Christensen J, et al. Natural agonists for aryl hydrocarbon receptor in culture medium are essential for optimal differentiation of Th17 T cells. J Exp Med. 2009;206:43–49. doi: 10.1084/jem.20081438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim SH, Henry EC, Kim DK, et al. Novel compound 2-methyl-2H-pyrazole-3-carboxylic acid (2-methyl-4-o-tolylazo-phenyl)-amide (CH-223191) prevents 2,3,7,8-TCDD-induced toxicity by antagonizing the aryl hydrocarbon receptor. Mol Pharmacol. 2006;69:1871–1878. doi: 10.1124/mol.105.021832. [DOI] [PubMed] [Google Scholar]
- 45.Gramatzki D, Pantazis G, Schittenhelm J, et al. Aryl hydrocarbon receptor inhibition downregulates the TGF-beta/Smad pathway in human glioblastoma cells. Oncogene. 2009;28:2593–2605. doi: 10.1038/onc.2009.104. [DOI] [PubMed] [Google Scholar]
- 46.Owens WE, Berg RD. Bacterial translocation from the gastrointestinal tract of athymic (nu/nu) mice. Infect Immun. 1980;27:461–467. doi: 10.1128/iai.27.2.461-467.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ghare S, Patil M, Hote P, et al. Ethanol inhibits lipid raft-mediated TCR signaling and IL-2 expression: potential mechanism of alcohol-induced immune suppression. Alcohol Clin Exp Res. 2011;35:1435–1444. doi: 10.1111/j.1530-0277.2011.01479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tilg H. Diet and intestinal immunity. N Engl J Med. 2012;366:181–183. doi: 10.1056/NEJMcibr1113158. [DOI] [PubMed] [Google Scholar]
- 49.Lee JS, Cella M, McDonald KG, et al. AHR drives the development of gut ILC22 cells and postnatal lymphoid tissues via pathways dependent on and independent of Notch. Nat Immunol. 2011;13:144–151. doi: 10.1038/ni.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kimura A, Naka T, Nohara K, et al. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc Natl Acad Sci U S A. 2008;105:9721–9726. doi: 10.1073/pnas.0804231105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li X, Rana SN, Kovacs EJ, et al. Corticosterone suppresses mesenteric lymph node T cells by inhibiting p38/ERK pathway and promotes bacterial translocation after alcohol and burn injury. Am J Physiol Regul Integr Comp Physiol. 2005;289:R37–R44. doi: 10.1152/ajpregu.00782.2004. [DOI] [PubMed] [Google Scholar]
- 52.Schwacha MG, Chaudry IH. The cellular basis of post-burn immunosuppression: macrophages and mediators. Int J Mol Med. 2002;10:239–243. [PubMed] [Google Scholar]
- 53.McGwin G, Jr, George RL, Cross JM, et al. Gender differences in mortality following burn injury. Shock. 2002;18:311–315. doi: 10.1097/00024382-200210000-00004. [DOI] [PubMed] [Google Scholar]
- 54.Akhtar S, Li X, Chaudry IH, et al. Neutrophil chemokines and their role in IL-18-mediated increase in neutrophil O2- production and intestinal edema following alcohol intoxication and burn injury. Am J Physiol Gastrointest Liver Physiol. 2009;297:G340–G347. doi: 10.1152/ajpgi.00044.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li X, Akhtar S, Choudhry MA. Alteration in intestine tight junction protein phosphorylation and apoptosis is associated with increase in IL-18 levels following alcohol intoxication and burn injury. Biochim Biophys Acta. 2012;1822:196–203. doi: 10.1016/j.bbadis.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kiss EA, Vonarbourg C, Kopfmann S, et al. Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science. 2011;334:1561–1565. doi: 10.1126/science.1214914. [DOI] [PubMed] [Google Scholar]
- 57.Li Y, Innocentin S, Withers DR, et al. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell. 2011;147:629–640. doi: 10.1016/j.cell.2011.09.025. [DOI] [PubMed] [Google Scholar]