

Abstract
This study examined the hypothesis that cardiomyocyte metabolism is inherently linked to the Ubiquitin Proteasome System. Rat neonatal ventricular cardiomyocytes were pulse-treated with 5 αM lactacystin for 30 min, resulting in 95% loss of proteasome activity, and then maintained in culture for up to 24 h. Pulse-treatment resulted in 36% decrease in cardiomyocyte mitochondrial reductase activity by 8 h which improved to 15% by 24 h. Bax proteins were increased 2.5-fold by 8 h but declined by 16 h. Similar effects were observed for ubiquitinated proteins suggesting recovery of proteasome function. Proteasome activity started to increase by 4 h and was back to baseline by 16 h. Multiple proteasome subunits, including α1, were upregulated with peak 2 to 2.5-fold increased protein levels at 8-16 h post-lactacystin which then declined. Incubating cardiomyocytes with 4 αM morpholino-antisense oligonucleotides to the α1-subunit for up to 24 h post-lactacystin diminished recovery of proteasome activity (45% at 24 h) and prevented the increase in α1 protein levels. Ubiquitinated proteins remained elevated and cardiomyocyte mitochondrial reductase activity was decreased 35% by 16 h. These results show that diminished function of the ubiquitin proteasome system decreases cardiomyocyte metabolism. If proteasome activity recovers, function improves, but preventing recovery diminishes metabolic function supporting the hypothesis that cardiomyocyte metabolism is inherently linked to the ubiquitin proteasome system.
Keywords: Ubiquitin proteasome system, apoptosis, cardiomyocyte, metabolic function
Introduction
The ubiquitin-proteasome system (UPS) represents the major nonlysosomal pathway for degradation of intracellular proteins. This pathway removes proteins that have been tagged with polyubiquitin at the e-NH2 group of a substrate lysine residue. Turnover of proteins by this system has been implicated in control of numerous cell functions, including the cell cycle and cell division [1,2], immune response and antigen presentation [3-5], apoptosis [6-8], and cell signaling [9-12]. In addition, the UPS plays critical roles in protein quality control by removal of damaged, oxidized and/or misfolded proteins [13-16]. Over the past several years, it has become increasingly clear that numerous diseases, such as Alzheimers’ disease, Huntingdons’ disease, and amyotrophic lateral sclerosis can be classified as proteasomal dysfunction disorders [17]. With regards to the cardiovascular and related systems, studies have shown that myocardial ischemia [18], various mutant protein-associated cardiomyopathies [16], atherosclerosis [19], and heart failure [20] are associated with some degree of UPS dysfunction. However, until recently, the prevailing belief was that the disease process caused the UPS dysfunction, and not that the dysfunction could cause the disease. Wang and co-workers [21,22] have provided convincing evidence that at least with respect to the heart, proteasomal dysfunction can not only worsen a disease process but may also be a direct cause.
Yet, despite these studies it has never been convincingly shown that overall metabolic function of a cell, let alone a cardiomyocyte, is dependent on proteasome function. This has always been inferred from the early yeast studies [23,24] which show that proteasome subunit knockouts are invariably lethal and from later studies that show that pharmacologic inhibition can result in apoptosis [7,8,25]. This study examined the hypothesis that cardiomyocyte metabolism is inherently linked to proteasome function. We present evidence that cardiomyocyte metabolic functions are diminished when proteasome is inhibited, yet will recover if inhibition is reversed.
Materials and methods
Animals
Female Sprague Dawley rats (19 days gravid) were obtained from Taconic Research Animal Breeders (Germantown, NY). All protocols were approved by the Institutional Animal Care and Utilization Committee and were in compliance with the NIH Guide for the Care and Use of Laboratory Animals (revised 1996).
Chemicals and reagents
Suc-LLVY-AMC, lactacystin (Lac), and the following antibodies: polyubiquitin (FK1) and proteasome subunits - β5 (poly), monoclonal α3 (MCP106), α6 (MCP257) and Rpt6 (P45-110) were obtained from Biomol Research Lab (Plymouth Meeting, PA). Polyclonal antibody for proteasome subunit, α1, was obtained from Abcam (Cambridge, MA). The morpholino-antisense oligonucleotide (morpholino-5’-TACTGGTTGCGAAACATAGCTCCGG-3’) directed against the α1 subunit of the proteasome (α1-MAO) was synthesized by Gene Tools LLC (Philomath OR) and was used in combination with the Endo-Porter® transport system (Gene Tools LLC). All other reagents were obtained from reputable sources.
Isolation and culture of neonatal cardiomyocytes
Ventricular cardiomyocytes were harvested from 1 to 2 day old neonatal rat pups as previously described [26] using established techniques [27,28]. Cardiomyocytes were maintained in DMEM/F-12 medium (Invitrogen, Carlsbad CA) containing 10% fetal bovine serum, penicillin/streptomycin, and Ara-C, 10 µmol/L.
Metabolic activity
Cell metabolic function was assessed using the MTT (dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) assay as described by Mosmann [29] using dimethyl sulfoxide to dissolve the crystals.
DNA content
Changes in DNA content was determined by assessing 5-bromo-2’-deoxyuridine (BrdU) incorporation into DNA using a commercially available kit (Cell Proliferation ELISA, BrdU (colorometric), Roche Applied Sciences, Indianapolis, IN).
26S-proteasome activity
Cardiomyocytes were disrupted using sonication into Hepes buffer (50 mmol/L) containing: KCl 20 mmol/L, MgCl2 5 mmol/L, DTT 1 mmol/L, pH 7.5, and then centrifuged at 10,000xg for 30 min, at 4°C. ATP-stimulated proteasome chymotryptic activity was determined in cell lysates using the method described by Reinheckel et al [30] as modified by Powell et al [31] over a range of ATP concentrations from 7-56 µmol/L.
Western blotting
To prepare lysate, cardiomyocytes were suspended in RIPA buffer containing (mmol/L): NaCl 150, Tris 50, 1% NP-40, 0.5% DOC, 0.1% SDS, pH 8.0, sheared, and then centrifuged at 10,000g for 5 min. Lysate proteins (10-50 µg) were separated on 4-20% Tris-HCl gels (Ready Gel® (Bio-Rad Laboratories, Hercules, CA)) using standard SDS-PAGE [32] with immuno-blotting carried out using standard techniques, and developed with an enhanced chemiluminescence (ECL) kit (Perkin Elmer Life Sciences, Boston MA).
Statistical analysis
All results are expressed as mean ± SEM. Statistical significance of differences between two populations with equal variance was with a Students t-test. Analysis of differences between multiple sample populations with equal variances was with one way ANOVA followed by Tukeys test for post-hoc analysis. Statistical differences of p<0.05 were considered to be significant. All statistics were performed using the SigmaStat statistical analysis package (Jandel Scientific (SSI), Richmond, CA).
Results
In these experiments two protocols, pulse-treatment with Lac, or pulse-treatment with Lac followed by sustained incubation with a morpholino-based antisense oligonucleotide directed against the α1 subunit of the proteasome, were assessed for their effects on cardiomyocyte metabolic function. These two protocols had decidedly different effects so results are described separately. However, because it is necessary to compare the two protocols with each other the results may be summarized in a single figure.
Effects of pulse-treatment with Lac
To examine the effect of short term inhibition of proteasome on cardiomyocyte metabolic function RNVCs were pulse-treated with Lac, 5 µmol/l, for 30 min, after which the inhibitor was washed out and the cells incubated for up to 24 h.
Recovery of proteasome activity
Pulse treatment with Lac resulted in 95% loss of 26S proteasome chymotryptic activity within 30 min (Figure 1A). Proteasome activity started to recover by 4 h post-treatment and was not different from baseline by 8 h. Initial inhibition of proteasome was accompanied by incremental increases in cardiomyocyte ubiquitinated proteins which peaked at 8 h, then decreased by 16 h, and by 24 h was back at baseline (Figure 1B - top) consistent with the changes in proteasome activity. Recovery of proteasome activity was accompanied by a concerted upregulation and increase in de novo synthesis of all proteasome subunits analyzed including α3, α6, β5 and Rpt 6 (Figure 2) and α-1 (Figure 3). All of the subunits showed a similar pattern of expression which typically was an incremental increase in protein content peaking at 8 to 16 h and then decreasing towards baseline by 24 h (Figure 2 - bottom panel presents semi-quantitative analysis of the α3 subunit).
Figure 1.
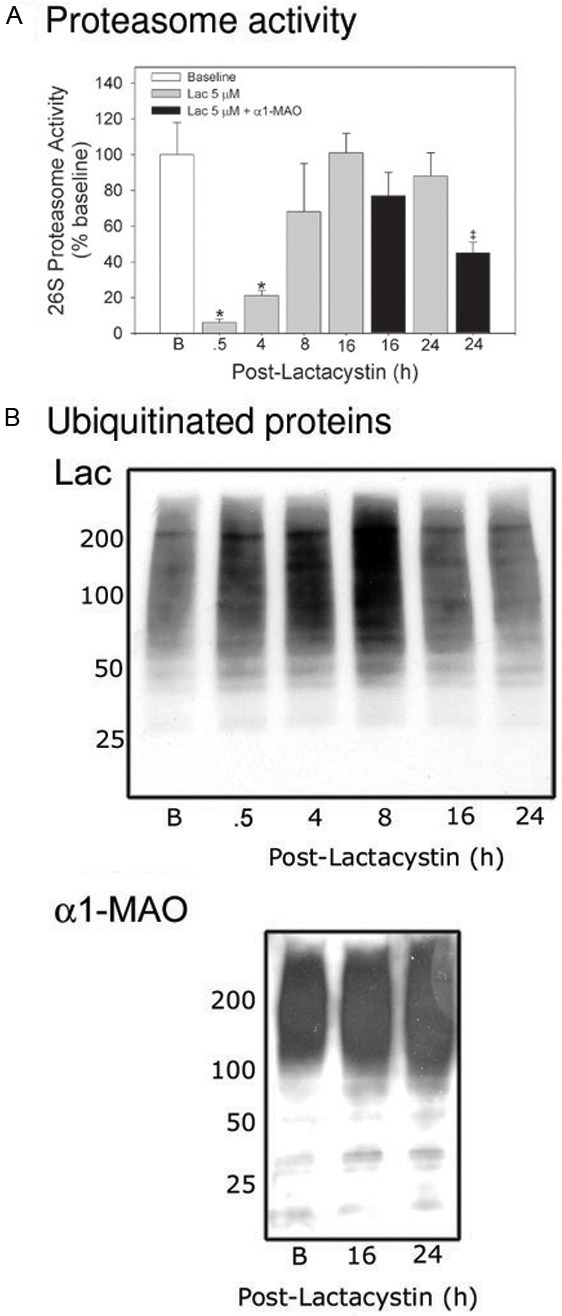
Cardiomyocyte proteasome activity recovers after pulse-treatment with Lac but is diminished when treatment is followed by α1-MAO. RNVCs were pulse-treated with Lac, 5 µmol/L, and then maintained in culture for up to 24 h. The times indicated in the figure represent elapsed time (h) after addition of Lac. In a separate set of experiments, RNVCs were pulse-treated with Lac, 5 µmol/l, for 30 min after which the cells were incubated with α1-MAO for 16 and 24 h. The cells were then harvested and (A) 26S-proteasome activity determined or (B) ubiquitinated proteins assessed. The values are expressed as percent of baseline (mean value = 6127 ± 1129 AFU/h/mg protein) and represent the mean ± SEM of 3 to 10 separate determinations. The images in panel b are representative of 3 separate experiments *P<0.05 (ANOVA) when compared with baseline values. ‡P<0.05 (t-test) when compared with corresponding Lac alone group only.
Figure 2.
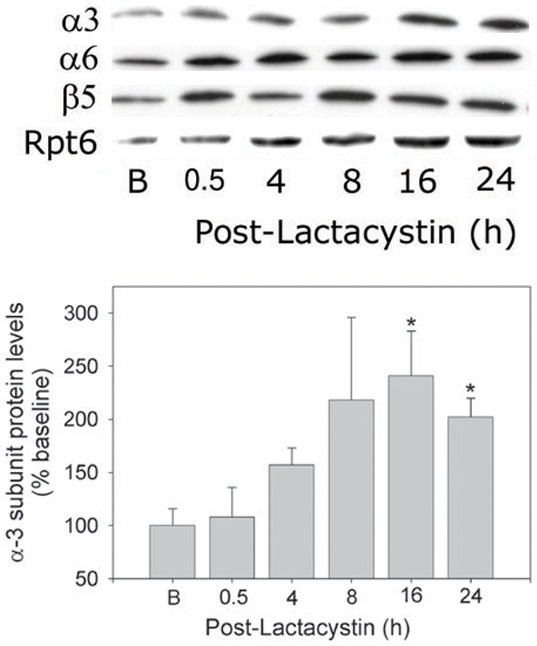
Concerted upregulation of 26S-proteasome subunits following pulse-treatment of cardiomyocytes with Lactacystin. RNVCs were pulse-treated with Lac, 5 µmol/l, for 30 min, and then maintained in culture for up to 24 h. The times indicated in the figure represent elapsed time (h) after addition of Lac. At the indicated time points, RNVCs were harvested and α3, α6, β5, and Rpt6 subunit protein levels determined using immunoblot methods. Top panel, Composite membrane depicting changes in protein levels (representative of 3 to 4 separate experiments). Bottom panel, Densitometry of changes in α3 subunit protein levels. The values are expressed as the % of baseline and represent the mean ± SEM of 3 to 4 separate experiments. *P<0.05 (ANOVA) when compared with baseline values.
Figure 3.
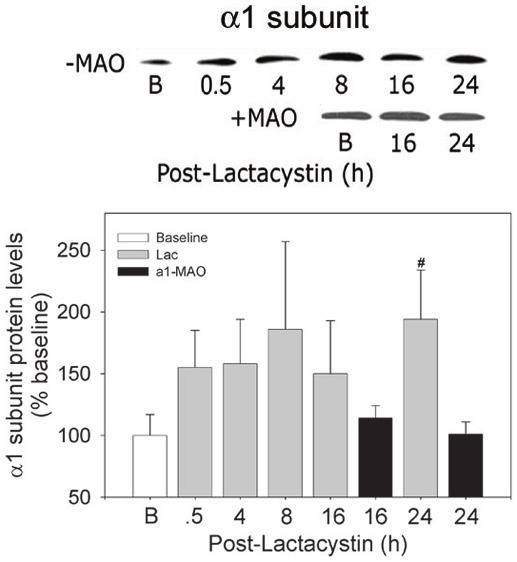
Diminished α1 subunit expression following treatment of cardiomyocytes with antisense oligonucleotides. RNVCs were pulse-treated with Lac, 5 µmol/L, for 30 min, and then maintained in culture for up to 24 h. In a separate set of experiments, RNVCs were pulse-treated with Lac, 5 µmol/l, for 30 min after which the cells were incubated with α1-MAO for 16 and 24 h. At the indicated time points, RNVCs were harvested and protein levels of the α1 subunit determined using immunoblot techniques. Top panel, composite depicting changes in α1 protein levels in the absence (-MAO) and presence (+MAO) of the antisense oligonucleotide. Bottom panel, densitometry of α1 subunit protein. The depicted membranes are representative of 3 to 4 separate experiments. The values are expressed as the percent of the baseline values and are expressed as the mean ± SEM of 3 to 4 separate experiments. #P<0.05 (ANOVA) when compared with baseline and corresponding + α1-MAO values.
Bax protein levels
We analyzed Bax protein levels as a representative proteasome regulated protein [33]. Bax was observed to be significantly (P<0.05) increased by about 2.5-fold at 8 h (Figure 4A - top). Yet, none of the changes in Bax protein content were accompanied by corresponding increases in Bax mRNA levels (Figure 4B) suggesting stabilization rather than increased expression of the protein.
Figure 4.
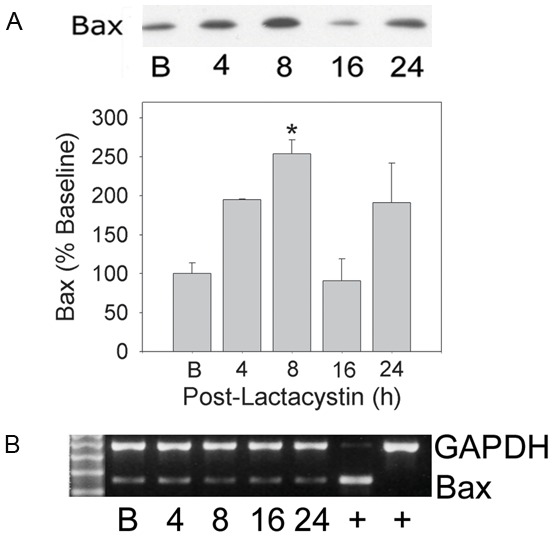
Changes in Bax protein in cardiomyocytes pulse-treated with Lac alone. RNVCs were pulse-treated with Lac, 5 µmol/L, for 30 min, and then maintained in culture for 24 h. At the indicated time points cells were harvested and Bax protein determined. Panel (A) (upper) Bax protein following pulse treatment with Lac alone; (lower) densitometry of Bax protein. Panel (B) Bax mRNA determination. Depicted images are representative of 3 to 4 separate experiments. *P<0.05 (ANOVA) when compared with control group.
Metabolic activity
The MTT assay was then used to measure overall cardiomyocyte metabolic activity through NADP(H)-dependent oxidoreductases. Pulse treatment of cardiomyocytes with Lac had a biphasic effect on metabolic activity as determined by the MTT assay (Figure 5A). Metabolic activity was depressed by 36% (P<0.05) by 8 h post-treatment, however recovered to a large degree and stabilized about 15% lower than baseline by 24 h. DNA content studies with BrdU (Figure 5B) indicated that the initial decrease in cardiomyocyte metabolic activity was not associated with cell death and the recovery was not associated with cell division.
Figure 5.
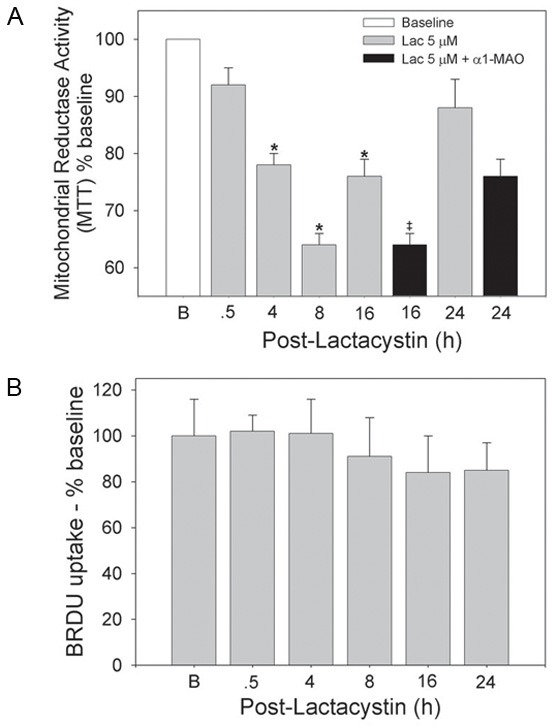
Reversible suppression of cardiomyocyte metabolic activity following pulse treatment of cardiomyocytes with Lac alone is more sustained when followed by α1-MAO. RNVCs were pulse-treated with Lac, 5 µmol/l, for 30 min and then maintained in culture for up to 24 h. In a separate set of experiments, RNVCs were pulse-treated with Lac, 5 µmol/l, for 30 min after which the cells were incubated with α1-MAO for 16 and 24 h. Cells were assessed for (A) metabolic activity and (B) BrdU uptake at the indicated times. The values represent the means ± SEM of 3 to 8 separate experiments. *P<0.05 (ANOVA) when compared with 0.5 h time point. ‡P<0.05 (t-test) when compared with corresponding Lac alone group only.
Effects of pulse-treatment with Lac followed by incubation with α1-MAO
To determine if the reversible effects observed with pulse-treatment with Lac could be prevented, experiments were conducted to inhibit recovery of proteasome activity. To be certain that the effects were due to proteasome inhibition, proteasome was knocked down using a gene silencing technique. Following pulse-treatment with Lac, RNVCs were incubated with α1-MAO (4 µmol/l plus Endo-Porter®, 6 µmol/l) for the remainder of the experiment. To allow time for endocytosis of the antisense oligo these experiments concentrated on the later time points, 16 h and 24 h post-Lac treatment.
Diminished recovery of proteasome activity
Unlike Lac alone, treatment with α1-MAO resulted in suppression of 26S proteasome activity which was most apparent at 24 h (P<0.05) which recovered to only 45% of baseline (Figure 1A). Diminished recovery of proteasome activity was accompanied by incremental increases in cardiomyocyte content of ubiquitinated proteins over baseline at 16 and 24 h (Figure 1B - bottom) in contrast to that observed with Lac alone (Figure 1B - top) at the same time points. Treatment with the α1-MAO almost completely suppressed the de novo synthesis of α1 subunit protein (Figure 3) to levels no greater than baseline).
Metabolic activity
Incubation with the α1-MAO resulted in sustained suppression of cardiomyocyte metabolic activity as indicated by depressed NADP(H) oxidoreductase activity (Figure 5A). Metabolic activity was significantly less than that observed with Lac alone (P<0.05) at the 16 h time point averaging a 30-35% decrease and was still depressed at 24 h. Incubation with the α1-MAO in the absence of the Endo-Porter had no effect on metabolic activity (data not shown).
Discussion
This study demonstrates that reversible inhibition of the UPS impairs cardiomyocyte metabolic function which improves upon recovery of UPS function. However, if steps are taken to diminish recovery of the UPS cardiomyocyte metabolic function remains depressed. While this may seem intuitive it has previously never been demonstrated. Proteasomal dysfunction has now been described in a variety of disorders. With respect to the cardiovascular system, myocardial ischemia [34] and heart failure [20,35] are two prominent cardiac pathologies possessing a component of proteasome dysfunction. Both of these pathologies have diminished cell function as a prominent component but it is unclear whether this can be directly related to the dysfunctional proteasome. The results of the current studies would suggest such a relationship and that cell metabolic function can be directly related to overall proteasome function.
To demonstrate this relationship it was necessary to devise a means of reversibly inhibiting proteasome since sustained inhibition of cardiomyocyte proteasome invariably leads to cell death [26]. This was accomplished by pulse-treatment of neonatal cardiomyocytes with a concentration of Lac that inhibited proteasome virtually completely. Following washout, proteasome activity recovered over a period of hours even though Lac is often thought to be irreversible. Recovery was due to a concerted upregulation of proteasome subunits with peak levels of most of the subunits occurring within 8 to 16 h consistent with a previous study in vascular smooth muscle cells [36]. Aside from the obvious focus on the cardiomyocyte, the current study differs from this previous study in that newly synthesized proteasome was demonstrated to be functional as increased cell content of ubiquitinated proteins diminished as function of the UPS returned.
Reversible dysfunction of the UPS was associated with reversible depression of mitochondrial NADP(H) oxidoreductase activity as measured by the MTT assay. The actual mechanism for this effect is not clear although it could be related to dysregulation of the multitude of signaling pathways regulated by the UPS. It is now recognized that many pro- and anti-apoptotic proteins are regulated by the UPS and include NFkB which is regulated at multiple levels [37], the JNK and JAK-STAT pathways [11], the Bcl-2 family, including Bax [38], and Smac/DIABLO [39]. Evidence for interference in degradation of such a protein can be garnered from the BAX experiments which demonstrated 2.5-fold increases in Bax protein levels which peaked at 8 h and then declined as proteasome activity recovered. That this small increase represents protein stabilization is very strongly supported by the lack of corresponding changes in mRNA levels for this protein. Moreover, there does not appear to be significant cell death as indicated by the BrdU results suggesting depression of metabolic function.
To show proof of concept recovery of the proteasome after pulse-treatment with Lac had to be prevented. Since upregulation of proteasome subunits during treatment with inhibitors involves expression of new protein [36], the easiest way to accomplish this would have been use of a protein synthesis inhibitor. Unfortunately, this involved incubation with cycloheximide for prolonged periods which invariably killed all of the cells (not shown). For this reason, an alternative and more specific approach was employed. Cardiomyocytes were incubated with a morpholino-based antisense oligonucleotide directed against the α1 subunit of the proteasome. The sequence of this oligo is based on a previous study showing that prolonged incubation of fibroblasts with the naked 15-mer antisense oligo can suppress proteasome activity [40]. Morpholino-based oligos are excellent substrates for endocytosis via the Endo-Porter® transport system [41] and have been used previously in cardiomyocytes [42]. Preliminary experiments with a fluorescent morpholino-based oligo showed rapid penetration into cardiomyocytes (not shown). Pulse-treatment with Lac followed by the α1-MAO resulted in a more sustained suppression of 26S proteasome activity which demonstrated about 50% recovery by 24 h. The upregulation of the α1 subunit associated with Lac pulse-treatment was virtually completely prevented. Suppression of proteasome activity was confirmed by the presence of increased content of ubiquitinated proteins. From a functional standpoint, this consecutive treatment protocol resulted in sustained irreversible suppression of cardiomyocyte NADP(H) oxidoreductase activity.
Conclusions
It is now becoming clear that many pathologies thought to involve primarily excessive activation of signaling pathways have as a component, dysfunction of the UPS. In its role as the major cellular degradative organelle, the proteasome would serve to degrade these signaling molecules resulting in cessation of their biological effect. However, in the presence of a severely dysfunctional UPS, activity of these pathways can not be terminated and what may have been an initial adaptive response becomes lethal. In the current study we demonstrate diminished cardiomyocyte NADP(H) oxidoreductase activity coincident with proteasome inhibition that is reversible if proteasome can recover activity within a short time frame. However, if UPS recovery is interfered with cardiomyocyte metabolic function does not recover. These observations support the hypothesis that cardiomyocyte viability is inherently linked to proteasome function and lend credence to the concept that proteasome dysfunction can initiate disease as suggested by the in vivo studies of Wang and co-workers [21,22].
Acknowledgements
These studies were supported by NIH R01-HL68936 to SRP.
Disclosure of conflict of interest
None.
Abbreviations
- BrdU
5-bromo-2’-deoxyuridine
- Lac
lactacystin
- MAO
morpholino-antisense oligonucleotide
- MTT
dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide
- RNVC
rat neonatal ventricular myocytes
- TUNEL
Terminal transferase-dUTP-Nick-End Labeling
- UPS
ubiquitin proteasome system
References
- 1.Castro A, Bernis C, Vigneron S, Labbe JC, Lorca T. The anaphase-promoting complex: a key factor in the regulation of cell cycle. Oncogene. 2005;24:314–325. doi: 10.1038/sj.onc.1207973. [DOI] [PubMed] [Google Scholar]
- 2.Hershko A. Roles of ubiquitin-mediated proteolysis in cell cycle control. Curr Opin Cell Biol. 1997;9:788–799. doi: 10.1016/s0955-0674(97)80079-8. [DOI] [PubMed] [Google Scholar]
- 3.Goldberg AL, Gaczynska M, Grant E, Michalek M, Rock KL. Functions of the proteasome in antigen presentation. Cold Spring Harb Symp Quant Biol. 1995;60:479–490. doi: 10.1101/sqb.1995.060.01.052. [DOI] [PubMed] [Google Scholar]
- 4.Goldberg AL, Cascio P, Saric T, Rock KL. The importance of the proteasome and subsequent proteolytic steps in the generation of antigenic peptides. Mol Immunol. 2002;39:147–164. doi: 10.1016/s0161-5890(02)00098-6. [DOI] [PubMed] [Google Scholar]
- 5.Kloetzel PM. Antigen processing by the proteasome. Nat Rev Mol Cell Biol. 2001;2:179–187. doi: 10.1038/35056572. [DOI] [PubMed] [Google Scholar]
- 6.Grimm LM, Osborne BA. Apoptosis and the proteasome. Results Probl Cell Differ. 1999;23:209–228. doi: 10.1007/978-3-540-69184-6_10. [DOI] [PubMed] [Google Scholar]
- 7.Jesenberger V, Jentsch S. Deadly encounter: ubiquitin meets apoptosis. Nat Rev Mol Cell Biol. 2002;3:112–121. doi: 10.1038/nrm731. [DOI] [PubMed] [Google Scholar]
- 8.Orlowski RA. The role of the ubiquitin-proteasome pathway in apoptosis. Cell Death Differ. 1999;6:303–313. doi: 10.1038/sj.cdd.4400505. [DOI] [PubMed] [Google Scholar]
- 9.Ciechanover A, Orian A, Schwartz AL. Ubiquitin-mediated proteolysis: biological regulation via destruction. Bioessays. 2000;22:442–451. doi: 10.1002/(SICI)1521-1878(200005)22:5<442::AID-BIES6>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 10.Desterro JM, Rodriguez MS, Hay RT. Regulation of transcription factors by protein degradation. Cell Mol Life Sci. 2000;57:1207–1219. doi: 10.1007/PL00000760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fuchs SY, Fried VA, Ronai Z. Stress-activated kinases regulate protein stability. Oncogene. 1998;17:1483–1490. doi: 10.1038/sj.onc.1202184. [DOI] [PubMed] [Google Scholar]
- 12.Haglund K, Di Fiore PP, Dikic I. Distinct monoubiquitin signals in receptor endocytosis. Trends Biochem Sci. 2003;28:598–603. doi: 10.1016/j.tibs.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 13.Divald A, Powell SR. Proteasome mediates removal of proteins oxidized during myocardial ischemia. Free Radic Biol Med. 2006;40:156–164. doi: 10.1016/j.freeradbiomed.2005.09.022. [DOI] [PubMed] [Google Scholar]
- 14.Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003;426:895–899. doi: 10.1038/nature02263. [DOI] [PubMed] [Google Scholar]
- 15.Hirsch C, Jarosch E, Sommer T, Wolf DH. Endoplasmic reticulum-associated protein degradation--one model fits all? Biochim Biophys Acta. 2004;1695:215–223. doi: 10.1016/j.bbamcr.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 16.Su H, Wang X. The ubiquitin-proteasome system in cardiac proteinopathy: a quality control perspective. Cardiovasc Res. 2010;85:253–262. doi: 10.1093/cvr/cvp287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Powell SR, Divald A. The ubiquitin-proteasome system in myocardial ischaemia and preconditioning. Cardiovasc Res. 2010;85:303–311. doi: 10.1093/cvr/cvp321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Herrmann J, Ciechanover A, Lerman LO, Lerman A. The ubiquitin-proteasome system in cardiovascular diseases-a hypothesis extended. Cardiovasc Res. 2004;61:11–21. doi: 10.1016/j.cardiores.2003.09.033. [DOI] [PubMed] [Google Scholar]
- 20.Predmore JM, Wang P, Davis F, Bartolone S, Westfall MV, Dyke DB, Pagani F, Powell SR, Day SM. Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation. 2010;21:997–1004. doi: 10.1161/CIRCULATIONAHA.109.904557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tian Z, Zheng H, Li J, Li YF, Su H, Wang X. Genetically induced moderate inhibition of the proteasome in cardiomyocytes exacerbates myocardial ischemia-teperfusion injury in mice. Circ Res. 2012;111:532–542. doi: 10.1161/CIRCRESAHA.112.270983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li J, Horak KM, Su H, Sanbe A, Robbins J, Wang X. Enhancement of proteasomal function protects against cardiac proteinopathy and ischemia/reperfusion injury in mice. J Clin Invest. 2011;121:3689–3700. doi: 10.1172/JCI45709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ghislain M, Udvardy A, Mann C. S. cerevisiae 26S protease mutants arrest cell division in G2/metaphase. Nature. 1993;366:358–62. doi: 10.1038/366358a0. [DOI] [PubMed] [Google Scholar]
- 24.Gordon C, McGurk G, Wallace M, Hastie ND. A conditional lethal mutant in the fission yeast 26 S protease subunit mts3+ is defective in metaphase to anaphase transition. J Biol Chem. 1996;271:5704–5711. doi: 10.1074/jbc.271.10.5704. [DOI] [PubMed] [Google Scholar]
- 25.Powell SR, Gurzenda EM, Mantell LL, Teichberg S, Maulik D. Association of increased ubiquinated proteins with cardiac apoptosis. Antioxid Redox Signal. 2000;2:103–112. doi: 10.1089/ars.2000.2.1-103. [DOI] [PubMed] [Google Scholar]
- 26.Powell SR, Wang P, Divald A, Teichberg S, Haridas V, McCloskey TW, Davies KJ, Katzeff H. Aggregates of oxidized proteins (lipofuscins) induce apoptosis through proteasome inhibition and dysregulation of pro-apoptotic proteins. Free Radic Biol Med. 2005;38:1093–1101. doi: 10.1016/j.freeradbiomed.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 27.Sun ZQ, Ojamaa K, Nakamura TY, Artman M, Klein I, Coetzee WA. Thyroid hormone increases pacemaker activity in rat neonatal atrial myocytes. J Mol Cell Cardiol. 2001;33:811–824. doi: 10.1006/jmcc.2001.1353. [DOI] [PubMed] [Google Scholar]
- 28.Engelmann GL, McTierman C, Gerrity RG, Samarel AM. Serum-free primary cultures of neonatal rat cardiomyocytes: cellular and molecular applications. Technique (philadelphia) 1990;2:279–291. [Google Scholar]
- 29.Mosmann T. Rapid colorometric assay for cellular growth and survival: application to proliferation and cytotoxicity. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 30.Reinheckel T, Sitte N, Ullrich O, Kuckelkorn U, Davies KJ, Grune T. Comparative resistance of the 20S and 26S proteasome to oxidative stress. Biochem J. 1998;335:637–642. doi: 10.1042/bj3350637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Powell SR, Davies KJ, Divald A. Optimal determination of heart tissue 26S Proteasome activity requires maximal stimulating concentrations of ATP. J Mol Cell Cardiol. 2007;42:265–9. doi: 10.1016/j.yjmcc.2006.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 33.Breitschopf K, Zeiher AM, Dimmeler S. Ubiquitin-mediated degradation of the proapoptotic active form of bid. A functional consequence on apoptosis induction. J Biol Chem. 2000;275:21648–21652. doi: 10.1074/jbc.M001083200. [DOI] [PubMed] [Google Scholar]
- 34.Powell SR. The ubiquitin proteasome system in cardiac physiology and pathology. Am J Physiol Heart Circ Physiol. 2006;291:H1–H19. doi: 10.1152/ajpheart.00062.2006. [DOI] [PubMed] [Google Scholar]
- 35.Wang X, Robbins J. Heart failure and protein quality control. Circ Res. 2006;99:1315–1328. doi: 10.1161/01.RES.0000252342.61447.a2. [DOI] [PubMed] [Google Scholar]
- 36.Meiners S, Heyken D, Weller A, Ludwig A, Stangl K, Kloetzel PM, Kruger E. Inhibition of proteasome activity induces concerted expression of proteasome genes and de novo formation of Mammalian proteasomes. J Biol Chem. 2003;278:21517–21525. doi: 10.1074/jbc.M301032200. [DOI] [PubMed] [Google Scholar]
- 37.Krappmann D, Scheidereit C. A pervasive role of ubiquitin conjugation in activation and termination of IkappaB kinase pathways. EMBO Rep. 2005;6:321–326. doi: 10.1038/sj.embor.7400380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marshansky V, Wang X, Bertrand R, Luo H, Duguid W, Chinnadurai G, Kanaan N, Vu MD, Wu J. Proteasomes modulate balance among proapoptotic and antiapoptotic Bcl-2 family members and compromise functioning of the electron transport chain in leukemic cells. J Immunol. 2001;166:3130–3142. doi: 10.4049/jimmunol.166.5.3130. [DOI] [PubMed] [Google Scholar]
- 39.MacFarlane M, Merrison W, Bratton SB, Cohen GM. Proteasome-mediated degradation of Smac during apoptosis: XIAP promotes Smac ubiquitination in vitro. J Biol Chem. 2002;277:36611–36616. doi: 10.1074/jbc.M200317200. [DOI] [PubMed] [Google Scholar]
- 40.Grune T, Reinheckel T, Joshi M, Davies KJ. Proteolysis in cultured liver epithelial cells during oxidative stress. Role of the multicatalytic proteinase complex, proteasome. J Biol Chem. 1995;270:2344–2351. doi: 10.1074/jbc.270.5.2344. [DOI] [PubMed] [Google Scholar]
- 41.Summerton JE. Endo-Porter: a novel reagent for safe, effective delivery of substances into cells. Ann N Y Acad Sci. 2005;1058:62–75. doi: 10.1196/annals.1359.012. [DOI] [PubMed] [Google Scholar]
- 42.Masaki M, Izumi M, Oshima Y, Nakaoka Y, Kuroda T, Kimura R, Sugiyama S, Terai K, Kitakaze M, Yamauchi-Takihara K, Kawase I, Hirota H. Smad1 protects cardiomyocytes from ischemia-reperfusion injury. Circulation. 2005;111:2752–2759. doi: 10.1161/CIRCULATIONAHA.104.490946. [DOI] [PubMed] [Google Scholar]