

Abstract
Previously, we demonstrated that Tim-1-Fc prevents acute cardiac graft rejection by inhibiting Th1 response. In the present report, we tackled the impact of Tim-1-Fc on Th17 cells in a model of cardiac chronic rejection. Administration of Tim-1-Fc did not result in a detectable impact on innate immunity and regulatory T cells, while it provided protection for Bm12-derive cardiac grafts against chronic rejection in B6 recipients, as manifested by the reduction of inflammatory infiltration along with less severity of vasculopathy. Studies in T-bet-/- recipients by implanting Bm12-derived cardiac grafts further revealed that Tim-1-Fc significantly protected cardiac grafts from chronic rejection along with attenuated production of IL-17 producing T cells. Depletion of CD4 and CD8 T cells or blockade of IL-17 in T-bet-/- recipients demonstrated that Tim-1-Fc selectively suppresses Th17 differentiation along with attenuated IL-17 secretion. Together, our data suggest that Tim-1-Fc protects cardiac grafts from chronic rejection by suppressing CD4 Th17 development and functionality. Therefore, Tim-1-Fc might be a potential immunosuppressive agent in the setting of cardiac transplantation.
Keywords: Th17, Tim-1, vasculopathy
Introduction
Cardiac transplantation is an effective treatment for congestive heart failure, particularly for those patients resistant to aggressive medical therapy [1,2]. Although short-term survival rate for patients after cardiac transplantation has been greatly improved [3,4], long-term survival is still challenged by chronic rejection, one of the major risk factors for such patients [5,6]. A characteristic feature for chronic rejection is the manifestation of coronary artery disease or cardiac vasculopathy, which is associated with coronary luminal occlusion and eventual graft failure [6,7]. Despite past extensive studies, the cellular and molecular mechanisms underlying chronic rejection, however, are yet to be fully elucidated [8-10].
Generally, Th1 mediated immune response along with activation of macrophages are thought to be responsible for allograft rejection, while Th2 response is considered beneficial to long-term allograft survival [11]. Nevertheless, fully mismatched cardiac allografts were also rapidly rejected in recipient mice deficient in IFN-γ or STAT4, the two key molecules essential for Th1 response [12,13]. Particularly, cardiac recipient mice lack of Th1 transcription factor T-bet, displayed exacerbated vasculopathy [14]. Together, these observations challenge the above described dogma. Indeed, there is ample evidence indicating a role for Th17 cells and IL-17 in allograft rejection, especially in the pathological remodeling during the course of chronic rejection [2]. IL-17 is a cytokine associated with inflammation, angiogenesis and fibrosis, which are characteristic features relevant to chronic cardiac rejection [14]. In line with these results, Th17 response has been noted with implication in the pathogenesis of chronic renal graft rejection in humans [15], and chronic cardiac rejection in mouse models [16,17].
Tim-1 belongs to the Tim protein family implicated in the regulation of Th1 and Th2 response. Ligation of Tim-1 on T cells with its ligand Tim-4 on antigen presenting cells provides co-stimulatory signals for T cell activation and proliferation [18-20]. However, the regulation of immune responses by Tim-1 and Tim-4 are much more complex than what we originally thought. For example, Tim-4-Ig can either stimulate or inhibit T cell proliferation based on the dose administered [18,20,21], while anti-Tim-1 mAbs are also found to serve as a double-edged sword in T cell activation given their differences in binding affinity [22,23]. Previously, we demonstrated evidence suggesting the existence of a novel Tim-1 ligand other than the aforementioned Tim-4, and by which Tim-1-Fc suppresses allograft acute rejection [24]. In the present report, we extended our studies of Tim-1-Fc to chronic cardiac vasculopathy. By transplantation of Bm12-derived cardiac grafts into B6 and T-bet-/- mice, we obtained evidence supporting that Tim-1-Fc attenuates chronic cardiac graft rejection by suppressing Th17 differentiation and functionality.
Materials and methods
Mice
C57BL/6 mice were purchased from Joint Ventures Sipper BK Experimental Animals Co. (Shanghai, China). B6-Bm12 (Bm12) mice and Tbx21-/- (the gene encodes T-bet) mice in B6 background were obtained from the Jackson’s Laboratory (Bar Harbor, ME, USA). The animal protocol of this study was approved by the Animal Care and Use Committee at the Second Military Medical University.
Antibodies and reagents
Anti-CD4 (L3T4), anti-CD8 (Ly2), anti-IFN-γ (XMG1.2), and anti-IL-17A (TC11-18H10) were purchased from BD Pharmalgen (San Diego, CA, USA). Mouse IL-17A mAb (MAB421) was from R&D Systems (Minneapolis, MN, USA). Recombinant mouse IL-17A was from BioLegend (San Diego, CA, USA). Tim-1-Fc was prepared as previously described [24].
Heart transplantation
Cardiac grafts from Bm12 donors were implanted into B6 or T-bet-/- mice as previously described [25]. Recipient mice were injected intraperitoneally every other day with 10 mg/kg of Tim-1-Fc or hIgG1 until day 14. In some cases, recombinant IL-17 (200 ng/mouse) or anti-IL-17 (0.1 mg/mouse) was also administered. The contraction of heart grafts was monitored daily by two independent observers without prior knowledge of the treatment protocol. The complete cessation of cardiac contraction was defined as the endpoint.
Histological analysis
Cardiac grafts were harvested on indicated days and fixed in 10% formalin and embedded in paraffin. Sections were cut at 4 mm, and were counterstained for 1 min with hematoxylin eosin. The severity of vasculopathy was graded according to the percentage of luminal occlusion by intimal thickening with a scoring system described previously [26,27]. Briefly, a vessel score of 0 indicated a normal artery; 1, <10% luminal occlusion; 2, 20 to 50% luminal occlusion; and 3, >50% luminal occlusion. Only vessels that were cut orthogonally and displayed a clear internal elastic lamina were scored. An examiner blinded to the groups scored all the samples.
Th17 cell differentiation
T cells were enriched from splenocytes using a mouse MACS CD4+ T cell kit (Miltenyi Biotec, Bergisch Gladbach, Germany). The cells were activated by plate-bound anti-CD3 (5 μg/ml) and anti-CD28 (5 μg/ml) (Biolegend, San Diego, CA, USA) for 3 days. For induction of Th17 differentiation with DCs, CD4 T cells were cocultured with DCs along with the addition of anti-CD3 into the cultures. For Th17 polarization, naïve CD4 T cells were cultured with IL-6 (10 ng/ml) and TGF-β1 (5 ng/ml) in the presence of anti-IFN-γ (10 μg/ml) and anti-IL-4 (10 μg/ml). All cytokines were obtained from R&D Systems (Minneapolis, MN, USA).
RNA isolation and real-time PCR
RNA was isolated using RNAfast200 (Fastagen, China) according to the manufacturer’s instructions. Intragraft expression of IL-2, IFN-γ, IL-4, IL-17, CD11b, CD3 and IL-6 were quantified by real-time RT-PCR. β-actin was used as an endogenous control. The 2-ΔΔCt method was used to calculate the fold change as reported [28]. Primer sequences used in this study are shown in Table 1.
Table 1.
Real time PCR Primers sequences
| Gene symbol | Primer sequence (5’ to 3’) |
|---|---|
| Il2 | Forward TGAGCAGGATGGAGAATTACAGG |
| Reverse GTCCAAGTTCATCTTCTAGGCAC | |
| Il17a | Forward TTTAACTCCCTTGGCGCAAAA |
| Reverse CTTTCCCTCCGCATTGACAC | |
| Il6 | Forward CCAAGAGGTGAGTGCTTCCC |
| Reverse CTGTTGTTCAGACTCTCTCCCT | |
| Il4 | Forward GCCGATGATCTCTCTCAAGTGAT |
| Reverse GCCGATGATCTCTCTCAAGTGAT | |
| IFNγ | Forward ATGAACGCTACACACTGCATC |
| Reverse CCATCCTTTTGCCAGTTCCTC | |
| Beta-actin | Forward GCTGCGTTTTACACCCTTTC |
| Reverse GCTGTCGCCTTCACCGTTC |
Flow cytometry
Surface staining was performed as described previously [24]. For intracellular cytokine staining, the cells were stimulated with 25 ng/ml PMA and 500 ng/ml ionomycin (Sigma-Aldrich, St. Louis, MO, USA) for 6 h at 37°C. Brefeldin A (10 mg/ml, eBioscience, San Diego, CA, USA) was added for the last 4 h of incubation. The cells were stained with the Cytofix/Cytoperm kit according to the manufacturer’s instructions (eBioscience, San Diego, CA, USA), followed by flow cytometry analysis as reported [29].
T cell depletion and IL-17A neutralization
To deplete CD4 or CD8 T cells, the mice were i.v. injected with 200 μg anti-CD4 (GK1.5, eBioscience) or anti-CD8 (2.43, eBioscience) mAb 3 days before transplantation and days 2, 7, and 12 after transplantation, and depletion of CD4 or CD8 T cells was confirmed by flow cytometry. For neutralization of IL-17A, 100 μg/mouse anti-mouse IL-17A mAb or rat IgG2a isotype control (eBioscience, San Diego, CA, USA) were injected into mice via tail vein every other day till day 15 after transplantation.
ELISA analysis of cytokine production
The IL-2, IL-4, IFN-γ and IL-17 levels in the serum and culture supernatants were assessed by ELISA using the kits from R&D Systems (Minneapolis, MN, USA) as previously described [30].
Statistical analysis
All data are presented as mean ± SD. Student’s t-test was used to compare two groups. For graft survival rate, the Kaplan-Meier graphs were constructed and log-rank comparison was used to calculate p values. Differences were considered significant when p<0.05.
Results
Tim-1-Fc alleviates chronic cardiac rejection by attenuating IL-17 secretion
Given Bm12 mice only manifest MHC II mismatch with B6 mice [31], we thus implanted Bm12-derived cardiac grafts into B6 mice to address the impact of Tim-1-Fc on chronic cardiac graft rejection. Interestingly, administration of Tim-1-Fc significantly attenuated chronic cardiac graft rejection, in which all grafts from Tim-1-Fc treated mice survived longer than 60 days, while only 60% of control IgG treated mice manifested graft survival >60 days (Figure 1A). Histological analysis of graft sections from recipient mice 5 weeks after transplantation revealed a significant reduction for the severity of inflammatory infiltration in Tim-1-Fc treated mice as compared with that of control mice (Figure 1B). The severity of cardiac allograft vasculopathy (CAV) was next assessed by vasculopathy scores as described, much lower CAV scores were noted in Tim-1-Fc treated mice than that of control mice (Figure 1C).
Figure 1.

Tim-1-Fc attenuates chronic cardiac rejection in MHC II mismatched cardiac grafts. A: Survival rate of Bm12-derived cardiac grafts in B6 recipients treated with either Tim-1-Fc or control IgG. Loss of graft function was defined as cessation of a palpable impulse. B: Hematoxylin and eosin (H&E) staining of cardiac graft sections harvested after day 35 of transplantation. C: Scores for the severity of vasculopathy in cardiac grafts after day 35 of transplantation. D: Intragraft expression of IL-2, IL4, IFN-γ, IL-17 and IL-6. The relative expression levels of cytokines within the grafts were assessed by real-time PCR. E: Administration of recombinant IL-17 abolished the protective effect conferred by Tim-1-Fc. Recombinant IL-17 was administrated along with Tim-1-Fc or control IgG after transplantation every other day until day 15. Histological data and real-time PCR data were obtained from studies of 3 mice.
Next, we analyzed the expression of inflammatory cytokines in the grafts. As shown in Figure 1D, a moderate reduction for cytokines IL-6, IFN-γ and IL-2 was noted in Tim-1-Fc treated grafts, while the expression of IL-17 was reduced by 1.1-fold as compared with that of control grafts. Given that IL-17 has been demonstrated to promote mesenchymal and CD4 T cells secretion of IL-6 and IFN-γ [32,33], we thus hypothesized that Tim-1-Fc attenuates chronic cardiac graft rejection by suppressing IL-17 expression. To address this question, recombinant IL-17 was administered into recipient mice along with Tim-1-Fc. Indeed, Administration of exogenous recombinant IL-17 accelerated allograft rejection and completely abolished the protective effect of Tim-1-Fc on cardiac graft rejection (Figure 1E).
To further address the above question, we transplanted Bm12-derived cardiac grafts into T-bet-/- mice, by which we were able to exclude the impact of IFN-γ. Treatment of T-bet-/- recipients with Tim-1-Fc significantly prolonged cardiac graft mean survival time (MST) as compared with that of IgG treated mice (18 ± 3.46 days vs. 14 ± 2 days, Figure 2A). Consistently, histological analysis revealed higher severity for vasculopathy in control mice as compared with that of Tim-1-Fc treated mice (Figure 2B). A remarkable reduction for CD11b (macrophages and neutrophils) and CD3 (CD4 and CD8 T cells) expression was observed in the grafts originated from Tim-1-Fc treated recipients (Figure 2C), indicating an attenuated inflammatory infiltration. No perceptible change for IL-2, IL-4 and IFN-γ expression in the grafts was noted between Tim-1-Fc treated and control mice, while the expression of IL-17 decreased by 1.3-fold in Tim-1-Fc treated mice (Figure 2C). In line with this result, a significant reduction for serum IL-17 was indentified in Tim-1-Fc treated recipients (Figure 2D). All together, our data support that administration of Tim-1-Fc protects cardiac grafts from rejection by suppressing IL-17 secretion.
Figure 2.
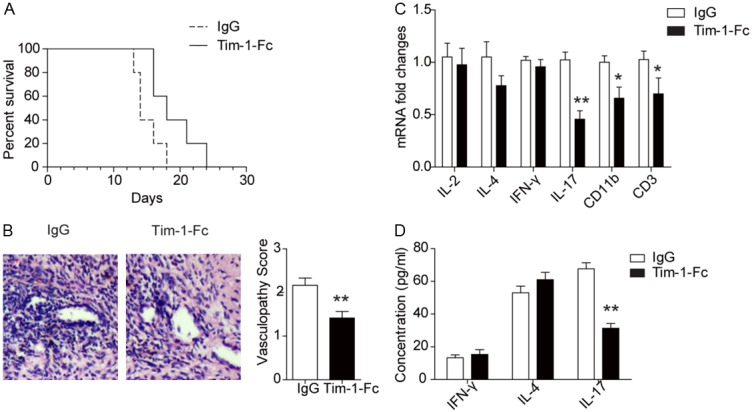
Tim-1-Fc protects Bm12-derived cardiac grafts from rejection in T-bet deficient recipients. A: Survival rate of Bm12-derived cardiac grafts in T-bet-/- recipients after treating with Tim-1-Fc or control IgG (n=5 for each study group). B: Results for H&E staining and vasculopathy scores of cardiac grafts after day 14 of transplantation. C: Relative expression levels for IL-2, IL-4, IFN-γ, IL-17, CD11b and CD3 in the grafts after 14 days of transplantation. D: Serum levels for cytokines IL4, IFN-γ and IL-17 in the recipient mice. All data are presented as means ± SD, and 3 replications were included for each assay.
Tim-1-Fc suppresses the number of effector T cells
Next, we assessed the impact of Tim-1-Fc on CD4 and CD8 T effector cell differentiation in recipient mice. Peripheral blood originated from recipient mice 2 weeks after transplantation was subjected to flow cytometry analysis. Interestingly, Tim-1-Fc treated recipients displayed less amount of effector or effector memory (CD44hiCD62Llow) CD4 T cells (9.7% vs. 15.4%) and CD8 T cells (12% vs. 19%) (Figure 3A). This result prompted us to investigate whether the reduction of effector cells was caused by the increase of regulatory T cells (Tregs). Unexpectedly, analysis of peripheral blood of recipient mice 2 weeks after transplantation revealed similar number of Tregs in total CD4 T cells between Tim-1-Fc treated and control mice (Figure 3B).
Figure 3.
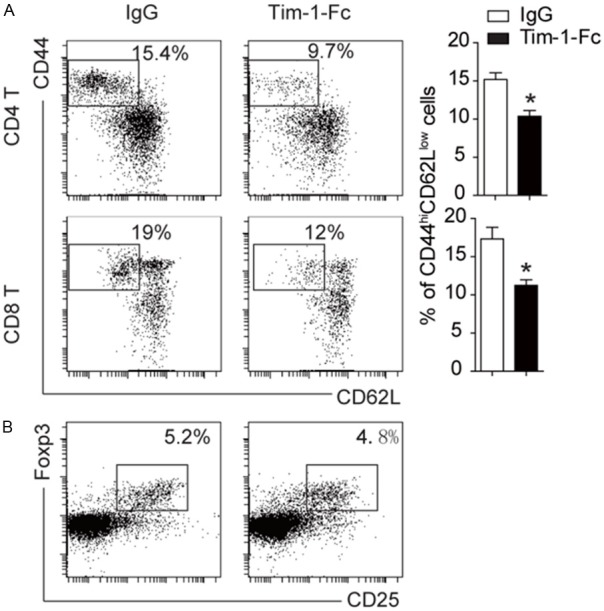
Tim-1-Fc inhibits the number of effector T cells. A: Flow cytometry analysis of lymphocytes in the periphery blood of T-bet-/- recipients. Tim-1-Fc treatment significantly reduced the number of CD44hiCD62low effecter T cells. B: Flow cytometry data for regulatory T cells. Addition of Tim-1-Fc did not affect the number of regulatory T cells. All experiments were conducted with 3 replications. *, P<0.01.
Administration of Tim-1-Fc does not affect DC functionality
Given that Tim-1-Fc administration may lead to DC depletion through activating complement or antibody dependent cytotoxicity, which then contributes to the reduced activation of CD4 and CD8 T cells, we thus further examined the impact of Tim-1-Fc on DC functionality. Splenic cells collected from recipient mice 2 weeks after transplantation was subjected to flow cytometry analysis of DC number and maturation status. Interestingly, we failed to detect a discernable difference for the number of CD11c+MHCIIhi DCs between Tim-1-Fc treated and control IgG treated recipients (Figure 4A). Similarly, no perceptible difference was noted for the expression of surface markers CD80 and CD86 between two groups of mice (Figure 4B). We further examined the number for macrophages, DCs, B cells, NK cells and neutrophils, and failed to detect a significant difference between two groups of mice (Figure 4C). Together, these data suggest that innate immune cells are not involved in Tim-1-Fc mediated cardiac graft protection.
Figure 4.
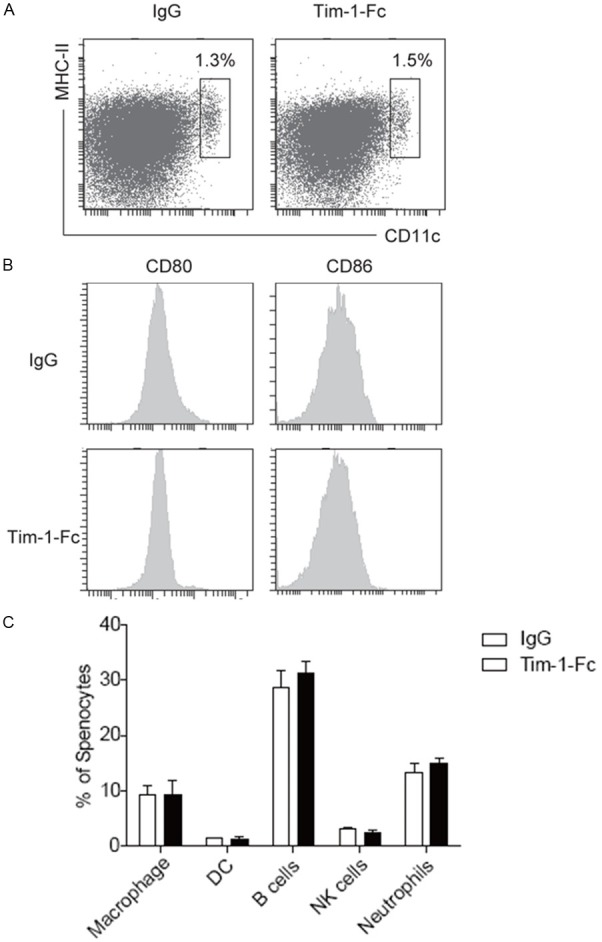
The impact of Tim-1-Fc administration on DC number and maturation. A: Tim-1-Fc treatment did not result in a significant change for the number of splenic DCs and MHC II expressions. B: Flow cytometry data for CD80 and CD86 expressions. Data are a representative of 3 independent experiments conducted.
Tim-1-Fc is potent to suppress Th17 cell differentiation
To address the impact of Tim-1-Fc on Th17 development, we specifically examined T-bet-/- recipients after receiving Bm12-derived cardiac grafts. Remarkably, the frequency of alloreactive CD4 T cells expressing Th17 associated cytokine IL-17 was markedly decreased in Tim-1-Fc treated mice as compared with that of control mice, while the number of IFN-γ positive Th1 cells and IL-4 positive Th2 cells was the same (Figure 5A). We next examined whether Tim-1-Fc affected IL-17 producing CD8 T cells, but failed to identify a detectable difference between two groups of mice (Figure 5B), suggesting that Tim-1-Fc specifically attenuates CD4 Th17 differentiation. To further address this issue, naïve CD4 T cells were cultured under Th17 condition in the presence of Tim-1-Fc or control IgG for 3 days and then subjected to analysis for the production of Th17 cells. Indeed, Tim-1-Fc dose-dependently suppressed the number of Th17 cells (Figure 5C). Similar results were also obtained in a system using DCs for induction of Th17 cells (Figure 5D).
Figure 5.
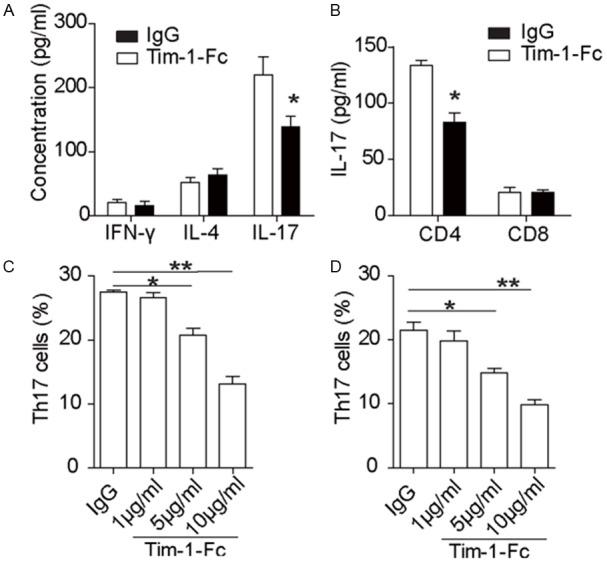
Tim-1-Fc suppresses Th17 cell differentiation. Bm12-derived cardiac grafts were implanted into T-bet-/- recipients, and Tim-1-Fc or control IgG were administered as described earlier. The recipient mice were sacrificed 14 day after transplantation, and splenic T cells were prepared for flow cytometry analysis of Th17 production. A: Results for IL-17 producing Th17 cells. Splenic T cells were stained for CD4 and then co-stained for IL-17, IL-4 and IFN-γ, respectively. A significant reduction for CD4 Th17 cells was noted in Tim-1-Fc treated recipient mice. B: Results for flow cytometry analysis of IL-17 producing CD8 T cells. The above prepared splenic T cells were first stained for CD8 and then co-stained for IL-17. Staining of CD4 Th17 cells was used as a control. No detectable change was noted for IL-17 producing CD8 T cells. C: Tim-1-Fc dose-dependently suppressed the production of CD4 Th17 cells. CD4 naive T cells were cultured under Th17 condition in the presence of different doses of Tim-1-Fc or control IgG for 3 days, and then subjected to flow cytometry analysis of Th17 production. D: Tim-1-Fc attenuated DC induced Th17 differentiation. CD4 naive T cells were induced for Th17 differentiation with DCs in the presence of Tim-1-Fc or control IgG as described. All data are shown as means ± SD of 3 independent experiments conducted.
Tim-1-Fc prevents cardiac graft rejection relying on its effect on CD4 Th17 cells
To further demonstrate that Tim-1-Fc protects cardiac grafts from chronic rejection by suppressing CD4 Th17 development, we depleted CD4 and CD8 T cells in T-bet-/- recipients and then implanted BM12-derived cardiac grafts. As expected, depletion of CD4 T cells provided protection for cardiac grafts against rejection. Interestingly, the protective effect conferred by Tim-1-Fc treatment was completely masked by the depletion of CD4 T cells (Figure 6A). In sharp contrast, depletion of CD8 T cells only manifested a mild protection, and more importantly, the protection conferred by Tim-1-Fc was still noted in CD8 T cell depleted recipient mice (Figure 6B). Inflammatory cytokine expression profiles in the grafts further supported the above observations (Figure 6C). Finally, IL-17 neutralizing antibodies were administered into T-bet-/- recipients along with control IgG or Tim-1-Fc. Remarkably, administration of IL-17 neutralizing antibodies completely abolished the protective effect conferred by Tim-1-Fc, and all grafts survived over 40 days (Figure 6D). Collectively, our data support that Tim-1-Fc protects cardiac grafts from chronic rejection dependent on its effect on suppression of CD4 Th17 development.
Figure 6.
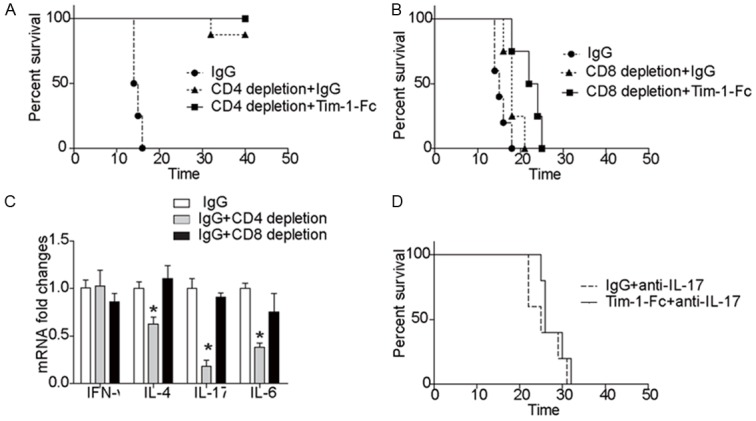
Tim-1-Fc confers protection against cardiac rejection relying on its effect on CD4 Th17 cells. A: Depletion of CD4 T cells diminished the protection conferred by Tim-1-Fc treatment. T-bet-/- recipients were depleted for CD4 T cells and then implanted with Bm12-derived cardiac grafts along with administration of Tim-1-Fc or control IgG as described (n=5 for each study group). B: Administration of Tim-1-Fc provided protection for cardiac grafts against rejection in CD8 depleted T-bet-/- recipients (n=5 for each study group). C: Results for intragraft cytokine expressions by real-time PCR. Data presented here are means ± SD of 3 independent experiments. D: Neutralization of IL-17 completely abolished the protection conferred by Tim-1-Fc (n=5 for each study group). IL-17 was neutralized with a mAb in T-bet-/- recipients after receiving Bm12-derived cardiac grafts along with treatment of Tim-1-Fc or control IgG as described.
Discussion
The manifestation of vasculopathy during chronic cardiac allograft rejection acts as a major contributing factor limiting long-term survival of cardiac grafts in the clinical settings [34,35]. In general, intimal thickening can be noted within the first year, while CAV development could occur up to 80% of cardiac grafts within the first 5-year of transplantation [36,37]. It is believed that Th1 cells are undisputedly involved in the development of chronic CAV, as manifested by that cardiac grafts are protected from chronic allograft vasculopathy in recipient mice deficient in STAT4 or IFN-γ [38,39]. However, discrepant results are also noted, in which cardiac grafts displayed accelerated rejection along with the presence of vasculopathy in Th1 transcription factor T-bet deficient recipients [40]. Indeed, more and more studies support that Th17 cells play a critical role in cardiac vasculopathy during the course of chronic rejection [16,17]. We thus proposed that Th17 cells play a pathogenic role during chronic cardiac rejection, while Th1 cells affect chronic rejection by modulating acute alloimmune responses. Transplantation of Bm12-derived cardiac grafts into B6 recipients was employed for the establishment of a model for chronic rejection. Previously, we demonstrated that Tim-1-Fc prevents acute cardiac rejection by inhibiting Th1 response [24]. We now in the present report tackled the impact of Tim-1-Fc on Th17 cells in a model of cardiac chronic rejection. We first demonstrated that administration of Tim-1-Fc provided protection for Bm12-derive grafts against chronic rejection. We next implanted BM12-derived cardiac grafts into T-bet-/- recipients, in which the impact of Th1 response on chronic rejection can be excluded. Interestingly, Tim-1-Fc significantly protected cardiac grafts from chronic rejection along with attenuated production of IL-17 producing T cells. Next, we depleted CD4 and CD8 T cells in T-bet-/- mice, and then implanted Bm12-derived cardiac grafts. Depletion of CD4 T cells completely masked the protective effect of Tim-1-Fc on chronic cardiac rejection, while the protective effect conferred by Tim-1-Fc was still noted in CD8 T cell depleted recipients. Finally, we blocked IL-17 in T-bet-/- recipient mice by administration of IL-17 neutralizing antibodies. Blockade of IL-17 completely diminished the protective effect on chronic cardiac graft rejection conferred by Tim-1-Fc. All together, we demonstrated ample evidence that Tim-1-Fc protects cardiac grafts from chronic rejection by attenuating CD4 Th17 development.
To exclude the involvement of innate immune cells in Tim-1-Fc mediated protection, we first examine the impact of Tim-1-Fc on DC functionality. Flow cytometry analysis revealed similar number and similar levels of surface marker expression on DCs originated from Tim-1-Fc treated and control IgG treated recipients, indicating that administration of Tim-1-Fc did not affect DC development and maturation. Other than DCs, macrophages, B cells, NK cells and neutrophils have also been recognized contributing to CAV pathogenesis [41-43]. We thus further examined those cells but failed to detect a perceptible difference between two groups of mice, indicating that Tim-1-Fc protection of cardiac grafts from chronic rejection is independent of innate immunity. We further extended our studies to regulatory T cells, and demonstrated that administration of Tim-1-Fc did not result in a detectable change for Tregs.
Other than CD4 Th17 cells, IL-17 can be also produced by CD8 T cells or γδT cells [16,44,45]. Nevertheless, analysis of Bm12-derived cardiac grafts in T-bet-/- mice revealed that those infiltrated T cells expressed αβ TCR [40], demonstrating that γδT cells were not involved in chronic rejection of cardiac allograft. On the other hand, depletion of CD8 T cells did not result in a similar protective effect on chronic cardiac graft rejection as that of depletion of CD4 T cells, and more importantly, administration of Tim-1-Fc further protected cardiac graft from rejection in CD8 T cell depleted recipients. Together, these data support that the production of IL-17 in cardiac grafts is likely originated from CD4 Th17 cells rather than CD8 T cells or γδT cells, and Tim-1-Fc attenuates chronic cardiac graft rejection by suppressing CD4 Th17 function.
Previous studies including ours suggested the existence of additional Tim-1 ligand other than the currently identified Tim-4 [24,46]. To address this possibility, we conducted immunoprecipitation of membrane proteins from activated T cells with Tim-1-Fc, and the resulting precipitates were next subjected to mass spectrometry analysis. Unfortunately, no informative data were resulted from this study. We then embarked on phosphatidylserine and leukocyte mono-Ig-like receptor 5 (LMIR5/CD300b), a potential Tim-1 ligand suggested by previous studies [47-49]. Unexpectedly, we noted that LMIR5 was only expressed in myeloid cells, not in naïve CD4 T cells or activated T cells. As such, we were unable to provide novel information in terms of new Tim-1 ligand in the present report. Clearly, additional studies are necessary to address this challenging question.
In summary, we demonstrated evidence supporting that Tim-1-Fc possesses the capability to prolong cardiac graft survival and prevents chronic cardiac vasculopathy. Administration of Tim-1-Fc did not result in a perceptible impact on innate immunity and regulatory T cells, while a significant reduction for the number of Th17 cells and the secretion of IL-17 was noted. Our studies in recipient mice by depleting CD4 and CD8 T cells or blocking IL-17 further revealed that Tim-1-Fc selectively attenuates the development and functionality of CD4 Th17 cells. Together, our data suggest that Tim-1-Fc might be a viable immunosuppressive agent in the setting of cardiac transplantation.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81001326, 81273253, 81072444, 31270957), the National Key Basic Research Program of China (2009CB522402), and the Shanghai Changzheng Hospital Foundation for Young Scientist (2012CZQN01, 09411951900).
Disclosure of conflict of interest
The authors declare no competing financial interests.
References
- 1.Booth AJ, Bishop DK. TGF-beta, IL-6, IL-17 and CTGF direct multiple pathologies of chronic cardiac allograft rejection. Immunotherapy. 2010;2:511–520. doi: 10.2217/imt.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Korewicki J. Cardiac transplantation is still the method of choice in the treatment of patients with severe heart failure. Cardiol J. 2009;16:493–499. [PubMed] [Google Scholar]
- 3.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB American Heart Association Statistics Committee and Stroke Statistics. Heart disease and stroke statistics--2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Stafford R, Thom T, Wasserthiel-Smoller S, Wong ND, Wylie-Rosett J American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Executive summary: heart disease and stroke statistics--2010 update: a report from the American Heart Association. Circulation. 2010;121:948–54. doi: 10.1161/CIRCULATIONAHA.109.192666. [DOI] [PubMed] [Google Scholar]
- 5.Taylor DO, Edwards LB, Boucek MM, Trulock EP, Aurora P, Christie J, Dobbels F, Rahmel AO, Keck BM, Hertz MI. Registry of the International Society for Heart and Lung Transplantation: twenty-fourth official adult heart transplant report--2007. J Heart Lung Transplant. 2007;26:769–781. doi: 10.1016/j.healun.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 6.Uretsky BF, Murali S, Reddy PS, Rabin B, Lee A, Griffith BP, Hardesty RL, Trento A, Bahnson HT. Development of coronary artery disease in cardiac transplant patients receiving immunosuppressive therapy with cyclosporine and prednisone. Circulation. 1987;76:827–834. doi: 10.1161/01.cir.76.4.827. [DOI] [PubMed] [Google Scholar]
- 7.Womer KL, Vella JP, Sayegh MH. Chronic allograft dysfunction: mechanisms and new approaches to therapy. Semin Nephrol. 2000;20:126–147. [PubMed] [Google Scholar]
- 8.Kwun J, Knechtle SJ. Overcoming Chronic Rejection-Can it B? Transplantation. 2009;88:955–961. doi: 10.1097/TP.0b013e3181b96646. [DOI] [PubMed] [Google Scholar]
- 9.Weiss MJ, Madsen JC, Rosengard BR, Allan JS. Mechanisms of chronic rejection in cardiothoracic transplantation. Front Biosci. 2008;13:2980–8. doi: 10.2741/2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hayry P, Aavik E, Savolainen H. Mechanisms of chronic rejection. Transplant Proc. 1999;31:5S–8S. [PubMed] [Google Scholar]
- 11.Waaga AM, Gasser M, Kist-van Holthe JE, Najafian N, Muller A, Vella JP, Womer KL, Chandraker A, Khoury SJ, Sayegh MH. Regulatory functions of self-restricted MHC class II allopeptide-specific Th2 clones in vivo. J Clin Invest. 2001;107:909–916. doi: 10.1172/JCI11427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou P, Szot GL, Guo Z, Kim O, He G, Wang J, Grusby MJ, Newell KA, Thistlethwaite JR, Bluestone JA, Alegre ML. Role of STAT4 and STAT6 signaling in allograft rejection and CTLA4-Ig-mediated tolerance. J Immunol. 2000;165:5580–5587. doi: 10.4049/jimmunol.165.10.5580. [DOI] [PubMed] [Google Scholar]
- 13.Saleem S, Konieczny BT, Lowry RP, Baddoura FK, Lakkis FG. Acute rejection of vascularized heart allografts in the absence of IFNgamma. Transplantation. 1996;62:1908–1911. doi: 10.1097/00007890-199612270-00039. [DOI] [PubMed] [Google Scholar]
- 14.Weaver CT, Hatton RD, Mangan PR, Harrington LE. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 15.Deteix C, Attuil-Audenis V, Duthey A, Patey N, McGregor B, Dubois V, Caligiuri G, Graff-Dubois S, Morelon E, Thaunat O. Intragraft Th17 infiltrate promotes lymphoid neogenesis and hastens clinical chronic rejection. J Immunol. 2010;184:5344–5351. doi: 10.4049/jimmunol.0902999. [DOI] [PubMed] [Google Scholar]
- 16.Itoh S, Nakae S, Axtell RC, Velotta JB, Kimura N, Kajiwara N, Iwakura Y, Saito H, Adachi H, Steinman L, Robbins RC, Fischbein MP. IL-17 contributes to the development of chronic rejection in a murine heart transplant model. J Clin Immunol. 2010;30:235–240. doi: 10.1007/s10875-009-9366-9. [DOI] [PubMed] [Google Scholar]
- 17.Faust SM, Lu G, Marini BL, Zou W, Gordon D, Iwakura Y, Laouar Y, Bishop DK. Role of T cell TGFbeta signaling and IL-17 in allograft acceptance and fibrosis associated with chronic rejection. J Immunol. 2009;183:7297–7306. doi: 10.4049/jimmunol.0902446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodriguez-Manzanet R, Meyers JH, Balasubramanian S, Slavik J, Kassam N, Dardalhon V, Greenfield EA, Anderson AC, Sobel RA, Hafler DA, Strom TB, Kuchroo VK. TIM-4 expressed on APCs induces T cell expansion and survival. J Immunol. 2008;180:4706–4713. doi: 10.4049/jimmunol.180.7.4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meyers JH, Chakravarti S, Schlesinger D, Illes Z, Waldner H, Umetsu SE, Kenny J, Zheng XX, Umetsu DT, DeKruyff RH, Strom TB, Kuchroo VK. TIM-4 is the ligand for TIM-1, and the TIM-1-TIM-4 interaction regulates T cell proliferation. Nat Immunol. 2005;6:455–464. doi: 10.1038/ni1185. [DOI] [PubMed] [Google Scholar]
- 20.Umetsu SE, Lee WL, McIntire JJ, Downey L, Sanjanwala B, Akbari O, Berry GJ, Nagumo H, Freeman GJ, Umetsu DT, DeKruyff RH. TIM-1 induces T cell activation and inhibits the development of peripheral tolerance. Nat Immunol. 2005;6:447–454. doi: 10.1038/ni1186. [DOI] [PubMed] [Google Scholar]
- 21.Mizui M, Shikina T, Arase H, Suzuki K, Yasui T, Rennert PD, Kumanogoh A, Kikutani H. Bimodal regulation of T cell-mediated immune responses by TIM-4. Int Immunol. 2008;20:695–708. doi: 10.1093/intimm/dxn029. [DOI] [PubMed] [Google Scholar]
- 22.Degauque N, Mariat C, Kenny J, Zhang D, Gao W, Vu MD, Alexopoulos S, Oukka M, Umetsu DT, DeKruyff RH, Kuchroo V, Zheng XX, Strom TB. Immunostimulatory Tim-1-specific antibody deprograms Tregs and prevents transplant tolerance in mice. J Clin Invest. 2008;118:735–741. doi: 10.1172/JCI32562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao S, Najafian N, Reddy J, Albin M, Zhu C, Jensen E, Imitola J, Korn T, Anderson AC, Zhang Z, Gutierrez C, Moll T, Sobel RA, Umetsu DT, Yagita H, Akiba H, Strom T, Sayegh MH, DeKruyff RH, Khoury SJ, Kuchroo VK. Differential engagement of Tim-1 during activation can positively or negatively costimulate T cell expansion and effector function. J Exp Med. 2007;204:1691–1702. doi: 10.1084/jem.20062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xiao L, Fu ZR, Liu F, Zhang LD, Shi XM, Shen XY, Ni ZJ, Fu H, Li RD, Cao XT, Ding GS, Wang QX. Suppression of allograft rejection by Tim-1-Fc through cross-linking with a novel Tim-1 binding partner on T cells. PLoS One. 2011;6:e21697. doi: 10.1371/journal.pone.0021697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Q, Liu Y, Li XK. Simplified technique for heterotopic vascularized cervical heart transplantation in mice. Microsurgery. 2005;25:76–79. doi: 10.1002/micr.20082. [DOI] [PubMed] [Google Scholar]
- 26.Yang J, Popoola J, Khandwala S, Vadivel N, Vanguri V, Yuan X, Dada S, Guleria I, Tian C, Ansari MJ, Shin T, Yagita H, Azuma M, Sayegh MH, Chandraker A. Critical role of donor tissue expression of programmed death ligand-1 in regulating cardiac allograft rejection and vasculopathy. Circulation. 2008;117:660–669. doi: 10.1161/CIRCULATIONAHA.107.741025. [DOI] [PubMed] [Google Scholar]
- 27.Lee RS, Womer KL, Yamada K, Maloney ME, Houser SL, Sayegh MH, Madsen JC. The role of indirect recognition of donor MHC class II peptides in cardiac transplantation in miniature swine. J Heart Lung Transplant. 2001;20:172. doi: 10.1016/s1053-2498(00)00342-9. [DOI] [PubMed] [Google Scholar]
- 28.Zhang S, Lv JW, Yang P, Yu Q, Pang J, Wang Z, Guo H, Liu S, Hu J, Li J, Leng J, Huang Y, Ye Z, Wang CY. Loss of dicer exacerbates cyclophosphamide-induced bladder overactivity by enhancing purinergic signaling. Am J Pathol. 2012;181:937–946. doi: 10.1016/j.ajpath.2012.05.035. [DOI] [PubMed] [Google Scholar]
- 29.Rao X, Zhong J, Zhang S, Zhang Y, Yu Q, Yang P, Wang MH, Fulton DJ, Shi H, Dong Z, Wang D, Wang CY. Loss of methyl-CpG-binding domain protein 2 enhances endothelial angiogenesis and protects mice against hind-limb ischemic injury. Circulation. 2011;123:2964–2974. doi: 10.1161/CIRCULATIONAHA.110.966408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Han J, Zhong J, Wei W, Wang Y, Huang Y, Yang P, Purohit S, Dong Z, Wang MH, She JX, Gong F, Stern DM, Wang CY. Extracellular high-mobility group box 1 acts as an innate immune mediator to enhance autoimmune progression and diabetes onset in NOD mice. Diabetes. 2008;57:2118–2127. doi: 10.2337/db07-1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li D, Tsang JY, Peng J, Ho DH, Chan YK, Zhu J, Lui VC, Xu A, Lamb JR, Tam PK, Chen Y. Adiponectin mediated MHC class II mismatched cardiac graft rejection in mice is IL-4 dependent. PLoS One. 2012;7:e48893. doi: 10.1371/journal.pone.0048893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu S, Han Y, Xu X, Bao Y, Zhang M, Cao X. IL-17A-producing gammadeltaT cells promote CTL responses against Listeria monocytogenes infection by enhancing dendritic cell cross-presentation. J Immunol. 2010;185:5879–5887. doi: 10.4049/jimmunol.1001763. [DOI] [PubMed] [Google Scholar]
- 33.Hwang SY, Kim JY, Kim KW, Park MK, Moon Y, Kim WU, Kim HY. IL-17 induces production of IL-6 and IL-8 in rheumatoid arthritis synovial fibroblasts via NF-kappaB- and PI3-kinase/Akt-dependent pathways. Arthritis Res Ther. 2004;6:R120–128. doi: 10.1186/ar1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stehlik J, Edwards LB, Kucheryavaya AY, Benden C, Christie JD, Dipchand AI, Dobbels F, Kirk R, Rahmel AO, Hertz MI International Society of Heart and Lung Transplantation. The Registry of the International Society for Heart and Lung Transplantation: 29th official adult heart transplant report--2012. J Heart Lung Transplant. 2012;31:1052–1064. doi: 10.1016/j.healun.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 35.Mehra MR. Contemporary concepts in prevention and treatment of cardiac allograft vasculopathy. Am J Transplant. 2006;6:1248–1256. doi: 10.1111/j.1600-6143.2006.01314.x. [DOI] [PubMed] [Google Scholar]
- 36.Weis M, von Scheidt W. Cardiac allograft vasculopathy: a review. Circulation. 1997;96:2069–2077. doi: 10.1161/01.cir.96.6.2069. [DOI] [PubMed] [Google Scholar]
- 37.Ewel CH, Foegh ML. Chronic graft rejection: accelerated transplant arteriosclerosis. Immunol Rev. 1993;134:21–31. doi: 10.1111/j.1600-065x.1993.tb00638.x. [DOI] [PubMed] [Google Scholar]
- 38.Koglin J, Glysing-Jensen T, Gadiraju S, Russell ME. Attenuated cardiac allograft vasculopathy in mice with targeted deletion of the transcription factor STAT4. Circulation. 2000;101:1034–1039. doi: 10.1161/01.cir.101.9.1034. [DOI] [PubMed] [Google Scholar]
- 39.Nagano H, Mitchell RN, Taylor MK, Hasegawa S, Tilney NL, Libby P. Interferon-gamma deficiency prevents coronary arteriosclerosis but not myocardial rejection in transplanted mouse hearts. J Clin Invest. 1997;100:550–557. doi: 10.1172/JCI119564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yuan X, Paez-Cortez J, Schmitt-Knosalla I, D’Addio F, Mfarrej B, Donnarumma M, Habicht A, Clarkson MR, Iacomini J, Glimcher LH, Sayegh MH, Ansari MJ. A novel role of CD4 Th17 cells in mediating cardiac allograft rejection and vasculopathy. J Exp Med. 2008;205:3133–3144. doi: 10.1084/jem.20081937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hirohashi T, Uehara S, Chase CM, DellaPelle P, Madsen JC, Russell PS, Colvin RB. Complement independent antibody-mediated endarteritis and transplant arteriopathy in mice. Am J Transplant. 2010;10:510–517. doi: 10.1111/j.1600-6143.2009.02958.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Graham JA, Wilkinson RA, Hirohashi T, Chase CM, Colvin RB, Madsen JC, Fishman JA, Russell PS. Viral infection induces de novo lesions of coronary allograft vasculopathy through a natural killer cell-dependent pathway. Am J Transplant. 2009;9:2479–2484. doi: 10.1111/j.1600-6143.2009.02801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Uehara S, Chase CM, Kitchens WH, Rose HS, Colvin RB, Russell PS, Madsen JC. NK cells can trigger allograft vasculopathy: the role of hybrid resistance in solid organ allografts. J Immunol. 2005;175:3424–3430. doi: 10.4049/jimmunol.175.5.3424. [DOI] [PubMed] [Google Scholar]
- 44.Yuan X, Ansari MJ, D’Addio F, Paez-Cortez J, Schmitt I, Donnarumma M, Boenisch O, Zhao X, Popoola J, Clarkson MR, Yagita H, Akiba H, Freeman GJ, Iacomini J, Turka LA, Glimcher LH, Sayegh MH. Targeting Tim-1 to overcome resistance to transplantation tolerance mediated by CD8 T17 cells. Proc Natl Acad Sci U S A. 2009;106:10734–10739. doi: 10.1073/pnas.0812538106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Burrell BE, Csencsits K, Lu G, Grabauskiene S, Bishop DK. CD8+ Th17 mediate costimulation blockade-resistant allograft rejection in T-bet-deficient mice. J Immunol. 2008;181:3906–3914. doi: 10.4049/jimmunol.181.6.3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lin J, Chen L, Kane LP. Murine Tim-1 is excluded from the immunological synapse. F1000Research. 2012;1:10. doi: 10.12688/f1000research.1-10.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamanishi Y, Kitaura J, Izawa K, Kaitani A, Komeno Y, Nakamura M, Yamazaki S, Enomoto Y, Oki T, Akiba H, Abe T, Komori T, Morikawa Y, Kiyonari H, Takai T, Okumura K, Kitamura T. TIM1 is an endogenous ligand for LMIR5/CD300b: LMIR5 deficiency ameliorates mouse kidney ischemia/reperfusion injury. J Exp Med. 2010;207:1501–1511. doi: 10.1084/jem.20090581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kobayashi N, Karisola P, Pena-Cruz V, Dorfman DM, Jinushi M, Umetsu SE, Butte MJ, Nagumo H, Chernova I, Zhu B, Sharpe AH, Ito S, Dranoff G, Kaplan GG, Casasnovas JM, Umetsu DT, Dekruyff RH, Freeman GJ. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity. 2007;27:927–940. doi: 10.1016/j.immuni.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamanishi Y, Kitaura J, Izawa K, Matsuoka T, Oki T, Lu Y, Shibata F, Yamazaki S, Kumagai H, Nakajima H, Maeda-Yamamoto M, Tybulewicz VL, Takai T, Kitamura T. Analysis of mouse LMIR5/CLM-7 as an activating receptor: differential regulation of LMIR5/CLM-7 in mouse versus human cells. Blood. 2008;111:688–698. doi: 10.1182/blood-2007-04-085787. [DOI] [PubMed] [Google Scholar]