

Abstract
Reactive oxygen species (ROS) contribute to alveolar cell death in Acute Respiratory Distress Syndrome (ARDS) and we previously demonstrated that NOX1-derived ROS contributed to hyperoxia-induced alveolar cell death in mice. The study investigates whether NOX1 expression is modulated in epithelial cells concomitantly to cell death and associated to STAT3 signaling in the exudative phase of ARDS. In addition, the role of STAT3 activation in NOX1-dependent epithelial cell death was confirmed by using a lung epithelial cell line and in mice exposed to hyperoxia. NOX1 expression, cell death and STAT3 staining were evaluated in the lungs of control and ARDS patients by immunohistochemistry. In parallel, a stable NOX1-silenced murine epithelial cell line (MLE12) and NOX1-deficient mice were used to characterize signalling pathways. In the present study, we show that NOX1 is detected in alveolar epithelial cells of ARDS patients in the exudative stage. In addition, increased alveolar epithelial cell death and phosphorylated STAT3 are observed in ARDS patients and associated with NOX1 expression. Phosphorylated STAT3 is also correlated with TUNEL staining. We also confirmed that NOX1-dependent STAT3 activation participates to alveolar epithelial cell death. Silencing and acute inhibition of NOX1 in MLE12 led to decreased cell death and cleaved-caspase 3 induced by hyperoxia. Additionally, hyperoxia-induced STAT3 phosphorylation is dependent on NOX1 expression and associated with cell death in MLE12 and mice. This study demonstrates that NOX1 is involved in human ARDS pathophysiology and is responsible for the damage occurring in alveolar epithelial cells at least in part via STAT3 signalling pathways.
Keywords: NOX1, ARDS, cell death, hyperoxia, STAT3, ROS
Introduction
Acute Respiratory Distress Syndrome (ARDS), the most severe form of Acute Lung Injury (ALI), is a progressive disease often associated with high mortality. ARDS is caused by direct injuries to the lung resulting from pneumonia, gastric aspiration or toxic inhalation, or by indirect injuries when associated with sepsis or severe burn. This syndrome is characterized by the exudative (acute) stage involving the disruption of the alveolar-capillary barrier and diffuse inflammation and a subsequent organizing stage characterized by alveolar pneumocyte hyperplasia and extensive lung fibrosis [1]. Most of ARDS patients require ventilation with high fractions of inspired oxygen responsible for oxygen toxicity [2].
The cellular and molecular mechanisms involved in the pathogenesis of ALI/ARDS remain unclear, although there is evidence that reactive oxygen species (ROS) generated by inflammatory cells as well as epithelial and endothelial cells contribute to alveolar damage, the inflammatory response and abnormal repair [2]. Indeed, increase in markers of oxidative stress and evidence of alveolar cell death has been observed in the lungs of patients with ALI/ARDS [3,4]. Among several ROS-generating enzymes, NADPH oxidase (NOX) enzymes are implicated in the main pathophysiological changes of ALI/ARDS [5]. NOX isoforms are expressed in a variety of lung cell types and participate in several physiological as well as pathological lung processes [6]. In a previous work, we demonstrated that NOX1, an isoform preferentially expressed in alveolar epithelial and endothelial cells, is an important contributor to acute lung injury induced by hyperoxia in mice [7], an established model of the exudative phase of ARDS [8]. This enzyme plays also an essential role in the death of primary mouse alveolar epithelial and endothelial cells [7]. In addition, in vitro studies have demonstrated that diphenyleneiodonium (DPI), a non-specific inhibitor of NOX enzymes, reduces ROS generation in a murine epithelial cell line (MLE12) [9] and in primary pulmonary type II cells [9,10] under hyperoxic condition.
Several redox-sensitive signalling pathways including signal transducer and activator of transcription (STAT), PI3K/Akt, mitogen-activated protein kinase (MAPK) pathways have been also shown to participate to cell death mediating acute lung injury [7,11-16]. We previously demonstrated that NOX1 contributed to hyperoxic lung damage in part through MAPK activation in mice [7], however, the role of NOX1 in STAT3 signaling-dependent alveolar epithelial cell death was not elucidated in ARDS/ALI.
In the present study, we first examined whether NOX1 is correlated to epithelial cell death in Acute Respiratory Distress Syndrome and associated with STAT3 signaling. In parallel, we confirm the role of STAT3 activation in NOX1-dependent epithelial cell death in hyperoxia by using a murine epithelial cell line and in mice.
Methods
Control and ARDS patients
Human lung biopsies of patient suffering from ARDS (n=10) in the exudative phases, and human control lungs (n=10) were obtained by thoracotomy in accordance to an approved protocol by the Institutional Ethical Committee of Geneva (Authorization N° NAC 10-052R). Control lungs were obtained from a pulmonary lobectomy removed for carcinoma. Parenchyma non adjacent to the tumor was used. The exudative phase was defined by the disruption of alveolo-capillary barrier, pulmonary edema, protein accumulation and inflammatory cell infiltration into the alveolar space.
Human immunohistochemistry
Paraffin-embedded sections of human lungs fixed with 4% paraformaldehyde were subjected to heat-induced epitope retrieval for 15 min in 0.01 mol/L citrate buffer (pH 6.0) and endogenous peroxidase was blocked by adding DAKO peroxidase block solution. After blocking in 10% normal goat serum and 1% bovine serum albumin in PBS solution, lung sections were stained with an anti-NOX1 polyclonal antibody (1:500; kindly provided by Pr. Lambeth [17] followed by an incubation with a biotinylated goat anti-rabbit Ig (1:100; Vector Laboratories, Servion, Switzerland) or with an antibody anti-digoxigenin-AP Fab fragments for 30 min at room temperature (1:500; Chemicon, Darmstadt, Germany) as described by the manufacturer (ApopTag® Peroxidase In Situ Apoptosis Detection Kit, Chemicon, Darmstadt, Germany), or with an anti-phospho-STAT3 monoclonal antibody (Tyr705, 1:200, Cell Signaling, Allschwil, Switzerland), anti-prosurfactant C polyclonal antibody (1:1000, Chemicon, Darmstadt, Germany.) or alternatively with the monoclonal antibody, M30 (M30 CytoDEATH, Roche, Basel, Switzerland) for 60 min. Negative controls were obtained by incubating the sections with a biotinylated goat anti-rabbit Ig only (1:100; Vector Laboratories, Servion, Switzerland) or alternatively with a IgG2a (1:50) in DAKO antibody dilution buffer. The detection of positive cells was made using Fast Red substrate system (Dako SA, Geneva, Switzerland) or horseradish peroxidase anti-mouse or rabbit Envision+ system with diaminobenzidine (DAB, Dako SA, Geneva, Switzerland). Sections were then counterstained with cresyl violet and mount with Ultrakitt. Quantification of positive staining was performed using Metamorph analysis software (10 images per subjects, 3-4 subjects per group).
Cell culture and hyperoxia experiments
Murine lung epithelial cells (MLE12) were grown in Dulbecco’s modified Eagle’s medium (DMEM, glucose 1000 mg/l, Sigma-Aldrich, Allschwil, Switzerland), supplemented with 1% Penicillin-Streptomycin (Gibco) and 2% fetal calf serum (FCS) and the medium was changed every two days. For hyperoxia experiments, cells plated at subconfluence (70%) were placed in sealed glass chambers filled with 95% O2-5% CO2 at 37°C, 24 h after plating up to 72 h. Normoxic cells were kept in normal air condition (21% O2-5% CO2) at 37°C. Medium and gases were replaced every two days.
Inhibition of NOX1 and pSTAT3
MLE12 were treated with GKT136901, a NOX1/NOX4 inhibitor (GenKyoTex, [18]) at 10 μm, or WP1066, a STAT3 inhibitor [19] at 1 μm or DMSO, 1 or 6 hours prior hyperoxia exposure (for GKT136901 and WP1066 respectively) and for 72 h. Culture medium containing inhibitor was replaced every day. ROS measurement and TUNEL staining were then performed for GKT136901 and cleaved-caspase-3 was measured by western blot for WP1066.
Silencing of mouse NOX1 in MLE12 cell line by shRNA interference
The following NOX1-shRNA encoding sequences (5’-CGCGTCCCCTCATTGCACACCTATTTAATTCAAGAGATTAAATAGGTGTGCAATGATTTTGGAAAT-3’ and 5’-CGATTTCCAAAAATCATTGCACACCTATTTAATCTCTTGAATTAAATAGGTGTGCAATGAGGGGA-3’) were synthesized by EUROGENTEC S.A. and subcloned into the lentiviral vector pLVTHM, which contains the GFP marker [20]. Recombinant lentiviruses containing NOX1-shRNA were produced by co-transfecting 293T cells with the following 3 plasmids: the packaging plasmid psPAX2, the vesicular stomatitis virus-G envelope protein expression plasmid pMD2G, and the pLVTHM containing NOX1-shRNA or scramble-shRNA control vector. Infection of MLE12 cell line was performed as described previously [21]. Transduction efficiency was determined by FACS analysis and sorting (Becton Dickinson, Allschwil, Switzerland). The downregulation of mouse NOX1 expression was further assessed under hyperoxia conditions by quantitative RT-PCR.
RT-PCR and quantitative RT-PCR
MLE12 mRNA was extracted as previously described [18]. Primers for NOX1, NOX2, NOX3, NOX4, DUOX1, DUOX2, NOXA1, NOXO1, p67phox, p47phox, p40phox, p22phox and the reference genes EEF1A1 (eukaryotic elongation factor 1A1) and HPRT (hypoxanthine-guanine phosphoribosyltransferase) were used. RNA was extracted by Total RNA Isolation NucleoSpin RNAII (Macherey-Nagel, Oensingen, Switzerland) and reverse transcribed using the superscript reverse transcriptase (Superscript Choice; Invitrogen). Reference genes were EEF1A1 (eukaryotic elongation factor 1A1) and HPRT (hypoxanthine-guanine phosphoribosyltransferase).
The following primers were used for qualitative RT-PCR: NOX1: NOX1-Forward 5’-CCC ATC CAG TCT CCA AAC ATG AC-3’, NOX1-Reverse 5’-CTC CCC TTA TGG TCA TCC CAC TC-3’; NOX2: NOX2-Forward 5’-TCA ACT ACT ATA AGG TTT ATG ATG ATG G-3’, NOX2-Reverse 5’-CAG ATA TCT AAA TTA TGC TCT TCC AAA-3’; NOX3: NOX3-Forward 5’-AGC TGC CTT ATG CCC TGT ACC TC-3’, NOX3-Reverse 5’-AGG CCT TCA ATA ACG CCT CTG TC-3’; NOX4: NOX4-Forward 5’-CAC AAC CAT TCC TGG TCT GAC G-3’, NOX4-Reverse 5’-AAA ACC CTC GAG GCA AAG ATC C-3’; DUOX1: DUOX2-Forward 5’-CCG ACT CAG AGC TGG AGA AG-3’, DUOX2-Reverse 5’-TTT CAG GGG CTG GTA GAC AC-3’; DUOX2: DUOX2-Forward 5’-CGA AGG AAG GTT CAG ACA GC-3’, DUOX2-Reverse 5’-CCA CCC AGG AGT AGT TCC AA-3’, Reverse; NOXO1: NOXO1-Forward 5’-GGT CCC CAC ATC CCT ATC TT-3’, NOXO1-Forward 5’-Reverse 5’-TTA TAC CTG CAC AGC CAC CA-3’; NOXA1: NOXA1-Forward 5’-ACG GRG GAT GTT CTG TGT GA-3’, NOXA1-Reverse 5’-AAG CAT GGC TTC CAC ATA GG-3’; p67phox: p67phox-Forward 5’-CAG CCA GCT TGC GAA CAT G-3’, p67phox-Reverse 5’-GAC AGT ACC AGG ATT ACA TC-3’; p47phox: p47phox-Forward 5’-GCC CAA AGA TGG CAA GAA TA-3’, p47phox-Reverse 5’- ATG ACC TCA ATG GCT TCA CC-3’; p40phox: p40phox-Forward 5’-GAG CAG AGG CCT TGT TTG AC-3’, p40phox-Reverse 5’-TCC CAC ATC CTC ATC TGA CA-3’; p22phox: p22phox-Forward 5’-AAA GAG GAA AAA GGG GTC CA-3’, p22phox-Reverse 5’-TAG GCT CAA TGG GAG TCC AC-3’.
The following primers were used for real time PCR: NOX1: NOX1-Forward 5’-CAG TTA TTC ATA TCA TTG CAC ACC TAT TT-3’, NOX1-Reverse 5’-CAG AAG CGA GAG ATC CAT CCA-3’; NOX4: NOX4-Forward 5’-CCG GAC AGT CCT GGC TTA TCT-3’ NOX4-Reverse 5’-TGC TTT TAT CCA ACA ATC TTC TTG TT-3’; NOX2: NOX2-Forward 5’-CAG GAA CCT CAC TTT CCA TAA GAT G-3’ NOX2-Reverse 5’-AAC GTT GAA GAG ATG TGC AAT TGT-3’; Reference genes: EEF1A1-Forward, 5’-TCC ACT TGG TCG CTT TGC T-3’ and EEF1A1-Reverse, 5’-CTT CTT GTC CAC AGC TTT GAT GA-3’; HPRT-Forward, 5’-GCT CGA GAT GTC ATG AAG GAG AT-3’, and HPRT-Reverse, 5’-AAA GAA CTT ATA GCC GCC CCC CTT GA-3’.
TUNEL staining
TUNEL detection was performed in MLE12 cells as described by the manufacturer (TUNEL assay fluorescent kit, Roche, Basel, Switzerland [18]). Briefly, after hyperoxia exposure, MLE12 cells were fixed with 4% PAF for 15 min at room temperature and then permeabilized during 2 min on ice with 0.1% Triton-X-100 in 0.1% sodium citrate freshly prepared. Cells were incubated with Tunel reaction buffer for 1 h at 37°. The nuclei were stained with 4,6-Diamidino-2-phenylindole (DAPI, 1:200; Roche Diagnostic, Basel, Switzerland). These slides were then mounted with fluosave as described previously. Images were acquired by confocal microscopy (LSM510 Meta, Zeiss) and quantified using Metamorph analysis software (>50 cells, 3 independent experiments).
Detection of reactive oxygen species
After hyperoxia exposure, MLE12 were stained with 10 μM of dihydroethidium (DHE, Invitrogen, Basel, Switzerland) diluted in PBS. Images were captured after 30 min with inverted microscope (Nipkow) and analyzed with Metafluor imaging software (Molecular Devices). Values were obtained by measuring fluorescence intensity on MLE12 cells (>50 cells) from 3 independent experiments [7].
DNA oxidation staining
After hyperoxia exposure, MLE12 cells were fixed with 4% PAF for 1 h at room temperature and then permeabilized during 2 min on ice with 0.1% Triton-100 in 0.1% sodium citrate freshly prepared. Cells were incubated with 8-hydroxy-2’-deoxyguanosine antibody (8-OHdG, 1:30; Oxis, Beverly Hills, US) for 1 h at 37°. As secondary antibody, a goat anti-rabbit Texas Red conjugated (dilution 1:250; Molecular Probe, Lucerne, Switzerland) was used. The nuclei were stained with (DAPI) (1:200; Roche Diagnostic, Basel, Switzerland). These slides were then mounted with fluosave (VWR, Nyon, Switzerland) as described previously. Images were acquired by confocal microscopy (LSM510 Meta, Zeiss, Feldbach, Switzerland) and quantified using Metamorph analysis software (>50 cells, 3 independent experiments).
Cell growth
Cells were seeded in 96-well plates and incubated for different times. Cell growth was stopped by addition of 50 μl of trichloroacetic acid (50% v/v) and protein content of each well was determined by staining with sulforhodamine B [22]. Absorbance was determined at 490 nm. The relationship between cell number (protein content per well) and absorbance is linear from 0 to 5.106 cells.
Western blot analysis
After hyperoxia exposure, cell proteins were extracted as previously described [18]. Proteins were blotted on nitrocellulose membrane and then incubated with a polyclonal anti-STAT3 (1:1000; Cell Signaling, Allschwil, Switzerland), or a polyclonal anti-pSTAT3 (1:250; Cell Signaling, Allschwil, Switzerland) or a polyclonal anti-actin (1:1000; Sigma, Buchs, Switzerland), or a polyclonal anti-cleaved caspase-3 (1:1000; Cell Signaling, Allschwil, Switzerland), or a monoclonal anti-PARP-1 (1:1000; BD Biosciences). The membranes were then washed and incubated with a horseradish peroxidase-conjugated anti-mouse antibody (1:3,000; Bio-Rad Laboratories, Reinach, Switzerland) or peroxidase-conjugated anti-rabbit antibody (1:10,000; Jackson Immunoresearch Laboratories, Rheinfelden, Switzerland). Proteins were detected by using ECL reagents (Amersham Pharmacia Biotech, Dübendorf, Switzerland). Densitometric evaluation was performed using Quantity One software (Bio-Rad Laboratories, Cressier, Switzerland).
Animals and immunohistochemistry
NOX1-deficient mice and wild-type (WT) mice inbred on the C57BL/6J background were exposed to room air or 100% O2 for 72 hours [7]. Animal were kept under specific pathogen-free conditions. The animal procedure was performed in accordance with the Ethical committee of the University of Geneva and the Cantonal veterinary Office (Authorization N° 31.1.1005/2218/II). Mice (female aged 8-10 weeks) were exposed to room air or 100% O2 for 72 hours. Frozen lung tissues were cryosectioned (6 μm) and collected onto SuperFrost Plus slides (Perbio Science, Lausanne, Switzerland). Lung sections from WT and NOX1-deficient mice were stained with a polyclonal anti-p-STAT3 (1:50, Cell Signaling, Allschwil, Switzerland) followed by fluorescein isothiocyanate-conjugated secondary antibody before counterstaining with DAPI. Slides were mounted with Fluor Save (Calbiochem, Darmstadt, Germany) and analyzed by confocal microscopy.
Statistical analysis
Results are expressed as mean ± SEM or ± SD as indicated and were analyzed either by Wilcoxon Rank test or by analysis of variance (ANOVA), as appropriate. Significance levels were set at P<0.05.
Results
NOX1 is highly expressed in alveolar epithelial cells of ARDS lungs
NOX1 expression was first studied using a specific anti-NOX1 antibody in lung sections of control and ARDS patients in the exudative phase (Figure 1A and 1B). Lung sections stained with the secondary antibody were used to verify the specificity of the anti-NOX1 antibody. In control lungs, NOX1 was detected in endothelial cells whereas epithelial cells were negative for NOX1. In ARDS lungs, NOX1 was present in endothelial cells (arrow) and at high levels in alveolar type II epithelial cells in the exudative of ARDS (Figure 1A and 1B, arrowhead). Due to technical difficulties to perform co-immunohistochemistry on human lung sections, serial ARDS lung sections stained with an anti-pro-surfactant C antibody (SPC), a specific marker of type II epithelial cells, and an anti-NOX1 antibody were used to confirm the localization of NOX1 in type II epithelial cells (Figure 1B). In addition, based on cell morphology and localization, we noted the presence of NOX1 in macrophages (Figure 1A, ampersand) during the exudative phase, but not in neutrophils (Figure 1A, asterisk).
Figure 1.
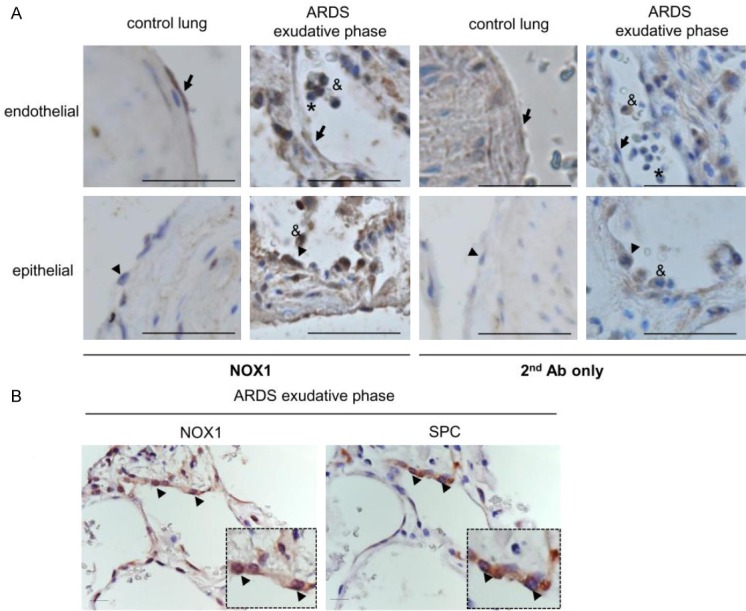
NOX1 is highly expressed in alveolar epithelium of ARDS patients. ARDS lung tissues were analyzed for NOX1 expression by immunohistochemistry. (A) Lung structures from control and ARDS in exudative phase were stained with anti-NOX1 antibody (NOX1 Ab) or with secondary antibody only (2nd Ab only). In control lungs, the anti-NOX1 antibody stained pulmonary endothelial cells (arrow) but not epithelial cells (type II, arrowhead). In the exudative stage of ARDS, both epithelial cells (type II, arrowhead) and endothelial cells (arrow) were positive. Note the presence of NOX1 in macrophages (ampersand), but not in neutrophils (asterisk) located inside the alveoli of ARDS lungs. Scale bars, 100 μm. (B) Serial lung sections of ARDS in the exudative phase were stained with NOX1 antibody and pro-surfactant C (a specific marker of Type II epithelial cells). Epithelial type II cells are positive for NOX1 in the exudative stage of ARDS (arrowhead, two different magnifications), Scale bars, 50 μm.
Thus, NOX1 was detected at high levels in the alveolar epithelium during the early phase of ARDS.
Increased alveolar epithelial cell death is partially associated to NOX1 expression in the exudative ARDS phase
As alveolar epithelial cell death was known to participate to the pathogenesis of ARDS [23], we analyzed cell death in control and ARDS lungs during the exudative phase by TUNEL staining (Figure 2A). The number of TUNEL-positive cells was significantly increased in the exudative phase of ARDS patients compared to control lungs (6.9 ± 3% ARDS exudative phase as compared to 1.9 ± 0.34% control lungs, p=0.04, Figure 2A).
Figure 2.
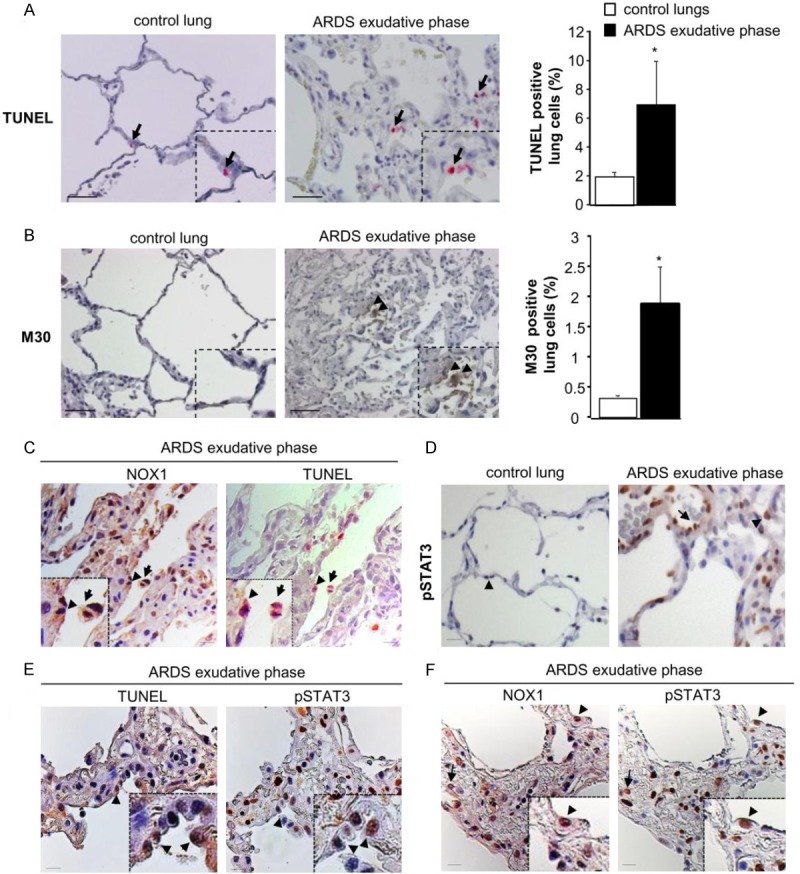
Epithelial cell death detected in early phase of ARDS is associated with NOX1 and pSTAT3. Cell death was analyzed in control and ARDS lungs during exudative phases by TUNEL and M30 staining. (A) Representative images of control and ARDS lungs sections stained with TUNEL and (B) M30. TUNEL-positive cells appear in pink (arrow) and M30-positive cells are in brown (arrowhead). Scale bars, 50 μm. The numbers of TUNEL- and M30-positive cells are expressed as percent of all nuclei counted in lung sections. Quantification of positive staining was performed using Metamorph analysis software (10 images per subjects, 3-4 subjects per group) P=NS, *P<0.05, ARDS versus control patients. (C) Immunostaining for NOX1 (brown) and TUNEL (pink) were done on serial lung sections of ARDS patients in the exudative phase (two different magnifications). Note the presence of NOX1 in the TUNEL-positive epithelial cells (arrowhead) and macrophages (arrow). Scale bars, 50 μm. (D) Control and ARDS lung tissues were analyzed for phosphorylated STAT3 by immunohistochemistry. In control lungs, STAT3 phosphorylation was not detected in epithelial (arrowhead) and endothelial (data not shown) cells whereas in ARDS lung sections, epithelial (arrowhead) and endothelial (arrow) cells were positive for phosphorylated STAT3 in the exudative phase. (E) Serial lung sections of ARDS exudative phase were stained with an anti-phosphorylated STAT3 antibody and TUNEL, or (F) with an anti-NOX1 antibody. Phosphorylated STAT3-positive cells were also positive for both TUNEL and NOX1 (arrowheads and arrow), Scale bars, 50 μm.
To determine whether epithelial cells were involved in the processes of cell death observed in the exudative phase of ARDS, we next evaluated the number of apoptotic epithelial cells by immunohistochemistry using the M30 antibody, which detects an epitope of caspase-dependent cleaved cytokeratin 18, a known marker of early apoptotic events in epithelial cells (Figure 2B). The number of M30-positive epithelial cells was increased in ARDS lungs in the exudative phase compared to healthy lung subjects (1.88 ± 0.61% ARDS exudative phase as compared to 0.33 ± 0.02% control lungs, p=0.03, Figure 2B). To study if alveolar cell death observed in ARDS patients during the exudative phase was associated to the expression of NOX1, we performed immunostaining on serial lung sections with an anti-NOX1 antibody and TUNEL staining. Based on cell localization and morphology, we showed the expression of NOX1 in TUNEL-positive epithelial cells (Figure 2C, arrowhead). We also noted the presence of NOX1 in some alveolar macrophages which were also TUNEL positive (Figure 2C, arrow).
Taken together, these data demonstrate that death of lung epithelial cells is in part, associated with NOX1 expression.
Phosphorylated STAT3 is correlated to NOX1 expression and cell death
As STAT3 signalling is known to be modulated by oxidative stress [24,25] and participates to cell death in ARDS [16], we then examined phosphorylated STAT3 in ARDS lungs. In control lungs, STAT3 phosphorylation was not detected in epithelial (arrowhead) and endothelial cells (data not shown), whereas in ARDS lung sections, epithelial (arrowhead) and endothelial (arrow) cells were positive for phosphorylated STAT3 (pSTAT3) in the exudative phase (Figure 2D). To determine if phosphorylated STAT3 staining was correlated to cell death and NOX1 expression, we performed immunostaining on serial lung sections with an anti-phosphorylated STAT3 antibody and TUNEL staining, or and an anti-NOX1 antibody. We observed that pSTAT3-positive cells were also stained for TUNEL (arrowhead, Figure 2E) and NOX1 (arrowhead and arrow, Figure 2F).
These data demonstrate that pSTAT3 is at least correlated to cell death and NOX1 expression.
Hyperoxia increases NOX1 mRNA expression and ROS-derived NOX1 in MLE12
To confirm the correlation between STAT3 and NOX1-dependent cell death observed in the exudative phase of ARDS patients, we generated stable silenced NOX1 murine epithelial cell line (MLE12) by shRNA interference and we exposed them to hyperoxia.
We first characterized the expression of NOX isoforms and their regulatory subunits by RT-PCR in MLE12. We found that MLE12 expressed NOX1 and DUOX1 and 2, and the regulatory subunits p22phox, p40phox, p67phox and NOXO1. The other members of the NOX family (NOX2, NOX3, and NOX4) were not detected (Figure 3A).
Figure 3.
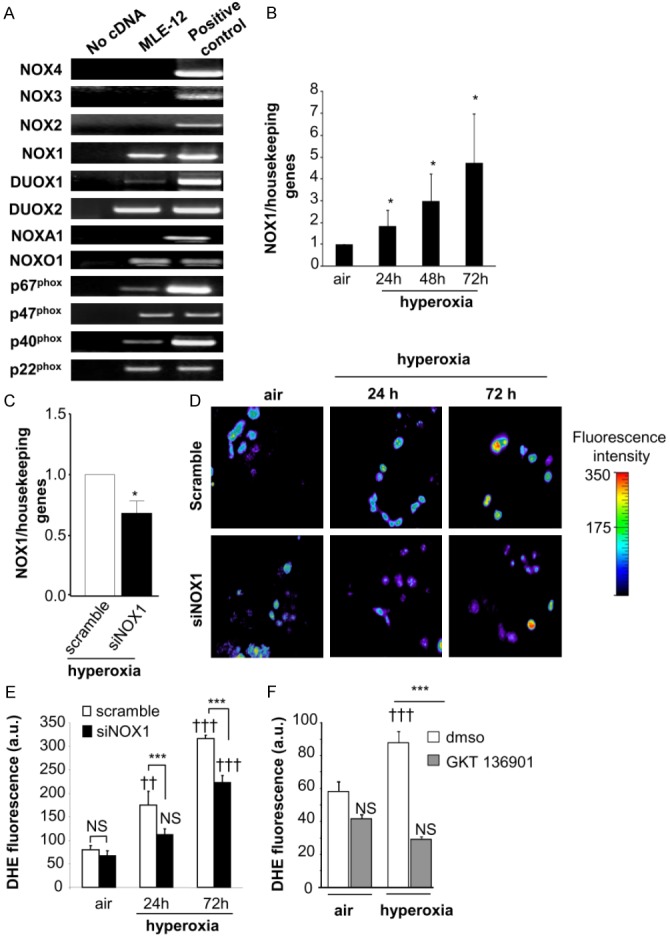
Hyperoxia increases NOX1 mRNA expression and ROS-dependent NOX1 production in MLE12. (A) Expression of NOX isoforms in MLE12. NOX1, DUOX1/2, and the regulatory subunits p22phox, p40phox, p67phox and NOXO1 were detected in MLE12 by RT-PCR. Lung tissues were used as positive control for the detection of NOX1, 2, 4, DUOX1/2, and the regulatory subunits. For the detection of NOX3 mRNA expression, ear tissue was used as positive control. Absence of cDNA was used as negative control for expression of NOX1 mRNA in MLE12 measured by real time PCR in air condition and (B) at 24, 48 and 72 h following hyperoxia exposure (n=3). *P<0.05. (C) NOX1 mRNA measured by qualitative RT-PCR in scramble- and NOX1-silenced MLE12 (siNOX1) at 72 h of hyperoxia. (D, E) Representative fluorescent images of scramble- and NOX1-silenced MLE12 loaded with DHE. ROS production was measured by analysing DHE staining (10 M) in scramble- and NOX1-silenced MLE12 in air or hyperoxia for 24 and 72 h, and visualized by confocal microscopy (pseudocolor). Original magnification, X40. Fluorescence intensity was quantified in MLE12, bars represent the mean ± SEM (n>50 cells for each group; P=NS, ***P<0.001, scramble-versus NOX1-silenced cells under hyperoxia; ††P<0.01, †††P<0.001, air versus hyperoxia). (F) Acute inhibition of NOX1 prevents hyperoxia-induced ROS production. MLE12 were treated with dmso, or GKT136901 (10 μm) 1 hour before hyperoxia exposure and for 72 h. ROS generation was measured by analysing DHE fluorescence intensity in treated MLE12 exposed to air or hyperoxia for 72 h. Bars represent the mean ± SEM (n>50 cells for each group; ***P<0.001 cells treated with NOX inhibitor compared to cells treated with DMSO exposed to hyperoxia; P=NS, †††P<0.001, air versus hyperoxia).
We then investigated the effect of hyperoxia exposure on NOX1 mRNA expression by RT-PCR and quantitative RT-PCR. Exposure to hyperoxia for 24 to 72 h increased the expression of NOX1 mRNA in MLE12 cells (Figure 3B). No expression of NOX2 and NOX4 mRNA was detected after 72 h of hyperoxia exposure (data not shown).
We then measured the effect of NOX1 shRNA transduction in MLE12 on NOX1 expression and ROS production in hyperoxia. Hyperoxia exposure of NOX1-silenced MLE12 for 72 h led to a 32% reduction (p<0.05) in NOX1 mRNA as compared to scramble-silenced control cells (Figure 3C). In the absence of a specific anti-mouse NOX1 antibody, we measured ROS-derived NOX1 production in hyperoxia. We observed that hyperoxia-induced ROS production was inhibited by 36% at 24 h and 30% at 72 h in NOX1-silenced cells compared to control cells (Figure 3D and 3E). These results were confirmed by using GKT136901, a NOX1/NOX4 inhibitor. Acute inhibition of NOX1 with GKT136901, reduced ROS production following 72 h of hyperoxia in MLE12 (Figure 3F).
These results demonstrated that hyperoxia regulates NOX1 mRNA expression in MLE12 and ROS production during hyperoxia was dependent on NOX1.
Inhibition of NOX1 reduces hyperoxia-induced epithelial cell death in MLE12
We have previously demonstrated that hyperoxia induced cell death in MLE12 [26], we then examined cell death in scramble and NOX1-silenced MLE12 cells in hyperoxia by using 8-hydroxy-2’-deoxyguanosine (8OH-dG) and TUNEL staining.
Hyperoxia led to an increase in 8OH-dG-positive cells during hyperoxia compared to air condition (p<0.05, Figure 4A). By contrast, DNA oxidation was abolished in NOX1-silenced cells (p<0.05, Figure 4A). The number of TUNEL-positive cells were increased after 72 h of hyperoxia in control cells, which was decreased in NOX1-silenced cells (p<0.01, Figure 4B). These results were confirmed by treating MLE12 with GKT136901, which decreased the number of TUNEL-positive cells during hyperoxia (p<0.001, Figure 4C).
Figure 4.
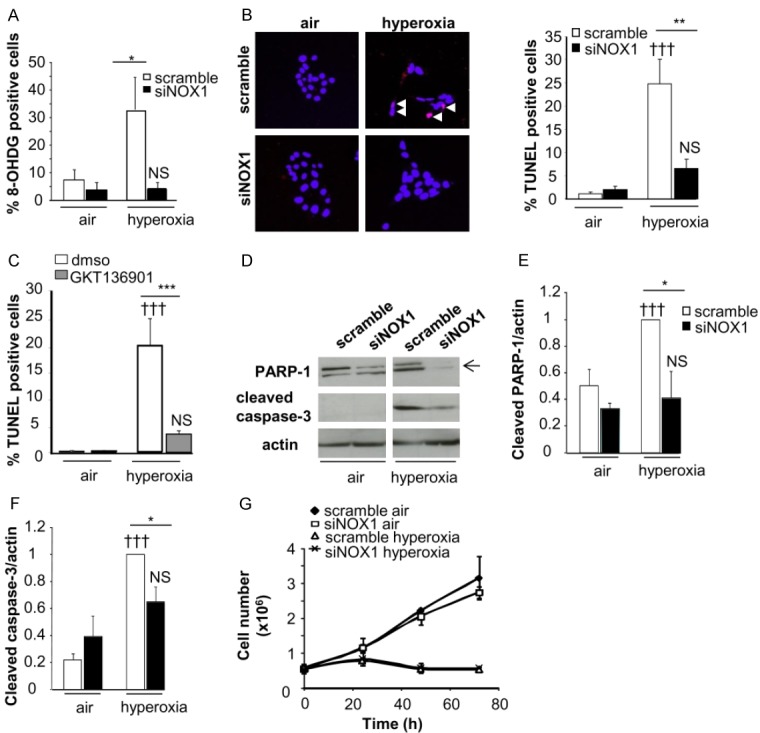
Acute and stable NOX1 inhibition decrease hyperoxia-induced death of MLE12. Cell death was evaluated in control and NOX1-silenced MLE12 in air or hyperoxic condition. A: Transduced MLE12 exposed to air or hyperoxia for 72 h were stained with 8-hydroxy-2’-deoxyguanosine antibody (8-OHdG, red) and DAPI (blue) and the number of 8-OHdG-positive cells is expressed as percent of all nuclei (n>50 for each group, 3 independent experiments). B: Representative images of transduced MLE12 stained with TUNEL (red) and DAPI (blue) at 72 h of air or hyperoxia. White arrows indicate TUNEL-positive cells which appear in pink. The number of TUNEL-positive cells is expressed as percent of all nuclei (n>50 for each group, 3 independent experiments). P=NS, †††P<0.001 air versus hyperoxia; ***P<0.001, **P<0.01, *P<0.05 scramble-versus NOX1-silenced cells in hyperoxia. C: MLE12 were treated with DMSO, or GKT136901 (10 m) and exposed to hyperoxia for 72 h. ***P<0.001 cells treated with NOX inhibitor compared to cells exposed to DMSO in hyperoxia. P=NS, †††P<0.001 air versus hyperoxia. D-F: Protein lysate of transduced MLE12 were blotted for cleaved caspase-3 and PARP-1 and quantified by densitometry (right panel; n=3). β-actin was used to control equal loading. P=NS, †††P<0.001 air versus hyperoxia; *P<0.05 scramble-versus NOX1-silenced cells in hyperoxia. G: Cell growth was measured by using sulforhodamine B for different times. Absorbance was measured at 490 nm and cell number was determined. The relationship between cell number (protein content per well) and absorbance is linear from 0 to 5.106 cells. No difference was observed between scramble-versus NOX1-silenced cells in hyperoxia.
Caspase-3/PARP-1 pathways are known to participate in the death of murine epithelial cells during hyperoxia [7]. We observed that hyperoxia-induced cleavage of caspase-3 and PARP-1 was decreased in NOX1-silenced cells compared to control cells (p<0.05, Figure 4D-F).
We also determined whether NOX1 inhibition modulated cell growth using sulforhodamine B staining. NOX1 silencing did not affect cell growth in air condition or cell growth arrest in hyperoxia (Figure 4G). In addition, we did not find any difference in the level of cyclin D1 between control- and NOX1-silenced cells exposed to hyperoxia (data not shown).
Thus, these results demonstrated that acute and stable inhibition of NOX1 led to decreased hyperoxia-induced epithelial cell death through direct DNA oxidation, as well as modulation of the caspase-3 and PARP-1 pathways, without modifying cell growth.
Hyperoxia-induced STAT3 phosphorylation participates to cell death and is dependent on NOX1
To confirm the involvement of pSTAT3 in NOX1-dependent epithelial cell death in hyperoxia, we analyzed the phosphorylation of STAT3 in scramble and NOX1-silenced cells during hyperoxia at different time points.
In scramble cells, hyperoxia changed the level of STAT3 phosphorylation after 6 h (p<0.05) which returned to a basal level at 24 h (Figure 5A). Interestingly, STAT3 phosphorylation was significantly inhibited in NOX1-silenced cells exposed to hyperoxia (p<0.05, Figure 5A), whereas no modification in total STAT3 protein level was noted. To confirm the role of STAT3 in cell death during hyperoxia, WP1066 (1 μm), a STAT3 inhibitor was then used. WP1066 was shown to inhibit STAT3 phosphorylation at low concentration [19]. Treatment with STAT3 inhibitor in this condition increased the basal level of cleaved caspase-3 in control cells; nevertheless, in hyperoxia, STAT3 inhibition decreased significantly cleaved caspase-3 (Figure 5B).
Figure 5.
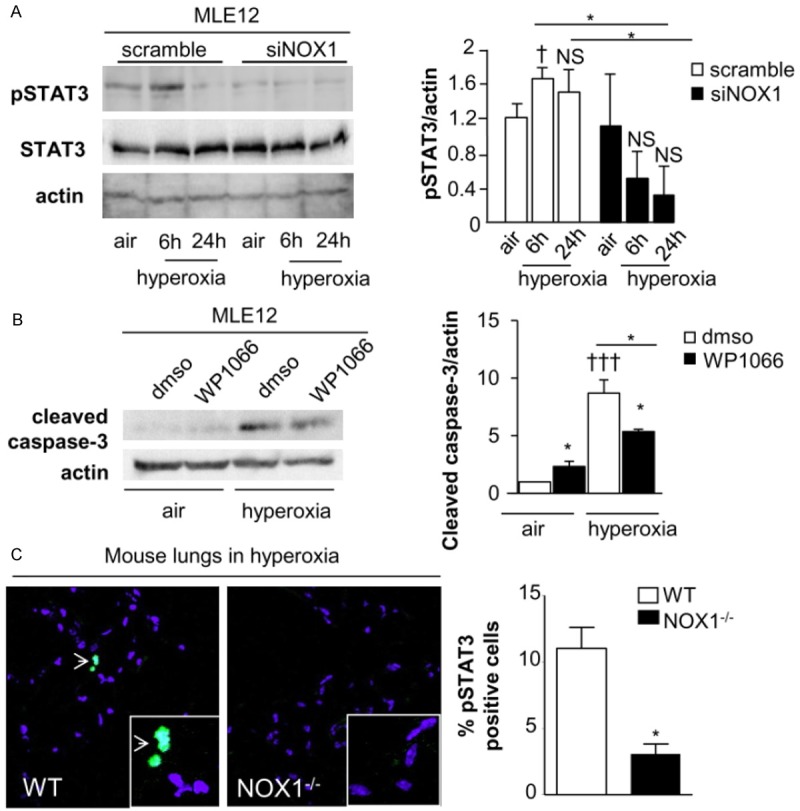
STAT3 phosphorylation is dependent on NOX1 and participates to cell death in hyperoxic condition. A: Western-blot of phosphorylated and total STAT3 in MLE12 were quantified by densitometry (n=3). β-actin was used to control equal loading. Phosphorylated forms of the respective proteins are indicated by the prefix “p”. P=NS, P=NS, †P<0.05 air versus hyperoxia *P<0.05 scramble-versus NOX1-silenced cells in hyperoxia at different time points. B: MLE12 was pre-treated with DMSO or WP1066 (1 m) 6 hours before hyperoxia and for 72 h. Western-blot of cleaved-caspase 3 was quantified by densitometry (n=3). β-actin was used to control equal loading †††P<0.001 cells treated with STAT3 inhibitor compared to cells exposed to DMSO in hyperoxia, *P<0.05 cells exposed to DMSO compared to cells treated to STAT3 inhibitor in air, and cells treated to STAT3 inhibitor in air compared to cells treated with STAT3 inhibitor in hyperoxia. C: Representative images of mouse lung sections from WT and NOX1-deficient mice stained with anti-pSTAT3 (green) and DAPI (blue). Original magnification, X 40. The number of pSTAT3-positive cells is expressed as percent of all nuclei (n=3 mice), *P<0.05 WT-versus NOX1-deficient mice exposed to hyperoxia for 72 h.
As we previously demonstrated that NOX1 contributed to epithelial cell death in mice exposed to hyperoxia [7] and to ensure the role of STAT3 in NOX1-dependent epithelial cell death in mice, we verified by immunostaining whether hyperoxia-induced STAT3 phosphorylation was modulated in NOX1-deficient mice. Hyperoxia was associated with increased pSTAT3 in wildtype mice whereas its expression was significantly lower in NOX1-deficient mice (p<0.05, Figure 5C).
Therefore, activation of STAT3 signalling-mediating cell death during hyperoxia is dependent on NOX1 in MLE12 and in mice.
Discussion
Epithelial cell death is known to be a crucial event in the development of ARDS [23]. Indeed, apoptosis of epithelial cells was observed in biopsies and bronchoalveolar lavage fluid from patients with ARDS [3], and the severity of lung injury has been correlated with the degree of alveolar epithelial damage [1]. Evidence pointed to the participation of ROS in the pathogenesis of human ARDS [25]. In particular, ROS generated by the NADPH oxidase complex were shown to contribute to the pathological mechanisms of ARDS, including alveolar epithelial cell death in mouse models [5,7]. Our previous studies have demonstrated that NOX1, a NADPH oxidase (NOX) isoform expressed in lung epithelial cells, plays an important role in mediating hyperoxic lung damage in mice through the modulation of epithelial and endothelial cell death [7]; however, to date, it remained unclear whether NOX1 also participates to the development of ARDS in humans and its specific signalling pathways have not been defined. For the first time, we demonstrated the presence of NOX1 in alveolar epithelial and endothelial cells of patients with ARDS during the exudative/acute phase. NOX1 was also observed in alveolar epithelial cells positive for cell death. In addition, we detected phosphorylated STAT3, a signal-transducing protein and transcriptional factor known to be activated by oxidative stress [27] and to participate in the cell cycle progression, alveolar cell proliferation and apoptosis during the acute phase of ARDS [16], in cells expressing NOX1 and positive for TUNEL staining. All these observations suggest that NOX1 could participate in alveolar epithelial cell death though in part the activation of STAT3 during the acute phase of ARDS. Nevertheless, since integrity of the alveolo-capillary barrier depends not only on the epithelium but also on the endothelium, we cannot exclude that NOX1 expressed in the endothelium could also contribute to the damage. Indeed, we detected NOX1 in endothelial cells of patients with ARDS in the exudative phase. In addition, our previous study demonstrated NOX1 expression in murine endothelial cells and its participation in hyperoxia-induced endothelial cell death [7]. To date, whether NOX1 restricted to the epithelium is sufficient to induce acute lung injury in hyperoxia, or/and whether endothelium is also required, remains an open question.
Direct genotoxic stress [24,25] and several redox-sensitive signalling pathways including MAPK [7,12,24,28,29] and STAT3 [13,16] have been shown to participate to epithelial cell death in experimental model of ARDS. We previously demonstrated that NOX1 contributes to hyperoxia-induced epithelial cell death through ERK signalling in mice [7]. In MLE12, ERK signalling was shown to be involved in cell death induced by hyperoxia [24,28,29]. The present study supports a role for ROS-derived NOX1 in hyperoxia-induced epithelial cell death through additional complementary mechanisms: a direct genotoxic effect and the activation of redox-sensitive STAT3 signalling. Abolishing NOX1 in MLE12 decreased DNA oxidation in hyperoxic condition concomitantly with reduced DNA fragmentation. Indeed, it has been proposed that DNA oxidation could lead to DNA fragmentation and cell death in ischemia-reperfusion-induced brain stroke [30] suggesting a direct genotoxic effect of ROS produced by NOX1 and its participation in cell death.
In addition, we demonstrated that NOX1-derived ROS led to cell death through the activation of redox-sensitive STAT3 signalling pathways in hyperoxia. We observed a transient increase in STAT3 phosphorylation after 6 h of hyperoxia, which was decreased by NOX1 silencing in MLE12. This is consistent with data reporting that hyperoxia exposure for 2 to 6h activates STAT3 phosphorylation in MLE12 [31], and with a recent study demonstrating the involvement of NADPH oxidase in STAT3 activation in pancreatic acinar cells stimulated with cerulean [32]. In addition, H2O2 is known to activate STAT3 in rat-1 fibroblasts [27] and antioxidant treatment of mice exposed to LPS prevents STAT3 activation [16]. In parallel, we also demonstrated that hyperoxia-induced phosphorylation of STAT3 was prevented in NOX1-deficient mice. The mechanism involving STAT3 activation by ROS is not known. It may be due to direct cysteine oxidation of tyrosine phosphatase (such as PTP1B) by ROS, leading to phosphatase inhibition [33]. STAT3 was shown to participate to the modulation of both cell survival and cell death by regulating pro- and anti-apoptotic factors, caspase and cell cycle regulators, in response to different stimuli [27]. In the context of hyperoxia, we confirmed that treatment of MLE12 with a STAT3 inhibitor lead to a decrease cleaved-caspase 3. Although the pro-apoptotic role of STAT3 in hyperoxic lung injury is controversial [34,35], our results are consistent with these of Ao et al. who reported that STAT3 phosphorylation was increased in hyperoxia and correlated with apoptosis rate [31]. In addition, increased STAT3 phosphorylation was also correlated with increased caspase-3 protein level in the heart of rats subjected to ischemia-reperfusion [36], suggesting that the pro-apoptotic effect of STAT3 is dependent on the type and duration of stimuli. In parallel, we observed an increased level of cleaved-caspase 3 and cleaved-PARP1 concomitantly with STAT3 phosphorylation in MLE12 under hyperoxic condition, which were both inhibited by NOX1 silencing. We found no modification of the level of cyclin D1 and cell proliferation in NOX1-silenced cells exposed to hyperoxia. All these data suggested that, in hyperoxia, NOX1 drives epithelial cell death through caspase3-dependent STAT3 activation in addition to direct genotoxicity.
In conclusion, our results indicate that ROS-derived NOX1 contribute to hyperoxia induced-epithelial cell death through direct DNA oxidation as well as through signaling pathways involving STAT3, caspase-3 and PARP-1. Thus, we speculate that decreased epithelial cell death through inhibition of NOX1 could be a potential therapeutic strategy in the early phase of ARDS.
Acknowledgements
This work was funded by the Swiss National Research Foundation Grant (CBA, WR, IDS, KHK) and SNSF Marie Heim-Vögtlin Programme (SC) and Swiss Pneumology Society (SC). The authors would like to thank, K. Hammad, L. Beer, P. Henchoz, C. Szyndralewiez and F. Stollar, for technical assistance. The anti-NOX1 polyclonal antibody was kindly given by J.D. Lambeth and the antibody cyclin D1 was a gift from I. Szanto. The dual NOX4/NOX1 inhibitor, GKT136901, was provided by Genkyotex SA (www.genkyotex.com), Plan-les-Ouates, Geneva, Switzerland.
Disclosure of conflict of interest
S.C., I.D.S, F.Z, G.S, Y.D, C.D, T.C., J.C.P, W.R, and C.B: no competing financial interests exist. K.H.K is a founding member of the start-up company Genkyotex which develops NOX inhibitors.
References
- 1.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 2.Kallet RH, Matthay MA. Hyperoxic acute lung injury. Respir Care. 2013;58:123–141. doi: 10.4187/respcare.01963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee KS, Choi YH, Kim YS, Baik SH, Oh YJ, Sheen SS, Park JH, Hwang SC, Park KJ. Evaluation of bronchoalveolar lavage fluid from ARDS patients with regard to apoptosis. Respir Med. 2008;102:464–469. doi: 10.1016/j.rmed.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 4.Lamb NJ, Gutteridge JM, Baker C, Evans TW, Quinlan GJ. Oxidative damage to proteins of bronchoalveolar lavage fluid in patients with acute respiratory distress syndrome: evidence for neutrophil-mediated hydroxylation, nitration, and chlorination. Crit Care Med. 1999;27:1738–1744. doi: 10.1097/00003246-199909000-00007. [DOI] [PubMed] [Google Scholar]
- 5.Carnesecchi S, Pache JC, Barazzone-Argiroffo C. NOX enzymes: potential target for the treatment of acute lung injury. Cell Mol Life Sci. 2012;69:2373–2385. doi: 10.1007/s00018-012-1013-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van der Vliet A. NADPH oxidases in lung biology and pathology: host defense enzymes, and more. Free Radic Biol Med. 2008;44:938–955. doi: 10.1016/j.freeradbiomed.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carnesecchi S, Deffert C, Pagano A, Garrido-Urbani S, Metrailler-Ruchonnet I, Schappi M, Donati Y, Matthay MA, Krause KH, Barazzone Argiroffo C. NADPH oxidase-1 plays a crucial role in hyperoxia-induced acute lung injury in mice. Am J Respir Crit Care Med. 2009;180:972–981. doi: 10.1164/rccm.200902-0296OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barazzone C, Horowitz S, Donati YR, Rodriguez I, Piguet PF. Oxygen toxicity in mouse lung: pathways to cell death. Am J Respir Cell Mol Biol. 1998;19:573–581. doi: 10.1165/ajrcmb.19.4.3173. [DOI] [PubMed] [Google Scholar]
- 9.Zhang X, Shan P, Sasidhar M, Chupp GL, Flavell RA, Choi AM, Lee PJ. Reactive oxygen species and extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase mediate hyperoxia-induced cell death in lung epithelium. Am J Respir Cell Mol Biol. 2003;28:305–315. doi: 10.1165/rcmb.2002-0156OC. [DOI] [PubMed] [Google Scholar]
- 10.van Klaveren RJ, Roelant C, Boogaerts M, Demedts M, Nemery B. Involvement of an NAD(P)H oxidase-like enzyme in superoxide anion and hydrogen peroxide generation by rat type II cells. Thorax. 1997;52:465–471. doi: 10.1136/thx.52.5.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herold S, Gabrielli NM, Vadasz I. Novel concepts of acute lung injury and alveolar-capillary barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2013;305:L665–681. doi: 10.1152/ajplung.00232.2013. [DOI] [PubMed] [Google Scholar]
- 12.Cornell TT, Fleszar A, McHugh W, Blatt NB, Le Vine AM, Shanley TP. Mitogen-activated protein kinase phosphatase 2, MKP-2, regulates early inflammation in acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2012;303:L251–258. doi: 10.1152/ajplung.00063.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Severgnini M, Takahashi S, Tu P, Perides G, Homer RJ, Jhung JW, Bhavsar D, Cochran BH, Simon AR. Inhibition of the Src and Jak kinases protects against lipopolysaccharide-induced acute lung injury. Am J Respir Crit Care Med. 2005;171:858–867. doi: 10.1164/rccm.200407-981OC. [DOI] [PubMed] [Google Scholar]
- 14.Tejera P, Wang Z, Zhai R, Su L, Sheu CC, Taylor DM, Chen F, Gong MN, Thompson BT, Christiani DC. Genetic polymorphisms of peptidase inhibitor 3 (elafin) are associated with acute respiratory distress syndrome. Am J Respir Cell Mol Biol. 2009;41:696–704. doi: 10.1165/rcmb.2008-0410OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yum HK, Arcaroli J, Kupfner J, Shenkar R, Penninger JM, Sasaki T, Yang KY, Park JS, Abraham E. Involvement of phosphoinositide 3-kinases in neutrophil activation and the development of acute lung injury. J Immunol. 2001;167:6601–6608. doi: 10.4049/jimmunol.167.11.6601. [DOI] [PubMed] [Google Scholar]
- 16.Severgnini M, Takahashi S, Rozo LM, Homer RJ, Kuhn C, Jhung JW, Perides G, Steer M, Hassoun PM, Fanburg BL, Cochran BH, Simon AR. Activation of the STAT pathway in acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1282–1292. doi: 10.1152/ajplung.00349.2003. [DOI] [PubMed] [Google Scholar]
- 17.Gianni D, Bohl B, Courtneidge SA, Bokoch GM. The involvement of the tyrosine kinase c-Src in the regulation of reactive oxygen species generation mediated by NADPH oxidase-1. Mol Biol Cell. 2008;19:2984–2994. doi: 10.1091/mbc.E08-02-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carnesecchi S, Deffert C, Donati Y, Basset O, Hinz B, Preynat-Seauve O, Guichard C, Arbiser JL, Banfi B, Pache JC, Barazzone C, Krause KH. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid Redox Signal. 2011;15:607–19. doi: 10.1089/ars.2010.3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yuan K, Huang C, Fox J, Gaid M, Weaver A, Li G, Singh BB, Gao H, Wu M. Elevated inflammatory response in caveolin-1-deficient mice with Pseudomonas aeruginosa infection is mediated by STAT3 protein and nuclear factor kappaB (NF-kappaB) J Biol Chem. 2011;286:21814–21825. doi: 10.1074/jbc.M111.237628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wiznerowicz M, Trono D. Conditional suppression of cellular genes: lentivirus vector-mediated drug-inducible RNA interference. J Virol. 2003;77:8957–8961. doi: 10.1128/JVI.77.16.8957-8961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salmon P, Trono D. Production and titration of lentiviral vectors. Curr Protoc Hum Genet. 2007;Chapter 12:Unit 12.10. doi: 10.1002/0471142905.hg1210s54. [DOI] [PubMed] [Google Scholar]
- 22.Carnesecchi S, Bradaia A, Fischer B, Coelho D, Scholler-Guinard M, Gosse F, Raul F. Perturbation by geraniol of cell membrane permeability and signal transduction pathways in human colon cancer cells. J Pharmacol Exp Ther. 2002;303:711–715. doi: 10.1124/jpet.102.039263. [DOI] [PubMed] [Google Scholar]
- 23.Galani V, Tatsaki E, Bai M, Kitsoulis P, Lekka M, Nakos G, Kanavaros P. The role of apoptosis in the pathophysiology of Acute Respiratory Distress Syndrome (ARDS): an up-to-date cell-specific review. Pathol Res Pract. 2010;206:145–150. doi: 10.1016/j.prp.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 24.Horowitz S. Pathways to cell death in hyperoxia. Chest. 1999;116:64S–67S. doi: 10.1378/chest.116.suppl_1.64s. [DOI] [PubMed] [Google Scholar]
- 25.Tasaka S, Amaya F, Hashimoto S, Ishizaka A. Roles of oxidants and redox signaling in the pathogenesis of acute respiratory distress syndrome. Antioxid Redox Signal. 2008;10:739–753. doi: 10.1089/ars.2007.1940. [DOI] [PubMed] [Google Scholar]
- 26.Metrailler-Ruchonnet I, Pagano A, Carnesecchi S, Ody C, Donati Y, Barazzone Argiroffo C. Bcl-2 protects against hyperoxia-induced apoptosis through inhibition of the mitochondria-dependent pathway. Free Radic Biol Med. 2007;42:1062–1074. doi: 10.1016/j.freeradbiomed.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 27.Simon AR, Rai U, Fanburg BL, Cochran BH. Activation of the JAK-STAT pathway by reactive oxygen species. Am J Physiol. 1998;275:C1640–1652. doi: 10.1152/ajpcell.1998.275.6.C1640. [DOI] [PubMed] [Google Scholar]
- 28.Zhuang S, Schnellmann RG. A death-promoting role for extracellular signal-regulated kinase. J Pharmacol Exp Ther. 2006;319:991–997. doi: 10.1124/jpet.106.107367. [DOI] [PubMed] [Google Scholar]
- 29.Pantano C, Anathy V, Ranjan P, Heintz NH, Janssen-Heininger YM. Nonphagocytic oxidase 1 causes death in lung epithelial cells via a TNF-RI-JNK signaling axis. Am J Respir Cell Mol Biol. 2007;36:473–479. doi: 10.1165/rcmb.2006-0109OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cui J, Holmes EH, Greene TG, Liu PK. Oxidative DNA damage precedes DNA fragmentation after experimental stroke in rat brain. FASEB J. 2000;14:955–967. doi: 10.1096/fasebj.14.7.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ao X, Fang F, Xu F. Vasoactive intestinal peptide protects alveolar epithelial cells against hyperoxia via promoting the activation of STAT3. Regul Pept. 2011;168:1–4. doi: 10.1016/j.regpep.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 32.Ju KD, Lim JW, Kim KH, Kim H. Potential role of NADPH oxidase-mediated activation of Jak2/Stat3 and mitogen-activated protein kinases and expression of TGF-beta1 in the pathophysiology of acute pancreatitis. Inflamm Res. 2011;60:791–800. doi: 10.1007/s00011-011-0335-4. [DOI] [PubMed] [Google Scholar]
- 33.Barrett WC, DeGnore JP, Keng YF, Zhang ZY, Yim MB, Chock PB. Roles of superoxide radical anion in signal transduction mediated by reversible regulation of protein-tyrosine phosphatase 1B. J Biol Chem. 1999;274:34543–34546. doi: 10.1074/jbc.274.49.34543. [DOI] [PubMed] [Google Scholar]
- 34.Lian X, Qin Y, Hossain SA, Yang L, White A, Xu H, Shipley JM, Li T, Senior RM, Du H, Yan C. Overexpression of Stat3C in pulmonary epithelium protects against hyperoxic lung injury. J Immunol. 2005;174:7250–7256. doi: 10.4049/jimmunol.174.11.7250. [DOI] [PubMed] [Google Scholar]
- 35.Xu Y, Ikegami M, Wang Y, Matsuzaki Y, Whitsett JA. Gene expression and biological processes influenced by deletion of Stat3 in pulmonary type II epithelial cells. BMC Genomics. 2007;8:455. doi: 10.1186/1471-2164-8-455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duan W, Yang Y, Yan J, Yu S, Liu J, Zhou J, Zhang J, Jin Z, Yi D. The effects of curcumin post-treatment against myocardial ischemia and reperfusion by activation of the JAK2/STAT3 signaling pathway. Basic Res Cardiol. 2012;107:263. doi: 10.1007/s00395-012-0263-7. [DOI] [PubMed] [Google Scholar]