

Abstract
The Six1 homeodomain protein is a developmental transcription factor that has been implicated in tumor onset and progression. Recently, it’s reported that overexpression of Six1 is sufficient to induce epithelial-to-mesenchymal transition (EMT) and metastasis of colorectal cancer. Moreover, its expression is significantly associated with poorer overall survival probability in advanced-stage colorectal cancer. To address whether Six1 could serve as a therapeutic target for human colorectal cancer, we used a lentivirus-mediated short hairpin RNA (shRNA) gene knockdown method to suppress the expression of Six1 in colorectal cancer cells. We showed that lentivirusmediated shRNA targeted to Six1 gene efficiently reduced its expression in colorectal cancer cells at both mRNA and protein levels. In vitro functional assays revealed that knockdown of Six1 significantly suppressed cell proliferation, and inhibited cell migration and invasion of colorectal cancer cells. Furthermore, tumor xenograft model demonstrated that downregulation of Six1 dramatically inhibited colorectal cancer growth in vivo. In conclusion, these findings suggest that lentivirus-mediated Six1 inhibition may represent a novel therapeutic approach for treatment of colorectal cancer.
Keywords: Six1, colorectal cancer, cell growth, invasion
Introduction
Colorectal cancer is one of the most common causes of cancer-related deaths in the world [1,2]. Great advances have been made in clinical treatment in the past few decades. Still, the prognosis of patients with colorectal cancer, especially those with advanced disease, is very poor due to the recurrence and distant metastasis [3]. Therefore, a better understanding of the molecular mechanisms involved in colorectal cancer tumorigenesis and malignant progression is essential for the management of hard-to-treat colorectal cancer.
Six1 is a member of the Six family of homeodomain transcription factors and is highly conserved from Drosophila to humans [4,5]. It is broadly expressed in many tissues and promotes the progenitor cell proliferation and survival during early mammalian development, while in most adult tissues Six1 expression is low or absent [4-6]. However, Six1 is aberrantly upregulated in a variety of human cancers, including breast cancer [7,8], cervical cancer [9,10], ovarian cancer [11], etc., where it leads to increased proliferation, survival and metastasis [12-14]. It was recently reported that Six1, which is aberrantly upregulated in human colorectal cancer, promotes epithelial-mesenchymal transition via ZEB1 activation, and its expression is significantly associated with poorer overall survival probability in advanced-stage colorectal cancer [15]. These evidences strongly support an essential role for Six1 in the tumorigenesis and progression of colorectal cancer; we therefore propose that blocking the expression of Six1 would be a rational therapeutic strategy for colorectal cancer.
In this study, we investigated the effects of Six1 silencing on the growth and invasion of colorectal cancer cells by constructing recombinant shRNA-expressing lentiviral vector targeting Six1. In addition, the effect of Six1 down-regulation on the tumor growth of colorectal cancer in vivo was examined as well. Our data suggest that lentivirus-mediated Six1 inhibition could be a novel approach for colorectal cancer therapy.
Material and methods
Cell culture
Human colorectal cancer cell lines HT29, SW620, SW480, LOVO, HCT15 and HCT116 were obtained from American Type Culture Collection ATCC (Rockville, MD, USA). All cell lines were cultured in Dulbecco’s modified Eagle’s medium (Hyclone, Logan, UT, USA) supplemented with 10% fetal bovine serum (Hyclone, Logan, UT, USA), 100 units/ml penicillin, and 0.1 mg/ml streptomycin (Invitrogen, California, USA) in 5% CO2 atmosphere at 37°C.
Lentivirus-mediated shRNA knockdown of Six1 expression
The following small interfering RNA (siRNA) target sequences in the human Six1 gene (NM_005982) were selected: #1, AGTTTGAGCTCCTGGCGTG; and #2, TTTCTATTTACAAGTGTCC. A scrambled sequence (TTCTCCGAACGTGTCA CGT) was used as negative control for RNA interference (RNAi), which had no significant homology to any human gene sequences. Inverted and self-complementary hairpin DNA oligos targeting Six1 mRNA were obtained from Genchem Biotechnology Company (Shanghai, China). The stem-loop oligonucleotides were synthesized and cloned into a lentivirus-based vector pGCSIL-GFP and resulting plasmids were named as scrambled shRNA, Six1-shRNA-1 and Six1-shRNA-2, respectively. Lentiviruses were generated in 293T cells by co-transfection of Six1-shRNA-1, Six1-shRNA-2, or scrambled shRNA, with pHelper plasmids. These plasmids were transfected into 80% confluent 293T cells using Lipofectamine 2000 (Invitrogen, USA). Then lentiviral particles were harvested from the media forty-eight hours after transfection, and purified with ultracentrifugation. Viral titer was determined by the method of end point dilution through counting the numbers of infected green cells at × 100 magnification under fluorescence microscope (Olympus, Tokyo, Japan) three days after infection to 293T cells. Titer in IU/ml = (the numbers of green fluorescent cells) × (dilution factor) / (volume of virus solution). For lentivirus transduction, SW620 and LOVO cells were subcultured at 5 × 104 cells/well into 6-well culture plates. After grown to 30% confluence, cells were transducted with scrambled shRNA, Six1-shRNA-1 and Six1-shRNA-2 lentivirus at a multiplicity of infection (MOI) of 50. Cells were harvested at 72 hours after infection and the knockdown efficiency of Six1 was evaluated by quantitative real-time RT-PCR and western blot analysis.
RNA isolation and quantitative real-time RT-PCR
Total RNA was isolated from patient specimens by the RNeasy mini kit according to the manufacturer’s instructions (Qiagen, Germany). Quantitative Real-time RT-PCR analysis was done as described. Primer sets used were as follows: for β-actin, 5’-CATGTACGTTGCTATCCAGGC-3’ and 5’-CTCCTTAATGTCACGCACGAT-3’; for Six1, 5’-AAGGAGAAGTCGAGGGGTGT-3’ and 5’-TGCTTGTTGGAGGAG GAGTT-3’.
Western blot analysis
Western blot analysis was performed as we previously described [16]. Briefly, cells were lysed in cold lysis buffer contaning protease inhibitor mixture. Proteins (10-25 μg) were resolved on SDS-PAGE, transferred onto nitrocellulose membranes (Amersham Biosciences, Piscataway, NJ, USA). The membrane was blocked in TBS-T buffer containing 5% (w/v) non-fat milk at room temperature for 1 hour and then probed with antibodies for Six1 and β-actin (all from Santa Cruz Biotech, Santa Cruz, CA, USA) at 4°C overnight. Detection was performed with the SuperSignal West Femto Maximum Sensitivity Substrate Trial Kit (Pierce, Rockford, IL, USA). The band images were digitally captured and quantified with a FluorChem FC2 imaging system (Alpha Innotech, San Leandro, CA, USA).
Cell count assay
Cell count assay was performed as we previously described [16]. Briefly, cells plated in 6-well plates were incubated at 37°C for different periods of time and then removed by trypsinization, and the number of viable cells was counted in a hemocytometer with the use of trypan blue staining. Every sample was measured in triplicate and repeated three times.
BrdU incorporation
Cells were exposed to 10 μM BrdU (BD Biosciences, Mountain View, CA) for 30 min and fixed in 70% ethanol, and then washed with PBS, resuspended, and incubated with 4N HCL and 0.5% Triton X-100 for 30 min at room temperature. After washing with PBS, cells were neutralized with 0.1 M sodium borate before being labeled with FITC-conjugated BrdU antibody (BD Biosciences, Mountain View, CA) and incubated with 50 μg/ml propidium iodide (Sigma Chemical Company, St. Louis, MO) according to the manufacturer’s protocol before being analyzed by a Becton Dickinson FACStar Plus flow cytometer.
Colony formation assay
Six hundred cells were seeded into a six-well plate. After 14 days, cells were stained by 0.5% crystal violet (Sigma-Aldrich, St. Louis, MO, USA) in methanol for 10 min. Colonies (more than 50 μm diameter) were counted directly on the plate. Statistical signifi cance was calculated from at least three independent experiments.
Wound healing and cell invasion assays
Cells (5 × 105) were seeded on a six-well plate and cultured for 24 hours. A scratch was made on the cell monolayer with a 200 μL pipette tip and monitored with a microscope every 12 hours. The migration was determined by the rate of cells filling the scratched area. The normalized wound area was calculated by the software TScratch [17]. The Boyden chamber and polycarbonate membrane precoated with matrigel (BD Biosciences) was used to evaluate invasion ability of the cancer cell. DMEM with 10% fetal bovine serum was added to the lower compartment as a chemoattractant. SW620 or LOVO cells (5 × 104) suspended in 50 μL DMEM with 0.5% BSA was loaded onto the upper compartment of each chamber. After incubation for 36 hours, Cells migrated through the chamber were stained by hematoxylin and eosin (H&E) and subsequently counted under the microscope. Statistical signifi cance was calculated from at least three independent experiments.
Tumor xenografts in nude mice
All procedures were conducted in accordance to Animal Care and Use Committee guidelines of Zhengzhou University. Female BALB/c athymic nude mice (6-week-old) were housed at five/cage in microisolator units under humidity and temperature controlled conditions with 12-hour light-dark cycles. The mice were randomly distributed into two groups (n=5). SW620 cells transduced with either scrambled shRNA or Six1-shRNA-1 were injected subcutaneously into the flank regions of nude mice. The tumor size was measured with calipers from week 1 to week 4. The tumor volume was calculated with the formula: (Length × Width2)/2. Four weeks following implantation, mice were euthanized by asphyxiation in a CO2 chamber and tumor weights were measured. The average values were expressed as mean ± SEM.
Statistical analysis
All data were expressed as mean ± standard error of the mean (SEM). Between groups and among groups comparisons were conducted with Student t test and ANOVA, respectively. Mann-Whitney U test is used for nonparametric variables. Statistical analysis was performed using GraphPad Prism software version 4.0 (PRISM4) (GraphPad Software Inc, LaJolla, CA), and P<0.05 was considered significant.
Results
Suppression of Six1 expression in colorectal cancer cells by lentivirus-mediated RNA interference
We first compared the expression of Six1 in several colorectal cancer cell lines by western blot analysis. As shown in Figure 1A, higher expression of Six1 was detected in SW620 and LOVO cells, therefore those two cell lines were selected in the following loss of function experiments. Next, both SW620 and LOVO cells were transduced with lentiviral vectors encoding either a scrambled control sequence (scrambled shRNA) or two different shRNAs targeting Six1 (Six1-shRNA-1/2). At 72 hours upon transduction, quantitative real-time RT-PCR and western blot analysis were performed to determine the knockdown efficiency of Six1. Results showed that both mRNA and protein levels of Six1 in cells transduced with Six1-shRNA-1/2 were significantly decreased in comparison with that in cells transduced with scrambled shRNA (Figure 1B and 1C; *, P<0.05). These results indicated that lentivirus-mediated RNA interference could efficiently and specifically suppress Six1 expression in both SW620 and LOVO cells.
Figure 1.
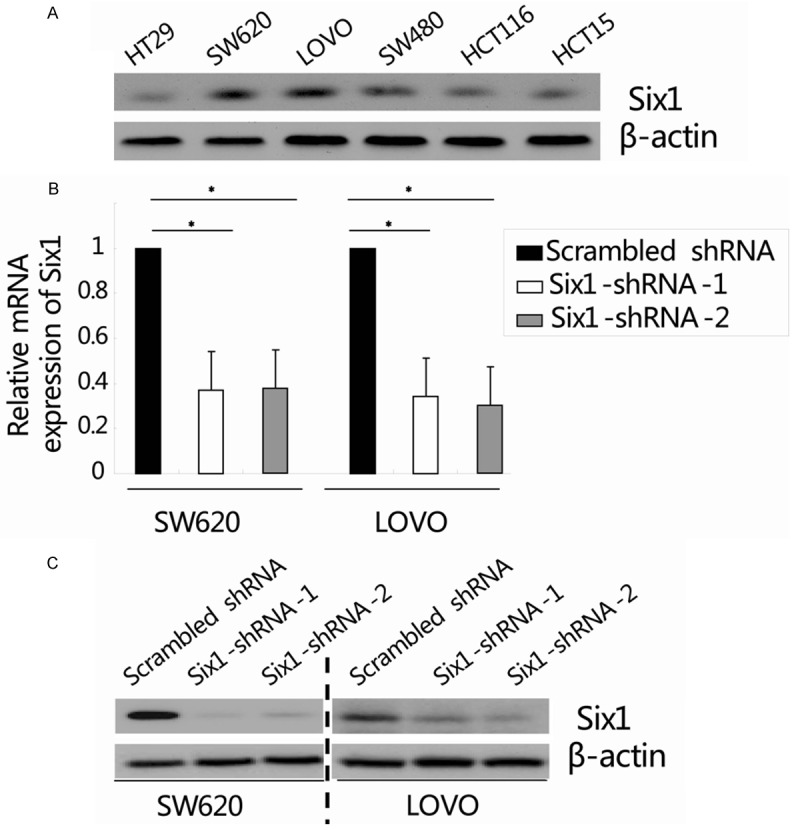
Suppression of Six1 expression in colorectal cancer cells by lentivirus-mediated RNA interference. (A) Western blot analysis showed the expression of Six1 in colorectal cancer cell lines. β-actin was used as an internal control. (B and C) Both SW620 and LOVO cells were transduced with lentiviral vectors encoding either a scrambled control sequence (scrambled shRNA) or two different shRNAs targeting Six1 (Six1-shRNA-1/2). 72 hours after transduction, the relative Six1 mRNA and protein expression was determined by quantitative real-time RT-PCR (B) and western blot analysis (C), respectively. β-actin was used as an internal control. Data represent the mean ± SEM of three independent experiments. *, P<0.05, compared with the scrambled shRNA group.
Knockdown of Six1 inhibits colorectal cancer cells growth in vitro
The effect of Six1 knockdown on the growth of colorectal cancer cells in vitro was assessed by cell count, BrdU incorporation and colony formation assay. Firstly, cell proliferation was determined using cell count assay once daily for 5 days. As shown in Figure 2A, Six1 silencing inhibited SW620 cell proliferation in a time-dependent manner. Compared with the scrambled shRNA group, the cell number in Six1-shRNA1/2 group was significantly reduced on day 5 (scrambled shRNA: (8.8 ± 0.8) × 105, Six1-shRNA-1: (5.7 ± 0.6) × 105 and Six1-shRNA-2: (5.5 ± 0.7) × 105; *, P<0.05). Similar results were found in LOVO cells, as shown in Figure 2B (scrambled shRNA: (7.6 ± 0.6) × 105, Six1-shRNA-1: (5.1 ± 1.1) × 105 and Six1-shRNA-2: (4.6 ± 0.5) × 105; *, P<0.05). Cell proliferation was then assessed by BrdU incorporation assay. A significant decrease the growth rate of SW620 cells in Six1-shRNA-1/2 group (29.3% ± 1.7% and 27.0% ± 2.2%, respectively) was detected compared to scrambled shRNA group (39.0% ± 1.9%) and that was further confirmed in LOVO cells (Figure 2C; scrambled shRNA: 36.3% ± 4.2%, Six1-shRNA-1: 24.6% ± 2.8%, Six1-shRNA-2: 25.0% ± 2.9%; *, P<0.05). Furthermore, the colony formation capacity in both SW620 and LOVO cells transduced with scrambled shRNA or Six1-shRNA1/2 lentivirus was estimated. As shown in Figure 2D, the numbers of SW620 cell colonies in the scrambled shRNA, Six1-shRNA1 and Six1-shRNA-2 group were 157 ± 14, 83 ± 14 and 76 ± 20, respectively (Figure 2D; *, P<0.05). Similar findings were observed in LOVO cells (Figure 2D; scrambled shRNA: 142 ± 20, Six1-shRNA1: 100 ± 15, and Six1-shRNA-2: 95 ± 7; *, P<0.05). Collectively, these results showed that knockdown of Six1 by lentivirus-mediated siRNA could inhibit colorectal cancer cells growth in vitro.
Figure 2.
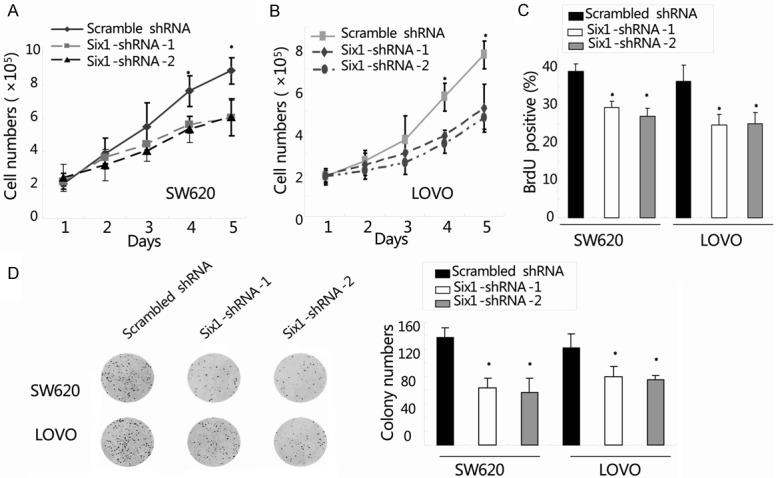
Knockdown of Six1 inhibited colorectal cancer cells growth in vitro. (A and B) Cell proliferation was determined by cell count assay once daily for 5 days in both SW620 (A) and LOVO cells (B). (C) DNA synthesis was measured by BrdU incorporation assay in the two cell lines after transduction. (D) Detection of cell proliferation by colony formation assay in both SW620 and LOVO cells. Representative photographs showed cell colony in 6-well plate. Cell colonies were scored visually and counted using a light microscopy. Data represent the mean ± SEM of three independent experiments. *, P<0.05, compared with the scrambled shRNA group.
Six1 silencing decreases cell motility and invasion of colorectal cancer cells
The cell motility of colorectal cancer cells was evaluated by wound healing assay. The migration was determined by the rate of cells filling the scratched area. The normalized wound area was calculated by the software TScratch. As shown in Figure 3A, compared to cells transduced with scrambled shRNA, the cells transduced with Six1-shRNA-1/2 showed a wider wound area 36 hours after wound generation. The normalized wound area was calculated by the software TScratch [17]. It indicated that knockdown of Six1 inhibits cell motility in colorectal cancer. We further investigated the cell invasiveness using matrigel-coated transwell assay, and the number of invaded cells was quantified in Figure 3B. Consistent with the findings in wound healing assay, cells transduced with Six1-shRNA-1/2 showed a significant reduction in cell invasion ability when compared with scrambled shRNA-treated cells. The number of invaded SW620 cells transduced with scrambled shRNA, Six1-shRNA-1 and Six1-shRNA-2 were 61 ± 14, 39 ± 9 and 36 ± 10, respectively (Figure 3B; *, P<0.05). Similar findings were observed in LOVO cells (Figure 3B; scrambled shRNA: 65 ± 8, Six1-shRNA-1: 40 ± 10, and Six1-shRNA-2: 52 ± 14; *, P<0.05). Taken together, these results indicated that silencing of Six1 decreased the invasive properties of colorectal cancer cells.
Figure 3.
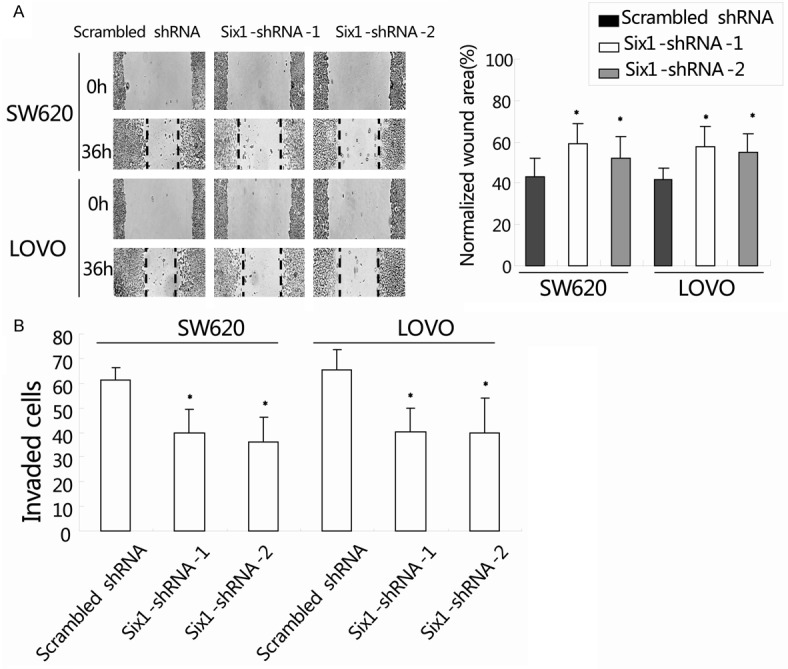
Six1 silencing decreased cell motility and invasion of colorectal cancer cells. A: Wound healing assay was used to evaluate the migration of both SW620 and LOVO cells after silencing Six1. Monolayers of cells were mechanically wounded with a pipette tip and monitored with a microscope every 12 hours. The migration was determined by the rate of cells filling the scratched area. The normalized wound area was calculated by the software TScratch (right panel). B: Cell invasion was determined by matrigel-coated transwell assay. Cells crossed the Matrigel-coated filter were fixed, stained and counted. Data represent the mean ± SEM of three independent experiments. *, P<0.05, compared with the scrambled shRNA group.
Knockdown of Six1 inhibits tumor growth in colorectal cancer xenografts
We have demonstrated that Six1 silencing can efficiently inhibit cell proliferation and suppress the migration and invasion of colorectal cancer cells in vitro. To confirm the above findings, an in vivo tumor xenograft model was used. SW620 cells transduced with either scrambled shRNA or Six1-shRNA-1 were injected subcutaneously into the flank regions of nude mice. As expected, the tumor growth curve showed that tumors derived from Six1-shRNA-1 group grew more slowly than those from scrambled shRNA group. Four weeks after injection, the tumor volume in scrambled shRNA group and Six1-shRNA-1 group were (0.65 ± 0.32) cm3 and (0.14 ± 0.12) cm3, respectively (Figure 4A; *, P<0.05). Then the mice were sacrificed and the weights of the tumors were recorded. Consistent with tumor volume results, the mean tumor weight of the Six1-shRNA-1 group (0.15 ± 0.07 g) was prominently reduced compared to the scrambled shRNA group (0.76 ± 0.23 g) (Figure 4B; *, P<0.05). These data indicated that knockdown of Six1 expression dramatically inhibits colorectal cancer growth in vivo.
Figure 4.
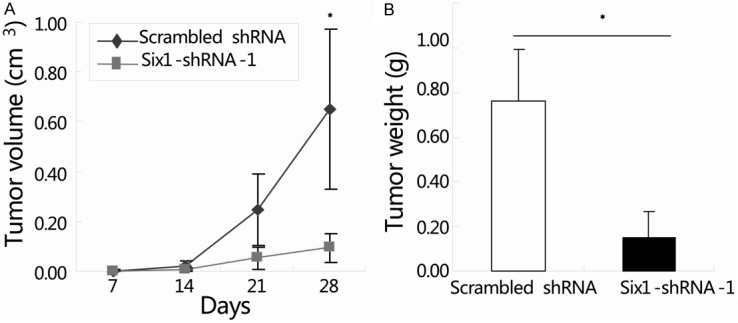
Knockdown of Six1 inhibited tumor growth in colorectal cancer xenografts. SW620 cells transduced with either scrambled shRNA or Six1-shRNA-1 were injected subcutaneously into two groups of nude mice (n=5). Tumor volume was determined on day 7, 14, 21 and 28. Growth curve of tumor xenografts was assessed by serial microcaliper measurements (A). Average Weights of tumor xenografts 28 days after inoculation were recorded (B). Data represent the mean ± SEM. of three independent experiments. *, P<0.05, compared with the scrambled shRNA group.
Discussion
RNA interference mediated gene silencing is already being tested as potential therapy in clinical trials for a number of diseases [18-20]. With the ability to transduce non-dividing and dividing cells, stable transgene expression and minimal toxicity, lentivirus vectors are considered one of the most promising vehicles to efficiently deliver a gene for basic research and gene therapy [21]. In the present study, we constructed recombinant shRNA-expressing lentiviral vector targeting Six1, and then evaluated the effects of Six1 silencing on the celluar growth and invasion in colorectal cancer. The results demonstrate that lentivirus-mediated RNAi which downregulate Six1 specifically might be an effective and convenient approach for the treatment of colorectal cancer. The decreased cell growth and invasion of colorectal cancer cells as well as the suppressive effect on xenografted tumors by silencing Six1 lend further support to the effectiveness of this treatment.
Six1 is a powerful developmental regulator affecting a variety of cellular processes including proliferation, survival, migration and invasion [5,22]. Deregulation of Six1 expression leads to an out of context activation of its developmental functions, contributing to tumor initiation and progression [23]. It is reported that overexpression of Six1 in mammary epithelial cells causes an inappropriate reactivation of the cyclin A1, promoting cell cycle progression, genomic instability and malignant transformation [7,8,24]. In colorectal cancer, overexpressed Six1 in tumor cell contributes to progression of cancer through induction of EMT [15]. Unlike the aberrantly upregulated level in tumor cells, Six1 is not normally expressed in most adult tissues [6,7,25]. Currently most cancer therapy strategies can not exclusively target cancer cells, leading to unwanted and frequently severe side effects. Our study provides evidence that knockdown of Six1 may serve as an effective therapeutic measure with specificity to inhibit cancer progression. It has been reported that overexpression of Six1 renders ovarian carcinoma cells resistant to tumor necrosis factor-related apoptosis inducing ligand (TRAIL)-mediated apoptosis [11,26]. Knockdown of endogenous Six1 in the TRAIL-resistant ovarian cancer cells dramatically sensitizes the cells to TRAIL [11]. We previously reported that Six1 might function as an important modifier of the paclitaxel response in breast cancer cells, and knockdown of Six1 by siRNAs in breast cancer cells sensitizes their response to paclitaxel treatments [27]. These results suggest that lentivirus-mediated knockdown of Six1 could also be used to circumvent drug resistance or re-sensitize the resistant cancer cells, and further studies are then needed to confirm it.
In summary, we established a lentivirus-mediated RNAi system to specifically suppress Six1 expression in colorectal cancer. Knockdown of Six1 could inhibit cell growth and invasion of colorectal cancer cells, and therefore provide a novel approach for colorectal cancer therapy.
Acknowledgements
This study was supported by funds from the National Natural Science Foundation of China (Grant No: 81301087), China Postdoctoral Science Foundation (Grant No: 2013M540574) and Youth innovation funds project of the first affiliated hospital of Zhengzhou university (to Zhaoming Li).
Disclosure of conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Huerta S. Recent advances in the molecular diagnosis and prognosis of colorectal cancer. Expert Rev Mol Diagn. 2008;8:277–288. doi: 10.1586/14737159.8.3.277. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011;61:212–236. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- 3.Chua YJ, Zalcberg JR. Progress and challenges in the adjuvant treatment of stage II and III colon cancers. Expert Rev Anticancer Ther. 2008;8:595–604. doi: 10.1586/14737140.8.4.595. [DOI] [PubMed] [Google Scholar]
- 4.Kumar JP. The sine oculis homeobox (SIX) family of transcription factors as regulators of development and disease. Cell Mol Life Sci. 2009;66:565–583. doi: 10.1007/s00018-008-8335-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Christensen KL, Patrick AN, McCoy EL, Ford HL. The six family of homeobox genes in development and cancer. Adv Cancer Res. 2008;101:93–126. doi: 10.1016/S0065-230X(08)00405-3. [DOI] [PubMed] [Google Scholar]
- 6.Ford HL, Kabingu EN, Bump EA, Mutter GL, Pardee AB. Abrogation of the G2 cell cycle checkpoint associated with overexpression of HSIX1: a possible mechanism of breast carcinogenesis. Proc Natl Acad Sci U S A. 1998;95:12608–12613. doi: 10.1073/pnas.95.21.12608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coletta RD, Christensen K, Reichenberger KJ, Lamb J, Micomonaco D, Huang L, Wolf DM, Müller-Tidow C, Golub TR, Kawakami K, Ford HL. The Six1 homeoprotein stimulates tumorigenesis by reactivation of cyclin A1. Proc Natl Acad Sci U S A. 2004;101:6478–6483. doi: 10.1073/pnas.0401139101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reichenberger KJ, Coletta RD, Schulte AP, Varella-Garcia M, Ford HL. Gene amplification is a mechanism of Six1 overexpression in breast cancer. Cancer Res. 2005;65:2668–2675. doi: 10.1158/0008-5472.CAN-04-4286. [DOI] [PubMed] [Google Scholar]
- 9.Wan F, Miao X, Quraishi I, Kennedy V, Creek KE, Pirisi L. Gene expression changes during HPV-mediated carcinogenesis: a comparison between an in vitro cell model and cervical cancer. Int J Cancer. 2008;123:32–40. doi: 10.1002/ijc.23463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng XH, Liang PH, Guo JX, Zheng YR, Han J, Yu LL, Zhou YG, Li L. Expression and clinical implications of homeobox gene Six1 in cervical cancer cell lines and cervical epithelial tissues. Int J Gynecol Cancer. 2010;20:1587–1592. [PubMed] [Google Scholar]
- 11.Behbakht K, Qamar L, Aldridge CS, Coletta RD, Davidson SA, Thorburn A, Ford HL. Six1 overexpression in ovarian carcinoma causes resistance to TRAIL-mediated apoptosis and is associated with poor survival. Cancer Res. 2007;67:3036–3042. doi: 10.1158/0008-5472.CAN-06-3755. [DOI] [PubMed] [Google Scholar]
- 12.Sehic D, Karlsson J, Sandstedt B, Gisselsson D. SIX1 protein expression selectively identifies blastemal elements in Wilms tumor. Pediatr Blood Cancer. 2012;59:62–68. doi: 10.1002/pbc.24025. [DOI] [PubMed] [Google Scholar]
- 13.Ng KT, Lee TK, Cheng Q, Wo JY, Sun CK, Guo DY, Lim ZX, Lo CM, Poon RT, Fan ST, Man K. Suppression of tumorigenesis and metastasis of hepatocellular carcinoma by shRNA interference targeting on homeoprotein Six1. Int J Cancer. 2010;127:859–872. doi: 10.1002/ijc.25105. [DOI] [PubMed] [Google Scholar]
- 14.Ng KT, Man K, Sun CK, Lee TK, Poon RT, Lo CM, Fan ST. Clinicopathological significance of homeoprotein Six1 in hepatocellular carcinoma. Br J Cancer. 2006;95:1050–1055. doi: 10.1038/sj.bjc.6603399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ono H, Imoto I, Kozaki K, Tsuda H, Matsui T, Kurasawa Y, Muramatsu T, Sugihara K, Inazawa J. SIX1 promotes epithelial-mesenchymal transition in colorectal cancer through ZEB1 activation. Oncogene. 2012;31:4923–34. doi: 10.1038/onc.2011.646. [DOI] [PubMed] [Google Scholar]
- 16.Li Z, Tian T, Lv F, Chang Y, Wang X, Zhang L, Li X, Li L, Ma W, Wu J, Zhang M. Six1 Promotes Proliferation of Pancreatic Cancer Cells via Upregulation of Cyclin D1 Expression. PLoS One. 2013;8:e59203. doi: 10.1371/journal.pone.0059203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gebäck T, Schulz MM, Koumoutsakos P, Detmar M. TScratch: a novel and simple software tool for automated analysis of monolayer wound healing assays. Biotechniques. 2009;46:265–274. doi: 10.2144/000113083. [DOI] [PubMed] [Google Scholar]
- 18.Petrocca F, Lieberman J. Promise and challenge of RNA interference-based therapy for cancer. J. Clin. Oncol. 2011;29:747–754. doi: 10.1200/JCO.2009.27.6287. [DOI] [PubMed] [Google Scholar]
- 19.Phalon C, Rao DD, Nemunaitis J. Potential use of RNA interference in cancer therapy. Expert Rev Mol Med. 2010;12:e26. doi: 10.1017/S1462399410001584. [DOI] [PubMed] [Google Scholar]
- 20.Wang Z, Rao DD, Senzer N, Nemunaitis J. RNA interference and cancer therapy. Pharm Res. 2011;28:2983–2995. doi: 10.1007/s11095-011-0604-5. [DOI] [PubMed] [Google Scholar]
- 21.Manjunath N, Wu H, Subramanya S, Shankar P. Lentiviral delivery of short hairpin RNAs. Adv Drug Deliv Rev. 2009;61:732–745. doi: 10.1016/j.addr.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Micalizzi DS, Farabaugh SM, Ford HL. Epithelial-mesenchymal transition in cancer: parallels between normal development and tumor progression. J Mammary Gland Biol Neoplasia. 2010;15:117–134. doi: 10.1007/s10911-010-9178-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abate-Shen C. Deregulated homeobox gene expression in cancer: cause or consequence? Nat Rev Cancer. 2002;2:777–785. doi: 10.1038/nrc907. [DOI] [PubMed] [Google Scholar]
- 24.Wang CA, Jedlicka P, Patrick AN, Micalizzi DS, Lemmer KC, Deitsch E, Casás-Selves M, Harrell JC, Ford HL. SIX1 induces lymphangiogenesis and metastasis via upregulation of VEGF-C in mouse models of breast cancer. J Clin Invest. 2012;122:1895–1906. doi: 10.1172/JCI59858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coletta RD, McCoy EL, Burns V, Kawakami K, McManaman JL, Wysolmerski JJ, Ford HL. Characterization of the Six1 homeobox gene in normal mammary gland morphogenesis. BMC Dev Biol. 2010;10:4. doi: 10.1186/1471-213X-10-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thorburn A, Behbakht K, Ford H. TRAIL receptor-targeted therapeutics: resistance mechanisms and strategies to avoid them. Drug Resist Updat. 2008;11:17–24. doi: 10.1016/j.drup.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Z, Tian T, Hu X, Zhang X, Nan F, Chang Y, Lv F, Zhang M. Six1 mediates resistance to paclitaxel in breast cancer cells. Biochem Biophys Res Commun. 2013;441:538–43. doi: 10.1016/j.bbrc.2013.10.131. [DOI] [PubMed] [Google Scholar]