

Significance
Molecular machines, such as the proteasome in eukaryotes and archaea as well as the HslU–HslV complex in bacteria, play critical roles in maintaining cellular homeostasis. Here, we have used methyl-Transverse Relaxation-Optimized Spectroscopy to study the 230 kDa HslV dodecamer that cleaves substrate polypeptides. The initial catalytic step is investigated by measuring the pKa of the terminal amine of catalytic residue T1. Furthermore, we show that single site mutations in key regions, which contact HslU or change on HslU binding, lead to propagated changes in structure along an allosteric pathway, affecting proteolysis rates. NMR relaxation experiments are consistent with a coupling of millisecond timescale dynamics throughout regions of HslV that link HslU and substrate binding with catalysis.
Keywords: protein NMR, protein dynamics, molecular machines
Abstract
The HslU–HslV complex functions as a bacterial proteasome, degrading substrate polypeptides to preserve cellular homeostasis. Here, we use methyl-Transverse Relaxation-Optimized Spectroscopy (TROSY) and highly deuterated, methyl-protonated samples to study the 230 kDa dodecameric HslV protease component that is structurally homologous to the stacked pair of β7-rings of the proteasome. Chemical shift assignments for over 95% of the methyl groups are reported. From the pH dependence of methyl chemical shifts, a pKa of 7.7 is measured for the amine group of the catalytic residue T1, confirming that it can act as a proton acceptor during the initial step in substrate proteolysis. Analyses involving a series of single site mutants in HslV, localized to HslU binding sites or regions undergoing significant changes on HslU binding, have identified hot spots whose perturbation leads to an allosteric pathway of propagated changes in structure and ultimately, substrate proteolysis efficiency. HslV plasticity is explored through methyl-TROSY 13C relaxation dispersion experiments that are sensitive to millisecond timescale dynamics. The data support a dynamic coupling between residues involved in both HslU and substrate binding and residues localized to the active sites of HslV that facilitate the allostery between these distal sites. An important role for dynamics has also been observed in the archaeal proteasome, suggesting a more generally conserved role of motion in the function of these barrel-like protease structures.
The 20S proteasome core particle (CP) is a molecular machine that plays a critical role in maintaining cell viability through protein degradation, leading to the removal of damaged or misfolded proteins to prevent formation of toxic aggregates. It is also involved in the regulation of the cell cycle and in generating peptides used in the immune response (1, 2). The proteasome barrel-like structure is comprised of a series of four heptameric rings stacked on top of each other and arranged as α7β7β7α7, with active site residues sequestered within the lumen of the proteasome (3, 4). Regulation of the 20S CP function is, in part, accomplished by binding additional large complexes. These partners include the 19S regulatory particle (RP) ATPase that recognizes ubiquitinated protein substrates, 11S RPs that control the sizes and relative distributions of product peptides that are produced, Blm10/PA200 whose binding stimulates the degradation of peptides, and AAA protein complexes, such as Cdc48/p97/VAT (5–7). Although proteasomes are ubiquitous in eukaryotes and archaea and present in some bacteria, most bacteria have what is considered to be a primitive proteasome system that degrades target proteins in an ATP-dependent manner (8). The protease component, HslV, is a double hexameric ring structure (Fig. 1A), with each of the concentrically arranged rings analogous to β7 in the proteasome. The proteolytic activity of HslV can be enhanced several orders of magnitude (8) through the binding of the hexameric ATPase HslU that unfolds protein substrates and translocates them through a central channel to HslV for degradation (9–11).
Fig. 1.

Near-complete I, L, M, T, and V methyl chemical shift assignments for HslV. (A) Surface model showing the dodecameric structure of HslV, comprising a pair of hexameric rings that are aligned coaxially (9) (Protein Data Bank ID code 1G3K) with the front four subunits removed to show the proteolytic chamber. All monomers of the hexamer are equivalent. Shown in Upper are ribbon diagrams for a pair of monomers, corresponding to the two front-facing subunits of the surface model, related to each other by a 180° rotation about an axis of sixfold symmetry. Helices are labeled H1, H2 (highlighted in cyan), H2A, H3, and H4, with catalytic residue T1 in red. Parts of the substrate binding pocket (S1, S3, and S3′ from an adjacent molecule) are also indicated. (B) Regions of 13C,1H HMQC spectra of [U-2H; Ileδ1-13CH3; Leu,Val-13CH3/12CD3; Met-13CH3]– or [U-2H; Ileδ1-13CH3; Met-13CH3; Thr-13CH3]–labeled HslV (18.8T and 40 °C).
The 20S CP and the HslV protease share many features. For example, both degrade proteins through active sites localized to the lumen of a barrel structure, with function controlled through binding of additional regulatory particles (4, 9). Structurally, the β-subunits of the 20S CP and each of 12 subunits of HslV are highly homologous, and it is believed that both the 20S CP and HslV cleave substrate using a similar catalytic mechanism involving an amino terminal threonine residue (9, 12). However, the HslV protease is simpler, because it is comprised of only a single polypeptide chain, whereas the 20S CP is made up of a pair of proteins, α and β, in the case of the archaeal version or seven different α/β-subunits for the eukaryotic complex (1, 2, 7). Some similarities also exist between the 19S and the HslU RPs that interact with the 20S CP and HslV, respectively. For example, the hexameric HslU RP consists of six identical AAA ATPases that are positioned on the top and bottom of the HslV donut structure (9). Although the 19S RP is much more complex, with 19 canonical subunits, it too includes a ring of six AAA ATPases, architecturally similar to HslU, that binds to the 20S CP α7-rings (1, 2, 7).
The inherent simplicity of the HslU–HslV system makes it attractive as a model for understanding the structure–dynamics–function paradigm in proteasome-like structures using solution NMR, a technique that has, until recently, been limited to much smaller protein systems. Furthermore, HslV from the malaria-causing parasite Plasmodium falciparum may be an attractive antimicrobial drug target (13), especially because the HslV protease is not present in humans. In this regard, a detailed understanding of the mechanism of substrate proteolysis and the general role of dynamics in enzyme function could be important for the design of effective pharmaceuticals. Here, we focus on the Haemophilus influenzae HslV protease because high-resolution X-ray structures are available (9, 11, 12) as well as a significant body of biophysical and biochemical data (14–16). This protease has an aggregate molecular mass of 230 kDa, with each equivalent monomer of ∼19 kDa.
The development of isotope labeling strategies, in which highly deuterated, methyl-protonated proteins are readily produced (17), along with NMR experiments that exploit the labeling to minimize the decay of NMR signals (18) have significantly increased the use of solution NMR for quantitative studies of supramolecular complexes (17, 19). Below, we use these NMR methods in a study of the HslV protease, with the results compared with data obtained on the 20S CP previously. Central to the motivation of the present work is the recent demonstration of an allosteric pathway of communication in the 20S CP that extends from the α-rings, where RPs bind, to the β-ring active sites that are 75 Å removed (20). The high degree of homology between the structures of the β-subunits from the 20S CP and the subunits of the HslV protease led us to wonder whether a pathway might be found in HslV as well and if present, whether it could regulate proteolytic activity. Here, we show that conservative single point mutations in the HslU binding interface of HslV lead to significant chemical shift changes of methyl probes extending to the active site/substrate binding regions of HslV, with concomitant changes in proteolysis rates, identifying an allosteric pathway. By contrast, mutations that do not result in shift changes have little effect on hydrolysis. Relaxation dispersion experiments establish that the regions most affected exchange between conformations on a millisecond timescale, suggesting that inherent plasticity is a key component in mediating the observed allostery.
Results
Methyl Group Assignments and Characterization of the 230 kDa HslV Complex.
Central to any NMR study is the assignment of key probes to specific sites in the primary amino acid sequence. Here, we have chosen I, L, V, M, and T methyl groups as spies of molecular structure and dynamics using [U-2H; Ileδ1-13CH3; Leu,Val-13CH3/12CD3; Met-13CH3]– or [U-2H; Ileδ1-13CH3; Leu,Val-13CH3/12CD3; Met-13CH3; Thr-13CH3]–labeled protein samples, which has been done in previous studies of the proteasome by our group (20–23). Near-complete assignments (91 of 95 methyl groups) were obtained through a combined approach involving mutagenesis (SI Appendix) and analysis of NOE datasets that exploited the availability of a high-resolution X-ray structure of H. influenzae HslV (9). Fig. 1B shows regions of 2D 13C-1H heteronuclear multiple-quantum coherence (HMQC) methyl correlation maps of HslV (40 °C and 18.8T) with assignments provided in SI Appendix, Fig. S1. Stereospecific assignments were achieved using a labeling strategy introduced by Gans et al. (24), where the proR(S) methyl of Leu and Val side chains is 13CH3(12CD3) (SI Appendix, Fig. S1).
We have also validated that the proteins in our NMR samples were of the size expected by recording pulsed-field gradient diffusion experiments (SI Appendix, Fig. S2) on a number of protein samples, including HslV (230 kDa), a single ring of the 20S CP (α7; 180 kDa), the half proteasome (α7α7; 360 kDa), and the 8.9 kDa protein ubiquitin (25 °C). The calculated diffusion rates are inversely proportional to the cube root of the molecular mass, assuming that each of the diffusing particles is spherical. The values for WT HslV and a series of point mutants (see below) are very similar (3.8 ± 0.4 × 10−7 cm2/s) and between those of α7 (4.5 ± 0.1 × 10−7 cm2/s) and α7α7 (2.6 ± 0.1 × 10−7 cm2/s) as expected, whereas ubiquitin diffuses much more rapidly (13.5 ± 0.1 × 10−7 cm2/s). The diffusion data, thus, provide strong evidence that the recombinantly produced samples of HslV, both WT and mutants (see below), have the expected molecular architecture of the intact complex.
NMR Titration Data Support Similar Proteolysis Mechanisms for HslV and the 20S CP.
As described in detail elsewhere, methyl groups can potentially provide detailed structural and dynamics information, even in studies involving very high-molecular mass protein complexes (17, 19). Most applications have focused on I, L, and V probes and sometimes, M (22, 25) and A (26) methyl groups. Although these residues remain important for much of the work described here, extending 13CH3 labeling to threonine is critical, because the catalytic residue in HslV is T1. Therefore, we have used the assignments of I, L, V, and M methyl groups (80 of 84 methyl groups) that were obtained in the initial phases of our work along with NOE data from 13C-edited 3D spectra (SI Appendix, Fig. S3) to obtain complete assignments for all 11 T methyl groups in highly deuterated samples of HslV prepared with 13CH3 label at I, L, V, M, and T sites. Key to the success of these experiments is the availability of [α, β-2H; γ-13C]-T, which has been produced using a biosynthetic scheme described previously (27). We have shown that deuteration at the Tα- and Tβ-positions is critical for recording high-quality datasets when applications to large complexes, such as HslV, are considered.
The availability of assignments for methyl group probes throughout the HslV complex, including for the catalytic residue T1, allowed us to address key mechanistic questions at an early stage in the analysis. For example, in analogy with the proteasome, the catalytic mechanism for peptide bond hydrolysis is thought to involve a nucleophilic attack of the substrate peptide bond by T1 Oγ1 after first removing the hydroxyl proton (SI Appendix, Fig. S4) (3, 28). In its neutral form, the T1 amino group can serve as a base in this first step of the catalytic cycle by accepting the proton that is released from the hydroxyl group (9). However, the ionization state of this site, and hence its ability to function as a base, remains to be confirmed. As we describe below, the titration of methyl group chemical shifts from T1, T2, and I3 as a function of pH facilitates the measurement of the T1 amino group pKa value and hence, the determination of the ionization state of the T1 amino moiety.
Fig. 2A shows a superposition of specific regions of 13C-1H correlation spectra focusing on T1, T2, and I3 recorded over a pH range extending from 6.0 to 11.0, where HslV is stable. Not surprisingly, the methyl groups of these three residues titrate over a much larger chemical shift range than other I and T methyls, providing confidence in the assignments. Fig. 2B plots chemical shift vs. pH profiles for methyl groups of these residues. Notably, none of the profiles could be well-fitted to the Henderson–Hasselbalch equation with only a single pKa. However, they were well-fit assuming a model involving a pair of protonation events each of which is thermodynamically independent but where the chemical shifts of the reporter nuclei depend on the ionization state of both titratable groups (29). Profiles from each of T1, T2, and I3 were fit independently, and the pairs of pKa values obtained (pKa1 = 7.7 ± 0.2 and pKa2 = 10.2 ± 0.3) are in excellent agreement with each other. These values are consistent with those for an unperturbed protein amino terminus (8.0) and a Lys amino group (10.5), respectively (30). X-ray structures of both HslV (9) and the β-subunit of the 20S CP (3) show that K33 is in close proximity to T1 (within 2.9 Å) (Fig. 2C) and functional assays involving mutations of K33 to A, H, or R in the archaeal 20S CP from Thermoplasma acidophilum (31) or to H or R in HslV reduce protease activity to <0.1% of the WT enzyme. Therefore, it is likely that the pKa values measured derive from the titration of the amino groups of T1 and K33. Notably, the activity vs. pH profile of HslV is shifted into a range where the amino terminus of T1 is neutral, and therefore, it can serve as a base, accepting T1 Hγ1 (Fig. 2D). The results from this analysis are in agreement with those from a previous study of the 20S CP, where it was shown that T1 of each β-subunit of the proteasome is also neutral at a pH that is optimal for activity (23).
Fig. 2.

Measurement of active site T1 amine pKa establishes the first step of the catalytic mechanism. (A) Superposition of selected regions of HMQC spectra as a function of pH, focusing on residues T1, T2, and I3. (B) Chemical shift titration data were fit on a per-residue basis to a model that involved ionization of two groups, which is described in the text. The 13C and 1H shifts of a given methyl group were fit simultaneously (e.g., T1γ2). Duplicate measurements at pH 7.5 and pH 10 were used to estimate errors in measured chemical shifts; the error bar is within the size of each dot. (C) Ribbon diagram of a region of a single HlsV monomer highlighting the active site (Protein Data Bank ID code 1G3K). Residues that are critical for the proposed peptidase activity are shown in ball-and-stick representation (SI Appendix). (D) pH profile of HslV peptidase efficiency based on hydrolysis rates of the substrate Z-GGL-AMC (N-carbobenzoxy-Gly-Gly-Leu-amido-4-methylcoumarin; 40 °C).
Allosteric Pathway.
In addition to providing information about the catalytic mechanism, methyl group probes can also be used to obtain insight into the relation between dynamics and function (17, 19). In a previous study of the 20S CP, we observed that mutations of key amino acids in regions forming the binding sites for RPs (α-subunits) resulted in chemical shift changes (hence, structural perturbations) that were propagated over a distance of 75 Å from the α-rings to the substrate binding pockets in the β-rings (20). The relation between allostery and dynamics was clarified by additional solution NMR studies showing that the 20S CP interconverts rapidly between a series of distinct conformers, whose relative populations are changed on introduction of point mutants at key sites in the enzyme. Given the structural similarities between the β-subunits of the proteasome and the subunits of HslV, we wondered whether an allosteric pathway might be present in HslV as well and what the role of dynamics would be in this case. As a first step, we constructed a series of HslV mutants that were localized to regions that either interact directly with HslU or change on HslU binding, with some of the sites of mutation indicated in Fig. 3A. For example, L71 and V76 (mutated to V and A, respectively) are part of helix H2 that, along with H1, binds the carboxyl-terminal tail of each of the subunits of HslU. Other sites include L88 and L91, which are part of a small four-residue helical structure, H2A, immediately after helix H2. A comparison of the X-ray structures of HslV, free and bound to HslU, shows that this region undergoes the largest conformational change on binding, with H2A becoming unstructured (9). In addition to mutations in the RP binding region, we also considered several mutations in H4 at the opposite end of the molecule that is effectively the symmetry-related counterpart of H2. The structure of each HslV monomer is approximately symmetric about a pseudotwofold axis that runs through the center of the protein (Fig. 3A). Shown in gray (cyan for H1/H2) is the structure of HslV using the same orientation as in Fig. 1A, whereas the structure obtained by applying a 180° rotation about the symmetry axis (blue arrow) is indicated in gold in Fig. 3A. Many of the sites of mutation considered in this analysis are labeled in Fig. 3A. Importantly, all mutants have the same oligomeric structure as WT, which was established by pulsed-field gradient diffusion measurements (SI Appendix, Fig. S2) and gel filtration chromatography protein elution profiles (SI Appendix, Fig. S5).
Fig. 3.

An allosteric pathway regulating the function of HslV. (A) The structure of each HslV monomer is approximately symmetric about a pseudotwofold axis that runs through the center of the protein, which is indicated by the blue arrow. Shown in gray (cyan for H1/H2) is the structure of an HslV monomer using the same orientation as in Fig. 1, right subunit, whereas the structure obtained by applying a 180° rotation about the symmetry axis (blue arrow) is indicated in gold. The positions of some of the mutations used are indicated. (B–E) Chemical shift changes relative to the WT protein are indicated in bar chart form and color-coded on the structure. Each methyl-containing residue is depicted in stick representation, and those methyls that undergo changes in shift ≥25 Hz are highlighted in orange. The mean chemical shift change (±1 SD) is indicated, excluding methyl groups ≤ 8 Å from the site of mutation. Note that the L88I mutation (H2) shows large changes in chemical shifts, whereas the mutation involving the symmetry-related position I159V shows little change. Where changes are observed, they extend from helices H1/H2 to regions surrounding the active site and/or substrate binding sites (S1, S3, and S3′) as indicated.
Fig. 3 and SI Appendix, Fig. S6 show ribbon diagrams of one of the subunits from the dodecameric HslV complex, highlighting all I, L, V, M, and T methyl groups that serve as probes. Methyl groups with changes in chemical shifts 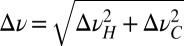 > 25 Hz on mutation of the WT protein, where Δνj,
j
> 25 Hz on mutation of the WT protein, where Δνj,
j [H,C] is the difference in shift in the j dimension of the 13C-1H HMQC spectrum, are color-coded orange on each structure. Immediately underneath each structure is a plot of Δν as a function of residue. Relatively large mean shift changes are observed for L71V (21 ± 29 Hz) (Fig. 3B), L88I (25 ± 25 Hz) (Fig. 3C), and L91I (28 ± 30 Hz) (Fig. 3D), excluding those methyl groups within 8 Å of the residue mutated. By contrast, very little shift difference is observed for I159V (5 ± 7 Hz) (Fig. 3E), where I159 (H4) is symmetry-related to L88 (H2) through a 180° rotation about the axis shown in Fig. 3A. This scenario, in which large changes in shifts are observed for residues at the HslU binding site (H1/H2) but not at symmetric positions (H3/H4), has also been noted for the L91I (H2)-V161I (H4) pair (Fig. 3 and SI Appendix, Fig. S6). Similar observations have also been made for other residues that are not at symmetric positions but nevertheless, brought into proximity on 180° rotation about the symmetry axis, such as L71V (H2)-V138I (H3) or V76A (H2)-V149A (H4).
[H,C] is the difference in shift in the j dimension of the 13C-1H HMQC spectrum, are color-coded orange on each structure. Immediately underneath each structure is a plot of Δν as a function of residue. Relatively large mean shift changes are observed for L71V (21 ± 29 Hz) (Fig. 3B), L88I (25 ± 25 Hz) (Fig. 3C), and L91I (28 ± 30 Hz) (Fig. 3D), excluding those methyl groups within 8 Å of the residue mutated. By contrast, very little shift difference is observed for I159V (5 ± 7 Hz) (Fig. 3E), where I159 (H4) is symmetry-related to L88 (H2) through a 180° rotation about the axis shown in Fig. 3A. This scenario, in which large changes in shifts are observed for residues at the HslU binding site (H1/H2) but not at symmetric positions (H3/H4), has also been noted for the L91I (H2)-V161I (H4) pair (Fig. 3 and SI Appendix, Fig. S6). Similar observations have also been made for other residues that are not at symmetric positions but nevertheless, brought into proximity on 180° rotation about the symmetry axis, such as L71V (H2)-V138I (H3) or V76A (H2)-V149A (H4).
Proteolytic activities of the mutants considered in this study were measured by monitoring changes in fluorescence resulting from hydrolysis of a large excess of the fluorogenic substrate peptide N-carbobenzoxy-Gly-Gly-Leu-amido-4-methylcoumarin (8). Fig. 4 plots the reaction velocity as a function of the concentration of substrate and lists kcat/KM values. All mutated residues are distal to the active site or substrate binding regions, with distances ≥13 Å for all sites, with the exception of I159 (10 Å) and V161 (7 Å). Nevertheless, some mutations result in changes in kcat/KM ratios by over an order of magnitude from the WT enzyme. Notably, mutations that lead to significant changes in chemical shifts are those that change proteolysis rates, such as L71V, L88I, and L91I (Fig. 3) as well as V76A (SI Appendix, Fig. S6). For the remaining mutants (I159V, V161I, V138I, V149A, and L72V), there is little change in either chemical shifts or kcat/KM.
Fig. 4.
Peptidase assay for the HslV mutants considered. A fluorogenic peptide Z-GGL-AMC was used as a substrate. The V–substrate curves were fitted to a simple Michaelis–Menten equation (solid lines) as indicated. Note that mutations showing large chemical shift perturbations (Fig. 3 and SI Appendix, Fig. S6) also give rise to an order of magnitude change in activity (either increase or decrease), whereas mutants (for example, I159V) producing small chemical shift changes have little effect on activity. Values of kcat/Km (M−1s−1) are indicated. The values, including errors, are WT = 18 ± 2; L71V = 422 ± 8; L72V = 20 ± 4; V76A = 151 ± 10; L88I = 136 ± 5; L91I = 1.40 ± 0.03; V138I = 47 ± 5; I159V = 37 ± 8; and V161I = 58 ± 2.
Spin Relaxation Experiments Confirm the Plasticity of HslV.
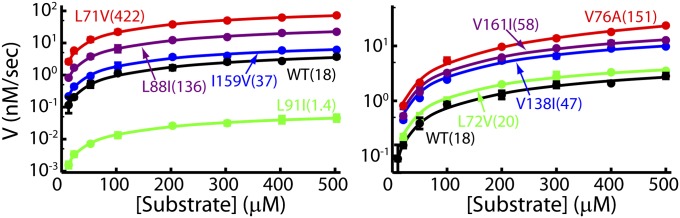
The structural plasticity of HslV, suggested by the mutational study described above, has been further examined by recording NMR spin relaxation experiments that are sensitive to motion on the millisecond timescale (32, 33). Conformational exchange in this time regime gives rise to stochastic changes of chemical shifts, leading to an increase in effective transverse relaxation rates of NMR spins, R2,eff. This effect can be modulated by the application of a series of chemical shift refocusing pulses, each separated by a delay-δ [νCPMG = 1/(2δ)], to produce dispersion profiles (R2,eff vs. νCPMG) that are then fit to extract rates of exchange between conformers and the fractional populations of each of the interchanging states. We have recorded 13C-1H multiple quantum (33) and 1H single quantum (34) relaxation dispersion profiles on samples of WT HslV (Materials and Methods). Fig. 5 shows a number of these curves (recorded at 18.8T and 40 °C). All 13C (18.8T and 14.0T) and 1H data (18.8T), with the exception of I3Cδ1, could be fit together reasonably well assuming a model of two-site exchange (Fig. 5, solid lines) (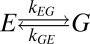 ), with kex = kEG + kGE = 620 ± 30 s−1 and a value for the fractional population of the excited state (E) of pE = 2.4 ± 0.1%. Residues for which methyl groups have dispersion profiles with
), with kex = kEG + kGE = 620 ± 30 s−1 and a value for the fractional population of the excited state (E) of pE = 2.4 ± 0.1%. Residues for which methyl groups have dispersion profiles with 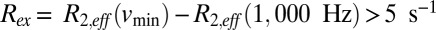 (νmin = 50 and 100 Hz for 13C and 1H dispersions, respectively) are plotted in orange on the ribbon structure of an HslV monomer in Fig. 5. Dynamic residues are found in all regions of the protein but particularly, helices H1/H2 and the β-sheet interface connecting them with H3/H4. Interestingly, residues that play an important role in catalysis (T1), are proximal to the active site (I3), are involved in substrate binding (L22 and I109), or form the binding sites for the carboxyl-terminal tails of HslU (L56, L72, V76, L104, and V112) are all dynamic on the millisecond timescale.
(νmin = 50 and 100 Hz for 13C and 1H dispersions, respectively) are plotted in orange on the ribbon structure of an HslV monomer in Fig. 5. Dynamic residues are found in all regions of the protein but particularly, helices H1/H2 and the β-sheet interface connecting them with H3/H4. Interestingly, residues that play an important role in catalysis (T1), are proximal to the active site (I3), are involved in substrate binding (L22 and I109), or form the binding sites for the carboxyl-terminal tails of HslU (L56, L72, V76, L104, and V112) are all dynamic on the millisecond timescale.
Fig. 5.

Representative single quantum 1H or 13C-1H multiple quantum CPMG relaxation dispersion profiles (R2,eff vs. νCPMG) for WT HslV (18.8T and 40 °C). Methyl-containing side chains are highlighted on the ribbon diagram and color-coded in orange in the case where Rex > 5 s−1.
Discussion
Of all the NMR parameters that can be measured, chemical shifts are obtained most accurately, and they can be exquisitely sensitive to changes in conformation. In this study, we have assigned over 95% of the I, L, V, M, and T methyl groups in the 230 kDa HslV complex (Fig. 1), and therefore, they can be used as probes of function and dynamics. Methyl groups are uniquely suited for studies of high-molecular mass complexes, because they can be readily 13C,1H-labeled in an otherwise highly deuterated background, they are localized to molecular interfaces and in the present case, to active sites, and they give rise to NMR spectra that can be quantified at a level of detail that is often reserved for studies of small- to medium-sized proteins using traditional labeling schemes (17, 19). Here, we have used the assigned methyl groups to test aspects of the reported mechanism of proteolysis involving catalytic residue T1 and to address the plasticity of the HslV protease.
We have used the methyl groups of T1, T2, and I3 as reporters of the ionization state of the amine group of T1, providing evidence that it functions as a base by abstracting the T1 hydroxyl proton in the first step of catalysis. If proton abstraction is to happen efficiently, then the amine must be neutral over the pH optimum activity range of HslV. Chemical shift titrations vs. pH have been fit with a pKa value ∼ 7.5, consistent with a neutral form for the terminal amine at pH 8–9, where HslV is optimally active (Fig. 2). Interestingly, from a similar analysis on the 20S CP, a pKa value of 6.3 was obtained for the amino terminal amine of T1 of the β-subunits (23).
Elegant X-ray studies of HslV and the HslU–HslV complex establish that HslV is an allosteric enzyme (9). On binding HslU, the apical helices of HslV (Fig. 1, cyan) shift substantially, leading to conformational changes that are propagated to residues proximal to the active site to increase proteolysis rates by two orders of magnitude (8). Moreover, it has been shown that, in an asymmetric complex in which only a single HslU was attached to the HslV double ring, the HslV ring bound with HslU is active in the sense that it could react with inhibitor, whereas the second is not (35). Although X-ray studies have not been able to explain the allosteric properties of the 20S CP that have been observed in biochemical assays (36), a recent methyl-Transverse Relaxation-Optimized Spectroscopy (TROSY) NMR study of the 20S CP from T. acidophilum shows an allosteric pathway extending from the sites of RP binding on the α-rings of the proteasome to residues involved in substrate binding localized to the β-rings. This pathway could be manipulated through mutations of residues that are critical for RP binding, leading to changes in the distribution of proteolysis products (20).
The similarities in structure between the β-rings of the 20S CP proteasome and the HslV protease as well as the crystallographic data described above suggest that HslV is likely to be inherently plastic, as observed for the proteasome in NMR studies (20), with this plasticity important for function. To explore this issue further, we have produced a series of single site mutants in HslV that are localized to HslU binding sites (apical helices H1/H2) or regions undergoing significant changes on HslU binding and quantified methyl chemical shift changes relative to the WT enzyme. In all cases where substantial chemical shift changes were observed, there were order of magnitude changes in kcat/KM (Figs. 3 and 4 and SI Appendix, Fig. S6), and where only very minor shifts were recorded, few changes to enzyme activity were noted. Moreover, mutations of residues that are related to amino acids on the apical helices by a 180° rotation of the structure about its pseudosymmetry axis (Fig. 3A) result in only small changes in chemical shifts and correspondingly, little effect on kcat/KM. Thus, the effects of the mutations on chemical shifts and hence, structure are unidirectional, extending from H1/H2 to the interior of the protein, ultimately leading to changes in activity. Despite the inherent symmetry of the molecule, it is not possible to effect the same changes in structure/function by mutations in the corresponding positions on H3/H4. Hot spots on H1/H2 are primed to sense perturbations through either HslU binding or mutation, which was noted for the 20S CP (20). By contrast, these hot spots are not found on H3/H4, because these helices carry out a function distinct from the role of H1/H2. Helices H3/H4 stabilize the HslV complex through packing interactions involving neighboring subunits, and therefore, there is no need for them to sense and respond to HslU binding. Furthermore, it may well be the case that the packing interactions involving adjacent stacked rings dampen the effects of mutations to H3/H4, providing a structural rational for the observed directionality of mutational effects from H1/H2 to the active site region of the protease, despite the inherent symmetry within a given HslV subunit.
To explore HslV plasticity in more detail, we have performed methyl-TROSY 13C and 1H relaxation dispersion experiments that are sensitive to millisecond timescale dynamics. Large dispersion profiles are found at many sites (Fig. 5), including residues L88 and L91, where mutation leads to a 10-fold increase (L88) or decrease (L91) in kcat/KM. Previous biochemical studies have shown that the proteolytic activity of HslV can be enhanced several orders of magnitude through the binding of HslU (8), suggesting that the major state of the isolated protease in solution corresponds to a proteolytically repressed conformation. We were, thus, interested in testing whether changes in activity observed for the various HslV mutants could be explained in terms of a shift of relative populations of conformers interconverting between repressed and derepressed states. To this end, we compared relaxation dispersion profiles from experiments recorded on WT, L71V (20-fold more active), and L91I (12-fold less active) HslV. As with the WT protein, a large number of dispersion profiles was also observed for both mutants, involving, for the most part, the same residues in all three proteins, and these profiles were fit to a two-site exchange model. Values of pE = 2.6 ± 0.3% and 1.6 ± 0.3% were obtained for L71V and L91I mutants, respectively, and were similar to 2.4 ± 0.1% for WT HslV. The dispersion data argue that the millisecond dynamics do not derive from exchange between inactive and active conformers, because very significant differences in pE would then be obtained for each of the HslV systems examined. More likely, the dispersion profiles are reporting a dynamic coupling between residues involved in HslU and substrate binding and amino acids localized to the active sites.
We have further extended the relaxation analysis to include a comparison of picosecond–nanosecond timescale motion of WT, L71V (20 times more active), L91I (12 times less active), and V138I (approximately the same as WT activity) HslV as described in SI Appendix. For each residue, an order parameter squared, 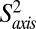 , that is related to the amplitude of motion of the methyl group symmetry axis was obtained (37). Values of
, that is related to the amplitude of motion of the methyl group symmetry axis was obtained (37). Values of 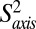 = 1 or 0 correspond to a methyl axis that is completely rigid or disordered, respectively, whereas values 0 <
= 1 or 0 correspond to a methyl axis that is completely rigid or disordered, respectively, whereas values 0 < 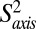 < 1 reflect intermediate levels of ordering. Values of
< 1 reflect intermediate levels of ordering. Values of 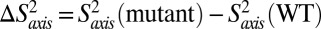 (SI Appendix, Fig. S7) are small (<0.2), indicating only small differences in methyl axis ordering between WT and mutant HslVs. Taken together, the relaxation data indicate that both picosecond–nanosecond and millisecond timescale dynamics are similar in each of the examined mutants of HslV; hence, the changes in activity do not derive from large global changes in motion but from structural rearrangements that propagate to active/substrate binding sites through the coupled dynamic network. This picture is consistent with and adds to the results from comparative X-ray studies (9), in which a shift in the position of G48 on binding HslU provides stabilization of the active form of HslV (SI Appendix, Fig. S4).
(SI Appendix, Fig. S7) are small (<0.2), indicating only small differences in methyl axis ordering between WT and mutant HslVs. Taken together, the relaxation data indicate that both picosecond–nanosecond and millisecond timescale dynamics are similar in each of the examined mutants of HslV; hence, the changes in activity do not derive from large global changes in motion but from structural rearrangements that propagate to active/substrate binding sites through the coupled dynamic network. This picture is consistent with and adds to the results from comparative X-ray studies (9), in which a shift in the position of G48 on binding HslU provides stabilization of the active form of HslV (SI Appendix, Fig. S4).
In summary, we have used methyl-TROSY NMR spectroscopy to show that the HslV protease is an inherently plastic molecule that is poised to undergo a conformational change on HslU binding, leading to activation. Key residues at the binding interface serve as hot spots in the sense that their mutation leads to changes in structure as reported by chemical shift perturbations and concomitantly, changes in proteolysis rates. Many of these residues participate in a concerted millisecond timescale dynamic process involving regions of structure linking HslU binding with substrate binding and proteolysis. Dynamics, thus, facilitate an allosteric pathway that communicates HslU binding to sites of proteolysis in HslV, leading to increased activity. The important role of plasticity that is established here and by earlier X-ray studies of HslV (9) is also seen with the 20S CP proteasome (20), suggesting that both structure and dynamics are critical components for the function of these barrel-like protease systems.
Materials and Methods
Proteins were expressed as described in SI Appendix. All NMR experiments were performed at 40 °C using 18.8T or 14.0T Varian Inova spectrometers equipped with room temperature (18.8T) or cryogenically cooled (14.0T) pulsed-field gradient triple resonance probes. Sample concentrations varied from 300 μM to 2 mM in HslV (subunit) concentration, depending on the application. HslV protein was dissolved in a buffer of 75 mM KCl, 1 mM EGTA, 5 mM MgCl2, 20 mM Hepes (pH 7.6), and 0.05% azide in 99.9% D2O (0.1% H2O, vol/vol) for all experiments except the pH titration, where a mixture of buffering reagents was used, including 25 mM monosodium citrate, 25 mM dipotassium phosphate, 25 mM Tris, 25 mM boric acid, 1 mM EGTA, 5 mM MgCl2, and 0.05% azide. Additional experimental details are provided in SI Appendix.
Supplementary Material
Acknowledgments
We thank Professor J. Forman-Kay for valuable discussions, A. Velyvis for the production of methyl-labeled threonine, R. Muhandiram for NMR support, and Professor R. Melnyk for use of the microplate reader. L.S. is a recipient of postdoctoral fellowships from the Province of Ontario and the Canadian Institutes of Health Research. L.E.K. holds a Canada Research Chair in Biochemistry. This work was funded by research grants from the Canadian Institutes of Health Research and Natural Sciences and Engineering Research Council (to L.E.K.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1318476111/-/DCSupplemental.
References
- 1.Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem. 1996;65:801–847. doi: 10.1146/annurev.bi.65.070196.004101. [DOI] [PubMed] [Google Scholar]
- 2.Baumeister W, Walz J, Zühl F, Seemüller E. The proteasome: Paradigm of a self-compartmentalizing protease. Cell. 1998;92(3):367–380. doi: 10.1016/s0092-8674(00)80929-0. [DOI] [PubMed] [Google Scholar]
- 3.Löwe J, et al. Crystal structure of the 20S proteasome from the archaeon T. acidophilum at 3.4 A resolution. Science. 1995;268(5210):533–539. doi: 10.1126/science.7725097. [DOI] [PubMed] [Google Scholar]
- 4.Bochtler M, Ditzel L, Groll M, Hartmann C, Huber R. The proteasome. Annu Rev Biophys Biomol Struct. 1999;28:295–317. doi: 10.1146/annurev.biophys.28.1.295. [DOI] [PubMed] [Google Scholar]
- 5.Barthelme D, Sauer RT. Identification of the Cdc48•20S proteasome as an ancient AAA+ proteolytic machine. Science. 2012;337(6096):843–846. doi: 10.1126/science.1224352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meyer H, Bug M, Bremer S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat Cell Biol. 2012;14(2):117–123. doi: 10.1038/ncb2407. [DOI] [PubMed] [Google Scholar]
- 7.Kish-Trier E, Hill CP. Structural biology of the proteasome. Annu Rev Biophys. 2013;42(2013):29–49. doi: 10.1146/annurev-biophys-083012-130417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yoo SJ, et al. Purification and characterization of the heat shock proteins HslV and HslU that form a new ATP-dependent protease in Escherichia coli. J Biol Chem. 1996;271(24):14035–14040. doi: 10.1074/jbc.271.24.14035. [DOI] [PubMed] [Google Scholar]
- 9.Sousa MC, et al. Crystal and solution structures of an HslUV protease-chaperone complex. Cell. 2000;103(4):633–643. doi: 10.1016/s0092-8674(00)00166-5. [DOI] [PubMed] [Google Scholar]
- 10.Bochtler M, et al. The structures of HsIU and the ATP-dependent protease HsIU-HsIV. Nature. 2000;403(6771):800–805. doi: 10.1038/35001629. [DOI] [PubMed] [Google Scholar]
- 11.Wang J, et al. Crystal structures of the HslVU peptidase-ATPase complex reveal an ATP-dependent proteolysis mechanism. Structure. 2001;9(2):177–184. doi: 10.1016/s0969-2126(01)00570-6. [DOI] [PubMed] [Google Scholar]
- 12.Bochtler M, Ditzel L, Groll M, Huber R. Crystal structure of heat shock locus V (HslV) from Escherichia coli. Proc Natl Acad Sci USA. 1997;94(12):6070–6074. doi: 10.1073/pnas.94.12.6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tschan S, Mordmüller B, Kun JF. Threonine peptidases as drug targets against malaria. Expert Opin Ther Targets. 2011;15(4):365–378. doi: 10.1517/14728222.2011.555399. [DOI] [PubMed] [Google Scholar]
- 14.Ramachandran R, Hartmann C, Song HK, Huber R, Bochtler M. Functional interactions of HslV (ClpQ) with the ATPase HslU (ClpY) Proc Natl Acad Sci USA. 2002;99(11):7396–7401. doi: 10.1073/pnas.102188799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kwon AR, Trame CB, McKay DB. Kinetics of protein substrate degradation by HslUV. J Struct Biol. 2004;146(1-2):141–147. doi: 10.1016/j.jsb.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 16.Park E, Lee JW, Eom SH, Seol JH, Chung CH. Binding of MG132 or deletion of the Thr active sites in HslV subunits increases the affinity of HslV protease for HslU ATPase and makes this interaction nucleotide-independent. J Biol Chem. 2008;283(48):33258–33266. doi: 10.1074/jbc.M805411200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruschak AM, Kay LE. Methyl groups as probes of supra-molecular structure, dynamics and function. J Biomol NMR. 2010;46(1):75–87. doi: 10.1007/s10858-009-9376-1. [DOI] [PubMed] [Google Scholar]
- 18.Tugarinov V, Hwang PM, Ollerenshaw JE, Kay LE. Cross-correlated relaxation enhanced 1H-13C NMR spectroscopy of methyl groups in very high molecular weight proteins and protein complexes. J Am Chem Soc. 2003;125(34):10420–10428. doi: 10.1021/ja030153x. [DOI] [PubMed] [Google Scholar]
- 19.Sprangers R, Velyvis A, Kay LE. Solution NMR of supramolecular complexes: Providing new insights into function. Nat Methods. 2007;4(9):697–703. doi: 10.1038/nmeth1080. [DOI] [PubMed] [Google Scholar]
- 20.Ruschak AM, Kay LE. Proteasome allostery as a population shift between interchanging conformers. Proc Natl Acad Sci USA. 2012;109(50):E3454–E3462. doi: 10.1073/pnas.1213640109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sprangers R, Kay LE. Quantitative dynamics and binding studies of the 20S proteasome by NMR. Nature. 2007;445(7128):618–622. doi: 10.1038/nature05512. [DOI] [PubMed] [Google Scholar]
- 22.Religa TL, Sprangers R, Kay LE. Dynamic regulation of archaeal proteasome gate opening as studied by TROSY NMR. Science. 2010;328(5974):98–102. doi: 10.1126/science.1184991. [DOI] [PubMed] [Google Scholar]
- 23.Velyvis A, Kay LE. Measurement of active site ionization equilibria in the 670 kDa proteasome core particle using methyl-TROSY NMR. J Am Chem Soc. 2013;135(25):9259–9262. doi: 10.1021/ja403091c. [DOI] [PubMed] [Google Scholar]
- 24.Gans P, et al. Stereospecific isotopic labeling of methyl groups for NMR spectroscopic studies of high-molecular-weight proteins. Angew Chem Int Ed Engl. 2010;49(11):1958–1962. doi: 10.1002/anie.200905660. [DOI] [PubMed] [Google Scholar]
- 25.Gelis I, et al. Structural basis for signal-sequence recognition by the translocase motor SecA as determined by NMR. Cell. 2007;131(4):756–769. doi: 10.1016/j.cell.2007.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Isaacson RL, et al. A new labeling method for methyl transverse relaxation-optimized spectroscopy NMR spectra of alanine residues. J Am Chem Soc. 2007;129(50):15428–15429. doi: 10.1021/ja0761784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Velyvis A, Ruschak AM, Kay LE. An economical method for production of 2H,13CH3-threonine for solution NMR studies of large protein complexes: Application to the 670 kDa proteasome. PLoS One. 2012;7(9):e43725. doi: 10.1371/journal.pone.0043725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marques AJ, Palanimurugan R, Matias AC, Ramos PC, Dohmen RJ. Catalytic mechanism and assembly of the proteasome. Chem Rev. 2009;109(4):1509–1536. doi: 10.1021/cr8004857. [DOI] [PubMed] [Google Scholar]
- 29.McIntosh LP, et al. Dissecting electrostatic interactions in Bacillus circulans xylanase through NMR-monitored pH titrations. J Biomol NMR. 2011;51(1–2):5–19. doi: 10.1007/s10858-011-9537-x. [DOI] [PubMed] [Google Scholar]
- 30.Lehninger AL, Nelson DL, Cox MM. Lehninger Principles of Biochemistry. 6th Ed. New York: Freeman; 2013. [Google Scholar]
- 31.Seemuller E, Lupas A, Baumeister W. Autocatalytic processing of the 20S proteasome. Nature. 1996;382(6590):468–471. doi: 10.1038/382468a0. [DOI] [PubMed] [Google Scholar]
- 32.Palmer AG, 3rd, Kroenke CD, Loria JP. Nuclear magnetic resonance methods for quantifying microsecond-to-millisecond motions in biological macromolecules. Methods Enzymol. 2001;339:204–238. doi: 10.1016/s0076-6879(01)39315-1. [DOI] [PubMed] [Google Scholar]
- 33.Korzhnev DM, Kloiber K, Kanelis V, Tugarinov V, Kay LE. Probing slow dynamics in high molecular weight proteins by methyl-TROSY NMR spectroscopy: Application to a 723-residue enzyme. J Am Chem Soc. 2004;126(12):3964–3973. doi: 10.1021/ja039587i. [DOI] [PubMed] [Google Scholar]
- 34.Baldwin AJ, Religa TL, Hansen DF, Bouvignies G, Kay LE. 13CHD2 methyl group probes of millisecond time scale exchange in proteins by 1H relaxation dispersion: An application to proteasome gating residue dynamics. J Am Chem Soc. 2010;132(32):10992–10995. doi: 10.1021/ja104578n. [DOI] [PubMed] [Google Scholar]
- 35.Kwon AR, Kessler BM, Overkleeft HS, McKay DB. Structure and reactivity of an asymmetric complex between HslV and I-domain deleted HslU, a prokaryotic homolog of the eukaryotic proteasome. J Mol Biol. 2003;330(2):185–195. doi: 10.1016/s0022-2836(03)00580-1. [DOI] [PubMed] [Google Scholar]
- 36.Kleijnen MF, et al. Stability of the proteasome can be regulated allosterically through engagement of its proteolytic active sites. Nat Struct Mol Biol. 2007;14(12):1180–1188. doi: 10.1038/nsmb1335. [DOI] [PubMed] [Google Scholar]
- 37.Sun H, Kay LE, Tugarinov V. An optimized relaxation-based coherence transfer NMR experiment for the measurement of side-chain order in methyl-protonated, highly deuterated proteins. J Phys Chem B. 2011;115(49):14878–14884. doi: 10.1021/jp209049k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.