

Significance
Strict laboratory precautions against present day human DNA contamination are standard in ancient DNA studies, but contamination is already present inside many ancient human fossils from previous handling without specific precautions. We designed a statistical framework to isolate endogenous ancient DNA sequences from contaminating sequences using postmortem degradation patterns and were able to reduce high-contamination fractions to negligible levels. We captured DNA sequences from a contaminated Neandertal bone from Okladnikov Cave in Siberia and used our method to assemble its mitochondrial genome sequence, which we find to be from a lineage basal to five of six previously published complete Neandertal mitochondrial genomes. Our method paves the way for the large-scale genetic analysis of contaminated human remains.
Keywords: paleogenomics, human evolution
Abstract
One of the main impediments for obtaining DNA sequences from ancient human skeletons is the presence of contaminating modern human DNA molecules in many fossil samples and laboratory reagents. However, DNA fragments isolated from ancient specimens show a characteristic DNA damage pattern caused by miscoding lesions that differs from present day DNA sequences. Here, we develop a framework for evaluating the likelihood of a sequence originating from a model with postmortem degradation—summarized in a postmortem degradation score—which allows the identification of DNA fragments that are unlikely to originate from present day sources. We apply this approach to a contaminated Neandertal specimen from Okladnikov Cave in Siberia to isolate its endogenous DNA from modern human contaminants and show that the reconstructed mitochondrial genome sequence is more closely related to the variation of Western Neandertals than what was discernible from previous analyses. Our method opens up the potential for genomic analysis of contaminated fossil material.
Retrieval and sequencing of DNA from fossil material has been revolutionized by the rapid surge of high-throughput sequencing technology (1). One of the advantages of the new generation of sequencing technologies over previous PCR-based approaches is the routine sequencing of molecules across their entire length in contrast to specific priming sites. Although this approach has been extended to ancient material from a number of organisms, there is particular interest in genomic analyses of ancient human populations (2–9). However, despite extensive precautions against contamination during laboratory sample preparation, many fossil samples show evidence of contamination from not only the microenvironment of the fossil, but also present day humans (2, 10–14). Although the former type of contamination can introduce statistical noise in comparisons of ancient DNA with modern populations, the latter type of contamination (from present day humans) can impose major biases on population genetic and phylogenetic analyses (15, 16).
Ancient DNA sequences show several features indicative of postmortem degradation (PMD) (17). The main diagnostic feature documented so far is a pattern of Cytosine (C) →Thymine (T) substitutions (18–20) that increases toward the 5′ end of the sequence reads (21), which in most applications, results in a complementary Guanine (G) →Adenine (A) pattern in the 3′ end caused by enzymatic repair (5). This pattern has been attributed to Cytosine deamination at single-stranded ends of the molecules (21) and shows a clear tendency of increase over time in contrast to other potential diagnostic features, such as fragment length and preferential fragmentation at purines (22, 23). However, although the observation of such a pattern suggests the presence of degraded DNA in a sequence dataset, it does not prove that modern contamination is absent or even at low frequency. For high-coverage data from extinct close relatives of modern humans, an efficient approach to estimate the contamination proportion is to leverage SNPs that are known to be fixed or nearly fixed in present populations (2, 24). If it can be assumed that contamination is minor relative to endogenous DNA, this approach can also be extended to high-coverage data from ancient modern humans by investigating whether more alleles are present than expected from the ploidy level of the chromosome (4, 24, 25) or making assumptions about the ancestry of both ancient and contaminating individuals (4). For low-coverage data, stratifying sequences based on whether a mismatch consistent with PMD degradation have occurred, and investigating the consistency of population genetic patterns across categories can be used to investigate whether contamination is present at such levels as to affect the conclusions of the analysis (8). However, these approaches only allow investigation of whether contamination is present. To our knowledge, no approach has been developed to overcome significant contamination levels, except for the case of targeted PCR-based sequencing of haploid regions, for which most of the informative termini of the fragments are lost (26). This situation has limited large-scale ancient DNA analyses of human specimens to those specimens with very low levels of present day DNA contamination, leaving the genetic information in potentially crucial fossil material often unstudied.
Here, we propose a likelihood approach that incorporates sequence errors and alignment to the reference genome to provide a score that is informative on whether a given sequence is likely to have arisen from a degraded template molecule. We show that this method allows reduction of high contamination rates down to levels that are negligible for most evolutionary purposes and give examples using both mitochondrial (mt) and genome-wide data from the literature. Finally, we produced a high-coverage mtDNA dataset from a Siberian Neandertal fossil excavated in Okladnikov Cave that is contaminated with a substantial amount of modern human DNA and use our method for separating endogenous sequences from contaminating sequences to reconstruct its near-complete mitochondrial genome sequence.
Results
Model Outline.
The diagnostic nucleotide misincorporation pattern that arises from ancient DNA damage can be detected only by observing matches or mismatches in an alignment with one or more reference sequences. In the 5′ end of a sequence fragment, as many as 30–40% of C residues can appear as T in DNA fragments from samples that are several thousand years old under typical preservation conditions (21, 23). This fraction reflects both Cs that are deaminated to Uracil, which are read as T by DNA polymerases, and methylated Cs that are deaminated to Ts. Whether such mismatches are observed, thus, provides information on the authenticity of a given DNA fragment. However, C→T and G→A mismatches appear not solely because of ancient DNA damage but also, because of biological polymorphisms and sequencing errors. We developed a likelihood framework that explicitly models these three processes (Material and Methods). To investigate authenticity of a given DNA fragment, we use this framework to evaluate two competing models, of which one assumes ancient DNA degradation and the other does not, arriving at a final log-likelihood ratio of the two models that we term a PMD score (PMDS). A positive PMDS for a DNA fragment indicates support for the ancient DNA model relative to the alternative model, and the greater the PMDS, then the greater the support for the DNA fragment having been subject to DNA degradation.
Empirical Distributions of PMD Scores in Ancient and Present Day Samples.
We computed PMD scores for 1 million randomly sampled sequences from the 100-y-old remains of an Australian individual (27), four ∼5,000-y-old Neolithic Scandinavian individuals (8), four 38,000- to 70,000-y-old Neandertal individuals (24), and four present day individuals (24). These individuals showed evidence of PMD roughly proportional to their age (Figs. S1 and S2), and four PMDS distributions from representative individuals are shown in Fig. 1. Most importantly, we find that sequences from ancient individuals have an excess of high PMD scores, implying that a set of ancient DNA sequences shows enough PMD to be unlikely to be mistaken for contaminants. For example, looking at the cumulative distribution of PMDS, we find that, for a threshold of PMDS > 5, we would retain ∼15–20% of sequences from Neandertals and Neolithic Scandinavians but only ∼0.01–0.02% from any present day individual and 0.27% from the 100-y-old remains of the Australian individual (note that this sample is derived from hair and thus, may have marginally decreased levels of DNA damage) (28) (Fig. S3). Thus, an empirical probability of a sequence being from a present day (contaminating) source could, in principle, be extrapolated and propagated in downstream genotyping inference, much like the common practice for sequence error estimates. However, this procedure is not straightforward, because the (usually unknown) initial level of contamination in the library affects the final contamination fraction associated with each given PMDS threshold. Thus, we will focus on imposing hard PMDS thresholds to contaminated datasets to be able to estimate the contamination fraction among the sequences downstream of genotyping.
Fig. 1.

PMDS distribution in ancient and present day human sequences. (A) A present day human. (B) Hundred-year-old remains of an Australian individual. (C) Five thousand-year-old remains of a Scandinavian individual. (D) A Pleistocene Neandertal. Note that the x axes in C and D are truncated, and PMD scores reach ∼26.
Artificial Contamination Experiments.
To investigate the efficiency of the PMDS approach for genotyping high-coverage data, we created mosaic datasets of mitochondrial sequences from a French present day individual and the Vindija 33.16 Neandertal individual. These contaminated data were filtered using PMDS thresholds of three or five and compared with the data with no filtering. We estimated contamination as the fraction of modern human-specific alleles using seven diagnostic transversion SNPs and found that the unfiltered data show increasing rates of modern human alleles with increasing contamination input (as expected). In contrast, a PMDS threshold of five results in no detectable contamination postfiltering (0.0%) for prefiltering contamination levels (per base pair) from 1% to ∼93% (Fig. 2A).
Fig. 2.
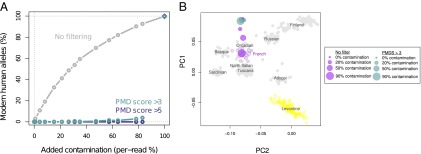
Artificial mtDNA and autosomal contamination experiments. (A) Artificial contamination represented by sequences from a present day human (French) was added to a sequence dataset from a Croatian Neandertal (Vindija 33.16). Final per base pair contamination was estimated using diagnostic mtDNA positions that differentiate Neandertals from modern humans. Up to 90% per base pair contamination levels can be reduced to negligible levels using the PMDS approach. (B) Artificial contamination represented by sequences from a present day human (French) was added to a sequence dataset from Neolithic Scandinavian hunter-gatherers. SNPs were extracted with and without filtering for PMDS, and a principal component analysis together with European and Levantine populations was performed for the different levels of contamination. The PCs were Procrustes transformed for each separate analysis (8).
To illustrate the use of the approach for autosomal low-coverage data, we performed a similar in silico contamination experiment using genome-wide sequence data from the Neolithic hunter-gatherers, successively adding French sequences to final contamination fractions of 20%, 50%, and 90%. We extracted SNPs typed in 504 individuals from Europe and the Levant (SI Materials and Methods) from these contaminated datasets as well as the uncontaminated dataset, performed principal component analysis for each dataset separately, and merged the obtained principal component 1 (PC1) –PC2 configurations using Procrustes transformation (8). As in the original study (8), we find that the unmodified hunter-gatherer data are outside the distribution of modern day populations but most similar to Northern European populations. As we artificially add French contamination, the ancient data are inferred to be successively more similar to the French population. When we apply a PMDS threshold of three to the sequences, no effect of contamination can be seen, with the PC1–PC2 configuration of all filtered datasets showing very similar projections as the uncontaminated (and unfiltered) data (Fig. 2B).
Contamination Reduction Using PMD Scores in Genuinely Contaminated Datasets.
Whereas the above examples are designed to test the efficiency of the method in common genetic analyses, the controlled nature of our artificial contamination experiments allows us to examine the contamination rate as a function of PMDS threshold directly. We confirm that present day French contamination in the Vindija 33.16 Neandertal will be negligible for PMDS ≥ 4 (Fig. 3A). Similarly, for data with a less extreme temporal difference (the 100-y-old remains of an Australian individuals as the contaminant and the 5,000-y-old remains of a Scandinavian hunter-gatherer as endogenous DNA), contamination levels as high as 95% can be reduced to negligible levels by increasing the threshold on the PMDS statistic (Fig. 3B).
Fig. 3.
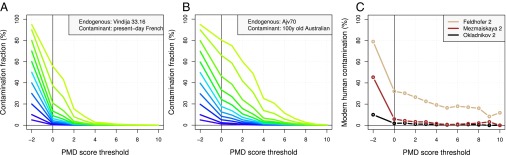
Reduction of artificial and genuine contamination using PMDSs. (A) Expected contamination fraction in the Vindija 33.16 Neandertal as a function of the PMDS threshold using sequence data from a present day French individual as the artificial contaminant. (B) Expected contamination fraction in the Ajv70 Scandinavian hunter-gatherer as a function of the PMDS threshold using sequence data from the 100-y-old remains of an Australian individual as the artificial contaminant. The different colored lines in A and B correspond to different contamination levels (before PMDS filtering, the initial contamination fraction is given at PMDS = −2). (C) Estimated modern human contamination in three genuinely contaminated Neandertal datasets from Feldhofer 2, Mezmaiskaya 2, and Okladnikov 2.
However, because the data used for our artificial contamination experiment may not perfectly represent genuine cases of contamination, we also investigated two genuinely contaminated Neandertal mtDNA datasets (10): Feldhofer 2 and Mezmaiskaya 2. In line with the artificial contamination experiment above, we find strong reductions from the initial contamination level of ∼79% (Feldhofer 2) and ∼45% (Mezmaiskaya 2) for both these datasets (Fig. 3C) (10). Moreover, we find that consensus sequences from the original (and contaminated) Mezmaiskaya 2 dataset falls within the variation of modern humans, whereas the consensus of sequences post-PMDS filtering falls within Neandertal variation (Fig. S4), similar to results from a less contaminated sample from the same individual (29).
Mitochondrial Genome Sequence of a Contaminated Neandertal Specimen from Okladnikov Cave, Siberia.
Because the PMDS approach provides a tool to substantially reduce contamination levels in ancient DNA, we used primer extension capture (29) to obtain 6,906 unique mtDNA sequences from a Neandertal sample from Okladnikov Cave (Okladnikov 2), in which contamination limited previous DNA analysis to a small mt region that could be amplified with Neandertal-specific primers sets (30). We found clear evidence of PMD consistent with endogenous DNA (Fig. S5), and 10.2% (95% confidence interval = 8.7–11.7%) of all fragments overlapping diagnostic positions displayed the modern day human allele, indicative of contamination. However, when we restricted the analysis to 3,908 sequences that had a PMDS ≥ 0, the point estimate of contamination was reduced to 1.3% (95% confidence interval = 0.7–1.9%) (Fig. 3C). We, thus, discarded sequences with PMDS < 0 and assembled the mitochondrion of Okladnikov 2 using an ancient DNA-aware iterative mapping assembler (31). Average sequence depth for this assembly was 15-fold compared with 25-fold in the unfiltered data, with a total of 96.0% of all positions covered by at least five fragments and 99.4% covered by at least two fragments (Fig. 4 and Fig. S6). There were 88 positions that were not reliably called for the PMDS ≥ 0 data (0.53%) compared with 24 positions in the unfiltered data (0.14%). However, we find that the seven sites that were gained when restricting to PMDS ≥ 0 were all polymorphic and thus, evolutionary informative (Table S1). In contrast, only 4 of 68 sites that were lost were polymorphic. This observation shows that PMDS filtering can resolve positions that are difficult to call because of different alleles in the contaminant and endogenous sequences (SI Results).
Fig. 4.

A mt genome sequence from a contaminated Neandertal sample from Okladnikov Cave, Siberia. (A) Coverage distribution and sequence features of the Okladnikov 2 mt genome. (B) Gene genealogy of 67 published complete hominid mitochondria and Okladnikov 2. Node support is only indicated for posterior probabilities ≥ 0.75. (C) Geographical sampling location of Neandertal and Denisovan individuals from which mtDNA genomes have been sequenced. Okladnikov Cave is marked by a red square. GC, Guanine-Cytosine content.
We aligned the Okladnikov 2 mt genome sequence to complete mt genomes from other archaic and modern humans and reconstructed a gene tree using a Bayesian approach (Material and Methods). We find that the Okladnikov 2 sequence falls outside all published complete Neandertal mt genomes from Central and Western Europe with high posterior probability but forms a cluster with these mt genomes to the exclusion of the mtDNA of Mezmaiskaya 1 from the Caucasus (Fig. 4). This topology is also obtained when a PMD filter is also applied to Mezmaiskaya 1, Vindija 33.16, Vindija 33.25, and Vindija 33.26 sequences (Fig. S7). The mtDNA genome sequence reconstructed here, thus, resolves the relationship between Okladnikov 2 and other Neandertal mt genomes, because analyses based on the control region have suffered from statistical uncertainty but indicated that the Okladnikov 2 mtDNA is basal to Mezmaiskaya 1 (30, 32), a result that we replicated by extracting a 304-bp region of the Okladnikov 2 mtDNA sequence assembly that overlapped with previous data (Fig. S8).
Discussion
Despite that modern human contamination has been shown to be present in a large proportion of archaeological human samples (2, 11, 33), large-scale DNA sequence retrieval has so far been limited to hominin specimens with very little contamination. Here, we developed a likelihood framework for large-scale ancient DNA studies that tries to gauge how likely a single sequence is to have originated from an ancient template molecule, incorporating base quality scores and biological polymorphism. We have shown that this method allows reduction of contamination in degraded ancient human sequence data to negligible levels. In contrast to a previously suggested computational approach for PCR-based amplification of targeted regions (26), our approach is particularly geared for the new generation of high-throughput sequencing approaches (1).
This framework offers many advantages over previous approaches, which aimed at detecting contamination by dividing sequences based on fragment length or observed mismatches (8, 15). Most notably, information is provided by not only mismatches typical of PMD (8) but also, matches to the reference. For example, observing several C-C matches in the terminus of a fragment provides support for the model without PMD, which could, for example, allow the exclusion of contaminant fragments that have a single C→T mismatch because of sequencing error or biological polymorphism. Furthermore, probabilistic weighting of each observed match or mismatch according to its position along the sequenced fragment allows balanced consideration of the evidence for PMD across the entire fragment. For example, observing a single C→T mismatch at the third 5′ position of the fragment may provide as much evidence for the sequence fragment being of ancient origin as observing several such mismatches at a more central position of the DNA fragment. Finally, explicit consideration of the probability of sequencing error at each informative position removes the need for strict base quality cutoffs. Accordingly, we found that simply conditioning on observing a C→T mismatch in the sequence (8) does not achieve as high of a degree of reduction in contamination rate as applying a PMDS threshold (SI Results and Fig. S9). We also note that we, for consistency, assumed a C deamination model corresponding to ∼30% C to T substitutions in the first 5′ positions throughout this study (Fig. S1), but in cases where endogenous DNA is more degraded, additional efficiency might be gained by modifying this model (optional in our software implementation PMDtools).
One cause of potential concern is if either contaminant or endogenous DNA is closer to the reference sequence than the other, in which case the number of mismatches caused by biological polymorphisms is not the same. In this paper, we assume equal divergence from the reference for the two models, because the observed biological polymorphism is, in most cases, much smaller than the rate of PMD; however, the model easily accommodates different assumptions on biological polymorphisms. As the catalog of genetic variation from, for example, humans becomes more complete, additional improvements to our approach might include masking positions with known polymorphisms, although it would also increase the computational expense. Another complication from enriching for postmortem damage is the errors that it contributes to downstream analyses, but we found that this could be alleviated by error correction (SI Results and Fig. S10).
We used the methodology described in this paper to retrieve a complete mtDNA sequence from a contaminated Siberian Neandertal sample. The Okladnikov 2 sequence is basal to all published complete Neandertal mtDNA sequences from Central and Western Europe, which may relate to it originating from the easternmost Neandertal sample available (30). In addition to the complete mitochondrion of the Mezmaiskaya 1 Neandertal from the Caucasus (29, 34), analyses of smaller fragments of the mt control region suggest that older Neandertals might contain mtDNA lineages basal to the Okladnikov 2 sequence (30, 32), but the discrepancy between the genealogy obtained using a smaller hypervariable section (Fig. S8) and the resolved phylogeny using the mtDNA genome sequence prompts additional large-scale studies of these remains.
Although chronological age seems to be the most important factor (23), PMD patterns in contaminant and endogenous sequences will likely be influenced by a complex range of factors, such as temperature, depositional conditions, postexcavation handling, and source tissue (17). Furthermore, it is possible that PMD patterns are also present in contaminant DNA. However, a recent study (23) showed that samples 50–100 y old all showed limited evidence of PMD (less than 10% nucleotide misincorporations in the 5′ end). Thus, while the relative levels of PMD in contaminant vs. endogenous sequences in a sample that contains a mixture of both will usually not be known, as long as substantial patterns of PMD are found in a sequence library (e.g., >10%), it seems reasonable to assume that PMD will be less extensive in the contaminating sequences under most circumstances. We suggest that, after identifying a set of sequences with high PMD scores that are likely to be endogenous, high- to medium-coverage data should be authenticated by analyzing whether the retained sequence reads are from a single individual (4, 10, 24). For low-coverage data, it may still be possible to investigate how many source individuals are represented using mtDNA, but because mtDNA/nuclear DNA ratios can vary greatly between samples (16), the most robust approach will likely be to stratify sequences based on the PMDS approach described here and assess whether population genetic inferences are consistent across different thresholds (cf. ref. 8 with Fig. 2). If the fraction of contamination can be estimated, we suggest that a PMDS threshold should be chosen such that contamination is negligible. If data are too sparse for such estimates, we suggest that main conclusions from the data should be robust for a range of PMDS thresholds.
Whereas we have focused on human population genetic examples, contamination from present day sources also affects nonhuman ancient DNA studies of pathogens, ancient microbiomes, animals, and plants, and thus, computational removal of contamination in ancient DNA data is likely to increase in importance as these fields move to high-throughput sequencing approaches.
Materials and Methods
Likelihood-Based Framework for Separating Ancient DNA from Present Day Contamination.
C deaminations in ancient DNA sequences can be identified by mismatches to the reference sequence of two specific types (18–20). We, therefore, restrict our model to two categories of sites in a pairwise alignment between a DNA fragment and a reference sequence: (i) where the reference has a C and the aligned fragment has either a C or a T and (ii) where the reference base is G and the aligned base is either a G or an A. Other positions in the alignment are not considered, except for providing the distance z to the 5′ (in the case of a C in the reference) and 3′ (in the case of a G in the reference) termini. For obtaining z, positions in the alignment where there is a gap in the sequenced DNA fragment are not considered. In SI Materials and Methods, we outline a modification of the model for libraries prepared using single-stranded methods (e.g., ref. 5).
There are three nonmutually exclusive events that can cause an observation of a C→T or a G→A mismatch at a given position in a sequence read where the reference sequence displays a C or a G: (i) a true biological polymorphism (occurring at rate-π), (ii) a sequence error (rate-ε), or (iii) in the case of degraded DNA, PMD (rate Dz), which we will assume varies according to a modified geometric distribution (plus a small constant) across the sequenced molecule with decreasing probability with distance z (measured in base pairs) from the relevant terminus (21) (Fig. S1). Alternatively, observing a C-C match could be caused by a sequence error having reverted a postmortem nucleotide misincorporation or a sequence error having reverted a biological polymorphism. The third combination of events that could be imagined—a nucleotide misincorporation having reverted a biological polymorphism—does not enter here, because nucleotide misincorporations only result in substitutions in one direction (i.e., C→T or G→A but not T→C or A→G).
Considering all three mutually exclusive possibilities together, we have
under a model of PMD (MPMD), which under a model of no PMD (MNULL, where Dz = 0 for all z), becomes P(Match|z) = (1 − π) × (1 − ε) + ε × π. The probability of a C→T mismatch (or a G→A mismatch) for a particular site is then any other event or combination of events, such that
In all applications here, we will assume that polymorphisms occur at a rate of π = 0.001 to approximate autosomal genetic variation between a pair of human chromosomes. We obtain ε from the phred-scaled base qualities Q, but because we restrict to a specific type of mismatch at each type of site (C→T or G→A), we divide the total probability of a sequence error given by the base qualities by three, such that ε = 1/3 × 10−Q/10. For the probability of postmortem nucleotide misincorporation, we use empirically observed patterns as the basis (21), and we approximate the probability by
where z is the distance from the relevant sequence terminus (the 5′ end for C→T mismatches and the 3′ end for G→A mismatches), and the first position has z = 1. In this study, we assumed P = 0.3 and C = 0.01, which are consistent with most sequence datasets that are several thousand years old (Fig. S1), but note that, for older specimens with pervasive damage, greater efficiency could be gained by increasing p. We set up a likelihood function of
where X is an indicator variable. Therefore, X = 1 if Si = Match, and X = 0 if Si = Mismatch; the probabilities are given by Eqs. 1 and 2. The likelihood function for model MNULL is set up in the same way, but in this case, Dz = 0 for all z. To investigate whether an alignment is more likely to have originated from a model with PMD (MPMD) or a model with no PMD (MNULL), we take the natural logarithm of the ratio of the likelihood for each model multiplied over all positions I in the sequence read S that satisfy our criteria. This log-likelihood ratio provides a PMDS,
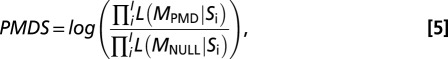 |
which is informative on how likely a sequence is to have originated from a degraded source. If I = 0, our test statistic is undefined for that particular sequence read, and in practice, such (extremely rare) reads are discarded.
Capture and Sequencing of Okladnikov 2 mtDNA.
DNA was extracted (35) from about 200 mg bone from a humerus shaft found in Okladnikov Cave in the Altai Mountains in the 1980s. Before mtDNA enrichment, 25 µL DNA extract were turned into a sequencing library using a modified 454 library preparation protocol as described (29, 36), with the exception that Illumina p5 and p7 adapters were used (37). The p7 adapter was modified to avoid contamination from other libraries carrying a unique 7-bp barcode. The primer extension capture mtDNA enrichment protocol was performed as described previously (29) (SI Materials and Methods) using the exact same four 144-plex and 143-plex mixes used to retrieve complete Neandertal, Denisovan, and Pleistocene modern human mtDNAs previously (10, 29, 38). The sequencing run was analyzed starting from raw images using the Illumina Genome Analyzer pipeline 1.3.2 and the base caller Ibis (39). Raw sequences obtained from Ibis for the two paired end reads of each sequencing cluster were merged (including adapter removal) as previously described (24, 40).
Assembly and Analysis of the Okladnikov 2 mtDNA Genome Sequence.
In total, 4,102,487 sequences were obtained after merging read pairs (unmerged reads were excluded). After collapsing PCR duplicates to consensus sequences (40), we obtained 6,906 sequences of 50,822 originally aligned sequences. We assembled the mtDNA sequence using an iterative mapping assembler (31), which takes position-specific PMD patterns into consideration in its scoring matrix. Convergence was observed in two rounds of assembly. For estimating contamination in the Okladnikov 2 sequence, we increased the number of informative sites by identifying positions (with minimum depth of 10) where the consensus base differed from 95% of 311 modern human mtDNAs and where the Okladnikov 2 consensus was not an A or T in a transition polymorphism. The seven fixed transversions between Neandertals and modern humans yielded a similar estimate of ∼10% contamination in Okladnikov 2. We inferred a gene tree relating 68 hominin mtDNA sequences (25, 29, 31, 38, 41) using mrBayes 3.2.0 (42). The Markov chain Monte Carlo was run for 5 million generations, with sampling every 1,000 generations and a burn-in of 1 million generations. A consensus tree was constructed implementing a 50% majority rule.
Supplementary Material
Acknowledgments
We thank Wieland Binzcik, Susanna Sawyer, Adrian Briggs, Philip Johnson, Allissa Mittnik, Ludovic Orlando, Amhed Velazquez, Per Sjödin, Lucie Gattepaille, Robert Ekblom, Ayça Omrak, Anders Götherström, and Love Dalén for discussions and technical assistance. PMDtools, a python program implementing PMDS computations, is available at www.ebc.uu.se/Research/IEG/evbiol/research/Jakobsson/software/pmdtools/. Computations were performed on resources provided by the Swedish National Infrastructure for Computing and Uppsala Multidisciplinary Center for Advanced Computational Science under Project b2010050. The laboratory work was financed by the Max Planck Society. Financial support was also provided by the Royal Swedish Academy of Sciences (P.S.) and the European Research Council (M.J.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The sequence reported in this paper has been deposited in the GenBank database (accession no. KF982693).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1318934111/-/DCSupplemental.
References
- 1.Stoneking M, Krause J. Learning about human population history from ancient and modern genomes. Nat Rev Genet. 2011;12(9):603–614. doi: 10.1038/nrg3029. [DOI] [PubMed] [Google Scholar]
- 2.Green RE, et al. Analysis of one million base pairs of Neanderthal DNA. Nature. 2006;444(7117):330–336. doi: 10.1038/nature05336. [DOI] [PubMed] [Google Scholar]
- 3.Noonan JP, et al. Sequencing and analysis of Neanderthal genomic DNA. Science. 2006;314(5802):1113–1118. doi: 10.1126/science.1131412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rasmussen M, et al. Ancient human genome sequence of an extinct Palaeo-Eskimo. Nature. 2010;463(7282):757–762. doi: 10.1038/nature08835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meyer M, et al. A high-coverage genome sequence from an archaic Denisovan individual. Science. 2012;338(6104):222–226. doi: 10.1126/science.1224344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fu Q, et al. DNA analysis of an early modern human from Tianyuan Cave, China. Proc Natl Acad Sci USA. 2013;110(6):2223–2227. doi: 10.1073/pnas.1221359110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keller A, et al. New insights into the Tyrolean Iceman’s origin and phenotype as inferred by whole-genome sequencing. Nat Commun. 2012;3:698. doi: 10.1038/ncomms1701. [DOI] [PubMed] [Google Scholar]
- 8.Skoglund P, et al. Origins and genetic legacy of Neolithic farmers and hunter-gatherers in Europe. Science. 2012;336(6080):466–469. doi: 10.1126/science.1216304. [DOI] [PubMed] [Google Scholar]
- 9.Sánchez-Quinto F, et al. Genomic affinities of two 7,000-year-old Iberian hunter-gatherers. Curr Biol. 2012;22(16):1494–1499. doi: 10.1016/j.cub.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 10.Krause J, et al. A complete mtDNA genome of an early modern human from Kostenki, Russia. Curr Biol. 2010;20(3):231–236. doi: 10.1016/j.cub.2009.11.068. [DOI] [PubMed] [Google Scholar]
- 11.Richards MB, Sykes BC, Hedges RE. Authenticating DNA extracted from ancient skeletal remains. J Archaeol Sci. 1995;22(2):291–299. [Google Scholar]
- 12.Handt O, Krings M, Ward RH, Pääbo S. The retrieval of ancient human DNA sequences. Am J Hum Genet. 1996;59(2):368–376. [PMC free article] [PubMed] [Google Scholar]
- 13.Kolman CJ, Tuross N. Ancient DNA analysis of human populations. Am J Phys Anthropol. 2000;111(1):5–23. doi: 10.1002/(SICI)1096-8644(200001)111:1<5::AID-AJPA2>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 14.Sampietro ML, et al. Tracking down human contamination in ancient human teeth. Mol Biol Evol. 2006;23(9):1801–1807. doi: 10.1093/molbev/msl047. [DOI] [PubMed] [Google Scholar]
- 15.Wall JD, Kim SK. Inconsistencies in Neanderthal genomic DNA sequences. PLoS Genet. 2007;3(10):1862–1866. doi: 10.1371/journal.pgen.0030175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Green RE, et al. The Neandertal genome and ancient DNA authenticity. EMBO J. 2009;28(17):2494–2502. doi: 10.1038/emboj.2009.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pääbo S. Ancient DNA: Extraction, characterization, molecular cloning, and enzymatic amplification. Proc Natl Acad Sci USA. 1989;86(6):1939–1943. doi: 10.1073/pnas.86.6.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hofreiter M, Jaenicke V, Serre D, von Haeseler A, Pääbo S. DNA sequences from multiple amplifications reveal artifacts induced by cytosine deamination in ancient DNA. Nucleic Acids Res. 2001;29(23):4793–4799. doi: 10.1093/nar/29.23.4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hansen AJ, Willerslev E, Wiuf C, Mourier T, Arctander P. Statistical evidence for miscoding lesions in ancient DNA templates. Mol Biol Evol. 2001;18(2):262–265. doi: 10.1093/oxfordjournals.molbev.a003800. [DOI] [PubMed] [Google Scholar]
- 20.Brotherton P, et al. Novel high-resolution characterization of ancient DNA reveals C > U-type base modification events as the sole cause of post mortem miscoding lesions. Nucleic Acids Res. 2007;35(17):5717–5728. doi: 10.1093/nar/gkm588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Briggs AW, et al. Patterns of damage in genomic DNA sequences from a Neandertal. Proc Natl Acad Sci USA. 2007;104(37):14616–14621. doi: 10.1073/pnas.0704665104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.García-Garcerà M, et al. Fragmentation of contaminant and endogenous DNA in ancient samples determined by shotgun sequencing; prospects for human palaeogenomics. PLoS One. 2011;6(8):e24161. doi: 10.1371/journal.pone.0024161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sawyer S, Krause J, Guschanski K, Savolainen V, Pääbo S. Temporal patterns of nucleotide misincorporations and DNA fragmentation in ancient DNA. PLoS One. 2012;7(3):e34131. doi: 10.1371/journal.pone.0034131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Green RE, et al. A draft sequence of the Neandertal genome. Science. 2010;328(5979):710–722. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reich D, et al. Genetic history of an archaic hominin group from Denisova Cave in Siberia. Nature. 2010;468(7327):1053–1060. doi: 10.1038/nature09710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Helgason A, et al. A statistical approach to identify ancient template DNA. J Mol Evol. 2007;65(1):92–102. doi: 10.1007/s00239-006-0259-8. [DOI] [PubMed] [Google Scholar]
- 27.Rasmussen M, et al. An Aboriginal Australian genome reveals separate human dispersals into Asia. Science. 2011;334(6052):94–98. doi: 10.1126/science.1211177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gilbert MTP, et al. Whole-genome shotgun sequencing of mitochondria from ancient hair shafts. Science. 2007;317(5846):1927–1930. doi: 10.1126/science.1146971. [DOI] [PubMed] [Google Scholar]
- 29.Briggs AW, et al. Targeted retrieval and analysis of five Neandertal mtDNA genomes. Science. 2009;325(5938):318–321. doi: 10.1126/science.1174462. [DOI] [PubMed] [Google Scholar]
- 30.Krause J, et al. Neanderthals in central Asia and Siberia. Nature. 2007;449(7164):902–904. doi: 10.1038/nature06193. [DOI] [PubMed] [Google Scholar]
- 31.Green RE, et al. A complete Neandertal mitochondrial genome sequence determined by high-throughput sequencing. Cell. 2008;134(3):416–426. doi: 10.1016/j.cell.2008.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dalén L, et al. Partial genetic turnover in neandertals: Continuity in the East and population replacement in the West. Mol Biol Evol. 2012;29(8):1893–1897. doi: 10.1093/molbev/mss074. [DOI] [PubMed] [Google Scholar]
- 33.Malmström H, Storå J, Dalén L, Holmlund G, Götherström A. Extensive human DNA contamination in extracts from ancient dog bones and teeth. Mol Biol Evol. 2005;22(10):2040–2047. doi: 10.1093/molbev/msi195. [DOI] [PubMed] [Google Scholar]
- 34.Ovchinnikov IV, et al. Molecular analysis of Neanderthal DNA from the northern Caucasus. Nature. 2000;404(6777):490–493. doi: 10.1038/35006625. [DOI] [PubMed] [Google Scholar]
- 35.Rohland N, Hofreiter M. Comparison and optimization of ancient DNA extraction. Biotechniques. 2007;42(3):343–352. doi: 10.2144/000112383. [DOI] [PubMed] [Google Scholar]
- 36.Margulies M, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437(7057):376–380. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meyer M, Kircher M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb Protoc. 2010;2010(6):t5448. doi: 10.1101/pdb.prot5448. [DOI] [PubMed] [Google Scholar]
- 38.Krause J, et al. The complete mitochondrial DNA genome of an unknown hominin from southern Siberia. Nature. 2010;464(7290):894–897. doi: 10.1038/nature08976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kircher M, Stenzel U, Kelso J. Improved base calling for the Illumina Genome Analyzer using machine learning strategies. Genome Biol. 2009;10(8):R83. doi: 10.1186/gb-2009-10-8-r83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kircher M. Analysis of High-Throughput Ancient DNA Sequencing Data. Ancient DNA. Berlin: Springer; 2012. pp. 197–228. [DOI] [PubMed] [Google Scholar]
- 41.Ingman M, Kaessmann H, Pääbo S, Gyllensten U. Mitochondrial genome variation and the origin of modern humans. Nature. 2000;408(6813):708–713. doi: 10.1038/35047064. [DOI] [PubMed] [Google Scholar]
- 42.Ronquist F, et al. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.