

Significance
Deletion of the E3 β-TrCP in the mouse gut epithelium deregulates enterocyte cell cycle, induces a DNA damage response (DDR), and abolishes the epithelium barrier function, resulting in a lethal mucosal inflammation. Epithelial-derived IL-1β, likely induced by DDR independently of NF-κB, is a major culprit, and initiates the pathology by compromising epithelial tight junctions (TJs). Anti–IL-1β treatment secures the TJs and prevents the fulminant mucosal inflammation. IL-1β secretion accompanies human mucositis, a severe mucosal inflammatory reaction caused by chemoradiation therapy-induced DNA damage, which often results in treatment suspension. We propose that anti–IL-1β preventive treatment may ameliorate mucositis, as well as multiple disorders associated with epithelial barrier permeability, including burn injuries, head and neck trauma, alcoholic intoxication, and graft-vs.-host disease.
Keywords: gut immunity, anakinra, anti–IL-1 preventive therapy, cytotoxic side effects, graft-vs.-host disease
Abstract
β-TrCP, the substrate recognition subunit of SCF-type ubiquitin ligases, is ubiquitously expressed from two distinct paralogs, targeting for degradation many regulatory proteins, among which is the NF-κB inhibitor IκB. To appreciate tissue-specific roles of β-TrCP, we studied the consequences of inducible ablation of three or all four alleles of the E3 in the mouse gut. The ablation resulted in mucositis, a destructive gut mucosal inflammation, which is a common complication of different cancer therapies and represents a major obstacle to successful chemoradiation therapy. We identified epithelial-derived IL-1β as the culprit of mucositis onset, inducing mucosal barrier breach. Surprisingly, epithelial IL-1β is induced by DNA damage via an NF-κB–independent mechanism. Tissue damage caused by gut barrier disruption is exacerbated in the absence of NF-κB, with failure to express the endogenous IL-1β receptor antagonist IL-1Ra upon four-allele loss. Antibody neutralization of IL-1β prevents epithelial tight junction dysfunction and alleviates mucositis in β-TrCP–deficient mice. IL-1β antagonists should thus be considered for prevention and treatment of severe morbidity associated with mucositis.
Only a few E3 ubiquitin ligases have been studied in vivo by gene disruption for the purpose of elucidating their functions and suitability as potential drug targets (1–3). Beta-transducin repeat containing protein (β-TrCP) is an E3 ubiquitin ligase that affects numerous major cell regulators (1, 4, 5), such as the effector of the Wnt pathway, β-catenin (6); the inhibitor of the NF-κB pathway, IκB (7); the cell cycle regulators CDC25A (8), WEE1 (9), Emi1 (10), and Bora (11); and DNA damage responsive proteins CDC25A and Claspin (12). β-TrCP is encoded by two paralog genes that are thought to be identical in their known biochemical features considering their high degree of sequence homology (77%) and virtually identical substrate recognition “pocket” (13). This redundancy may explain the mild phenotype observed upon in vivo ablation of β-TrCP1; the two best characterized substrates of β-TrCP, β-catenin and IκBα, do not accumulate in β-TrCP1–deficient MEFs (MEFβ-TrCP1−/−) (10). Furthermore, residual expression of ∼20% β-TrCP2 activity on a β-TrCP1–null background is sufficient for preserving homeostasis in most tissues (2). To gain a better understanding of in vivo β-TrCP functions, we created a floxed β-TrCP2 allele and crossed it to the β-TrCP1–null mouse (10).
Here we show that the primary cellular assault following β-TrCP KO is DNA damage, which, when occurring in the gut, ignites a fatal colitis process that is IL-1β–dependent. This pathological process resembles the one that occurs in human patients following chemotherapy or radiation therapy-induced intestinal mucositis, and will therefore be referred to here as mucositis. We suggest that IL-1β is expressed and secreted by epithelial cells following DNA damage via an unknown mechanism. Expression of IL-1β is NF-κB–independent as observed in different experimental systems following DNA damaging treatments. Our study may have implications for human mucositis; we propose the possibility of using anti–IL-1β treatments [e.g., anakinra, an IL-1 receptor antagonist (IL-1Ra) (14)] as a preventive means for chemoradiation therapy-induced mucositis.
Results
Mitotic Arrest and DNA Damage in Embryo Fibroblasts Following Ablation of both β-TrCP Paralogs.
The deletion of both β-TrCP paralogs [i.e., double KO (DKO)] was first studied in MEFs. Deletion of the floxed β-TrCP2 alleles in MEFs was achieved by using adeno-Cre transduction (SI Appendix, Fig. S1A). As expected, the deletion resulted in stabilization of various substrates (SI Appendix, Fig. S1B). β-TrCP1–deficient MEFs show delayed mitotic progression (10), and DKO MEFs showed accumulation of cells with a 4N DNA content (SI Appendix, Fig. S1C), aberrant mitoses (SI Appendix, Fig. S1D), and DNA damage, as shown by the presence of γ-H2AX (SI Appendix, Fig. S1B).
Inducible β-TrCP KO in the Gut Results in Substrate Accumulation and NF-κB Inhibition.
β-TrCP is a major NF-κB regulator, but, unlike other pathway regulators, its role in NF-κB signaling and homeostasis has hardly been studied in vivo (5). To study its physiological role in gut homeostasis, we crossed the conditional β-TrCP2 KO mouse with β-TrCP1–null and the tamoxifen-regulated Villin-Cre-ERT2 deleter mouse (15) to achieve inducible β-TrCP2 deletion (i.e., an inducible DKO mouse). β-TrCP2 was deleted specifically in the gut epithelium 24 h after treatment with tamoxifen, as evident at the DNA and RNA levels (SI Appendix, Fig. S2 A and B). As no specific antibody is available for detecting endogenous mouse β-TrCP1 or β-TrCP2 protein, we validated functional β-TrCP deletion through accumulation of phosphorylated β-catenin 48 h following tamoxifen treatment (SI Appendix, Fig. S2C). Another substrate predicted to accumulate is IκBα, whose stabilization should inhibit NF-κB activation (5). Indeed, immunofluorescence studies showed no nuclear p65 in DKO or WT gut epithelium, whereas considerable levels of nuclear p65 were detected in the single active allele (SAA) mouse model, which retains one of the four β-TrCP alleles (Fig. 1A and see Fig. 6 and SI Appendix, Fig. S6). Likewise, and in accordance with previous reports (16), higher sensitivity methods showed considerable basal NF-κB activity in WT gut epithelium (Fig. 1B, control mice) but silencing of NF-κB target genes in DKO mice (Fig. 1B, D1, D2, and D3 DKO mice).
Fig. 1.

β-TrCP double KO in the gut results in NF-κB inhibition. (A) Immunofluorescence of colons from mice with the indicated genotypes. p65 (red), an NF-κB protein, is present in epithelial cell cytoplasm of control mice, with no NF-κB stimulus, and in the nuclei of inflamed mice (SAA) that have residual β-TrCP activity and normal, unstable IκB. In spite of the severe inflammation in the DKO mice, p65 is cytoplasmic, probably because of stabilized IκB. E-cadherin (green)-negative immune cells infiltrating the epithelial layer signify the inflammation in DKO colons. Blue color indicates Hoechst, a nuclear marker. (Scale bar: 50 μm.) (B) NF-κB target genes expression analysis in enterocytes from the indicated mice. Unlike control mice that show basal NF-κB target gene expression, in DKO mice, NF-κB transcriptional activity is totally inhibited starting at day 1 (IκBα; P = 0.16563; MMP9, P < 0.0001; Ciap2, P < 0.0001; P100, P < 0.0001; P values calculated by unpaired two-tailed t test, controls vs. all DKO samples regardless of the time of harvest).
Fig. 6.

Mucositis mouse model implies IL-1β as an optional therapeutic target for the treatment of mucositis. (A) H&E staining of SAA colons at days 5 and 21. SAA mice recover from the pathologic condition induced by β-TrCP2 KO induction and present nearly normal gut histology 3 wk after KO induction. (B) qPCR analysis of the endogenous IL-1β antagonist IL-Ra, a well-characterized NF-κB target gene, 1, 2, 3, and 4 d after β-TrCP KO induction. No up-regulation is observed in the DKO mice (n = 2 for each day, P = 0.2374 vs. control at day 1, P = 0.1068 vs. control at day 2, P = 0.2586 vs. control at day 2, and P = 0.6802 vs. control at day 4), which have no NF-κB activation as a result of stabilized IκB. However, in SAA mice (n = 3 for each day; P = 0.4165 vs. control at day 1, P = 0.0089 vs. control at day 2, P = 0.0551 vs. control at day 3, and P = 0.0008 vs. control at day 4), with intact NF-κB, IL-RA is up-regulated following β-TrCP KO induction. (C) FITC levels in the plasma of the indicated mice 6 h after FITC-dextran force-feeding. SAA mice show fourfold increase in FITC levels, indicating high gut permeability. However, SAA treated with anti–IL-1β neutralizing antibody (daily i.p. injections, 400 mg/kg body weight) showed reduced levels of plasma FITC, comparable with the control mice, revealing the critical role of IL-1β in the barrier disruption process of these mice even in the presence of intact NF-κB signaling (P values are indicated).
Gut-Specific Deletion of β-TrCP Results in Severe Colitis and Lethality Within 5 d.
The phenotype observed in β-TrCP-deleted mice is dramatic; 3 d after β-TrCP2 ablation, inflammation is evident in the small and large intestines, with immune cells infiltrating the tissue (Fig. 2A, Upper Center). Gut inflammation is provoked in the absence of NF-κB activity, and similar to a previous report, it is associated with considerable apoptosis (16). Many enterocytes are positive for the apoptosis marker cleaved caspase 3 (Fig. 2A, Lower), and, 2 d later, most of the epithelial cells in the large intestine are lost and the remaining mucosa is heavily infiltrated with immune and inflammatory cells (Fig. 2A, Upper Right). At this stage, the mice have severe diarrhea and rectal bleeding, and die 1 d later.
Fig. 2.

Barrier disruption and severe colitis in β-TrCP DKO intestines. (A) (Upper) H&E staining of colon sections of control mouse 5 d after tamoxifen injection (n = 21; Left), DKO mouse 3 d after injection (n = 19; Middle; tissue is abnormal with high numbers of mitotic and apoptotic cells and small immune cell clusters), and DKO mouse 5 d after injection (n = 34; Right). The epithelial layer is extremely thin, and the majority of cells are immune cells infiltrating the mucosa, which is hardly functional at this stage. (Lower) Cleaved caspase 3, an apoptotic marker (red) is abundant in day 3 DKO colons. Hoechst, a nuclear marker, is shown in blue. (Scale bars: 100 μm.) (B) Relative levels of FITC-dextran in the blood 6 h after FITC-dextran force-feeding of the indicated mice (Ctrl, negative control; β-TrCP1+/−, β-TrCP2f/f, no Cre). DSS is positive control. DSS-treated mice received 4% daily dosage for 5 d. DKO mice at day 1 show high intestinal permeability that is similar to the DSS-treated positive control (P = 0.0370 for day 1 DKO mice vs. controls, unpaired t test). (C) H&E staining of representative colons from DKO mice at day 5 (n = 3; Left) and DKO mice at day 5 treated for 15 d with antibiotic at day 5 (n = 3; Right). A significant alleviation of the pathologic process following antibiotic treatment; in contrast to DKO colons, in which hardly any epithelial cells are left at day 5, in the antibiotic-treated DKO colons, the epithelial layer is still present and is functional, although abnormal. (Scale bar: 100 μm.)
The observed inflammation could be the result of intestinal barrier disruption (17, 18), and, to this end, we tested barrier continuity in β-TrCP–deleted mice by the FITC-dextran permeability assay (19). Remarkably, the barrier was already disrupted as early as 24 h after KO induction (Fig. 2B), well ahead of the inflammatory burst, suggesting it is a cause rather than the result of the mucosal inflammation.
To test the role of the intestinal flora in the developing phenotype, we treated the mice with an antibiotic mixture beginning 10 d before KO induction, until the animals were euthanized. The treatment significantly alleviated the pathologic process in deleted mice (Fig. 2C), supporting an important role for barrier disruption and microbial penetration in the pathological process following β-TrCP deletion.
IL-1β, but Not TNF or IL-1α Activation Is the Primary Cause for Intestinal Barrier Disruption.
TNF is the primary factor known to regulate intestinal barrier function upon inflammation and is a pathogenic component in inflammatory bowel disease (20). It is also particularly destructive for enterocytes lacking NF-κB activity (16). Similarly, IL-1α secretion was shown to play a key role in inflammation induced by damaged cells (21), and is known to be expressed in epithelial cells (22); thus IL-1α might contribute to barrier opening. However, ELISA analysis proved that TNF and IL-1α are induced only 3 d following KO induction, 2 d after barrier disruption and appearance of the inflammatory infiltrate (SI Appendix, Fig. S3A). This implies that TNF and IL-1α expression is likely an outcome rather than the source of inflammation. In accordance, pretreatment of the mice with etanercept, a soluble TNF decoy molecule, did not prevent the destructive inflammatory process following β-TrCP deletion (SI Appendix, Fig. S3B).
IL-1β is also known to affect intestinal barrier function (23), and, if induced following β-TrCP KO induction, could be the trigger for inflammation. However, IL-1β is known to be regulated by NF-κB, and, accordingly, should not be induced in β-TrCP–deficient cells. Surprisingly, IL-1β and its receptor IL-1R were up-regulated in epithelial cells already 24 h after KO induction (Fig. 3A). This mRNA increase is likely NF-κB–independent, as β-TrCP depletion should block NF-κB activation in enterocytes. Indeed, other NF-κB target genes were not induced at this time point (Fig. 1B), demonstrating two important points; the first is the high purity of the enterocytes sample, as the observed inhibition of NF-κB genes would have been impaired by any immune cells present in the samples. The second point is the NF-κB–independency of the IL-1β mRNA elevation. We confirmed IL-1β induction at the protein level by immunohistochemistry (Fig. 3B) and ELISA (Fig. 3C). To test the putative functional role of IL-1β in inducing barrier disruption, we pretreated the mice with a neutralizing IL-1β antibody, and maintained the treatment throughout the experiment, and then assayed barrier function as described earlier. Strikingly, DKO mice treated with the neutralizing antibody showed lower permeability, comparable to control mice (Fig. 3D), and significantly improved pathology at day 5 (Fig. 3E), as evidenced by the relatively high numbers of surviving epithelial cells in treated DKO mice (Fig. 3F). EM analysis of anti–IL-1β–treated and untreated mice at day 1 revealed impaired tight junctions (TJs) for DKO enterocytes, but mostly normal junctions in treated DKO cells (Fig. 3G and SI Appendix, Fig. 3C). We conclude that the aberrant activation of IL-1β in the gut epithelium is the primary cause of early barrier disruption in DKO mice and is likely a critical factor in the early onset of the pathologic condition in these mice.
Fig. 3.
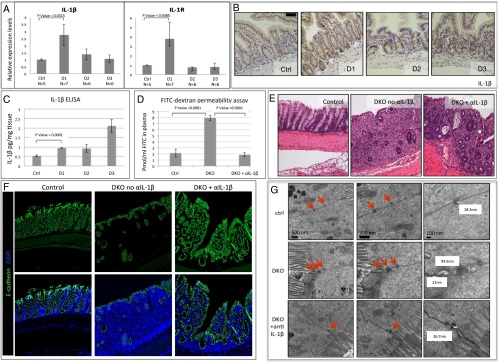
IL-1β is the primary cause of the barrier disruption. (A) Quantitative PCR (qPCR) analysis of IL-1β and IL-1R mRNA levels in enterocytes isolated from the indicated mice. Unlike other NF-κB target genes, IL-1β and IL-1R, which are commonly coregulated, are up-regulated at day 1 (IL-1β, P = 0.0315; IL-1R, P = 0.0385 by unpaired two-tailed t test). (B) IL-1β immunohistochemistry of control and DKO intestines at the indicated days. IL-1β is expressed in Paneth cells at day 1 and in enterocytes at days 1, 2, and 3, mainly in the crypt compartment. (Scale bar: 100 μm.) (C) IL-1β ELISA of intestinal samples. Unlike TNF, IL-1β is up-regulated starting at day 1, with additional increase at day 3 when general inflammation emerges (n = 3 for all groups; P = 0.0001 by unpaired two-tailed t test for day 1 DKO vs. control mice). (D) FITC levels in the plasma of the indicated mice, 24 h after KO induction and 6 h after FITC-dextran force-feeding. DKO mice show fourfold increase in FITC levels, indicating high gut permeability. However, DKO treated with anti–IL-1β neutralizing antibody (daily i.p. injections, 400 mg/kg body weight) showed reduced levels of plasma FITC, comparable with the control mice, revealing the critical role of IL-1β in the barrier disruption process of these mice. Neutralizing antibody dose was a daily injection of 4 mg/kg (46). (Control, n = 7; DKO, n = 5; DKO plus anti–IL-1β, n = 7; DKO vs. control mice, P < 0.0001; DKO vs. DKO plus anti–IL-1β–treated mice, P < 0.0001 by unpaired two-tailed t test.) (E) H&E large intestine staining of control mouse (Left), Untreated DKO (Middle), and DKO treated with neutralizing anti–IL-1β antibody (Right) at day 5 after KO induction. DKO mice at day 5 show severe colitis with scarce recognizable crypt structures and large numbers of immune cells. In contrast, following anti–IL-1β neutralizing antibody treatment, although the colon is still severely inflamed, the pathologic condition is significantly ameliorated. (F) Immunofluorescence of colons from the indicated mice. E-cadherin (green) is an epithelial cell marker, emphasizing the relatively high numbers of surviving enterocytes in the DKO treated with neutralizing αIL-1β antibody (Right) compared with untreated DKO (Center). (Left) Control Het1 mice. Blue indicates DAPI, a nuclear marker. (G) Intestinal TJs captured by EM. TJs (red arrows) appear normal in control (Top) and abnormal with wide gaps in untreated DKO mice (Middle). However, DKO mice treated with neutralizing anti–IL-1β antibody have normal TJs (Bottom). (Magnification: Left, ×20,000; Middle, ×50,000; Right, ×100,000.) All mice were killed at day 1. Distances between membranes in the TJs were measured, and the measurements appear next to the junction.
DNA Damage Is an Early Event Following β-TrCP Deletion.
The known transcriptional regulator of IL-1β expression, NF-κB, is inactive in the DKO enterocytes (Fig. 1), and therefore cannot be responsible for IL-1β expression in these cells. β-TrCP deletion results in DNA damage (10) (SI Appendix, Fig. S1) and up-regulates IL-1β expression (Fig. 3). DNA damage is known to induce IL-1β secretion, albeit via NF-κB activation and inflammasome-dependent secretion. We therefore looked for signs of DNA damage response (DDR) following β-TrCP deletion and observed intense γ-H2AX staining following β-TrCP KO, which increased with time (Fig. 4A). This was confirmed by Western blot analysis (SI Appendix, Fig. S4A), as well as a later increase in p53 and p21 (SI Appendix, Fig. S4B). This time course correlates with IL-1β transcriptional up-regulation (Fig. 3A). The reason for the observed DDR could be chromosomal instability, a result of prolonged mitosis (Fig. 4B), aberrant spindles (Fig. 4C, Lower), and abnormal centrosome number (Fig. 4C, Upper), all of which are specifically observed in the DKO enterocytes reflecting abnormal stabilization of β-TrCP substrates (1). In summary, following β-TrCP deletion, a DDR is strongly induced, temporally corresponding to IL-1β up-regulation.
Fig. 4.

DNA damage is an early event following β-TrCP deletion. (A) Enterocytes positive for γ-H2AX, a DNA damage marker (red), are present in DKO intestines starting at day 1, and their numbers increase in later days. Hoechst, a nuclear marker, is shown in blue. (Scale bar: 100 μm.) (B) (Upper) High numbers of pHH3 (mitotic marker, green)-positive cells in DKO intestines compare with control as seen by immunofluorescence. Blue represents Hoechst, a nuclear marker. (Lower) IHC shows similar quantities of BrdU-positive cells (S-phase marker) in control and DKO intestines. The ratio between mitotic cells (i.e., pHH3-positive) and cells in S phase (i.e., BrdU-positive) is extremely high, indicating prolonged mitosis rather than high proliferation rate. (Scale bar: 100 μm.) (C) (Upper) Centrosome staining (γ-tubulin, red) pointing to more than two centrosomes per cell in DKO intestines (n = 3). No cells with more than two centrosomes were found in control mice (n = 3). Nuclei are stained by Hoechst (blue). (Scale bar: 10 μm.) (Lower) Mitotic cells (pHH3, red) with abnormal spindles (α-tubulin, green) in DKO mice. No abnormal spindles were found in control mice. Nuclei are stained by Hoechst (blue). (Scale bar: 20 μm.)
DNA Damage Up-Regulates IL-1β Independently of NF-κB.
Observing NF-κB–independent IL-1β up-regulation, which overlaps with a DDR following β-TrCP KO, raises the possibility that DNA damage could induce IL-1β independently of NF-κB. To test this assumption, we γ-irradiated WT mice and monitored IL-1β levels by immunohistochemistry and ELISA. IL-1β protein levels and release [measured by immunohistochemistry (IHC) and ELISA, respectively] were induced as early as 2 h after irradiation (Fig. 5 A and B). Similarly to DKO, WT γ-irradiated mice showed early increased gut permeability (Fig. 5C), temporally corresponding to increased IL-1β. In addition, an intestinal epithelial cell line (SW480) treated with the DNA damage inducer doxorubicin showed increased expression of IL-1β and IL-1R (the latter is coregulated with IL-1β; Fig. 5D, Left). Similar results were obtained in primary bone marrow-derived macrophages treated with doxorubicin (Fig. 5D, Right). Like in β-TrCP–depleted enterocytes, induction of DNA damage in SW480 cells up-regulated IL-1β independently of NF-κB, as it was not affected by proteasome inhibitor treatment or IKKβ knockdown (SI Appendix, Fig. S5 A and B). We next tested whether mice lacking NF-κB activity in enterocytes (IKKα and IKKβ enterocyte-specific DKO, henceforth called IKK DKO) also induce IL-1β following γ-irradiation–induced DNA damage. Consistent with previous results from Greten et al. (24), in the absence of NF-κB activation, IL-1β was secreted at the same time, and to similar levels, in γ-irradiated IKK DKO and WT mice (Fig. 5E). However, compared with WT mice, IKK DKO mice had a sharper decrease in body weight (Fig. 5F), and worse signs of inflammation and tissue damage (Fig. 5G). These results suggest that, as in β-TrCP deletion, the secretion of IL-1β following irradiation is an early NF-κB–independent event and that IKK, likely via NF-κB activation, has a radioprotective role (25).
Fig. 5.
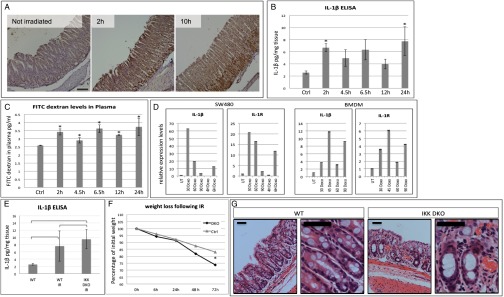
DNA damage can induce IL-1β up-regulation in an NF-κB–independent manner. (A) IL-1β IHC demonstrates IL-1β up-regulation in WT mice in response to DNA damage induced by 4 Gy γ-irradiation. The times indicated are number of hours after irradiation. (Scale bar: 100 μm.) (B) IL-1β ELISA on intestinal supernatants of WT irradiated mice. As soon as 2 h after 6 Gy γ-irradiation, IL-1β secretion is elevated more than twofold compared with nontreated mice (n = 3 for all groups; 2 h, P = 0.0109; 4.5 h, P = 0.1166; 6.5 h, P = 0.1361; 12 h, P = 0.2500; 24 h, P = 0.0915; P values calculated by unpaired two-tailed t test). (C) FITC-dextran permeability assay on WT irradiated mice. Gut permeability is increased following 6 Gy γ-irradiation (n = 3 in all groups; 2 h, P = 0.0017; 4.5 h, P = 0.0291, 6.5 h, P = 0.0019; 12 h, P = 0.0002; 24 h, P = 0.0212; P values calculated by unpaired two-tailed t test). (D) qPCR results provide evidence to IL-1β mRNA up-regulation shortly after doxorubicin treatment (5 μg/mL) in SW480 intestinal cell line (Left) and primary bone marrow-derived macrophages (Right). DOXO was given in a 15-min treatment at 1 μg/mL Time points are in minutes unless otherwise indicated. (E) IL-1β ELISA on intestinal supernatants of untreated WT, γ-irradiated WT, and γ-irradiated IKKα/IKKβ DKO mice. γ-Irradiation at 9 Gy induced similar elevation in IL-1β secretion in intestines with intact NF-κB or lacking any NF-κB activity, indicating an NF-κB–independent IL-1β secretion. P = 0.2027, WT vs. gamma-irradiated WT mice (WT IR). P = 0.0174, WT vs. IKK DKO IR. P = 0.4424, WT IR vs. IKK DKO IR. P values were calculated by unpaired two-tailed t test and are indicated in the chart. (F) Weight loss of WT and IKK DKO mice following 9 Gy γ-Irradiation. IKK DKO mice lost more weight than their WT littermates. P = 0.0031 at the day of euthanasia. (G) H&E staining of WT and IKK DKO large intestines 3 d after 9 Gy γ-irradiation. The pathology in IKK DKO mice is more severe, with increased inflammatory infiltrates. (Right) Higher-magnification image of each genotype. (Scale bar: 50 μm.)
IL-1β Inhibition Limits the Inflammatory Reaction and the Pathologic Process in a Mouse Model of Mucositis.
IL-1β up-regulation has been observed in radiation- and chemotherapy-treated patients with mucositis (26), a severe side effect that occurs in 70% to 90% of all patients treated with standard chemotherapy and radiation therapy (27). The development of mucositis often leads to the termination of therapy, and hence mucositis is of major clinical relevance (28). Mice with no gut β-TrCP are incapable of activating NF-κB in the gut epithelium and thus cannot model in full the human disease. To test whether IL-1β neutralization prevents mucositis in a translational-relevant model, we used our SAA model mice, which present gut pathologic conditions similar to human mucositis (Fig. 6A) and, like the human disease, features NF-κB activation (Fig. 1A, Middle, and SI Appendix, Fig. S6A). SAA mice, unlike DKO mice, recover spontaneously, and the intestinal mucosa is restored 3 wk after KO induction (Fig. 6A). As IL-1β expression levels in SAA and DKO are comparable, and considering the detrimental role of the cytokine in epithelial barrier function, we hypothesized that the resolution of SAA mucositis could result from an endogenous mechanism that antagonizes IL-1β activity, specifically in SAA. Indeed, unlike DKO mice, SAA mice showed incremental up-regulation of IL-1Ra following β-TrCP ablation (Fig. 6B). IL-1Ra transcription is dependent on NF-κB (29), offering an additional protective role of NF-κB activity in DNA-damaged epithelium.
This endogenous protective mechanism commences only several days after the onset of DNA damage (Fig. 6B), thereby exposing the animal to severe morbidity, albeit transiently, similarly to patients with mucositis; late-onset NF-κB–mediated transcription of IL-1Ra may restrict the mucositis phase in SAA mice. To appreciate the protective importance of IL-1Ra expression in SAA mice, we examined a pretreatment effect of neutralizing anti–IL-1β: SAA mice treated with the neutralizing antibody before β-TrCP ablation showed no intestinal barrier loss (Fig. 6C) and had EM evidence of preserved TJ function (SI Appendix, Fig. S6 B and C) and milder mucosal pathology with lessened inflammatory infiltrates (SI Appendix, Fig. S6 D and E). We therefore conclude that IL-1β inhibition, endogenously or with exogenous means, can enhance barrier function, thereby reducing DNA damage-associated mucosal inflammation.
Discussion
We found that full deletion of β-TrCP in the mouse intestine is lethal as a result of fast destruction of the colon epithelial layer. Destruction is promoted by the presence of bacteria in the gut lumen, likely responsible for the severe inflammation that develops in the gut 3 d after β-TrCP KO. We propose that intestinal barrier disruption following β-TrCP deletion is the primary cause of the pathology in β-TrCP DKO gut, as intestinal permeability is abnormally high already within 24 h of β-TrCP KO induction. We show that the cause of the high penetrability is the aberrant induction of IL-1β at the gut epithelium proper, differently from the reported effects of IL-1β secreted by mucosal inflammatory cells (30). More importantly, we were able to attribute to epithelial IL-1β a role of a doorkeeper, opening epithelial TJs, whereas anti–IL-1β treatment seals the TJs and tightens the epithelial barrier, thus preventing the pathological permeability. IL-1β is induced in the β-TrCP mouse model by an unresolved molecular mechanism, yet the upstream inducer of IL-1β expression is likely DNA damage caused by dysregulation of cell cycle machinery following β-TrCP deletion. Accordingly, we showed that DNA damage induces IL-1β expression in other systems, such as the gut epithelium of γ-irradiated WT and IKK DKO mice, doxorubicin-treated SW480 epithelial cell line, and doxorubicin-treated primary bone marrow-derived macrophages. DNA damage-induced IL-1β transcription is NF-κB–independent, as NF-κB activity is completely repressed in β-TrCP and IKK-depleted enterocytes and is proteasome inhibition-insensitive in cell culture. We therefore speculate that DNA damage may induce a defensive inflammatory response, which is initiated by IL-1β induction, yet is NF-κB–independent.
Unlike the current dogma that attributes this offensive inflammatory response primarily to NF-κB activation (28), the present study reveals an early immune reaction, based on IL-1β expression and secretion, independently of NF-κB. IL-1β expression may be endogenously counterbalanced by an NF-κB–dependent expression of the IL-1β natural antagonist IL-1Ra. Such protective NF-κB reaction may function in addition and possibly ahead of the well-known apoptosis protection function of the transcription factor (31). This proposed homeostatic mechanism is presumably fast and effective in local injuries, or low-grade insults (e.g., single, low-dose irradiation dose irradiation), enabling a short period of IL-1β action and its rapid termination by the antagonist. However, upon repeated doses of radiation or chemotherapy, the induction of IL-1Ra is probably too slow to counteract the immediate disruption of the barrier by IL-1β, resulting in mucositis; prophylactic anti–IL-1β treatment is likely to prevent the TJ dysfunction and may therefore ameliorate mucositis. Anakinra, an IL-1Ra, is currently in use for the treatment of rheumatoid arthritis, gout, and other inflammatory diseases (14). The drug has minimal side effects, and, unlike NF-κB inhibitors, will not hazardously compromise the patient’s immune system. We therefore propose testing the use of anakinra for the treatment of patients with mucositis as a prophylactic treatment.
Based on our results, and contrary to the conclusions of some previous studies (32), NF-κB inhibition may be deleterious in patients with mucositis. We show that pro–IL-1β can be produced independently of NF-κB, and its processing and secretion will be augmented by the inflammasome activation via NF-κB inhibition (24). Moreover, NF-κB inhibition will compromise the expression of IL-1Ra, which will further enhance the barrier-breach effect of IL-1β. We thus maintain that direct IL-1β inhibition would be the preferred means for preventing and ameliorating mucositis.
IL-1β is known to play a key role in inflammation (33) and has specific relevance in intestinal inflammation (34). Whereas most studies (35–38) addressed the role of inflammatory-cell IL-1β, the present study focused on epithelial-derived IL-1β, which probably has short-range effects, mostly within the intestinal epithelium itself. When an epithelial-born IL-1β has induced a barrier breach, it likely promotes an inflammatory cascade recalling multiple innate and adaptive immune and inflammatory cells rapidly into the intestinal mucosa. Then, inflammatory cell-derived IL-1β might cooperate with multiple innate and adaptive effector pathways (39) to propagate an inflammatory reaction. In humans, IL-1β up-regulation following DNA damage is observed in cancer patients with mucositis (28), a severe pathologic condition affecting most patients treated with standard chemotherapy or radiation therapy (28, 40). Anti–IL-1β treatment may thus ameliorate mucositis as well as multiple disorders associated with barrier permeability, including burn injuries (33), head and neck trauma (41), cardiogenic shock (42), alcoholic intoxication (43), and graft-vs.-host disease (40). Furthermore, preventive anti–IL-1β treatment may obviate the common need for chemoradiation therapy suspension as a result of mucositis and improve patients’ quality of life during treatment.
Methods
Generation of KO Mice.
β-TrCP targeting vector was constructed on the basis of pGEM 11 Zf (+), to which an XbaI/SaI1 fragment of Neomycin cassette flanked by two loxP sites was inserted from pL2neo expression vector for positive selection. Exon 4 of murine FbxW11 gene (encoding mouse β-TrCP2) was cloned into the vector, flanked by loxP sites using a third loxP site. Short (1 kb) and long (5 kb) homology sequences were cloned upstream and downstream of the targeted exon, respectively, to facilitate homologous recombination of the construct to the genome. All genomic fragments were amplified by PCR from 129/SVJ mouse DNA. The vector was linearized with SalI and purified by using phenol-chloroform and ethanol precipitation methods. Electroporation of the linear vector was performed with a BioRad electroporator using electroporation buffer (Sigma) into R1 (129/SVJ-derived) embryonic stem (ES) cells. ES cell culture was grown on feeder layer of MEFs using DMEM supplemented with 15% (vol/vol) ES-tested FBS and 1,000 U/mL of ESGRO (Chemicon). Neomycin selection was done in 0.2 mg/mL G-418 (Sigma). Validation of genomic modulations was performed by PCR. Positive ES clone was used to generate conditional β-TrCP2 KO mice. Morula aggregation method was used to establish chimeric mice from each of the positive ES cell clones, using ICR-derived morulas. ICR mice were used to check for germ-line transmission of the 129/SVJ-derived ES clones.
Production of MEFs.
MEFs were prepared from 12.5 d postcoitum, embryos and seeded onto 10-cm plates precoated with 0.5% gelatin. Culture was maintained in DMEM (Gibco) supplemented with 10% (vol/vol) FCS (Beit Ha’emek).
RNA and Protein Extraction and Quantitative PCR Analyses.
RNA was recovered from tissues by using Sigma Tri-Reagent according to manufacturer instructions. cDNA was prepared by using MMLV Reverse Transcriptase (Invitrogen). Quantitative real-time PCR analysis was performed by using Platinum SYBR green reagent (Invitrogen) and an ABI Prism 7900 Real Time Thermocycler. Protein extracts were prepared in protein lysis buffer (50 mM Tris, 420 mM NaCl, 5 mM EDTA) supplemented with protease and phosphatase inhibitors. Gene expression results were normalized to UBC, HPRT, and PPIA for mouse tissue samples and to UBC and HPRT for cell culture, from mouse or human origin.
Quantitative PCR Primers.
Quantitative PCR primers were as follows: β-TrCP1, forward, AAA TAA CCC AAC ACT GGC CTC A; reverse, TCA TCC TGG TCT TGG CAG GT; β-TrCP2, forward, TGG CGC CTA TGA TGG GAA; reverse, GTC AAG AGC AGC CTG CAA GTC; IκBα, forward, CTC ACG GAG GAC GGA GAC TC; reverse, TGA CTT CCA TGG TCA GCG G; Ciap2, forward, AGT ACT TGC TCA GAA TCA AAG GC; reverse, CGT CTG CAT TCT CAT CTT CTG; MMP9, forward, AAC CTC CAA CCT CAC GGA CAC; reverse, CTG CTT CTC TCC CAT CAT CTG G; P100, forward, CAG CGA GGC TTC AGA TTT CG; reverse, CAC CTG GCA AAC CTC CAT G; IL-1β, forward, ACC TTC CAG GAT GAG GAC ATG A; reverse, CTA ATG GGA ACG TCA CAC ACC A; and IL-1R, forward, TAC CTG CCA AGG TGG AGG AC; reverse, AGG TGG CCT GTG TGC TGT AA.
Antibodies and Immunohistochemistry.
The following antibodies were used: phosphorylated β-catenin (Ser33, Ser37, Thr41), β-catenin (Cell Signaling), E-cadherin (BD Transduction Laboratories), phospho-histone H3 (Upstate), tubulin (Sigma), p65 (SC-109; Santa Cruz), cleaved caspase 3 (Cell Signaling), p21 (Santa Cruz), p53 (CM5; Novocastra), IL-1β (AF-401-NA; R&D Systems), γ-H2AX (Millipore), GAPDH (Fitzgerald), BrdU (Neomarkers), γ-tubulin (T5326; Sigma), and α-tubulin (Sigma). For Western blot analysis, HRP-linked goat anti-mouse, goat anti-rabbit, goat anti-rat, and rabbit anti-goat (1:10,000; Jackson Laboratories) were used as secondary antibodies. Blots were developed by using ECL (GE Healthcare).
Immunohistochemical analyses were performed using standard procedures. Antigens were retrieved from formaldehyde or Bouin-fixed paraffin-embedded 5-μm sections in a decloaking chamber (Biocare Medical) in 20 mM citrate buffer (pH 6) or EDTA buffer (pH 8). Antibodies were hybridized in CAS-BLOCK (Zymed). HRP conjugated secondary antibodies were goat anti-rabbit (Nichirei Bioscience) and goat anti-mouse (Dako). Chromogen substrate for HRP reaction was DAB plus (Thermo Scientific). Fluorescent secondary antibodies were Alexa 488 or 647 goat Ig conjugates (Invitrogen). Counterstaining was done with Hoechst (Rhenium H1399).
All immunofluorescence and IHC results were blind tested by two different testers. In many cases the tester was Eli Pikarsky (Pathology Department, Hadassah Medical Center).
Animal Procedures.
All procedures were approved by the animal care and use committee of the Hebrew University of Jerusalem or Columbia University. Animals were provided food and water ad libitum. All mice used in the study were male, age 2–3 mo.
KO induction.
Tamoxifen (Sigma) was dissolved in corn oil (Sigma) to a stock solution of 20 mg/mL. Mice were injected twice i.p. with 120 mg/kg of tamoxifen for a subsequent 2 d. Mice were killed 1–5 d after the first injection.
Intestine analysis.
Small and large intestines were removed and flushed with ice-cold PBS solution. Proximal (jejunum) and distal (ileum) small intestine fractions and the entire large intestine were cut open longitudinally, rolled and fixed in 4% (vol/vol) formaldehyde for 12–24 h, and paraffin-embedded. Small pieces of jejunum and ileum were embedded in optimum cutting temperature compound (Sakura) and frozen at −80 °C. Enterocytes (intestinal epithelial cells) were isolated from the middle part of the small intestine as described previously (44), with slight modifications: intestine was washed and minced into small pieces in ice-cold PBS solution/1 mM DTT, then dispersed into single cells in Ca2+/Mg2+-free HBSS containing 10 mM Hepes, 5 mM EDTA, and 0.5 mM DTT, at 37 °C for 15 min, with slow agitation. Tissue pieces were mixed vigorously, and cell suspension was centrifuged. Cell pellets were divided into aliquots and frozen in liquid N2 and stored at −80 °C.
BrdU labeling.
Two hours before the animals were euthanized, mice were injected i.p. with 100 μL/kg of BrdU reagent (GE Healthcare).
Etanercept treatment.
Etanercept was injected s.c., 10 mg/kg body weight.
Antibiotic treatment.
Mice were treated for 15 d with metronidazole, imipenem, and vancomycin in drinking water. Dosage of each was 100 mg/kg body weight (45).
FITC-dextran permeability assay.
Mice were force-fed (i.e., gavage) 2 mg/kg body weight FITC-Dextran (4 kDa; FD4; Sigma). Six hours later, blood was collected from the tail vein and immediately drained into two duplicate test tubes and flushed with blood dilution buffer (50 mM of Tris base plus 150 mM sodium chloride, pH 10) for a 20-fold dilution. The tubes were then centrifuged at 600 × g for 10 min. Standard concentrations of the FITC-D (0, 9.4, 18.75, 37.5, 75, 150, and 300 pmol/mL) were prepared. Sample supernatants (200 µL) and standard solutions were pipetted into duplicate wells of a black microtiter plate, and the fluorescence was read on a FLUOstar OPTIMA plate reader (BMG Biotechnologies) with wavelengths at 485 nm excitation and 520 nm emission.
Transmission EM.
Animals were killed as described, and intestines were removed and immediately fixed by Karnovsky fixative. Following cacodylate buffer washes and postfixation with 2% (wt/vol) OSO4, samples were dehydrated in ethanol gradient and transferred to propylene oxide. Embedding was done in beam capsules by EMbed 812 Resin (no. 14120; EMS) at 60° for 48 h. Thin sections (50–70 nm) were stained by uranyl acetate and lead citrate and examined under a Philips EM 12P electron microscope (voltage 100 KV). All EM results were blind-tested by Kristy Brown (EM facility, Columbia University).
ELISA on Intestinal Samples.
After mice were killed, a piece (∼0.5 cm) of the intestine was taken for overnight incubation in 1 mL RPMI medium (Gibco) supplemented with penicillin/streptomycin (Beit Ha’emek) at 37° and 5% CO2. The next morning, the medium was collected and divided into aliquots, and samples were weighed for normalization. ELISA for TNF was done with a BD ELISA kit (cat no. 560478). IL-1β ELISA was done with a BD ELISA kit (cat no. 559603) according to manufacturer instructions.
Supplementary Material
Acknowledgments
We thank Dr. Pekka Katajisto (Institute of Biotechnology, University of Helsinki) and Dr. Avi Levin (Hadassah Medical Center, Jerusalem) for their helpful contribution. This work was supported by grants from Dr. Miriam and Sheldon G. Adelson Medical Research Foundation and Israel Science Foundation–Centers of Excellence (to Y.B.-N. and E.P.); European Research Council Grants 294390 PICHO (to Y.B.-N.) and LIVERMICROENV (to E.P.); Israel Cancer Research Fund (Y.B.-N. and E.P.); Israeli Centers of Research Excellence (Y.B.-N. and E.P.); National Institutes of Health Grants R01-GM057587, R37-CA076584, and R21-CA161108 (to M.P.); Columbia University Medical Center Schaeffer Scholars Program (N.K., Y.B.-N., and S.G.); National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases Award K99/R00 (to S.I.G.); and Crohn's and Colitis Foundation of America Career Developing Award 2693 (to S.I.G.). M.P. is an Investigator with the Howard Hughes Medical Institute.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1322691111/-/DCSupplemental.
References
- 1.Frescas D, Pagano M. Deregulated proteolysis by the F-box proteins SKP2 and beta-TrCP: Tipping the scales of cancer. Nat Rev Cancer. 2008;8(6):438–449. doi: 10.1038/nrc2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kanarek N, et al. Spermatogenesis rescue in a mouse deficient for the ubiquitin ligase SCFbeta-TrCP by single substrate depletion. Genes Dev. 2010;24(5):470–477. doi: 10.1101/gad.551610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adams J. Proteasome inhibition: A novel approach to cancer therapy. Trends Mol Med. 2002;8(4) suppl:S49–S54. doi: 10.1016/s1471-4914(02)02315-8. [DOI] [PubMed] [Google Scholar]
- 4.Fuchs SY, Spiegelman VS, Kumar KG. The many faces of beta-TrCP E3 ubiquitin ligases: Reflections in the magic mirror of cancer. Oncogene. 2004;23(11):2028–2036. doi: 10.1038/sj.onc.1207389. [DOI] [PubMed] [Google Scholar]
- 5.Kanarek N, Ben-Neriah Y. Regulation of NF-kappaB by ubiquitination and degradation of the IkappaBs. Immunol Rev. 2012;246(1):77–94. doi: 10.1111/j.1600-065X.2012.01098.x. [DOI] [PubMed] [Google Scholar]
- 6.Jiang J, Struhl G. Regulation of the Hedgehog and Wingless signalling pathways by the F-box/WD40-repeat protein Slimb. Nature. 1998;391(6666):493–496. doi: 10.1038/35154. [DOI] [PubMed] [Google Scholar]
- 7.Yaron A, et al. Identification of the receptor component of the IkappaBalpha-ubiquitin ligase. Nature. 1998;396(6711):590–594. doi: 10.1038/25159. [DOI] [PubMed] [Google Scholar]
- 8.Busino L, et al. Degradation of Cdc25A by beta-TrCP during S phase and in response to DNA damage. Nature. 2003;426(6962):87–91. doi: 10.1038/nature02082. [DOI] [PubMed] [Google Scholar]
- 9.Watanabe N, et al. M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc Natl Acad Sci USA. 2004;101(13):4419–4424. doi: 10.1073/pnas.0307700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guardavaccaro D, et al. Control of meiotic and mitotic progression by the F box protein beta-Trcp1 in vivo. Dev Cell. 2003;4(6):799–812. doi: 10.1016/s1534-5807(03)00154-0. [DOI] [PubMed] [Google Scholar]
- 11.Seki A, et al. Plk1- and beta-TrCP-dependent degradation of Bora controls mitotic progression. J Cell Biol. 2008;181(1):65–78. doi: 10.1083/jcb.200712027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mamely I, et al. Polo-like kinase-1 controls proteasome-dependent degradation of Claspin during checkpoint recovery. Curr Biol. 2006;16(19):1950–1955. doi: 10.1016/j.cub.2006.08.026. [DOI] [PubMed] [Google Scholar]
- 13.Fuchs SY, Chen A, Xiong Y, Pan ZQ, Ronai Z. HOS, a human homolog of Slimb, forms an SCF complex with Skp1 and Cullin1 and targets the phosphorylation-dependent degradation of IkappaB and beta-catenin. Oncogene. 1999;18(12):2039–2046. doi: 10.1038/sj.onc.1202760. [DOI] [PubMed] [Google Scholar]
- 14.Fleischmann RM, et al. Safety of extended treatment with anakinra in patients with rheumatoid arthritis. Ann Rheum Dis. 2006;65(8):1006–1012. doi: 10.1136/ard.2005.048371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.el Marjou F, et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39(3):186–193. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- 16.Nenci A, et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature. 2007;446(7135):557–561. doi: 10.1038/nature05698. [DOI] [PubMed] [Google Scholar]
- 17.Schulzke JD, et al. Epithelial tight junctions in intestinal inflammation. Ann N Y Acad Sci. 2009;1165:294–300. doi: 10.1111/j.1749-6632.2009.04062.x. [DOI] [PubMed] [Google Scholar]
- 18.Mankertz J, Schulzke JD. Altered permeability in inflammatory bowel disease: Pathophysiology and clinical implications. Curr Opin Gastroenterol. 2007;23(4):379–383. doi: 10.1097/MOG.0b013e32816aa392. [DOI] [PubMed] [Google Scholar]
- 19.Yoshikawa H, Takada K, Muranishi S. Molecular weight dependence of permselectivity to rat small intestinal blood-lymph barrier for exogenous macromolecules absorbed from lumen. J Pharmacobiodyn. 1984;7(1):1–6. doi: 10.1248/bpb1978.7.1. [DOI] [PubMed] [Google Scholar]
- 20.Turner JR. Molecular basis of epithelial barrier regulation: from basic mechanisms to clinical application. Am J Pathol. 2006;169(6):1901–1909. doi: 10.2353/ajpath.2006.060681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cohen I, et al. Differential release of chromatin-bound IL-1alpha discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc Natl Acad Sci USA. 2010;107(6):2574–2579. doi: 10.1073/pnas.0915018107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berda-Haddad Y, et al. Sterile inflammation of endothelial cell-derived apoptotic bodies is mediated by interleukin-1α. Proc Natl Acad Sci USA. 2011;108(51):20684–20689. doi: 10.1073/pnas.1116848108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Al-Sadi RM, Ma TY. IL-1beta causes an increase in intestinal epithelial tight junction permeability. J Immunol. 2007;178(7):4641–4649. doi: 10.4049/jimmunol.178.7.4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Greten FR, et al. NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell. 2007;130(5):918–931. doi: 10.1016/j.cell.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Egan LJ, et al. IκB-kinaseβ-dependent NF-κB activation provides radioprotection to the intestinal epithelium. Proc Natl Acad Sci USA. 2004;101(8):2452–2457. doi: 10.1073/pnas.0306734101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sultani M, Stringer AM, Bowen JM, Gibson RJ. Anti-inflammatory cytokines: Important immunoregulatory factors contributing to chemotherapy-induced gastrointestinal mucositis. Chemother Res Pract. 2012;2012:490804. doi: 10.1155/2012/490804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rodríguez-Caballero A, et al. Cancer treatment-induced oral mucositis: A critical review. Int J Oral Maxillofac Surg. 2012;41(2):225–238. doi: 10.1016/j.ijom.2011.10.011. [DOI] [PubMed] [Google Scholar]
- 28.Logan RM, et al. The role of pro-inflammatory cytokines in cancer treatment-induced alimentary tract mucositis: Pathobiology, animal models and cytotoxic drugs. Cancer Treat Rev. 2007;33(5):448–460. doi: 10.1016/j.ctrv.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 29.Antin JH, et al. Recombinant human interleukin-1 receptor antagonist in the treatment of steroid-resistant graft-versus-host disease. Blood. 1994;84(4):1342–1348. [PubMed] [Google Scholar]
- 30.Ong ZY, et al. Pro-inflammatory cytokines play a key role in the development of radiotherapy-induced gastrointestinal mucositis. Radiat Oncol. 2010;5:22. doi: 10.1186/1748-717X-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen LW, et al. The two faces of IKK and NF-kappaB inhibition: prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nat Med. 2003;9(5):575–581. doi: 10.1038/nm849. [DOI] [PubMed] [Google Scholar]
- 32.Jung C, et al. Yersinia pseudotuberculosis disrupts intestinal barrier integrity through hematopoietic TLR-2 signaling. J Clin Invest. 2012;122(6):2239–2251. doi: 10.1172/JCI58147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dinarello CA. A clinical perspective of IL-1β as the gatekeeper of inflammation. Eur J Immunol. 2011;41(5):1203–1217. doi: 10.1002/eji.201141550. [DOI] [PubMed] [Google Scholar]
- 34.Asquith M, Powrie F. An innately dangerous balancing act: Intestinal homeostasis, inflammation, and colitis-associated cancer. J Exp Med. 2010;207(8):1573–1577. doi: 10.1084/jem.20101330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bersudsky M, et al. Non-redundant properties of IL-1α and IL-1β during acute colon inflammation in mice. Gut. 2013 doi: 10.1136/gutjnl-2012-303329. 10.1136/gutjnl-2012-303329. [DOI] [PubMed] [Google Scholar]
- 36.Elinav E, Henao-Mejia J, Flavell RA. Integrative inflammasome activity in the regulation of intestinal mucosal immune responses. Mucosal Immunol. 2013;6(1):4–13. doi: 10.1038/mi.2012.115. [DOI] [PubMed] [Google Scholar]
- 37.Apte RN, et al. The involvement of IL-1 in tumorigenesis, tumor invasiveness, metastasis and tumor-host interactions. Cancer Metastasis Rev. 2006;25(3):387–408. doi: 10.1007/s10555-006-9004-4. [DOI] [PubMed] [Google Scholar]
- 38.Krelin Y, et al. Interleukin-1beta-driven inflammation promotes the development and invasiveness of chemical carcinogen-induced tumors. Cancer Res. 2007;67(3):1062–1071. doi: 10.1158/0008-5472.CAN-06-2956. [DOI] [PubMed] [Google Scholar]
- 39.Coccia M, et al. IL-1β mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4(+) Th17 cells. J Exp Med. 2012;209(9):1595–1609. doi: 10.1084/jem.20111453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johansson JE, Ekman T. Gut toxicity during hemopoietic stem cell transplantation may predict acute graft-versus-host disease severity in patients. Dig Dis Sci. 2007;52(9):2340–2345. doi: 10.1007/s10620-006-9404-x. [DOI] [PubMed] [Google Scholar]
- 41.Morganti-Kossmann MC, Rancan M, Stahel PF, Kossmann T. Inflammatory response in acute traumatic brain injury: A double-edged sword. Curr Opin Crit Care. 2002;8(2):101–105. doi: 10.1097/00075198-200204000-00002. [DOI] [PubMed] [Google Scholar]
- 42.Mitroulis I, Skendros P, Ritis K. Targeting IL-1beta in disease; the expanding role of NLRP3 inflammasome. Eur J Intern Med. 2010;21(3):157–163. doi: 10.1016/j.ejim.2010.03.005. [DOI] [PubMed] [Google Scholar]
- 43.Li X, Akhtar S, Choudhry MA. Alteration in intestine tight junction protein phosphorylation and apoptosis is associated with increase in IL-18 levels following alcohol intoxication and burn injury. Biochim Biophys Acta. 2012;1822(2):196–203. doi: 10.1016/j.bbadis.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Greten FR, et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004;118(3):285–296. doi: 10.1016/j.cell.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 45.Miyazaki S, et al. Development of systemic bacteraemia after oral inoculation of vancomycin-resistant enterococci in mice. J Med Microbiol. 2001;50(8):695–701. doi: 10.1099/0022-1317-50-8-695. [DOI] [PubMed] [Google Scholar]
- 46.Shchors K, et al. The Myc-dependent angiogenic switch in tumors is mediated by interleukin 1beta. Genes Dev. 2006;20(18):2527–2538. doi: 10.1101/gad.1455706. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.